
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mineralization pathways of organic matter deposited in a river–lake transition of the Rhone River Delta, Lake Geneva
Marie-Eve
Randlett
ab,
Sebastien
Sollberger
ab,
Tonya
Del Sontro
ab,
Beat
Müller
a,
Juan Pablo
Corella
c,
Bernhard
Wehrli
ab and
Carsten J.
Schubert
*a
aEawag, Swiss Federal Institute of Aquatic Science and Technology, 6047 Kastanienbaum, Switzerland. E-mail: carsten.schubert@eawag.ch
bETH, Swiss Federal Institute of Technology, Institute of Biogeochemistry and Pollutant Dynamics, 8092 Zurich, Switzerland
cMuseo Nacional de Ciencias Naturales (MNCN/CSIC), Serrano 115bis, 28006 Madrid, Spain
First published on 1st December 2014
Abstract
During the éLEMO endeavour (a research project in which the Russian MIR submersibles were used for studying Lake Geneva) four sediment cores were retrieved on a transect from the delta of the Rhone River towards the profundal part of the lake. The degradation pathways of organic material (OM) were investigated considering different electron acceptors. Essentially, OM at the delta sites had a higher fraction of terrestrial material than the lake sites indicated by higher C/N ratios, and higher long-chain n-alkane and alcohol concentrations. The concentrations of chlorins were higher at the distant sites indicating more easily degradable OM in the sediments. However, the chlorin index that was used to determine the degradation state of the OM material indicated that pigment derived OM of deltaic sediments was less degraded than that of the profundal sediments. The fluxes of reduced species from the sediments decreased from the delta to the profundal for CH4 (from 2.3 to 0.5 mmol m−2 d−1) and NH4+ (from 0.31 to 0.13 mmol m−2 d−1). Fluxes of Fe(II) and Mn(II), however, increased although they were generally very low (between 9 × 10−5 and 7.6 × 10−3 mmol m−2 d−1). Oxygen concentration profiles in the pore waters revealed lower fluxes close to the river inflow with 4.3 and 4.1 mmol m−2 d−1 compared to two times higher fluxes at the profundal sites (8.8 and 8.2 mmol m−2 d−1). The rates for totally mineralized OM (Rtotal) at the shallower sites (4.7 mmol C m−2 d−1) were only half of those of the deeper sites (9.7 mmol C m−2 d−1). Accordingly, not only the rates but also the mineralization pathways differed between the shallow and profundal sites. Whereas only 0–6% of the OM was mineralized aerobically at the shallow sites (since almost all O2 was used to oxidize the large flux of CH4 from below) the situation was reversed at the deeper sites and the fraction of aerobically degraded OM was 72–78%. We found a better efficiency in CH4 production per carbon equivalent deposited at the deeper sites as a result of the higher degradability of the mainly autochthonous OM in spite of the lower deposition rate and the higher degradation state of the OM compared to the delta sites.
Environmental impactAlthough lakes cover only 2–3% of the earth's surface they contribute up to 16% of all natural global methane emissions. We studied organic matter degradation and subsequent methane emission from the delta and profundal part of Lake Geneva. Early diagenetic processes were traced and quantified from pore water concentration gradients of various electron acceptors (oxygen, nitrate, Fe, and Mn) and methane production was measured. 72–78% of the OC was mineralized aerobically at the deeper sites but only 0–6% at the shallow sites leading to higher CH4 production per sedimented OC and higher methane fluxes. Hence, at deltas high methane emissions are expected in lacustrine systems, whereas in the deeper more central parts little methane is produced and hardly reaches the atmosphere. |
1 Introduction
Whereas countless studies on the transformation of organic material (OM) during degradation have been performed in the marine environment,1–3 studies in lakes on the same topic are rather rare.4,5 The understanding of preservation of OM that has been formed during photosynthesis and subsequently has been moved to and buried into the sediments is crucial for at least two reasons: first, we need to understand how OM is preserved or degraded on its way to the formation of oil or gas depending on the environment and redox conditions during burial/diagenesis. Second, and this might be a more important question at this moment of time, how much of the aquatic OM primarily built from atmospheric carbon dioxide (CO2) is finally buried in the sediments. The increase of CO2 and methane (CH4) in the atmosphere since the last ice age and, even more dramatically, over the last 150 years6–8 is naturally balanced only (i.e., not considering man made possibilities like carbon storage in the ocean or deeper terrestrial environments) by the uptake of CO2 by oceans/lakes and/or terrestrial biomass. The fixation of CO2 in the biomass and the subsequent burial in soils and sediments diminishes CO2 concentrations in the atmosphere and hence is crucial to understand the conditions that eventually decide about either mineralization or burial of OM. The burial efficiency of OM in lakes was highlighted recently.4 These authors found high organic carbon (OC) burial efficiency (ratio between OC buried to OC deposited) in lakes in general (0.48) and especially in lakes influenced by a high allochthonous input. A previous study estimated the OC buried in lake sediments to be 50% of what is stored in the ocean9 which is a huge number when comparing the ocean area (71%) to the integrated global lake area (2–3%).10 In addition to the fact that sediments receiving a high input of suspended particulate matter have high OC burial efficiencies a closely linked factor was the oxygen (O2) exposure time.4 However, it is not only O2 that is used as an electron acceptor when OC is degraded but also other oxidants like nitrate, iron, manganese, and sulfate can play a substantial role.11 In a study of the meromictic Lake Zug, sediments in which the input of allochthonous material can be neglected the distinction between aerobic and anaerobic degradation of OM showed that in the shallower part of the lake with oxic bottom water around 50% of the OM was degraded with O2.12 Hence, it is important to consider other electron acceptors besides O2 that might play a role in OM degradation. The study presented here focuses on the diagenetic degradation pathways and the factors controlling the burial efficiency of OM in Lake Geneva sediments. Lipids and pigments were studied in the sediments as indicators of the origin of sediment OM and electron acceptors in the sediment pore waters when moving from the delta of the main tributary, the Rhone River (higher river inflow, deltaic sites) to the deeper part of the lake (less river influence, profundal sites).2 Materials and methods
2.1 Sampling site
Lake Geneva is the largest western European lake with a surface area of 582 km2, a volume of 89 km3 and a maximum depth of 309 m. With a catchment area of 5220 km2, the Rhone River makes up 68% of the input to the lake.13 The annual discharge ranged between 127 and 227 m3 s−1 for the period 1935–2008.14 The annual sediment load transported by the Rhone River into Lake Geneva varies between 2 and 9 × 106 tonnes per year15 providing large amounts of allochthonous OM supply mainly to the large delta.2.2 Sampling
Sediment cores and peeper samples16 were collected at 4 locations from the northeastern part of Lake Geneva in front of the inflow of the Rhone River with increasing distance from the shore using the MIR submersibles17 and piston cores from a boat (Fig. 1 ESI;†Table 1). For each site, 3 sediment cores (inner diameter = 59 mm) were collected: one for CH4 analysis, one for porosity and bulk parameters and one for biomarker analysis. Sediment cores were sliced in 1 cm sections, frozen and freeze dried. Pore water equilibrators (peepers;18,19) were used and consisted of a 50 × 15 cm solid, plexiglass frame into which cells (1.5 × 3 cm) were milled at a spacing of either 0.5 or 1.5 cm. The cells were filled with distilled, deionized water, and a 0.2 μm filter (HT 200, Tuffryn polysulfone membrane; Gelman Inc.) was laid over the surface of the frame and held in place by an outer frame of plexiglass.16 To remove O2 from the water in the cells, the peeper was immersed in a tank of deionized water that was then purged for 24 hours with nitrogen.20 When the peeper is placed in the sediments, solutes from pore water in contact with the filter diffuse into the cells such that, after equilibrium is reached, concentrations inside each cell equal those at the corresponding depth in the pore water. A one week period was allowed for equilibration which allows e.g. over 90% of sulfate (which has a low diffusion coefficient) to be equilibrated (after 5.6 days; a more than 99% equilibration would need about 130 days;19). Peepers were mounted on a steel tripod that was positioned on the sediment surface. Exposure of the peeper to lake water following retrieval from the sediments was minimized and lasted no more than 3–5 minutes. Sampling of the cells was done immediately upon retrieval within less than an hour (preferable between 15 and 30 min) after recovery. Water from within the cells was sampled with pipettes and immediately transferred into vials containing appropriate fixative agents (see below for different species).| Site # | Date | Coordinates | Water depth | Sed. depth | TOC | Sed. rate (SR) | Porosity | Accumulation rate AR | Efficiency | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| dd.m.yy | WGS 84 | m | cm | % | cm per year | kg m−2 per year | gC m−2 per year | mmol CH4 per gC | |||
| 1 | 17.6.13 | N 46°24.416′ | E 6°50.667′ | 130 | 21 | 0.43 | 2.3 | 0.57 | 26.8 | 131.7 | 5.4 |
| 2 | 17.6.13 | N 46°24.321′ | E 6°50.975′ | 90 | 44 | 0.45 | 2.3 | 0.53 | 28.1 | 129.8 | 6.6 |
| 4 | 14.7.13 | N 46°26.231′ | E 6°49.364′ | 210 | 27 | 0.74 | 0.6 | 0.75 | 4.1 | 31.2 | 8.1 |
| 5 | 14.7.13 | N 46°27.048′ | E 6°47.933′ | 240 | 30 | 0.83 | 0.4 | 0.77 | 2.4 | 19.8 | 9.1 |
2.3 Sedimentation rates
To determine sedimentation rates (SRs) in the profundal area, core #5 was dated by gamma spectrometry using 137Cs on freeze-dried and ground sediment with a HPGe detector (Canberra GCW-3523) using an energy of 661.7 keV. SRs and accumulation rates (ARs) were calculated based on the 137Cs peaks from the Chernobyl accident and nuclear bomb tests assuming a constant rate of supply (CRS) according to the model of Niessen and Appleby.21,22 The SR of core #4, was estimated by correlation using the TOC profile with the nearby core #5 (see Fig. 2A, ESI†). Since cores #1 and #2 could not be dated through neither 210Pb nor 137Cs due to low activities we correlated the cores via the TOC profile to the nearby core LP (ESI Fig. 1,† N 46°24.190′; E 6°50.493) from 103 m water depth which was 14C and 137Cs dated (see Fig. 2B, ESI,†c.f.23).2.4 Bulk parameters
Total carbon (TC) and total nitrogen (TN) were measured in freeze-dried samples with a Thermo Quest CE Instrument NC 2500. Total organic carbon (TOC) was measured on decalcified samples.24 In brief, sediment samples were acid treated with 10% HCl until no further CO2 evolved and dried at 40 °C before weighing for elemental analysis. Total inorganic carbon (TIC) was calculated as the difference between TC and TOC. The precision for TC and TOC was ±0.1 wt% and ±0.2 wt% for TN. δ13C and δ15N of the OM were obtained with a GV Instruments IsoPrime isotope ratio mass spectrometer (IRMS). The precision of δ13C in ‰ vs. Vienna Peedee Belemnite (VPDB) and δ15N in ‰ vs. air was lower than ±0.3‰. The chlorin index (CI) and total chlorin concentration were determined with an analytical precision of ±5% according to Schubert et al.252.5 Accumulation rates
Whereas the SR only includes linear sedimentation in a specific core, bulk accumulation rates (ARbulk) also take compaction into account and hence sediment accumulation per time and area could be estimated. We used the following formulae:21| ARbulk = SR × dry density × (1 − porosity/100) | (1) |
| ARTOC = (TOC/100) × AR bulk | (2) |
2.6 Lipid biomarker analysis
The sediment samples were extracted in methanol/dichloromethane using a microwave system (MLS) to obtain the total lipid extracts. Aliquots of the extracts were saponified after addition of internal standards and separated into fatty acid and neutral (non-saponifiable) lipid fractions. The neutral fractions were silylated (BSTFA, Sigma) and analyzed by gas chromatography and gas chromatography-mass spectrometry for the quantification and identification of biomarkers, respectively. For a detailed description of lipid extraction see ref. 26.2.7 Methane concentrations
Sediment samples (2 cm3) were taken from the sediment core by sub-sampling the sediment from the side of the plastic tube through pre-drilled holes that were covered by tape during sampling. The sediment was filled in a 20 mL glass vial with 4 mL of 1 M NaOH. Subsequently the glass was covered with a butyl stopper and sealed with an aluminum crimp. Dissolved CH4 concentrations were measured in the headspace under controlled temperature conditions with an automated system (Joint Analytical Systems, Germany). In short, samples were introduced via an automated pumping system into a gas chromatograph (GC, Agilent) at constant temperature. The GC was equipped with a Carboxen 1010 column (30 m, Supelco) and a flame ionization detector. The oven temperature was 40 °C. CH4 standards were prepared by dilution of pure CH4 (99.9%) and calibrated against commercial references of 100 ppm, 1000 ppm and 1% (Scott, Supelco).2.8 pH, alkalinity, and DIC
pH was measured on the boat within minutes following the retrieval of the peepers using a portable pH meter (model 704, Metrohm) and a glass electrode (Minitrode, Hamilton) calibrated with 2 buffer reference solutions (Merck) of pH 6 and 8. Alkalinity was determined by end-point titration using a titration device (Titrino, Metrohm) and hydrochloric acid (HCl, 0.01 M). Dissolved inorganic carbon (DIC) concentrations were estimated using the measurements of pH and alkalinity, and equilibrium constants at 20 °C.272.9 Stable isotopic composition of carbon in DIC (δ13CDIC)
An aliquot of pore water was transferred without air bubbles onsite as fast as possible into a 2 mL glass vial, sealed with a Teflon cap using a crimper and kept at 4 °C. Before analysis, 1.5 mL of the water sample was transferred into an exetainer® and the headspace was flushed with helium. Concentrated phosphoric acid was added to convert all DIC to CO2. 100 μL of headspace was withdrawn from the vials with a GasTight Hamilton syringe and injected into a GC (Hewlet Packard 5890) interfaced to a ConFlo IV and a Delta V Plus IRMS (Thermo Fisher Scientific, Bremen, Germany). The GC was equipped with a GS-Carboplot column (Agilent, 30 m × 0.32 mm × 1.5 micron) to separate CO2 from the interfering gases.2.10 Oxygen concentrations
Oxygen at the sediment–water interface was measured within an hour after the sampling. An oxygen micro-optode operated with a temperature compensated system (Microx TX3, PreSens) and a custom made step motor with a vertical resolution of 0.05 mm was used. The micro-optode was calibrated with oxygen-free and air saturated solutions. The oxygen-free calibration solution was prepared by dissolving 1 g of sodium sulfite (Na2SO3) in 100 mL of nanopure water and the air saturated calibration solution was prepared by purging with air for 20 minutes using a glass frit following the recommendations of the manufacturer. The micro-optode was positioned in the water overlying the sediments, and slowly brought close to the sediment–water interface until the oxygen values (% air saturation) decreased. From this point on, the oxygen values (% air saturation) were recorded with a vertical resolution of 0.05 to 0.1 mm with an equilibration time between measurements of 20 s. Oxygen was measured until values of 0% air saturation were obtained two times in a row. Up to nine oxygen profiles per site were recorded.2.11 Determination of ammonium, sulfate, nitrate, iron and manganese
NH4+ concentrations were determined within hours following the sampling, using a standard photometric method following the protocol of GSM, 2002 (DIN 38 406-E5). Pore water aliquots were kept frozen until analysis of SO42− and NO3− concentrations by ion chromatography (ICS 1000 Ion Chromatography System, Dionex). For Fe(II) and Mn(II) concentrations, pore water aliquots (2 mL) were acidified onsite with 45 μL ultrapure concentrated nitric acid (65%, VWR) within minutes after sampling and then kept frozen until analysis using inductively coupled plasma optical emission spectroscopy (ICP-OES, Spectro Analytical Instruments). Samples were diluted 25 and 125 times (pore water samples from the center of the lake and from the delta region, respectively).2.12 Data analysis and calculation of mineralization rates
Areal fluxes across the sediment–water interface from sediment pore water data were determined with a model considering diffusion and reaction of dissolved compounds.28 The model was fitted to measured pore water concentration profiles by adjustment of reaction rates. The concentration change, dC, of a compound at depth z below the sediment–water interface with time dt is: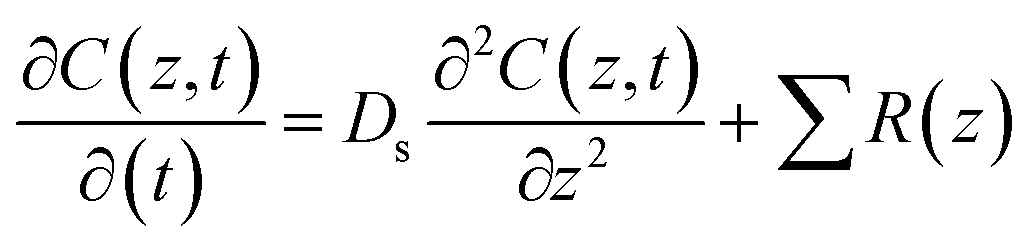 | (3) |
The mathematics of the model are detailed in Epping and Helder (1997).29 Diffusion coefficients at 4 °C were taken from Li and Gregory (1974).30 The porosity of the sediment (ϕ) was measured and the resulting formation factor (F = 1.02 × ϕ−1.81 = 1.23) was used as suggested by Maerki et al. (2004).31
3 Results and discussion
The two regions chosen for our investigation showed clear quantitative differences in the measured parameters. Sedimentation rates of the two cores close to the river mouth were much higher (SR = 2.3 cm per year) than the ones in the deeper part of the lake (SR = 0.4–0.6 cm per year). This already showed that sites on the Rhone River delta received more particulate material than the ones further out in the lake. The ARs amount to 26.8 and 28.4 kg m−2 per year at sites #1 and #2, and to 4.1 and 2.4 kg m−2 per year at sites #4 and #5 (Table 1). The values from the delta are in the range of published values in front of the Rhone river of 45.9 kg m−2 per year.32 The lowest OC contents (0.3–0.6%, Fig. 1) as a result of dilution by mineral particles delivered by the Rhone River were found at site #2, which together with site #1 is located closest to the river entrance. High OC values of ∼1% and up to 1.35% in the deeper sediment were detected at site #5, the core furthest away from the delta. This resulted in an ARTOC of 131.7 and 129.8 gC m−2 per year (sites #1 and #2) and 31.2 and 19.8 gC m−2 per year (sites #4 and #5), respectively for the delta and profundal sites (Table 1). | ||
| Fig. 1 Total organic carbon (TOC) and total nitrogen (TN) concentrations, the carbon isotopic composition of the OM (13Corg), and the chlorin index (CI) of the sediments at sites #2, #1, #4, and #5. | ||
C/N ratios are a way to characterize the OM as autochthonous, i.e. locally produced in the lake, and allochthonous, i.e. originating from outside the aquatic system. Autochthonous OM consists of algae and bacteria and hence is rich in N (proteins), whereas terrestrial OM (leaves, grass, etc.) is depleted in N and enriched in O-containing compounds (e.g. lignins).33 Therefore, C/N values for aquatic plants lie between 4 and 10, whereas terrestrial vascular plants can have values >20.34 In this study C/N ratios decreased continuously from 10.1 close to the river mouth to 9.1 and 7.9, and finally 7.4 at the most profundal site #5 (Table 2). Although TOC contents increased towards the open lake due to decreasing dilution by mineral particles transported by the river, the increase of N was even higher leading to lower C/N ratios. C/N ratios (Table 2) indicated a higher terrestrial influence at the delta sites contrary to the deeper sites where autochthonous OM was prevalent.
| Parameter | Site 1 | Site 2 | Site 4 | Site 5 |
|---|---|---|---|---|
| O2 | 4.3 ± 0.8 (n = 9) | 4.1 ± 1.1 (n = 6) | 8.2 ± 1.2 (n = 3) | 8.8 ± 1.3 (n = 2) |
| NO3− | Nd | 0.036 | 0.06 | 0.10 |
| SO42− | 0.32 | 0.15 | 0.62 | 0.54 |
| CH4 | 1.96 | 2.34 | 0.69 | 0.50 |
| NH4+ | 0.31 | 0.14 | 0.16 | 0.13 |
| Fe(II) | 1.1 × 10−4 | 9.0 × 10−5 | 3.8 × 10−4 | 3.3 × 10−4 |
| Mn(II) | 4.3 × 10−4 | 9.2 × 10−4 | 2.7 × 10−3 | 7.6 × 10−3 |
| C/N ratio | 10.1 | 9.1 | 7.9 | 7.4 |
|---|---|---|---|---|
| R aerobic | 0.3 (6%) | 0 (0%) | 6.8 (72%) | 7.8 (78%) |
| R anaerobic | 4.6 (94%) | 5.0 (100%) | 2.7 (28%) | 2.2 (22%) |
| R total | 4.9 (100%) | 5.0 (100%) | 9.5 (100%) | 10.0 (100%) |
The carbon isotopic composition of the OM (δ13Corg values) is used frequently in the marine environment to distinguish marine from terrestrial OM.35 However, in lacustrine systems the δ13Corg values of those two sources are rather indistinguishable since autochthonously produced OM is isotopically very similar to terrestrial OM. The reason is that the δ13CDIC in lakes in general and in the water column of Lake Geneva in particular is already very depleted (around −10 to −15‰, see the uppermost peeper values in Fig. 2), which leads to isotopically very light autochthonous OM compared to marine plankton. Carbon isotope values of OM in sediments of Lake Geneva were between −24.8 and −29.0‰ (Fig. 1) and hence were difficult to interpret with respect to differentiation of the OM as mentioned above. Nevertheless, our δ13Corg results showed that OM isotopically was increasingly heavier towards the bottom of the cores indicating the more labile character of the isotopically light aquatic OM. Based on this finding one could argue that values above −28‰ are indicative of a more autochthonous material since these values are unusual for an allochthonous material and hence the profundal cores have a higher contribution of autochthonous, i.e., algal material.
 | ||
| Fig. 2 Methane (CH4) and dissolved inorganic carbon (DIC) concentrations in the pore waters of the four sediment cores, the carbon isotopic composition of the DIC (13CDIC), and the alkalinity (ALK) at the four sites. | ||
One parameter used quite frequently in sediments is the chlorin index (CI) together with chlorin concentrations.25,36 Whereas the chlorin concentration gives a measure of how much pigment related OM is present in a sediment sample, the CI indicates how refractory is the OM. Relatively fresh OM has CI values of ∼0.6 (fresh chlorophyll has a value of 0.2), whereas more degraded OM shows values between 0.8 and 1.0. The sediments of Lake Geneva showed CI values between 0.5 and 0.9 with lower values on the delta and higher values at the distant sites #4 and #5 (Fig. 1). Hence, the material closer to the river mouth was fresher than the material deposited far away from the river mouth. Chlorin concentrations normalized to TOC, however, were higher for the distant sites #4 and #5 with 1226 and 1358 μg g−1 TOC compared to the delta sites #1 and #2 with 1032 and 822 μg g−1 TOC. Combined we see higher amounts of degraded pigment material in the distant sites.
Further characterization of OM in terms of autochthonous vs. allochthonous sources was done using lipids.34,37–39 Especially long chain n-alkanes and alcohols have been used as indicators for terrestrial OM. These compounds are constituents of surface waxes covering the leaves of higher plants.40 The sum of the long chain n-alkanes (C27, C29, C31) and long-chain alcohols (C24, C26, C28) was calculated. The n-alkanes showed contents of 277 to 429 μg per gC (#1 and #2) and 126 to 175 μg per gC (#4 and #5) with around 2 times higher concentrations closer to the river mouth. Similar results were obtained for the long-chain alcohols with 804 to 1128 μg per gC and 368 to 538 μg per gC, respectively. Calculating the AR for long chain n-alkanes and alcohols revealed an even more drastic difference and showed that the deltaic sites received 6 to 25 (n-alkanes) and 8–16 (alcohols) times more terrestrial OC than the profundal sites. In conclusion, a higher terrestrial input prevailed towards the river mouth supporting the above interpretation based on C/N ratios.
The final products of OM degradation are CH4 and CO2 (quantified as DIC dissolved in the pore water, neglecting the DIC produced by calcite dissolution in the sediment). Both constituents were released from Lake Geneva sediments into the water column, as suggested by the shape of their concentration profiles (Fig. 2), which decreased towards the sediment surface. CH4 fluxes varied spatially and were more intense in the river delta (1.96 and 2.34 at sites #2 and #1) compared to the deeper part of the lake (0.69 and 0.50 at sites #4 and #5, Table 2). The DIC fluxes were 2.0 and 1.4 mmol m−2 d−1 at sites #1 and #2 and 1.0 and 0.9 mmol m−2 d−1 at sites #4 and #5, respectively. Although DIC fluxes at the deltaic sites were not 4 to 5 times higher than those at the profundal sites, as was observed for CH4, they were still 1.5 to 2 times higher hinting to more pronounced fluxes of the degradation products of OM at the deltaic sites.
The main focus of this study was to investigate the difference in electron acceptors used for the degradation of OM depending on its concentration and origin, i.e. lacustrine or terrestrial, in a lacustrine system. Hence, fluxes of O2, SO4, and NO3 were calculated from the concentration profiles determined from peepers and sediment cores (Fig. 3, O2 concentration profiles not shown but up to 9 profiles per site were combined to estimate fluxes).
 | ||
| Fig. 3 Concentrations of sulfate, nitrate, manganese (triangles), iron (circles), and ammonium measured in the pore water and overlaying water of the sites #1, #2, #4, and #5. | ||
The O2 consumption of the sediment was similar for the profundal cores with 8.8 (site #4) and 8.2 mmol m−2 d−1 (site #5), whereas only half of this flux was measured at sites #1 and #2 close to the river inflow with 4.3 and 4.1 mmol m−2 d−1, respectively (Table 2). O2 concentrations in the deep waters were around 4 mg L−1 during the time of sampling (The Commission Internationale pour la Protection des Eaux du Léman, CIPEL). As described before for lacustrine sediments12 O2 penetrated only the uppermost mm of the sediment with the deepest penetration of 1.8 mm at site #1. The other two electron acceptors NO3− and SO42− showed very small fluxes compared to O2. The NO3− flux could not be determined at site #1 due the large scatter of the data. At site #2 the NO3− flux was lower (0.04 mmol m−2 d−1) than that at the deeper sites #4 and #5 where 0.06 and 0.1 mmol m−2 d−1 were measured. Similarly, the SO42− flux into the sediment was lower at the shallower sites #1 and #2 (0.15 and 0.32 mmol m−2 d−1) than that at the deeper sites #4 and #5 (0.62 and 0.54 mmol m−2 d−1).
The fluxes of reduced species from the sediments (Table 2) decreased from the delta to the sites in the open lake for CH4 (from 2.3 to 0.5 mmol m−2 d−1) and NH4+ (from 0.31 to 0.13 mmol m−2 d−1). Fluxes of Fe(II) and Mn(II), however, increased although they were generally very low (between 9 × 10−5 and 7.6 × 10−3 mmol m−2 d−1). The CH4 fluxes were comparable to values reported by Sollberger et al. (2014)41 who showed that diffusive CH4 fluxes were more intense in the active canyon of the Rhone delta than in the central and eastern regions. The sites with lower OC concentrations (sites #1 and #2) had higher CH4 fluxes, whereas sites #4 and #5 comprised higher OC contents but lower CH4 fluxes. However, one has to take into account that the AR of OC was about five times higher at the delta sites than that at the deep sites and the low content is a ‘dilution’ by non organic, i.e. minerals, delivered by the river.
If the production rate of CH4 is related to the deposition rate of OC we receive an efficiency for the methanogenesis from total OC (in mmol CH4 per gC) that is now clearly higher at the deeper sites than in the delta (Table 1). Hence, it seems that the AROC is not solely responsible for the degree of CH4 production, but that beside the accumulation of OC additional parameters like freshness/degradability and composition of the OM play a role.
The set of available pore water data allowed estimating the mineralization rates of OM Rtotal at the four sites, and the fraction of OM degraded aerobically (Raerobic) and anaerobically (Ranaerobic). As exemplified by Maerki et al. (2009)12 oxic mineralization was calculated from the flux of O2 to the sediment subtracting reduced compounds diffusing up from the deeper sediments thus contributing to the consumption of O2:
| Raerobic = JO2 − 2 × JCH4 − 0.5 × JMn(II) − 0.25 × JFe(II) | (4) |
The stoichiometric factors define the equivalents of O2 consumed per equivalent of reduced compounds. The areal mineralization of OM is expressed in mmol C m−2 d−1 (Table 2) assuming the mineralization of one equivalent of OM per equivalent of O2, thus neglecting nitrification. The fraction of OM mineralized with electron acceptors other than O2 (Ranaerobic) is calculated as:
| Ranaerobic = 1.25 × JNO3− + 0.25 × JFe(II) + 0.5 × JMn(II) + 2 × JSO42− + 2 × JCH4 | (5) |
Since we could not account for compounds that were potentially removed from the pore water and buried such as precipitates of Fe(II)/Mn(II) sulfides, vivianite, siderite, etc., those sinks were neglected.
The rates for totally mineralized OM (Rtotal = Raerobic + Ranaerobic) presented in Table 2 at the deeper sites #4 and #5 (average of 9.7 mmol C m−2 d−1) were two times higher than those at the shallower sites #1 and #2 (4.7 mmol C m−2 d−1). Not only the rates were different but also the mineralization pathway differed between the shallow sites #1 and #2 in the Rhone River delta, and the profundal sites #4 and #5. Only 0–6% was mineralized aerobically at the shallow sites, since almost all O2 was used to oxidize the large flux of CH4 from below (Fig. 4). At the deeper sites, the situation was reversed and the fraction of aerobically degraded OM was between 72 and 78% (Fig. 4). Also, the contribution of SO42− to Ranaerobic was very distinct between both areas with 44–49% at the deeper sites and only 6–14% at the shallower sites. The contributions of NO3− (1–5.5%), and of Fe and Mn (<0.2%) to the mineralization of OM in the sediments were negligible.
 | ||
| Fig. 4 Aerobic and anaerobic degradation rates of the river delta sites and of the profundal sites. Whereas sites #1 and #2 show a degradation that is mainly triggered by anaerobic degradation (94 and 100%), sites #4 and #5 show a dominance of aerobic degradation with an anaerobic fraction of only 28 and 22%. | ||
The increased generation of alkalinity observed in the sediment pore water of the shallow sites (Fig. 2) lessens up to 10% by the dissolution of calcite (determined from Ca2+ pore water profiles, data not shown), and even less at the deeper sites. However, the reduction of SO42− with both, OM or CH4 produces one equivalent of alkalinity, which for sites 1 and 2 amounts to 11–16% of the HCO3− flux but to ∼60% for sites 4 and 5. Similarly, alkalinity generated by denitrification is ∼3% at site 2, but 6–11% at sites #4 and #5. Contributions from Fe and Mn reduction are negligible. The remaining HCO3− originates from the production of CO2 by mineralization and the subsequent decrease of pH.
In conclusion, a consistent picture emerged that the mineralization pathways and rates in the sedimentary depositions in the Rhone delta area were significantly different from those out in the profundal parts of the lake. The much higher CH4 fluxes in the delta compared to the open lake sites are explained by the high deposition rate of OC in this area where OC accumulation was approximately 5 times higher in the delta at sites #1 and #2 (132 and 130 gC m−2 per year) than at sites #4 and #5 (31.2 and 19.8 gC m−2 per year, Table 1). Bulk deposition rates differed even by a factor of 8 (Table 1) demonstrating the high load of mineral particles transported by the river and diluting the OC. These “diluting” conditions led to almost two times higher content of OC (in percent %) at the profundal than at the delta sites. Estimating the efficiency of CH4 formation per equivalent of OC (the annual flux of CH4 per annually deposited C) therefore resulted in 5.5 to 6.4 μmol CH4 per gC and of 8.0 to 9.1 μmol CH4 per gC for the delta sites and open lake sites, respectively (Table 1). This supports our findings that a significant part of the OC deposited in the delta contained more refractory and thus microbially less degradable, terrestrial material. The higher CH4 flux on the delta must be recognized as caused by the high AR and the small fraction of aerobic mineralization of OC, which was qualitatively different from the more autochthonous material at the deeper sites and thus could be converted to CH4 only with a lower efficiency. This study supports earlier findings that have claimed that burial in lakes is high9 and it is especially high when high amounts of allochthonous material are delivered.4 It also points out that it is necessary not only to measure the OM concentration but to characterize the origin and the degradation state of OM to estimate how OM is transformed and which share is emitted as CO2 and/or CH4.
Similar to river deltas in the coastal ocean, river deltas of large lakes are active players in the carbon cycle as OM is buried, transformed or emitted from bottom sediments. However, an accurate estimate of the amount of carbon cycled in these active zones remains relatively unconstrained, largely because the fractions of various carbon sources are unresolved. Our results suggest that carbon sources and degradation pathways can differ widely in some freshwater systems, further complicating the situation. While similar analyses as those presented here would be necessary to accurately identify OC sources and degradation pathways, the clear trends in our data indicate that river deltas are regions of substantial carbon cycling, and that any system resembling our study site, particularly if it is an obvious hot spot for CH4 production (i.e., presence of ebullition), will exhibit the same redox conditions and OM degradation patterns we observed in the Rhone delta.
Acknowledgements
This publication is part of the international, interdisciplinary research project éLEMO for the investigation of the deep-waters of Lake Geneva using two Russian MIR submarines (http://www.elemo.ch). Funding for retrieving samples was provided by the ‘‘Fondation pour l’Etude des Eaux du Léman’’ (FEEL; in particular by the Ferring Pharmaceuticals, St. Prex). Laboratory and personnel was funded by Eawag. We thank the Russian MIR crew members (http://www.elemo.ch/mir-team) for their excellent performance and the SAGRAVE team who provided and operated the platform from which the dives were carried out. We also thank Ulrich Lemmin and Jean-Denis Bourquin for project coordination. The service of Mikhail Kranoperov (Russian Honorary Consulate) as liaison is greatly appreciated. We also thank C. Dinkel, M. Schurter, and A. Zwyssig for fieldwork assistance. R. Stierli and late G. Nobbe are thanked for the lab assistance. The Commission Internationale pour la Protection des Eaux du Léman (CIPEL) is thanked for providing O2 concentrations in the bottom water of Lake Geneva. Finally we thank all scientists of the éLEMO project for the excellent collaboration and two reviewers who really helped to improve this manuscript.Notes and references
- J. I. Hedges and R. G. Keil, Mar. Chem., 1995, 49, 81–115 CrossRef CAS.
- H. E. Hartnett, R. G. Keil, J. I. Hedges and A. H. Devol, Nature, 1998, 391, 572–575 CrossRef CAS.
- D. J. Burdige, Chem. Rev., 2007, 107, 467–485 CrossRef CAS PubMed.
- S. Sobek, E. Durisch-Kaiser, R. Zurbrugg, N. Wongfun, M. Wessels, N. Pasche and B. Wehrli, Limnol. Oceanogr., 2009, 54, 2243–2254 CrossRef.
- S. Sobek, R. Zurbrugg and I. Ostrovsky, Aquat. Sci., 2011, 73, 355–364 CrossRef CAS.
- J. Chappellaz, J. Barnola, D. Raynaud, Y. Korotkevich and C. Lorius, Nature, 1990, 345, 127–135 CrossRef CAS.
- Climate Change. The IPCC Scientific Assessment, ed. J. T. Houghton, G. J. Jenkins and J. J. Ephraums, Cambridge University Press, Cambridge, 1990 Search PubMed.
- D. J. Wuebbles and K. Hayhoe, Earth-Sci. Rev., 2002, 57, 177–210 CrossRef CAS.
- W. E. Dean and E. Gorham, Geology, 1998, 26, 535–538 CrossRef.
- J. A. Downing, Y. T. Prairie, J. J. Cole, C. M. Duarte, L. J. Tranvik, R. G. Striegl, W. H. McDowell, P. Kortelainen, N. F. Caraco, J. M. Melack and J. J. Middelburg, Limnol. Oceanogr., 2006, 51, 2388–2397 CrossRef.
- D. E. Canfield, B. B. Jorgensen, H. Fossing, R. Glud, J. Gundersen, N. B. Ramsing, B. Thamdrup, J. W. Hansen, L. P. Nielsen and P. O. J. Hall, Mar. Geol., 1993, 113, 27–40 CrossRef CAS PubMed.
- M. Maerki, B. Mueller, C. Dinkel and B. Wehrli, Limnol. Oceanogr., 2009, 54, 428–438 CrossRef CAS.
- P. Burkard, in Lausanne: Le Léman Synthèse 1957–1982, 1984, pp. 43–48 Search PubMed.
- OFEV, Annuaire hydrologique de la Suisse 2009, Office fédéral de l’environnement, Berne, Switzerland, 2013, p. 617 Search PubMed.
- D. Burrus, R. Thomas, J. Dominik and J. Vernet, Hydrol. Processes, 1989, 65–74 CrossRef.
- N. R. Urban, C. Dinkel and B. Wehrli, Aquat. Sci., 1997, 59, 1–25 CrossRef CAS.
- S. Girardclos, M. Hilbe, J. P. Corella, J. L. Loizeau, K. Kremer, T. Delsontro, A. Arantegui, A. Moscariello, F. Arlaud, Y. Akhtman, F. S. Anselmetti and U. Lemmin, Arch. Sci., 2012, 65, 103–118 Search PubMed.
- R. H. Hesslein, Limnol. Oceanogr., 1976, 21, 912–914 CrossRef CAS.
- H. Brandl and K. W. Hanselmann, Aquat. Sci., 1991, 53, 55–73 CrossRef.
- R. Carignan, S. S. Pierre and R. Gachter, Limnol. Oceanogr., 1994, 39, 468–474 CrossRef CAS.
- F. Niessen, L. Wick, G. Bonani, C. Chondrogianni and C. Siegenthaler, Aquat. Sci., 1992, 54, 257–276 CrossRef.
- P. G. Appleby and F. Oldfield, Catena, 1978, 5, 1–8 CrossRef CAS.
- J. P. Corella, A. Arantegui, J. L. Loizeau, T. DelSontro, N. le Dantec, N. Stark, F. S. Anselmetti and S. Girardclos, Aquat. Sci., 2014, 76(suppl. 1), 73–87 CAS.
- C. J. Schubert and B. Nielsen, Mar. Chem., 2000, 72, 55–59 CrossRef CAS.
- C. J. Schubert, J. Niggemann, G. Klockgether and T. G. Ferdelman, Geochem., Geophys., Geosyst., 2005, 6, 1–12 CrossRef.
- S. Naeher, R. H. Smittenberg, A. Gilli, E. P. Kirilova, A. F. Lotter and C. J. Schubert, Org. Geochem., 2012, 49, 86–95 CrossRef CAS.
- W. Stumm and J. J. Morgan, Aquatic Chemistry, An Introduction Emphasizing Chemical Equilibria in Natural Waters, Wiley, J., New York, 1981 Search PubMed.
- B. Müller, Y. Wang, M. Dittrich and B. Wehrli, Water Res., 2003, 37, 4524–4532 CrossRef.
- E. H. G. Epping and W. Helder, Cont. Shelf Res., 1997, 17, 1737–1764 CrossRef.
- Y. H. Li and S. Gregory, Geochim. Cosmochim. Acta, 1974, 38, 703–714 CrossRef CAS.
- M. Maerki, B. Wehrli, C. Dinkel and B. Müller, Geochim. Cosmochim. Acta, 2004, 68, 1519–1528 CrossRef CAS.
- J.-L. Loizeau, Arch. Sci., 1998, 51, 13–25 CAS.
- J. I. Hedges, W. A. Clark, P. D. Quay, J. E. Richey, A. H. Devol and U. d. M. Santos, Limnol. Oceanogr., 1986, 31, 717–738 CrossRef CAS.
- P. A. Meyers and R. Ishiwatari, Org. Geochem., 1993, 20, 867–900 CrossRef CAS.
- C. J. Schubert and S. E. Calvert, Deep Sea Res., Part I, 2001, 48, 789–810 CrossRef CAS.
- A. N. Meckler, C. J. Schubert, G. L. Cowie, S. Peiffer and M. Dittrich, Limnol. Oceanogr., 2004, 49, 2023–2033 CrossRef CAS.
- L. Westerhausen, J. Poynter, G. Eglinton, H. Erlenkeuser and M. Sarnthein, Deep-Sea Res., 1992, 1–53 Search PubMed , in press.
- C. J. Schubert and R. Stein, Org. Geochem., 1996, 24, 421–436 CrossRef CAS.
- S. Bourgeois, A. M. Pruski, M. Y. Sun, R. Buscail, F. Lantoine, P. Kerhervé, G. Vétion, B. Rivière and F. Charles, Biogeosciences, 2011, 8, 3107–3125 CrossRef CAS.
- G. Eglinton and R. J. Hamilton, Science, 1967, 156, 1322–1335 CAS.
- S. Sollberger, J. P. Corella, S. Girardclos, M. E. Randlett, C. J. Schubert, D. B. Senn, B. Wehrli and T. DelSontro, Aquat. Sci., 2014, 76(suppl. 1), S89–S101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4em00470a |
| This journal is © The Royal Society of Chemistry 2015 |