
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An evaluation of the “GGP” personal samplers under semi-volatile aerosols: sampling losses and their implication on occupational risk assessment
George C.
Dragan
ab,
Dietmar
Breuer
c,
Morten
Blaskowitz
c,
Erwin
Karg
ad,
Jürgen
Schnelle-Kreis
*ad,
Jose M.
Arteaga-Salas
ad,
Hermann
Nordsieck
e and
Ralf
Zimmermann
abd
aJoint Mass Spectrometry Centre, Cooperation Group “Comprehensive Molecular Analytics”, Helmholtz Zentrum München, D-85758 Neuherberg, Germany. E-mail: juergen.schnelle@helmholtz-muenchen.de
bJoint Mass Spectrometry Centre, Chair of Analytical Chemistry, Institute of Chemistry, University of Rostock, D-18057 Rostock, Germany
cGerman Social Accident Insurance, Institute for Occupational Safety and Health, Alte Heerstr. 111, D-53757 Sankt Augustin, Germany
dHelmholtz Virtual Institute of Complex Molecular Systems in Environmental Health – Aerosol and Health (HICE), Germany Web: http://www.hice-vi.eu
eBifa Environmental Institute, Am Mittleren Moos 46, D-86167 Augsburg, Germany
First published on 13th October 2014
Abstract
Semi-volatile (SV) aerosols still represent an important challenge to occupational hygienists due to toxicological and sampling issues. Particularly problematic is the sampling of hazardous SV that are present in both particulate and vapour phases at a workplace. In this study we investigate the potential evaporation losses of SV aerosols when using off-line filter-adsorber personal samplers. Furthermore, we provide experimental data showing the extent of the evaporation loss that can bias the workplace risk assessment. An experimental apparatus consisting of an aerosol generator, a flow tube and an aerosol monitoring and sampling system was set up inside a temperature controlled chamber. Aerosols from three n-alkanes were generated, diluted with nitrogen and sampled using on-line and off-line filter-adsorber methods. Parallel measurements using the on-line and off-line methods were conducted to quantify the bias induced by filter sampling. Additionally, two mineral oils of different volatility were spiked on filters and monitored for evaporation depending on the samplers flow rate. No significant differences between the on-line and off-line methods were detected for the sum of particles and vapour. The filter-adsorber method however tended to underestimate up to 100% of the particle mass, especially for the more volatile compounds and lower concentrations. The off-line sampling method systematically returned lower particle and higher vapour values, an indication for particle evaporation losses. We conclude that using only filter sampling for the assessment of semi-volatiles may considerably underestimate the presence of the particulate phase due to evaporation. Thus, this underestimation can have a negative impact on the occupational risk assessment if the evaporated particle mass is no longer quantified.
Environmental impactSemi-volatile (SV) aerosols still represent an important challenge to occupational hygienists due to toxicological and sampling issues. Particularly problematic is the sampling of hazardous SV that are present in both particulate and vapour phases at a workplace. The present paper focuses on the sampling of SV aerosols and investigates the particle evaporation losses that might bias the occupational risk assessment when using off-line filter-adsorber personal samplers. Comparison of the GGP inhalable sampler to on-line direct-reading instruments shows that filter sampling can significantly underestimate SV particles. It is therefore recommended that occupational exposure limits should be considered for the total aerosol mass, i.e. for the sum of particle and vapour mass concentration instead of either one of them separately. |
Introduction
The sampling of hazardous semi-volatile (SV) compounds in workplace air can prove to be difficult for occupational hygienists when confronted with substances that are present as aerosols, partitioned between the particulate and the gaseous phases (oil mists, PAHs, inorganic acids, alkanolamines, bitumen etc.).1–5 The European norm EN 13936 defines semi-volatiles as substances with vapour pressures between 100 and 0.001 Pa at room temperature.6 The compounds with vapour pressures within this range are expected to be found as mixed phase aerosols in workplaces. More attention needs to be given to these dynamic aerosol systems as the observed particle–vapour distribution is considerably dependent on the sampling conditions1,7 and the aerosols' real phase distribution can get biased by evaporation of collected particles (blow-off) or condensing of vapours onto collected particles or filtration substrate (blow-on).Mineral oils are commonly used in diverse industrial processes including offshore drilling,8,9 metallurgic10,11 and automotive industries.12,13 In machining workshops mineral oils are frequently applied as metalworking fluids (MWF) to simultaneously lubricate, cool, remove debris from metal pieces and prevent corrosion.14–18 Although MWFs improve the quality of machining,11 health risks are often associated with the inhalation of oil aerosols either due to toxic compounds within the fluid or because of microbial contamination.19–23 The usage of mineral oils in workplaces can lead to the formation of hazardous oil mists/aerosols through physical dispersion and evaporation followed by re-condensation.24,25
Occupational exposure to oil mists can be associated with adverse respiratory effects and dermatitis.26 In addition, there are concerns relating to occupational asthma, allergic alveolitis and other lung diseases.27 The adverse effects caused by hazardous aerosols mainly depend on the amount of substance present in the inhaled air, but also on the physical state of the inhaled compounds. From the toxicological point of view, particles are considered to have a greater threat to human health, as they can penetrate deeper into the respiratory tract and deposit into the lungs according to the particles' aerodynamic diameter.28–30 Due to the higher health concern posed by the particulate phase, some countries have adopted aerosol occupational exposure limits for particles only. However, having exposure limits for particles only can eventually lead to an underestimation of the health risks involved, as filter sampling of SV aerosols often loses particle mass by evaporation.31
Aerosols can be defined as dispersions of solid particles and/or liquid droplets in air or other gases. The measurement of workplace aerosol concentrations is currently carried out using a wide range of different instruments in the various countries of the European Union.32 Filters, photometers, optical particle counters (OPCs), impactors and electrostatic precipitators are usually employed to measure the particulate phase. Flame ionization detectors (FID), photoionization detectors (PID) and Fourier transform infrared (FTIR) spectrometers preceded by filters are used to monitor the organic vapours, while denuders and adsorbers can reversibly trap the compounds from the gas phase. As previous studies have shown33–38 the results obtained with each method can turn out to be biased by a series of factors. Sampler type, sampling flow rate, particle size distribution and substance volatility can have a significant influence on the observed particle–vapour ratio. Although most methods can be used with good accuracy for the sampling of low-volatile oil mists, issues may arise when sampling aerosol particles originating from semi-volatile compounds.
Previous studies33,39,40 have focused on the evaporation of collected droplets from filters and mist collectors when clean ambient air is passed through them. The authors used nebulizers to apply a polydisperse mist on the filters while the evaporated material was assessed gravimetrically. Svendsen et al.34 compared four off-line filter-adsorber samplers and found significant differences between them for the collected particle mass but no significant differences for the sum of particles and vapours. Simpson et al.27 spiked 13 different oils on filters and aspirated clean air at 2 l min−1 through. They found evaporative losses ranging from 1% up to 71% after 6 hours. In addition to the previously mentioned studies, we also investigated the flow rate influence on the evaporation of mineral oils spiked on filters.
Volckens and co-authors35,36 performed laboratory and field measurements of oil mists where they compared several off-line sampling methods (3 filter types, electrostatic precipitator, cascade impactor) to two direct-reading photometers. The authors noted that the filter sampling method may not be appropriate for semi-volatiles as the evaporative losses can lead to an exposure underestimation. Unfortunately, these studies did not provide a non-biased method that accurately differentiates between particles and vapours but compared the existing measurement techniques between themselves without being able to measure the actual particle–vapour distribution. We propose in our paper a more accurate reference method to assess the particle–vapour distribution.
Our earlier work7 has focused on the evaporation dynamic of a monodisperse particle population inside a flow tube. Within this study it was shown that the evaporation of a particle population can be predicted with good accuracy using a simple diffusion based model. Also shown was that particles of volatile compounds (C14, C16) can completely evaporate within one minute if the gas phase doesn't reach vapour saturation. In the case where vapour saturation is reached no further mass transfer takes place. Sutter et al.41 on the other hand focused on the evaporation of hexadecane particles trapped on filters under a clean air flow. They observed that the vapour concentration downstream of the clogged filters was nearly identical to the vapour saturation concentration, if sufficient particle mass was initially deposited.
The method used as reference in this paper (FID for total hydrocarbon mass and white light particle sizer for particle mass)7 is not influenced by evaporation as the aerosol characteristics are not changed in the sensor of the OPC and as all droplets are vaporized in the transfer line to the FID. This setup minimizes artefacts and allows it to be used to determine particle–vapour fractionation while sampling oil mists. Further improvements to previous studies are the use of monodisperse particles which are measured by optical sensors more accurately as well as the very stable Sinclair-La Mer aerosol generator (less than 5% variation over each 2 hour measurement) which considerably improves general stability. The temperature controlled chamber where the measurements took place eliminated any possible bias caused by temperature fluctuations.
Three n-alkanes (tetradecane, hexadecane, octadecane) were selected as test substances as they are of relevant volatility and are known to be main constituents of the mineral oils usually used as MWFs.11,42 Aerosols from the test substances were generated, diluted with nitrogen inside a flow tube and sampled in parallel with on-line (FID + OPC) and off-line methods (filter + adsorber). The on-line method served as a reference for the measured aerosol and thus shifts in the aerosols' real phase distribution could be observed for the off-line samplers by comparison with the direct-reading reference. Additionally, two mineral oils were spiked on the off-line GGP (Gesamtstaub-Gas-Probenahme = inhalable dust/gas sampling) samplers in order to assess the evaporation loss dependence on sampling flow rates.
The focus of the present study was to accurately quantify the sampling bias due to volatilization of oil mist from a “GGP” filter-adsorber personal sampler, which is used for oil mist sampling at workplaces in Germany, under temperature-controlled laboratory conditions.
Materials and methods
Part I: aerosol generation and comparison between on-line instruments and off-line GGP filter-adsorber samplers
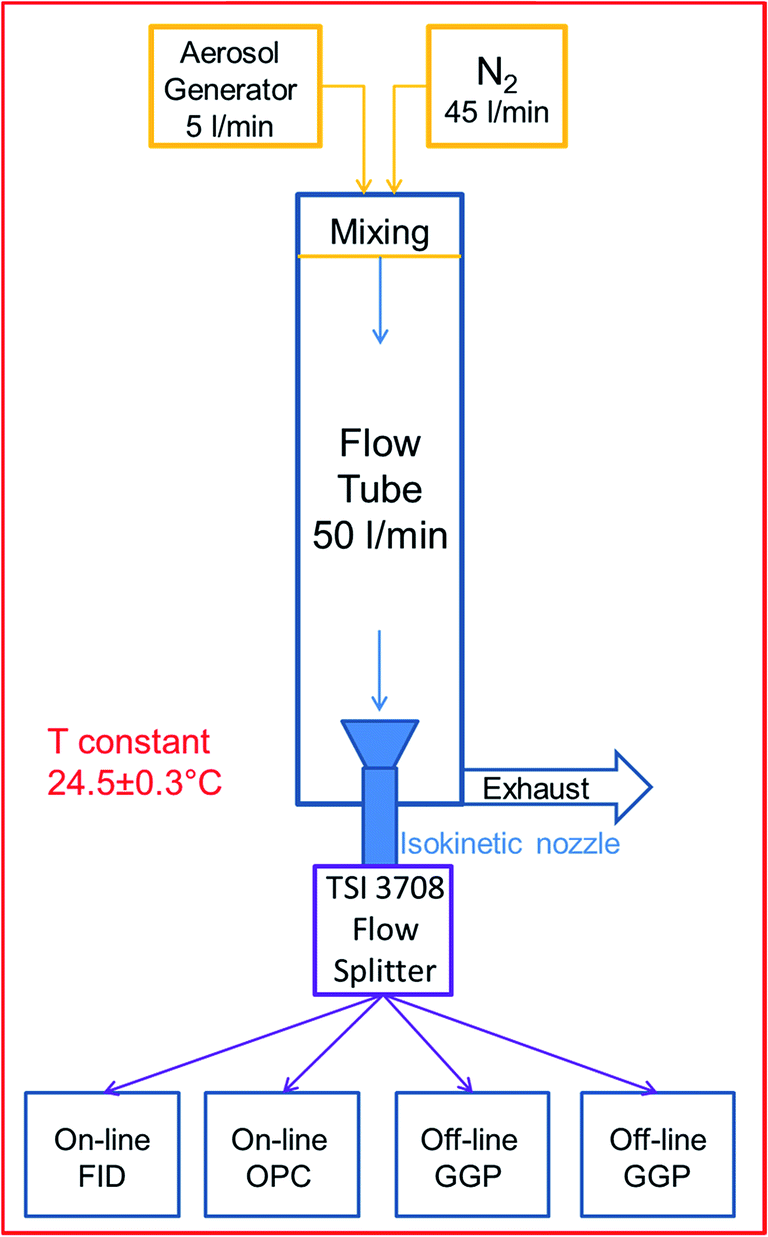 | ||
| Fig. 1 Experimental setup within the temperature controlled lab. | ||
A Sinclair-La Mer type aerosol generator (SLG 270, Topas GmbH, Germany) was chosen to produce monodisperse aerosol droplets (mass geometric standard deviation < 1.2) from three n-alkanes of different volatilities: n-tetradecane, n-hexadecane, n-octadecane (99% purity, Merck GmbH, Germany). The generator produced the monodisperse droplets by condensing super-saturated vapour onto separately produced NaCl nuclei. The produced aerosol proved to be stable throughout the experiments, with less than 5% mass concentration deviation during each 2 hour experiment. The monodisperse droplets' mass mean diameter (MMD) generated for this study was in the range of 1 to 4 μm. The generator was operated at a flow rate of 5 l min−1.
A vertical poly(methyl methacrylate) (PMMA) flow tube (FT) preceded by a dilution and mixing unit was employed to allow the aerosol to mix with the particle- and humidity free dilution gas (N2) at a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio, for a total flow of 50 l min−1. The dilution unit was introduced to adjust both the particle number concentration to the desired level and to mimic the mixing of emitted oil mist with ambient air at a workplace. The aerosols' dilution disturbs the initial particle–vapour equilibrium, thus triggering the evaporation of particles and thereby the particle to vapour mass transfer.
:10 ratio, for a total flow of 50 l min−1. The dilution unit was introduced to adjust both the particle number concentration to the desired level and to mimic the mixing of emitted oil mist with ambient air at a workplace. The aerosols' dilution disturbs the initial particle–vapour equilibrium, thus triggering the evaporation of particles and thereby the particle to vapour mass transfer.
A flow splitter (Model 3708, TSI Inc., USA) preceded by an isokinetic nozzle was used to provide four identical aerosol samples to the on-line and off-line test devices. Prior experiments using particle counters have shown no significant differences between the flow splitters' four channels. For equal distribution purposes, the sampling flow rates were kept similar for all devices so that comparability of samples was ensured for every channel.
The FID was used to continuously monitor the total concentration of the semi-volatile substance present in the aerosol at a flow rate of 4 l min−1. A transfer line heated to 180 °C was coupled to the FID inlet in order to evaporate all particles within the aerosol.
A OPC was used as a particle sizer to determine the aerosols' particle size distribution and calculate the PM. It uses a white light lamp to illuminate the aerosol particles that enter the instruments' sampling head. The light scattered by the passing particles is measured and converted into a particle size using a calibration curve. The calibration was verified and proved for accuracy using certified polystyrene latex (PSL) spheres of various diameters. The OPC software also allows adjusting the calibration curve for a refractive index of 1.45. Density corrections of 0.773; 0.777 and 0.781 (kg m−3) were used for C14, C16 and C18 respectively in order to calculate the particle mass. OPC measurements were taken with a 10 minute time resolution and at a flow rate of 5 l min−1.
The filters and XAD-2 were extracted directly after sampling, following the standard IFA procedure in 10 ml tetrachloroethylene (C2Cl4, PER, Merck KGaA, Germany) each and left to elute for more than 24 h. During the method validation for the sampling of oil mists, recovery rates between 91% and 107% were observed, with an average above 98%. Therefore no further recovery correction was calculated for the analysed samples. The extracts were analysed with a Nicolet Avatar 370 DTGS (Thermo-Scientific Inc., USA) Fourier transform infrared spectrometer by measuring the IR absorbance at the 3000–2800 cm−1 spectral region. The FTIR measurements were averaged over 16 scans at a resolution of 4 cm−1 and were conducted at the Institute for Occupational Safety and Health (DGUV-IFA) in Sankt Augustin, Germany.
Hexadecane and octadecane extract samples were also analysed with a HP 5980 gas chromatograph with flame ionization detection (GC-FID). The GC consisted of a 25 m DB5 column with a diameter of 0.2 mm and was set to increase the oven temperature from 90 to 240 °C, at a rate of 10 K min−1.
Part II: evaporation of semi-volatile oil from fibrous filters flushed with clean air at different flow rates
The flow rate at which air is being drawn through the GGP filter-adsorber sampler can influence the amount of evaporated material collected on filters.1 To check the influence of the flow rate on evaporative losses during sampling, two mineral oil mixtures of different flash points (1st oil “semi-volatile” = Superfin 160, Petrofer, Hildesheim, flash point 136 °C, boiling range C13 to C17 see Fig. 2; 2nd oil “low volatile” = special oil MS 15 EP, Beku-Oil, Deisslingen, flash point >195 °C, boiling range > C18) were spiked as a solution (4.2 mg oil solved in 100 μl C2Cl4) by means of a microliter syringe onto the glass fibre filter of a GGP sampler. Clean air was drawn at six different flow rates (1; 2; 3.5; 5; 7 and 10 l min−1) through the samplers for 2 hours. The filters and XAD-2 cartridges were processed and analysed as previously described. All experiments were repeated three times.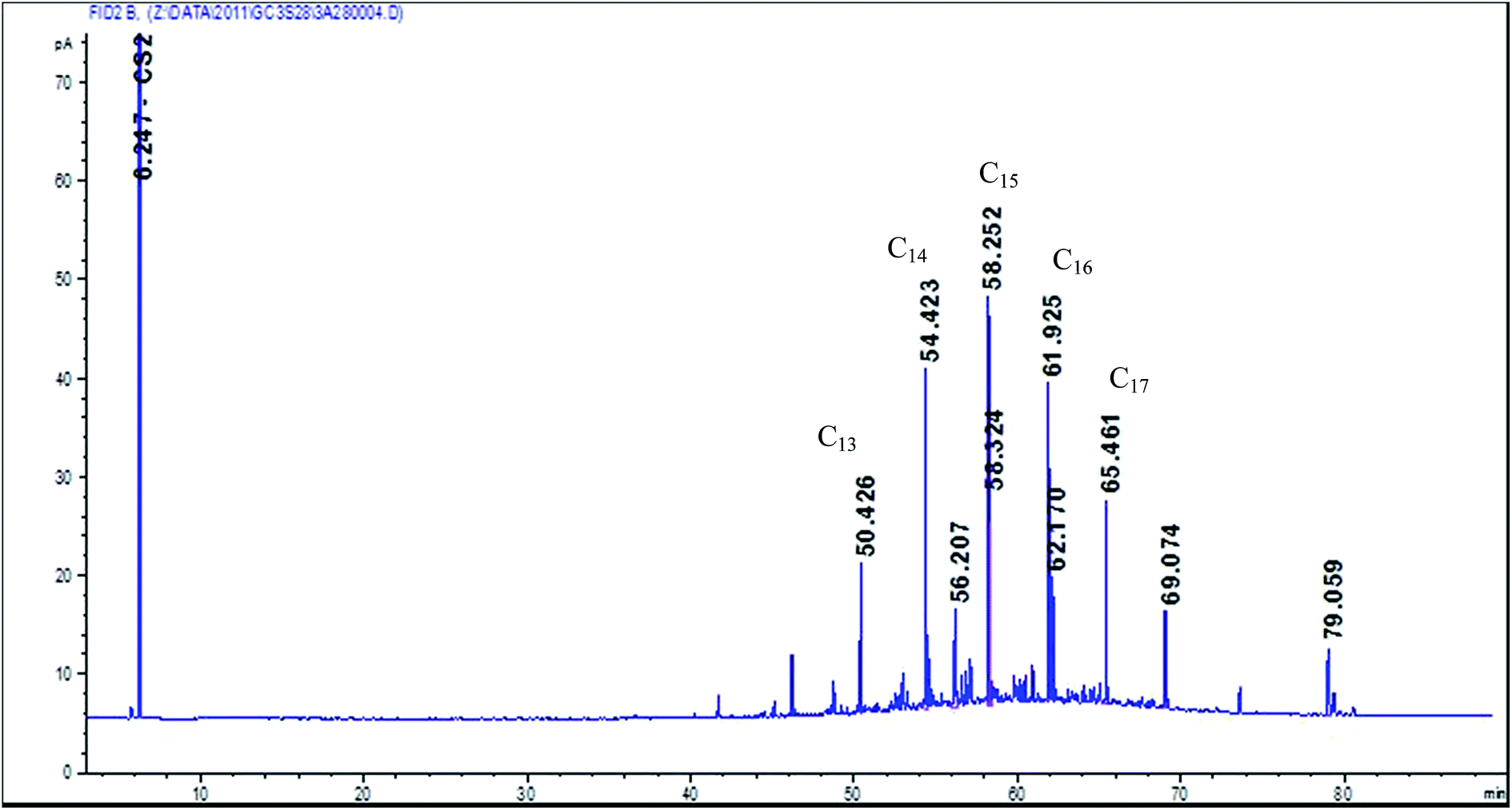 | ||
| Fig. 2 Gas chromatogram of the semi-volatile mineral oil Superfin 160. | ||
Two sided Student's t-tests were used as a tool to determine the statistical significance of the measured data.
Results and discussion
Parallel measurements with on-line and off-line techniques
For this study, the 1:10 dilution of the initial aerosol was able to trigger a mass transfer from particles to the vapour phase. The extent of evaporation losses is related to the vapour saturation degree, with the evaporation flux being highest when no vapours are present in the gas phase and at the lowest level when the gas phase is reaching vapour saturation. At the sampling point, the gas phase was not fully saturated with vapour and therefore particles trapped on the filter substrates continued to evaporate. The on-line measurements on the other hand present a “snapshot” of the particle–vapour distribution at the moment at which the measurement took place. Nevertheless, while the particles are collecting on the filter to a certain threshold, the evaporation flux from the filter can be capped by the ability of the passing gas to transfer away vapour. Therefore, the vapour concentration downstream of the filters for the high PM values will be close to that of the saturation concentration.
Furthermore, during the 2 hours sampling period, each particle trapped on the GGP samplers' filter remains in contact with the incoming air stream for a period ranging from two hours (the first particles that are trapped on the filter) down to about one minute (time required to verify the pump flow rates after sampling). Therefore, the long residence time on the filter surface will lead to the evaporation of the volatile particles, until the incoming gas stream will reach the vapour saturation. Thus, assuming that particles collected on the filters will evaporate until the passing gas reached the vapour saturation, we were able to predict the off-line particle–vapour distribution, as shown in Fig. 3 and 4.
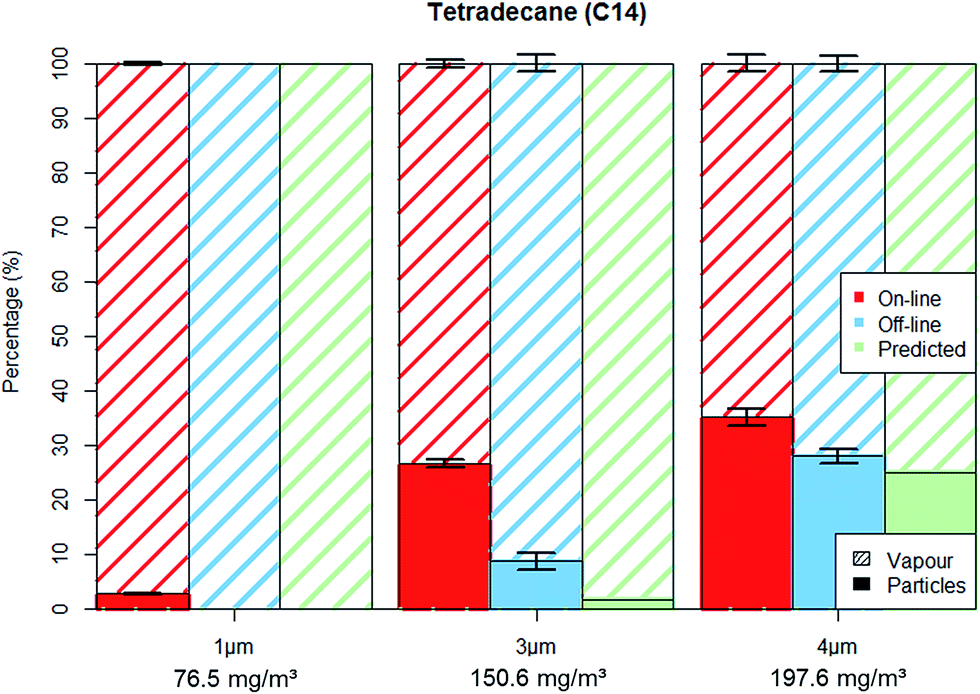 | ||
| Fig. 3 C14 aerosol fractionation between vapour and particle phase for on-line and off-line methods as well as the predicted off-line fractionation. Error bars represent one standard deviation. | ||
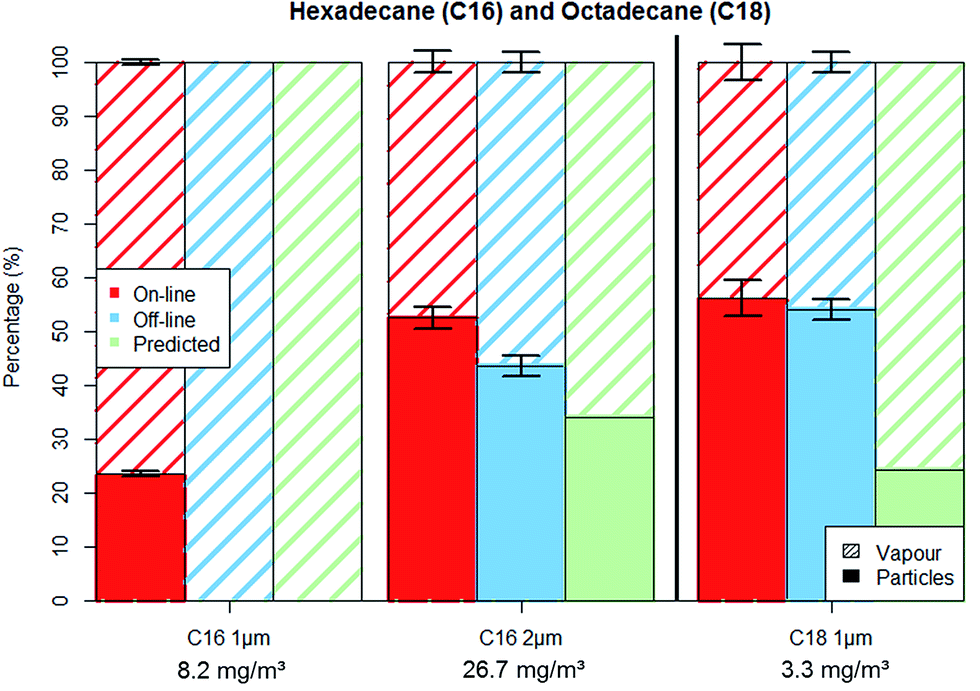 | ||
| Fig. 4 C16 and C18 aerosol fractionation between vapour and particle phase for on-line and off-line methods as well as the predicted off-line fractionation. Error bars represent one standard deviation. | ||
Tetradecane experiments were carried out on three particle sizes: 1 μm; 3 μm; 4 μm with number concentrations of about 4000 particles per cm3 (±10%). The particle mass concentration for these experiments (as measured by the OPC) was 2; 40 and 70 mg m−3 respectively. We note that although the total hydrocarbon mass for tetradecane may seem high (ca. 75, 150 and 200 mg m−3) it is still below the German Occupational Exposure Limit (OEL) for C9–C15 n-alkanes of 600 mg m−3. As the TM did not vary significantly throughout the experiments (coefficient of variation < 5%) the results for each set of measurements (particle size dependent) were averaged. Fig. 3 shows the aerosols' fractionation between the particulate and vapour phase as determined by the on-line and off-line methods for the different particle sizes. Table 1 shows both methods being in good agreement of less than 7% deviation for the total hydrocarbon mass. Also shown in Fig. 3 and Table 1 is the “predicted” off-line particle–vapour fractionation under the assumption that particles will evaporate from filters until the passing gas flow is fully saturated with vapours.
| On-line | Off-line | Predicted Off-line | |
|---|---|---|---|
| C14 1 μm | 76.5 ± 0.3 | 71.7 ± 1.0 | 76.5 |
| C14 3 μm | 150.6 ± 6.7 | 152.7 ± 2.4 | 150.6 |
| C14 4 μm | 197.6 ± 5.1 | 203.2 ± 7.3 | 197.6 |
| C16 1 μm | 8.2 ± 0.0 | 8.5 ± 0.2 | 8.2 |
| C16 2 μm | 26.7 ± 0.5 | 28.3 ± 0.5 | 26.7 |
| C18 1 μm | 3.3 ± 0.1 | 3.2 ± 0.2 | 3.3 |
Contrary to the good agreement found for TM (Table 1), the on-line and off-line methods showed different values when analysing the PM and VM separately (Tables 2 and 3). As shown in Fig. 3, the on-line OPC-FID method reported considerably higher PM concentrations, indicating that the filter sampling method loses a substantial amount of PM due to evaporation in all experiments. In the case of C14 1 μm experiments the off-line method returned no PM at all, although about 2 mg m−3 were measured in parallel with the particle sizer. Differences between the methods were also found for higher PM loads. The C14 3 μm experiments indicate a transfer of about 28 mg m−3 from particulate to vapour phase. Also, a filter underestimation of 13 mg m−3 was observed for the 4 μm measurements, with the missing PM being found in the XAD 2 adsorber cartridge as VM. The predicted particle–vapour distribution was also accurate, as shown in Fig. 3, the difference between predicted and measured fractionation being less than 10%.
| On-line | Off-line | Predicted Off-line | |
|---|---|---|---|
| C14 1 μm | 2.0 ± 0.1 | 0 | 0 |
| C14 3 μm | 39.9 ± 0.9 | 13.2 ± 2.2 | 2.4 |
| C14 4 μm | 69.3 ± 1.6 | 56.8 ± 3.1 | 49.4 |
| C16 1 μm | 1.9 ± 0.0 | 0 | 0 |
| C16 2 μm | 14.0 ± 0.3 | 12.3 ± 0.7 | 9.1 |
| C18 1 μm | 1.9 ± 0.1 | 1.7 ± 0.1 | 0.8 |
| On-line | Off-line | Predicted Off-line | |
|---|---|---|---|
| C14 1 μm | 74.4 ± 0.4 | 71.7 ± 1.0 | 76.5 |
| C14 3 μm | 110.7 ± 5.9 | 139.5 ± 4.0 | 148.2 |
| C14 4 μm | 128.4 ± 6.1 | 146.4 ± 6.4 | 148.2 |
| C16 1 μm | 6.3 ± 0.1 | 8.5 ± 0.2 | 8.2 |
| C16 2 μm | 12.7 ± 0.8 | 16 ± 0.4 | 17.6 |
| C18 1 μm | 1.5 ± 0.2 | 1.5 ± 0.1 | 2.5 |
Hexadecane (C16) experiments were conducted for aerosols with PM loads of approx. 2 and 14 mg m−3 (for the particle sizes of 1 μm and 2 μm). Particle number concentration was kept constant at around 5000 particles per cm3 (±10%) and therefore the PM increased proportionally with the particles' diameter to the third power. Fig. 4 shows the aerosols' percental distribution between particulate and vapour phases.
The C16 1 μm experiments showed similarities to the C14 measurements, with approximately 2 mg m−3 (100%) of PM being lost from the filter and found as vapour in the adsorber cartridge. The 2 μm experiments show a relatively small loss of 2 mg m−3, (14%) compared to the 12 mg m−3 which remained on the filters during sampling. The 2 μm C16 aerosol analysed by on-line method reported more than 50% of the aerosol mass as PM while the off-line analysis reported slightly over 40%. As was the case for C14 measurements, the predicted particle–vapour fractionation is similar to the actual measurements.
The C14 and C16 “1 μm” measurements have shown that the particles will completely evaporate from the filter substrate if the particulate phase is present at a concentration (around 2 mg m−3) that is considerably lower than the vapour saturation concentration (148 and 17.5 mg m−3) as the evaporated particle mass is insufficient to affect the vapour saturation degree. As a result, even if particles are present in a workplace it is likely that the GGP samplers will only register vapours. On the other hand, PM can still be found on filters when the particle concentration is high enough to partially or completely saturate the carrier gas with vapour and decrease the evaporation flux as a consequence.
Octadecane (C18) experiments were conducted with a particle size of 1 μm and PM load of about 2 mg m−3. Fig. 4 shows that both on-line and off-line methods yield similar results for the particle–vapour fractionation. This shows that the octadecane particles did not evaporate from the filters even though the vapour saturation degree was below 60%.
Substance volatility proved to have an important impact concerning the exactness of the filter PM measurements. The “1 μm” experiments performed with similar PM and comparable vapour saturation degrees for all three test substances showed that C14 and C16 particles evaporated completely while C18 particle concentration remained nearly unchanged on the filter matrix. Our “1 μm” experiments show that having a “particle only” occupational exposure limit could be effective for substances with volatilities similar or lower than octadecane. However, the sampling of compounds with volatilities similar to those of hexadecane and tetradecane will lead to evaporation of particles and possibly to significant errors in risk assessment. The PM also influenced the accuracy of the off-line measurements; the difference between the on-line and off-line results got smaller with increasing PM.
A good agreement was found between the predicted and the actual particle–vapour distribution found on filters for most measurements. This shows that the assumption of particle evaporation from filters until vapour saturation of the passing gas is accurate for the case of steady-state sampling, where the incoming aerosol is constant. A substantial difference was however observed for the C18 measurements, where the experimental off-line data shows that vapour saturation was not reached. Sutter et al.41 noted in his study that if the PM sampled on filter is too small, the vapour saturation of the passing gas cannot be reached. We believe that this effect combined with the longer particle lifetimes of C18 particles observed in our previous work7 can explain the higher difference between prediction and actual measurement.
Evaporation of oils spiked on filters
To mimic the evaporation bias dependency on sampling flow rate, experiments were carried out using mineral oils spiked on the of GGP samplers' filters.The particle–vapour fractionation of the semi-volatile mineral oil was strongly affected by the volume of air passing the filter. With a low air flow rate passing through the samplers, the main portion of the spiked semi-volatile oil remained on the filters while at a high flow rate nearly 90% of the oil evaporated and was transferred to the XAD-2 adsorption cartridge (Fig. 5). Significant differences (p < 0.001) were observed between the initial spiked amount and the mass found after 2 h.
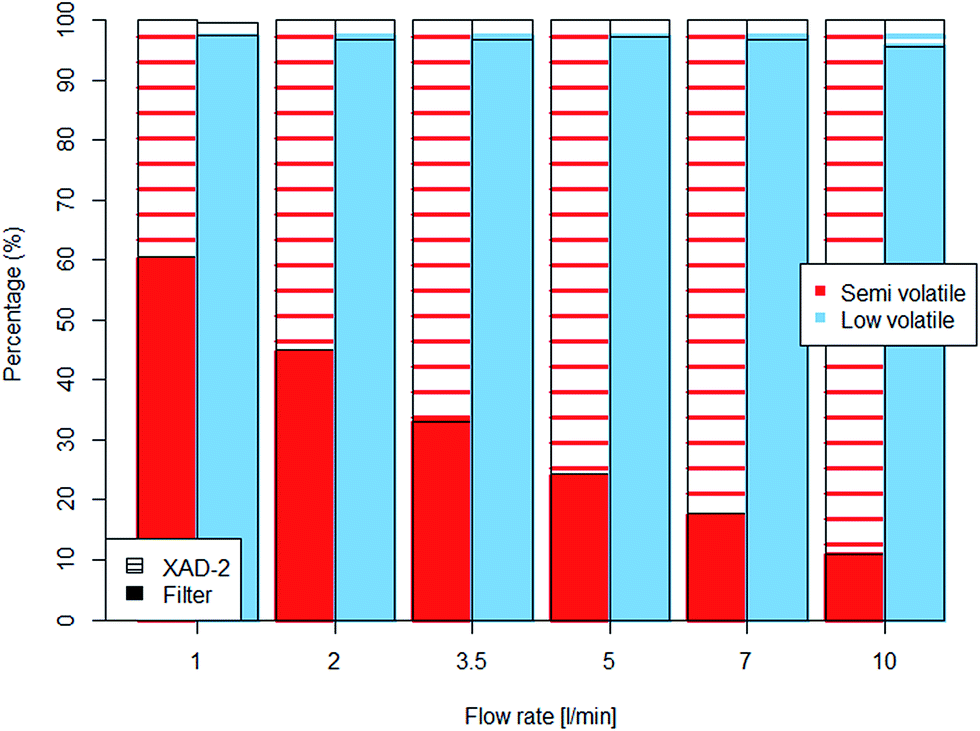 | ||
| Fig. 5 Percental mass distribution of the spiked mineral oils between filter and XAD-2 adsorber found after 2 hours of sampling with different flow rates. | ||
The low volatile mineral oil only evaporated slightly and the influence of the air flow rate was less clear, but nevertheless the portion of evaporated material doubled from 2% to 4% at higher air flow rates (Table 4). Although the evaporation doubled with the highest flow rate, the difference between the initial amount and the one found after 2 hours was not statistically significant.
| Flow rate | Semi volatile mixture | Low volatile mixture | ||||
|---|---|---|---|---|---|---|
| [l min−1] | Filter (liquid) [mg] | XAD-2 (vapour) [mg] | Total hydrocarbons [mg] | Filter (liquid) [mg] | XAD-2 (vapour) [mg] | Total hydrocarbons [mg] |
| 1 | 2.46 | 1.61 | 4.07 | 4.16 | 0.09 | 4.27 |
| 2 | 1.87 | 2.29 | 4.16 | 4.19 | 0.14 | 4.33 |
| 3.5 | 1.39 | 2.82 | 4.21 | 4.14 | 0.14 | 4.28 |
| 5 | 1.02 | 3.19 | 4.21 | 4.10 | 0.12 | 4.22 |
| 7 | 0.73 | 3.39 | 4.12 | 4.11 | 0.14 | 4.25 |
| 10 | 0.46 | 3.78 | 4.24 | 4.16 | 0.19 | 4.35 |
The spiking experiments have shown, analogue to the aerosol experiments, that semi-volatiles should be sampled by a combination of filter and adsorber. Filter sampling alone will underestimate the particulate phase due to evaporation. The measurements with the low-volatile oil showed similarities to the C18 aerosol measurements. In both cases almost no evaporation losses were observed.
Conclusions and recommendations
The results presented in this study have shown that the differences in observed phase distribution between the on-line and off-line methods are significant for compounds of high volatility. The total hydrocarbon mass was nonetheless similar for on-line and off-line results, independent of aerosol substance, mass load or size distribution. Beside the 1 μm droplet concentrations (2 mg m−3 as PM) that can be frequently found at workplaces we also compared the GGP filter-adsorber sampling to our reference method for higher concentrations. As it can be seen from our data, the larger particles (higher particle mass at constant number concentration) have enough mass to saturate the gas phase upon their evaporation and prevent the total vaporization of the PM from filters as a consequence. As a result, even volatile compounds like tetradecane can still be sampled on filters to some extent, though with sampling bias.Occupational exposure limits and sampling of “particles only” can turn out to be inappropriate for risk assessment as semi-volatiles can easily evaporate and cause an exposure underestimation. For instance, evaporation of aerosols with exposure limits for particles only may lead to an underestimation of health risks caused by the inhalation of the specified hazardous aerosol.
The authors strongly recommend that occupational exposure limits should be considered for the sum of particle and vapour mass concentration instead of either one of them separately since the TM is less affected by sampling bias. The Commission for the Investigation of Health Hazards of Chemical Compounds in the Work Area (MAK commission) in Germany already followed this recommendation and introduced a new chapter in the “List of MAK and BAT Values 2013”43 for semi-volatile substances and marked 58 substances that can be present as particulate/vapour mixtures at workplaces. The presented results were also considered during the development of the European standard EN 13936: workplace exposure – procedures for measuring a chemical agent present as a mixture of airborne particles and vapour – requirements and test methods.6
Acknowledgements
This study was financially supported by the German Social Accident Insurance (DGUV), research contract FP-299. Support from the Virtual Helmholtz Institute HICE – Aerosol and Health (Helmholtz Association, Berlin, Germany) is also acknowledged.References
- D. Breuer, J. Environ. Monit., 1999, 1, 299–305 RSC.
- A. Howe, D. Musgrove, D. Breuer, K. Gusbeth, A. Moritz, M. Demange, V. Oury, D. Rousset and M. Dorotte, J. Occup. Environ. Hyg., 2011, 8, 492–502 CrossRef CAS PubMed.
- D. Breuer, J.-U. Hahn, D. Hober, C. Emmel, U. Musanke, R. Ruhl, A. Spickenheuer, M. Raulf-Heimsoth, R. Bramer, A. Seidel, B. Schilling, E. Heinze, B. Kendzia, B. Marczynski, P. Welge, J. Angerer, T. Bruning and B. Pesch, Arch. Toxicol., 2011, 85(suppl 1), S11–S20 CrossRef PubMed.
- D. Breuer and A. Howe, J. Environ. Monit., 2006, 8, 120–126 RSC.
- D. Breuer, P. Heckmann, K. Gusbeth, G. Schwab, M. Blaskowitz and A. Moritz, J. Environ. Monit., 2012, 14, 440–445 RSC.
- European Committe for Standardisation, Editon edn, 2014.
- G. C. Dragan, E. Karg, H. O. Nordsieck, J. Schnelle-Kreis, D. Breuer, J. M. Arteaga-Salas, G. A. Ferron and R. Zimmermann, Environ. Eng. Manage. J., 2014, 13, 1793–1804 Search PubMed.
- K. Steinsvag, M. Bratveit and B. E. Moen, Ann. Occup. Hyg., 2006, 50, 109–122 CrossRef PubMed.
- K. S. Galea, A. Searl, A. Sánchez-Jiménez, T. Woldbæk, K. Halgard, S. Thorud, K. Steinsvåg, K. Krüger, L. MacCalman, J. W. Cherrie and M. van Tongeren, Ann. Occup. Hyg., 2011, 56, 1–9 Search PubMed.
- E. Menichini, Ann. Occup. Hyg., 1986, 30, 335–348 CrossRef CAS.
- C. Huynh and H. Herrera, J. Phys.: Conf. Ser., 2009, 151, 012040 CrossRef , 2008, 2–17.
- T. Robins, N. Seixas, A. Franzblau, L. Abrams, S. Minick, H. Burge and M. A. Schork, Am. J. Ind. Med., 1997, 31, 510–524 CrossRef CAS.
- D. K. Verma, D. S. Shaw, M. L. Shaw, J. A. Julian, S. A. McCollin and K. des Tombe, J. Occup. Environ. Hyg., 2006, 3, 53–66 CrossRef CAS PubMed.
- P. W. Wilsey, J. H. Vincent, M. J. Bishop, L. M. Brosseau and I. A. Greaves, Am. Ind. Hyg. Assoc. J., 1996, 57, 1149–1153 CrossRef CAS PubMed.
- W. A. Heitbrink, J. M. Yacher, G. J. Deye and A. B. Spencer, Am. Ind. Hyg. Assoc. J., 2000, 61, 275–281 CAS.
- G. M. Piacitelli, W. K. Sieber, D. M. O'Brien, R. T. Hughes, R. A. Glaser and J. D. Catalano, Am. Ind. Hyg. Assoc. J., 2001, 62, 356–370 CAS.
- M. Stear, Ann. Occup. Hyg., 2005, 49, 279–281 CrossRef PubMed.
- H. Wang, T. Reponen, S.-A. Lee, E. White and S. A. Grinshpun, J. Occup. Environ. Hyg., 2007, 4, 157–165 CrossRef CAS PubMed.
- G. M. Calvert, E. Ward, T. M. Schnorr and L. J. Fine, Am. J. Ind. Med., 1998, 33, 282–292 CrossRef CAS.
- P. Fischer, K. Hansen and D. Breuer, Appl. Occup. Environ. Hyg., 2003, 18, 226–231 CrossRef CAS PubMed.
- S. L. Gauthier, Appl. Occup. Environ. Hyg., 2003, 18, 818–824 CrossRef CAS PubMed.
- BBC, in BBC News (UK Edition) 1 October 2004. Available from URL http://news.bbc.co.uk/1/hi/england/west_midlands/3707234.stm, Editon edn, 2004.
- H. Cohen and E. M. White, J. Occup. Environ. Hyg., 2006, 3, 501–507 CrossRef CAS PubMed.
- A. Atmadi, D. A. Stephenson and S. Y. Liang, Int. J. Adv. Des. Manuf. Technol., 2001, 17, 238–243 CrossRef.
- D. Park, P. A. Stewart and J. B. Coble, Ann. Occup. Hyg., 2009, 53, 271–288 CrossRef CAS PubMed.
- L. Lillienberg, E. M. Andersson, B. Jarvholm and K. Toren, Ann. Occup. Hyg., 2010, 54, 403–411 CrossRef CAS PubMed.
- A. Simpson, J. A. Groves, J. Unwin and M. Piney, Ann. Occup. Hyg., 2000, 44, 165–172 CAS.
- E. Menichini, Ann. Occup. Hyg., 1986, 30, 349–363 CrossRef CAS.
- E. C. Gunderson and C. C. Anderson, Am. Ind. Hyg. Assoc. J., 1987, 48, 634–638 CrossRef CAS.
- A. Simpson and M. Wright, Ann. Occup. Hyg., 2008, 52, 249–257 CrossRef CAS PubMed.
- D. Breuer, C. Friedrich, C. Möhlmann and G. C. Dragan, Gefahrstoffe–Reinhalt. Luft, 2014, 74, 129–134 CAS.
- L. C. Kenny, R. Aitken, C. Chalmers, J. F. Fabriès, E. Gonzalez-Fernandez, H. Kromhout, G. Lidén, D. Mark, G. Riediger and V. Prodi, Ann. Occup. Hyg., 1997, 41, 135–153 CrossRef CAS.
- D. Leith, F. A. Leith and M. G. Boundy, Am. Ind. Hyg. Assoc. J., 1996, 57, 1137–1141 CrossRef CAS PubMed.
- K. Svendsen, O. Bjørseth and E. Børresen, Am. Ind. Hyg. Assoc. J., 1996, 57, 537–541 CrossRef CAS.
- J. Volckens, M. Boundy, D. Leith and D. Hands, Am. Ind. Hyg. Assoc. J., 1999, 60, 684–689 CrossRef.
- J. Volckens, M. Boundy and D. Leith, Appl. Occup. Environ. Hyg., 2000, 15, 370–379 CrossRef CAS PubMed.
- A. T. Simpson, Appl. Occup. Environ. Hyg., 2003, 18, 865–876 CrossRef CAS PubMed.
- A. Thorpe and P. T. Walsh, Ann. Occup. Hyg., 2013, 57, 824–841 CrossRef CAS PubMed.
- J. J. McAneny, D. Leith and M. G. Boundy, Appl. Occup. Environ. Hyg., 1995, 10, 783–787 CrossRef CAS.
- S. J. Cooper, P. C. Raynor and D. Leith, Appl. Occup. Environ. Hyg., 1996, 11, 1204–1211 CrossRef CAS.
- B. Sutter, D. Bémer, J.-C. Appert-Collin, D. Thomas and N. Midoux, Aerosol Sci. Technol., 2010, 44, 395–404 CrossRef CAS.
- P. Raynor and D. Leith, Ann. Occup. Hyg., 1999, 43, 181–192 CrossRef CAS.
- Commission for the Investigation of Health Hazards of Chemical Compounds in the Work Area, ed. A. Hartwig, Wiley-VCH Verlag GmbH & Co. KGaA, 2013, pp. I–XXIV Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |