
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel phenazine crystals enable direct electron transfer to methanogens in anaerobic digestion by redox potential modulation†
Sabrina
Beckmann
*a,
Cornelia
Welte‡
b,
Xiaomin
Li
a,
Yee M.
Oo§
a,
Lena
Kroeninger
b,
Yooun
Heo
c,
Miaomiao
Zhang
a,
Daniela
Ribeiro
a,
Matthew
Lee
a,
Mohan
Bhadbhade
d,
Christopher E.
Marjo
d,
Jan
Seidel
c,
Uwe
Deppenmeier
b and
Mike
Manefield
*ae
aSchool of Biotechnology and Biomolecular Science, University of New South Wales, High Street, 2052, Sydney, NSW, Australia. E-mail: s.beckmann@unsw.edu.au; manefield@unsw.edu.au
bUniversity of Bonn, Institute for Microbiology and Biotechnology, Meckenheimer Allee 168, 53115, Bonn, Germany
cSchool of Materials Science and Engineering, University of New South Wales, High Street, 2052, Sydney, NSW, Australia
dMark Wainwright Analytical Centre, University of New South Wales, High Street, 2052, Sydney, NSW, Australia
eUrban Water Systems Engineering, Technical University Munich, Am Coulombwall, Garching, 85748, Germany
First published on 16th December 2015
Abstract
With one billion tons of methane produced annually by microorganisms, biogas production can be appreciated both for its role in global organic matter turnover and as an energy source for humankind. The importance of electron transfer from electrically conductive surfaces or from bacteria to methanogenic Archaea has been implicated in widespread commercial anaerobic digestion processes, though a mechanism for reception of electrons from conductive surfaces or pili by methanogens has never been demonstrated. Here we describe a novel crystalline form of the synthetic phenazine neutral red that harvests electrons from reduced inorganic and organic microbial sources in anaerobic environments and makes them available to methanogenic Archaea. The novel crystalline form is so effective at harvesting reducing equivalents because it displays a potential for reduction 444 mV higher than the soluble form (E′ = 70 mV). Neutral red molecules solubilised in the reduced state by protonation at the point of methanogen cell contact with the crystal surface deliver electrons to methanogens at a negative midpoint potential (E′ = −375 mV). We demonstrate that soluble neutral red delivers reducing equivalents directly to the membrane bound HdrED heterodisulfide reductase of Methanosarcina, replenishing the CoM-SH and CoB-SH pool for methanogenesis and generating proton motive force. An order of magnitude increase in methane production is recorded in pure acetate fed Methanosarcina and coal and food waste fed mixed cultures in the laboratory. The phenomenon is also demonstrated at field scale in a sub-bituminous coal seam 80 m below ground level.
Broader contextBiogas production by methanogenic Archaea has a large role to play in meeting the energy needs and energy security of the human race into the future whilst reducing greenhouse gas emissions. Current operations tapping unconventional gas deposits generated by microbes in coal seams or harvesting methane from anaerobic waste digestion facilities are examples where the capacity to enhance biogas production is desirable. Recent progress in the field has revealed that methanogenic Archaea dependent on interspecies electron transfer are not limited to H2 or formate as a source of reducing equivalents but can access electrons directly from organic or inorganic surfaces and directly from bacteria. This conceptually broadens the scope of sources of energy in the environment from which biogenic methane can be produced. In this study we report a novel crystalline form of the synthetic phenazine neutral red that can harvest electrons from organic and inorganic electron carriers common in anaerobic environments and deliver them to acetoclastic methanogens. Application of neutral red crystals is shown to enhance methane production by an order of magnitude in anaerobic coal and food waste fed cultures and in a non-gassy coal seam 80 m below ground level. |
Introduction
Significant research effort is currently invested internationally to improve methane yields from the anaerobic digestion of renewable or non-renewable organic resources. Whilst large-scale, economically viable methanogenic treatment of renewable resources, such as wastewater, lignocellulosic crops and food waste, is widespread, additional gains in efficiency and expanded application are still anticipated.1–3 Effort has also been directed towards enhanced biogas production from non-renewable resources such as coal, but while extraction of biogenic coalbed methane is reducing international energy supply dependencies, only incremental improvements in real time biogas yields from coal have been observed.4Methane is a by-product of the means by which methanogenic Archaea generate proton motive force (PMF). Protons are extruded across the cytoplasmic membrane of methanogens by the heterodisulfide reductase enzyme.5 The energy for proton translocation is incumbent in the redox potential difference between reduced electron carriers in the membrane and the terminal electron accepting CoB-S-S-CoM heterodisulfide molecule. CoB-S-S-CoM is produced through reduction of methyl-S-CoM by the action of the methyl-S-CoM reductase, which releases methane.6
In acetoclastic methanogens such as Methanosaeta and Methanosarcina species, responsible for most methane production on Earth, the source of reducing equivalents for the heterodisulfide reductase enzyme is acetate, derived from fermentation of organic matter or from homoacetogenic bacteria. Acetate is converted to acetyl-CoA, which is cleaved to reduce ferredoxin, with the methyl moiety of acetate being converted to CH4via methyl-H4SPT and methyl-S-CoM, and the carboxyl group is ultimately released as CO2.7 Ferredoxin then delivers electrons directly or indirectly to membrane bound heterodisulfide reductase enzymes for reduction of CoB-S-S-CoM. In Methanosaeta species ferredoxin simply delivers electrons directly to the heterodisulfide reductase. In Methanosarcina species ferredoxin is used to produce H2 that is used to reduce a membrane bound phenazine that delivers electrons to the heterodisulfide reductase.6
Methanogenesis can also occur through the serial reduction of CO2 on C1 carriers converging on the acetoclastic pathway with production of methyl-H4SPT and methyl-S-CoM that is oxidised by CoB-SH to produce CoB-S-S-CoM. In this case, the reducing equivalents for reduction of CO2 and for the reduction of CoB-S-S-CoM by heterodisulfide come from H2 or formate, also derived from fermentation of organic matter.6
Recent convincing evidence has been presented suggesting that Methanosarcina species also capable of hydrogenotrophic methanogenesis and Methanosaeta species incapable of hydrogenotrophic methanogenesis can produce methane via CO2 reduction by exploiting direct electron transfer from elemental iron,8 iron oxides,9 activated carbon10 and other microorganisms.3 These exciting findings broaden the previously appreciated sources of reducing equivalents that could potentially be converted to methane, supplementing traditionally appreciated organic sources. For example, the experiments of Kato et al.9 suggest that energy stored in reduced iron oxides, which are enormously abundant in nature, could be used to generate methane. Scherson et al.11 also recently appreciated this general concept with a demonstration of energy recovery from waste nitrogen as nitrous oxide in wastewater treatment in addition to waste carbon as methane. Certainly, the possibility that other abundant inorganic sources of reducing equivalents in the environment (e.g. soluble and mineral sulphides) could be tapped for methane production is an intriguing one. Additional research is required to create adaptors enabling methanogens to access these reducing equivalents.
The direct biological conversion of electrical current to methane has also been examined extensively.12 In this context organic electron carriers or shuttles such as the synthetic phenazine neutral red have been exploited to transfer electrons from electrodes to microorganisms including methanogens. For example, Park et al.13 demonstrated that H2 could be replaced with neutral red in the cathode of a bioelectrochemical system to power methane production by a mixed species methanogenic inoculum. Beyond bioelectrochemical systems, neutral red and other electron shuttles have been examined extensively in biodegradation reactions for organochlorine dechlorination and azo dye reduction.14,15
Curiously, electron shuttles have never been tested for the ability to enhance methane production from renewable or non-renewable organic resources. Preliminary tests with cyanocobalamin, anthraquinone-2,6-disulfonate and neutral red, indicated that neutral red alone possessed properties that enhanced methane production in mixed species methanogenic cultures. Further testing with coal and food waste fed cultures revealed that under anaerobic physiological conditions neutral red assembles into a previously unobserved crystalline form that dramatically enhances biogas production.
Results and discussion
Neutral red crystal formation enhances biogas production
The addition of 250–500 μM neutral red (2-amino-8-dimethylamino-3-methylphenazine) to anaerobic coal and food waste fed microbial communities substantially (10 and 18 fold respectively) increased methane production over 10 months incubation (Fig. 1). Neutral red had no effect on methane production in the absence of cells, coal or food waste, as opposed to a proportionally reduced effect observed at a concentration of 100 μM indicating neutral red was not serving as an oxidisable substrate for fermentation and/or methanogenesis (expect 2.5 fold reduced methane production). Complete anaerobic digestion to methane of the 20 μmoles of neutral red added to the 80 ml cultures would yield 224 μmoles of methane in the 120 ml flasks. If neutral red was completely oxidised to methane, it would account for approximately half the methane generated by coal fed cultures and one percent of the methane observed in food waste fed cultures, supporting the assertion that the effect is catalytic.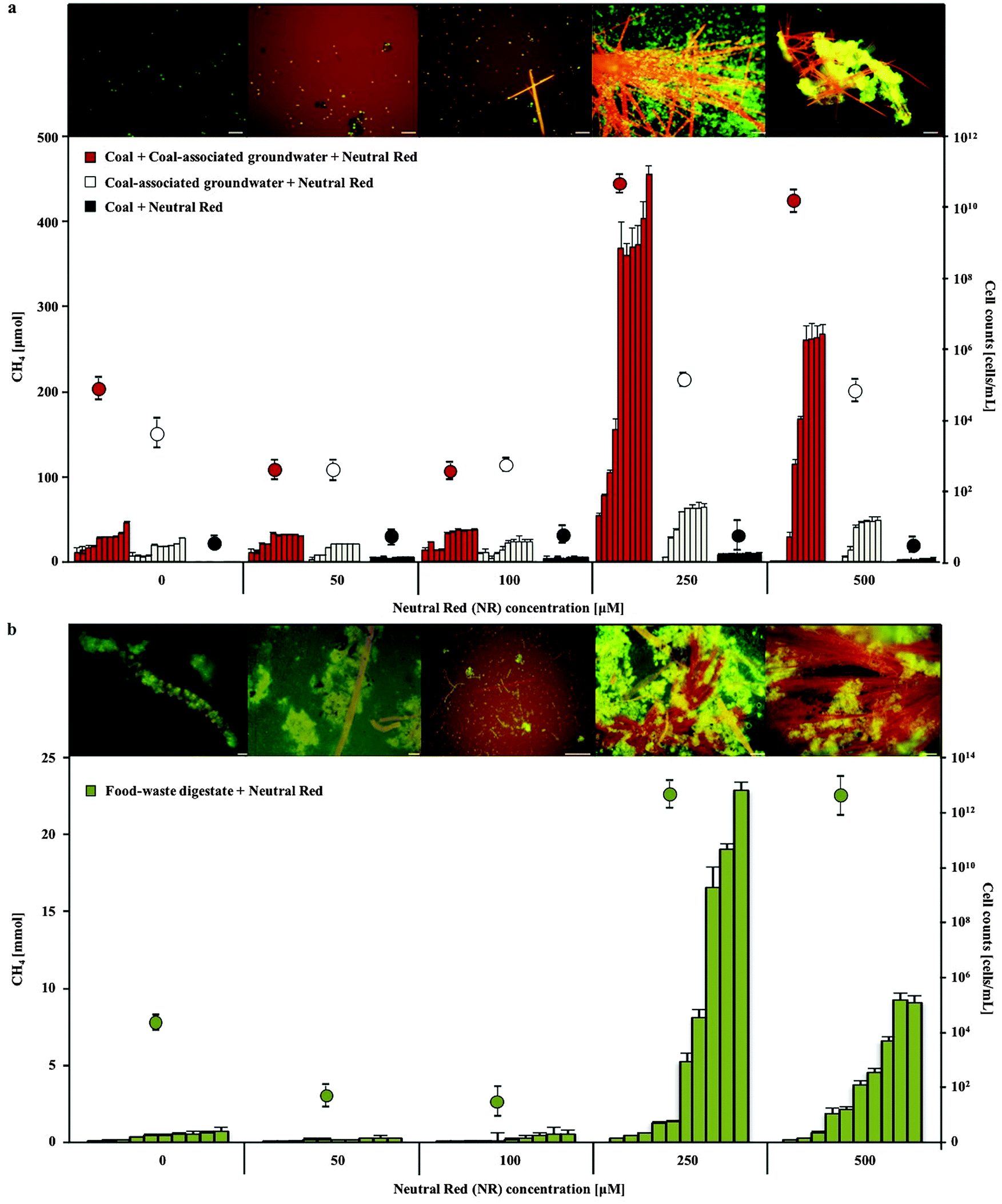 | ||
| Fig. 1 Methane production and cell concentration over time in 0–500 μM neutral red amended coal fed (a) and food waste fed (b) mixed species microbial cultures. The highest methane production (consecutive bars represent monthly readings) and cell proliferation (circles) were observed in 250 μM neutral red amended cultures, coinciding with the formation of neutral red crystals (orange) and microbial biomass aggregation (green). Scale bars 5 μM. Error bars represent standard deviation (n = 5). | ||
Enhanced biogas production in the presence of 250 μM neutral red was associated with the formation of insoluble needle like crystals (100–1500 μm length and 1–5 μm diameter) and rapid cell proliferation (Fig. 1 and 2a–c). The majority (>90%) of cells in the cultures adhered directly to the crystals. The average concentration of soluble neutral red after crystal formation was determined by spectroscopy to be ∼10 μM, with the remaining 240 μM neutral red in crystal formation (6.9 mg in 100 mL). An equilibrium exists between the soluble and crystalline form as determined by resuspension of crystals in fresh media, with dissolution to ∼10 μM again observed. Below 250 μM neutral red crystal formation was not observed (50 μM) or exceedingly rare (100 μM) and cell proliferation or methane production was not enhanced, indicating that the crystalline form rather than the soluble form of neutral red was responsible for enhanced biogas production. There was no evidence of neutral red biodegradation, with the crystals remaining intact throughout the 10 months incubation again suggesting their role is catalytic and enduring, serving as a concentrated environmental repository of electrons (macromolecular electronophore) that methanogens can interface with and use for metabolism and growth.
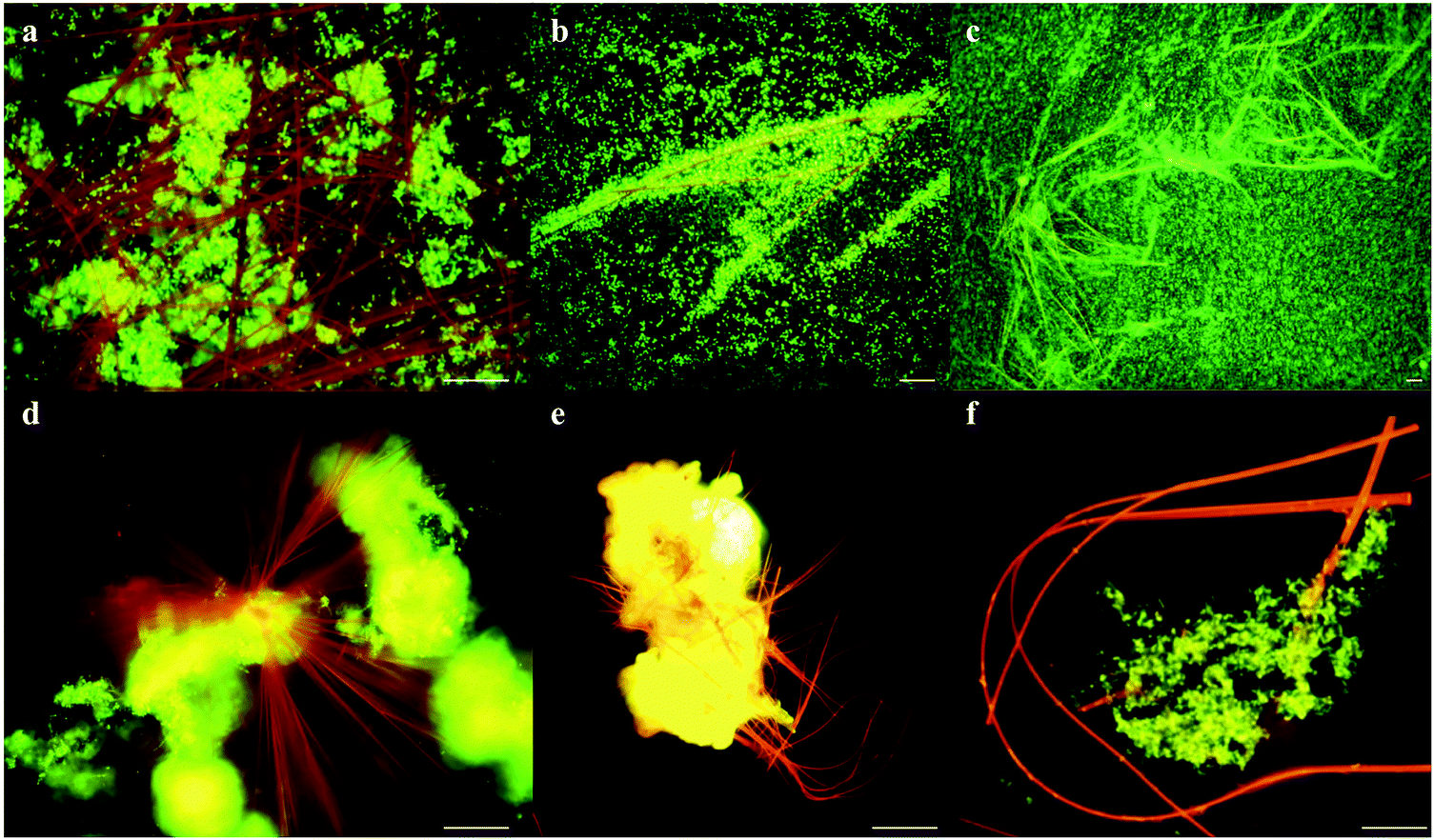 | ||
| Fig. 2 Formation of flexible needle like crystals. Fluorescence microscopy shows (a–c) rapid cell proliferation (green) around neutral red crystals (red-orange), (d and e) neutral red crystals nucleating from biomass (yellow-green) and (f) flexibility of neutral red crystals. Scale bars 5 μM. | ||
Neutral red crystal (NRC) characterisation
Neutral red crystal (NRC) assembly occurs in neutral to basic anaerobic solutions rich in particulate organic materials like coal and food waste, with NRCs often nucleating from the organic matter (Fig. 2d and e). Acidic pH (<4) and organic solvents (e.g. ethanol) immediately dissolve the crystals. At pH 9, an amorphous precipitate forms. Below pH 4, 99.8% of neutral red (pKa 6.7) is in the more water soluble protonated form whilst at neutral pH, 67% is in the less soluble non-protonated form, resulting in crystal formation. The structure of the newly discovered NRCs was determined after desiccation in an oxidative atmosphere, using synchrotron X-Ray crystallography (Fig. 3a). The NRC structure displays pi orbital stacking of the phenazine molecules in the oxidized state held together with long chains of hydrogen bonded water molecules.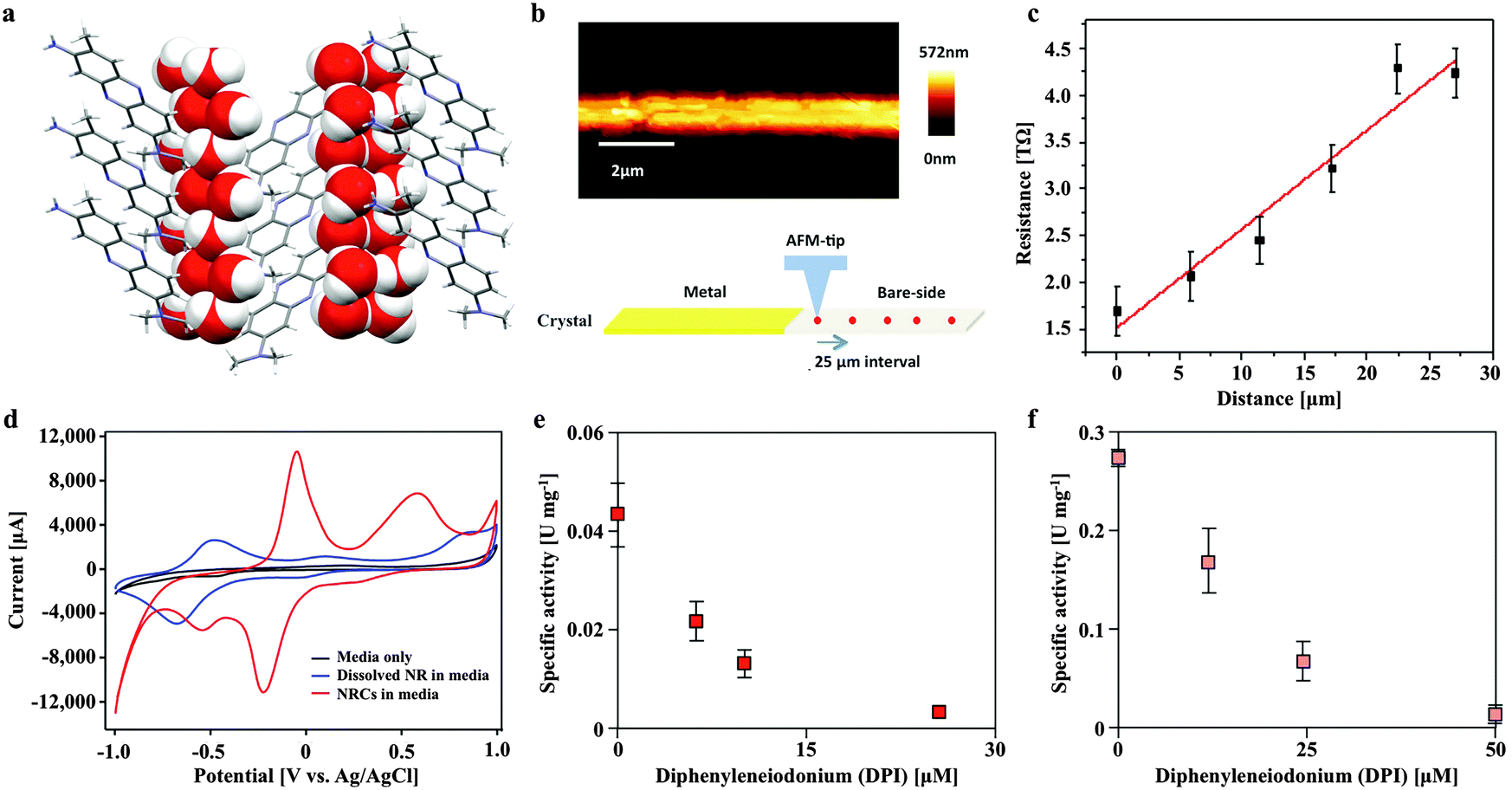 | ||
| Fig. 3 The novel neutral red crystal structure displays pi orbital stacking enabling long range electron transport and possesses a higher midpoint potential than the soluble form that delivers electrons to the membrane bound heterosulfide reductase of Methanosarcina mazei. (a) Neutral red crystal structure showing a stacked arrangement of neutral red molecules consisting of nitrogen containing heterocyclic rings (grey) held together by hydrogen bonding with long chains of water molecules (oxygen in red and hydrogen in white). (b) Topography of a single neutral red crystal by scanning probe microscopy. (c) Resistance as a function of distance along a single crystal. (d) Cyclic voltammogram of soluble (blue) and crystalline (red) neutral red in association with the working electrode in an electrochemical cell. (e) Neutral red reduction activity by Methanosarcina mazei membrane fractions in the presence of H2 and inhibition by a competitive inhibitor of methanophenazine-mediated reactions (diphenyleneiodonium chloride). (f) Neutral red oxidation activity by Methanosarcina mazei membrane fractions in the presence of the CoM-S-S-CoB heterodisulfide and inhibition by diphenyleneiodonium chloride. | ||
Other observations of electron transfer from solid surfaces to methanogens have been dependent on material conductivity (Fig. S1, ESI†). To test if the crystals were conductive Scanning Probe Microscopy based resistivity measurements were carried out, indicating that the structure is only very weakly semi-conductive, several orders of magnitude lower than activated carbon,10 conductive iron oxides9 or pili.3 The measured specific resistivity of micron sized NRCs was found to be ρ = 2.386 × 103 Ω m (Fig. 3b and c). This suggests NRCs enhance methanogen growth and methane production by a mechanism distinct from that observed with conductive surfaces.
We also compared the electron transfer properties of NRCs with soluble neutral red by cyclic voltammetry. This revealed the crucial point that NRCs accept electrons more efficiently than the soluble electron transfer mediator (Fig. 3d). The measured midpoint potential (vs. H2) of soluble neutral red in culture media was −375 mV, close to E0′ literature value of −325 mV, whilst the crystalline form was 444 mV more positive at +69 mV with the oxidative and reductive peak revealing 4.2 and 8.2 fold higher current responses (CVs with multiple scan rates are presented in Fig. S2, ESI†). This suggests the crystalline form is more amenable to reduction than the soluble form with an enhanced ability to attract reducing equivalents from reduced organic or inorganic electron carriers in the environment. Fig. S3 (ESI†) shows reduction of neutral red crystals by soluble sulfide, iron sulfide and acetate fed activated sludge, confirming acceptance of both abiotic and biological sources of reducing equivalents.
Microbial community dynamics in response to neutral red crystals
Methanogenic Archaea belonging to the order Methanosarcinales were over-represented in the rapid biomass increase observed in cultures in response to NRC formation as determined by 16S rRNA gene amplicon sequencing (Fig. 4). Prominent and abundant cell-aggregates of Methanosarcinales, directly attached to NRCs, were detected by Fluorescence In Situ Hybridization (FISH) using a Methanosarcinales specific probe (MSMX 860). Within the Methanosarcinales order known acetoclastic Methanosarcina spp. (up from 13 to 27% relative abundance) and Methanosaeta spp. (up from 4 to 12% relative abundance) were enriched in response to treatment with 250 μM neutral red after 10 months. Methermicoccus (from 1 to 45% relative abundance; fam. Methermicoccaceae) sequences were also enriched. To date, only one species of this genus, Methermicoccus shengliensis, has been described16 and whilst the isolate is strictly methylotrophic it is closely related to acetoclastic methanogens belonging to the family Methanosaetaceae and similar to Methanosaeta3 its genome contains all genes required for hydrogenotrophic methanogenesis (Gp0013046, DOE Joint Genome Institute). Methane production in coal fed groundwater cultures was partially inhibited by methyl fluoride,17 further suggesting neutral red stimulates acetoclastic methanogenesis (Fig. S4, ESI†).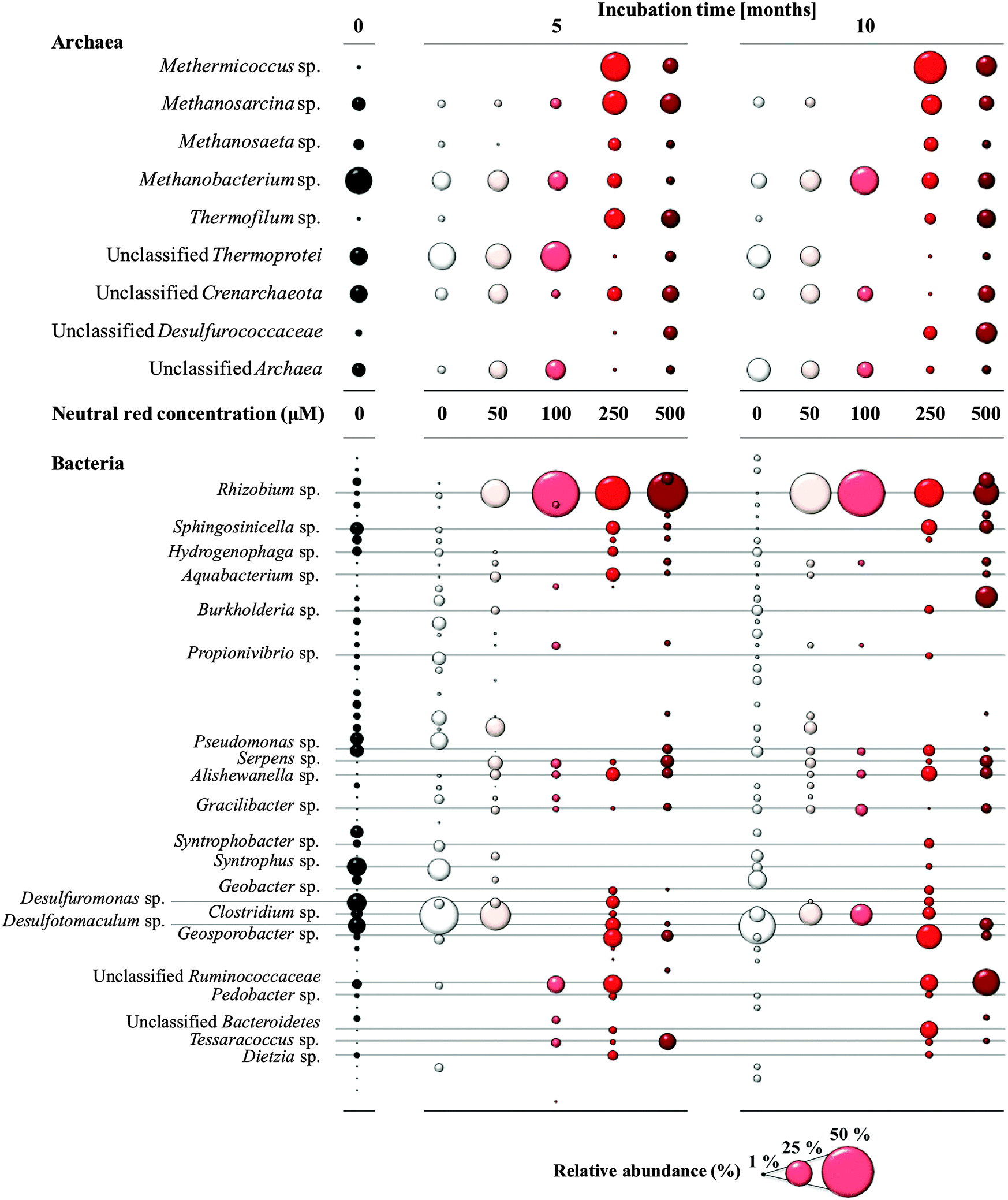 | ||
| Fig. 4 Changes in the archaeal and bacterial community composition in response to neutral red based on ribosomal RNA gene sequencing. Methanogenic Archaea of the order Methanosarcinales increased in relative abundance when the neutral red crystals were present (250–500 μM treatments). Bubble size reflects relative abundance. | ||
Neutral red also generated changes in the bacterial community along the 0–500 μM concentration gradient (Fig. 4) though the causes and consequences of the stimulatory or inhibitory effects are not known. Lineages of Rhizobium (up from 2 to 73%), Alishewanella (1 to 7%), Aquabacterium (0.5 to 7%), Clostridium (7 to 58%), Bacteroidetes (0.5 to 15%) and unclassified Ruminococcaceae (from 4 to 33%) were enriched. In contrast, some sulfate and sulfur reducing bacteria (SRB) such as Desulfuromonas and Desulfotomaculum sp. showed a decrease in relative abundance (down from 18 and 17% respectively to 0%). The observation that SRB were not favoured in the presence of neutral red is interesting in itself with SRB responsible for competition for reducing equivalents with methanogens, souring of hydrocarbon deposits and corrosion of municipal and industrial infrastructure through sulfide production.18,19
Neutral red enhances methane yield in acetate fed Methanosarcina mazei cultures
In pure culture incubations, methane production was enhanced by 250 μM neutral red in hydrogen free, carbonate buffered, acetate fed acetoclastic Methanosarcina mazei cultures (Fig. 5) but not in methanol fed methylotrophic Methanococcoides burtonii cultures where crystal formation was observed in both cases. In both pure cultures soluble sulfide was present chemically reducing the NRCs. In the M. mazei cultures the quantity of methane produced (∼2.52 ± 0.75 mmoles after 20 weeks) represented a five fold increase in methane output per cell (accounting for maintenance energy, the same quantity of methane can be produced by 2.7 fold fewer cells) and was approximately twice as high as the expected yield from a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stoichiometry for conversion of the methyl group of acetate to methane (∼1.10 ± 0.02 mmoles acetate consumed after 20 weeks). This indicates that neutral red enables reduction of the carbonyl group from acetate to methane. The lack of activity in cultures without acetate suggests neutral red does not act as a substrate for methanogenesis or enable reduction of CO2 to methane. We propose the shortfall in the electron balance is made up by reduced inorganic material in the media (e.g. sulfide) converted to accessible reducing equivalents by NRCs. This is possible with neutral red (E′ = −375 mV) acting as an electron donor for proton reduction to molecular hydrogen via Vho (E′ = −296 mV under H2 partial pressures in methanogenic environments20) or reduction of other unknown electron carriers (Fig. 6).6
:1 stoichiometry for conversion of the methyl group of acetate to methane (∼1.10 ± 0.02 mmoles acetate consumed after 20 weeks). This indicates that neutral red enables reduction of the carbonyl group from acetate to methane. The lack of activity in cultures without acetate suggests neutral red does not act as a substrate for methanogenesis or enable reduction of CO2 to methane. We propose the shortfall in the electron balance is made up by reduced inorganic material in the media (e.g. sulfide) converted to accessible reducing equivalents by NRCs. This is possible with neutral red (E′ = −375 mV) acting as an electron donor for proton reduction to molecular hydrogen via Vho (E′ = −296 mV under H2 partial pressures in methanogenic environments20) or reduction of other unknown electron carriers (Fig. 6).6
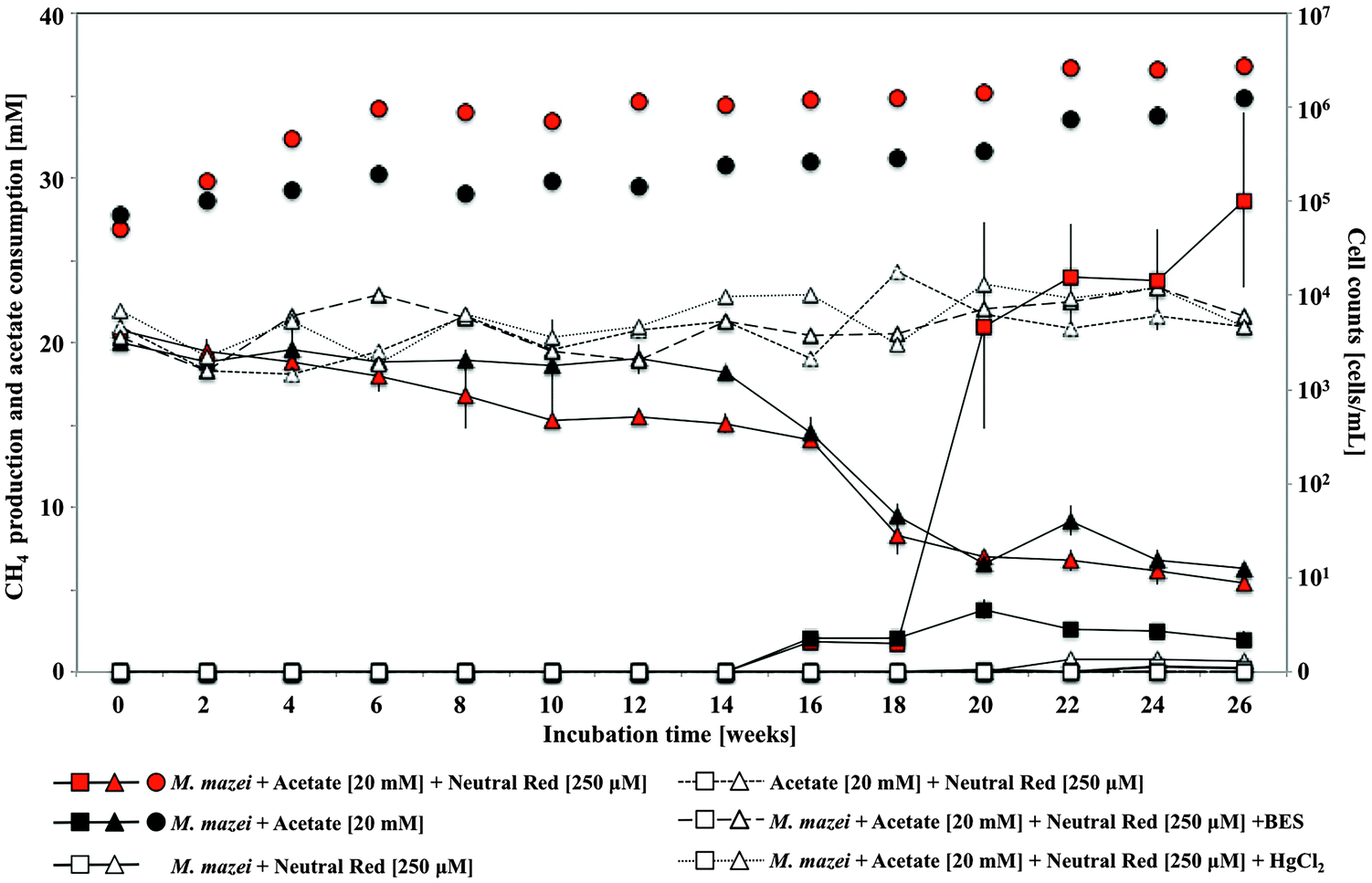 | ||
| Fig. 5 Methane production (squares), acetate consumption (triangles) and total cell counts (circles) by Methanosarcina mazei amended with 250 μM neutral red and acetate. In pure acetoclastic Methanosarcina mazei cultures methane production was enhanced by 250 μM neutral red (red square) compared to acetate fed cultures without neutral red amendment (black square). Error bars represent standard deviation (n = 5). | ||
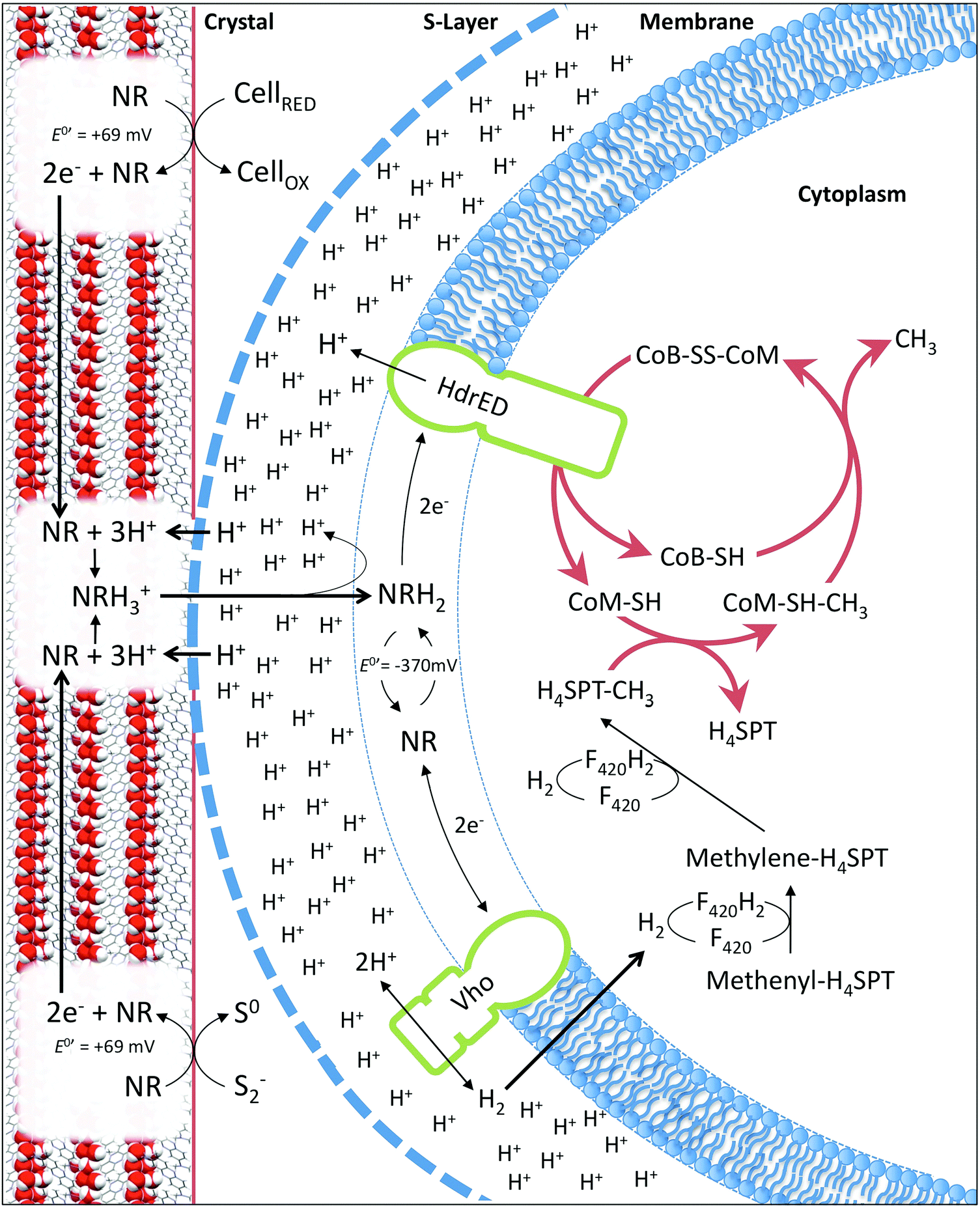 | ||
| Fig. 6 Diagram (not to scale) representing proposed model of neutral red crystal enhanced biological methane production based on Methanosarcina mazei. Crystals harvest electrons from reduced organic (cells) or inorganic (sulfide) material in anaerobic environments. Electrons relocate through the crystal lattice. At the point of contact between the methanogen S-layer and the crystal surface translocated protons solubilise reduced neutral red enabling entry into the cell membrane. Reduced neutral red delivers electrons to the heterodisulfide reductase (HdrED) in the membrane resulting in proton translocation and liberation of CoM-SH and CoB-SH enhancing the rate of methanogenesis. Reduced neutral red delivers electrons to the Vho dehydrogenase producing hydrogen enhancing methane yield via CO2 or CO reduction. | ||
Neutral red delivers electrons to the heterodisulfide reductase of Methanosarcina mazei
In Methanosarcina mazei electrons are transferred through the membrane from hydrogenases to the heterodisulfide reductase by the lipophilic electron shuttling metabolite methanophenazine. As neutral red and methanophenazine are structurally related phenazines (Fig. S5, ESI†) with similar mid-point potentials (E0′ −300 to −400 mV)21 we investigated whether neutral red could substitute methanophenazine in in vitro electron transport assays with isolated membrane fractions of the acetoclastic methanogen Methanosarcina mazei. The data revealed that neutral red in soluble form serves both as electron donor and acceptor to the membrane-bound respiratory chain of Methanosarcina mazei (Fig. 3e and f, Fig. S5, ESI†). In the presence of membrane fractions, neutral red was reduced when excess H2 was the electron donor (by Vho dehydrogenase) and oxidized when CoB-S-S-CoM heterodisulfide was the electron acceptor (by heterodisulfide reductase) with rates of 44 ± 8 and 303 ± 37 mU (mg membrane protein)−1, respectively. These rates are 40 and 10 fold lower respectively than the electron transport rates obtained with 2-hydroxy-phenazine, a water-soluble analogue of methanophenazine.22 Whilst these data suggest the activity of the heterodisulfide reductase is rate limiting for methanogenesis it is important to recognise that this may not be the only mechanism by which neutral red stimulates methane production. Electrochemically reduced neutral red has, for example, also been shown to reduce NAD+ to NADH and play the role of menaquinone in the fumarate reductase of Actinobacillus succinogenes.23 These or other unknown biochemical activities may influence methanogenesis.Some artificial electron carriers interact only with the peripheral subunits of electron-transporting membrane complexes and thus do not generate proton motive force. To investigate whether neutral red can occupy methanophenazine binding-sites, and therefore allow membrane complexes to undergo their complete catalytic cycle including proton translocation, we added the specific inhibitor diphenyleneiodonium (DPI) that competes with methanophenazine for enzyme binding sites in the in vitro assays.22 DPI inhibited both the hydrogen-dependent neutral red reduction and the heterodisulfide-dependent neutral red oxidation with IC50 values of 85 and 200 nmol DPI (mg membrane protein)−1, respectively, being in the same range as reported previously in experiments using 2-hydroxy-phenazine (60 and 125 nmol DPI per mg membrane protein) (Fig. 3e and f). Neutral red is therefore able to mimic the membrane integrated methanophenazine pool of Methanosarcina species, increasing rates of proton translocation and regeneration of the methanogenic cofactors CoM-SH and CoB-SH. The fact that neutral red serves as a substrate for the Vho dehydrogenase when H2 is supplied in excess supports the contention that at low H2 partial pressures (<100 kPa) neutral red could drive H2 production via this enzyme complex, enabling reduction of the carbonyl group from acetate to methane (Fig. 6).
In the membrane fraction experiments described, soluble neutral red was used because the concentrations of mediators typically applied are an order of magnitude lower than those required for crystal formation. It was not possible to test the crystalline form alone, because of its equilibrium with the soluble form. This raises the question of how neutral red molecules can interact with enzymes in cytoplasmic membranes whilst in crystalline form. The most parsimonious explanation is that neutral red molecules shuttle between the S-layer in direct contact with the crystal surface and the cytoplasmic methanogenic membrane (Fig. 6). We propose two mechanisms by which the state of equilibrium between soluble and crystalline neutral red at the surface of the crystals can be pushed towards the soluble form. The first involves the reduction of neutral red molecules at the surface of the crystals (2e− transfer to the azobenzene ring) at neutral pH resulting in protonation of the two nitrogen atoms.24 Additional protons cannot be accommodated spatially in the crystal structure described here, resulting in release of neutral red molecules. The second involves the elevated proton concentration associated with the proton motive force generated by the methanogen cell protonating the methylamine moiety of neutral red molecules at the surface of the crystal (pH < 6.3).25 Protonation of the methylamine moiety creates a positively charged molecule with altered bond lengths that would absolve it from the lattice. The oxidised form of neutral red generated through reduction of CoB-S-S-CoM via the heterodisulfide reductase is susceptible to protonation at a more neutral pH (6.3 < pH < 6.7),24 so oxidation of the crystal by soluble neutral red oxidised by the heterodisulfide reductase will further support production of a soluble pool of neutral red at the point of cell contact. Additionally, as shown experimentally, the reduction of the heterodisulfide reductase by neutral red bolsters the accumulation of protons in the extracellular space (Fig. 6). It is also plausible that unknown mechanisms involved in direct electron uptake by methanogens from activated carbon, iron oxides or bacteria are active.3,9,10
Neutral red addition to coal associated groundwater enhances biogas production in situ
Because neutral red can be delivered as a solution with ensuing crystal formation, it represents an attractive amendment for enhancing biogas production in a variety of applications. To examine the utility of the neutral red crystals in enhancing real time biogas production from a coal seam, 250 μM neutral red was introduced into coal seam associated groundwater in triplicate wells at an abandoned coalmine 80 m below ground level. NRCs were observed within a week of introducing the amendment and 5–10 fold increases in methane production were recorded over those obtained with nutrient amendments ((NH4)3PO4) over a 12 month period (Fig. 7). Enhanced biogas production occurred despite a background of 265 mg L−1 sulphate in the groundwater. Sulphate reduction rates were unaffected. Additionally, lag times for methane production were reduced and the longevity of methane production was increased. Ecotoxicology experiments have shown that the small quantities of neutral red remaining in solution after crystal formation are benign to sensitive ecological receptors.15 This field demonstration of enhanced biogas production in a non-gassy coal seam showcases the applicability of the technology at large scale in the subsurface environment.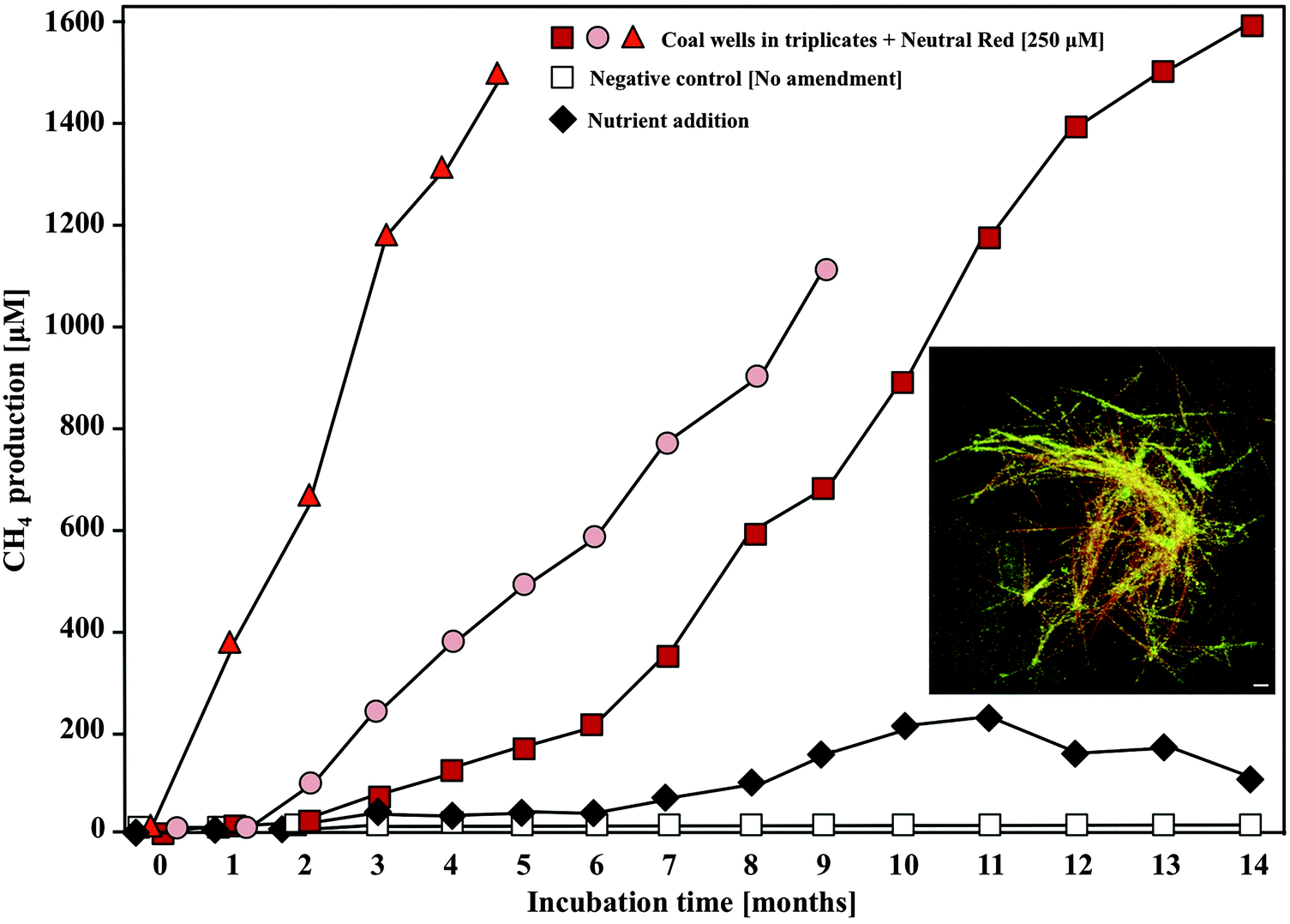 | ||
| Fig. 7 In situ methane formation from coal associated groundwater wells in response to 250 μM neutral red amendment. Increases in methane concentration in the headspace of the three neutral red amended wells were 5–10 fold faster than in a nutrient amended well (NH3 + PO43−). Inset image: Neutral red crystals (orange) formed in groundwater were rapidly colonised by microorganisms (green). Scale 5 μM. | ||
Conclusions
NRCs are novel crystalline structures with the ability to concentrate reducing equivalents from mineral and organic sources in the environment and make them available to methanogenic Archaea thereby enhancing biogas production by an order of magnitude. The process is demonstrated in mixed species microbial assemblages fed with renewable (food waste) and non-renewable (coal) reduced organic substrates and in acetate fed cultures of Methanosarcina mazei. A detailed molecular mechanism behind the phenomenon is presented along with a field scale demonstration in an abandoned coalmine. The crystals appear to stimulate methanogenesis not by virtue of conductivity but as a consequence of the equilibrium and the difference in redox potentials between the crystalline and soluble form of neutral red and its ability to mimic the methanogenic respiratory metabolite methanophenazine in interaction with the key respiratory enzyme heterodisulfide reductase. The discovery has immediate specific implications for unconventional gas production26 but more significantly demonstrates the possibility of constructing solid electrochemical adaptors for engineering the fate of reducing equivalents in microbial communities.Methods
Enrichment cultures
Coal-associated groundwater and coal were collected from a non-gassy subbituminous coal seam in NSW, Australia. Digested food-waste (digestate) was sampled from a food waste-to-energy facility (EarthPower Technologies, Sydney, Australia). Both sampling campaigns were conducted in 2013. Subsequent processing of all samples occurred under anaerobic conditions using an anaerobe chamber under N2 atmosphere and degassing techniques restricting the exposure of the samples to oxygen. Small coal pieces (1.5 cm3, 2.5 g total wet weight per flask) with 10 mL of coal-associated groundwater or 1 mL of digestate slurry were distributed into serum flasks containing 100 mL of sulfate-free mineral medium and a 40 mL N2 headspace sealed with silicon septa.27 Coal and food-waste cultures had a final pH of 7.5 and 5.5, respectively. Controls were supplemented either with 10 mM 2-bromoethanesulfonate to exclude abiotic degassing by inhibiting methanogenesis or sodium azide (NaN3) as an abiotic control. In neutral red amended cultures, a 250 mM neutral red (3-amino-7-dimethylamino-2-methylphenazine hydrochloride, C15H17ClN4, Sigma-Aldrich) solution was injected to final concentrations of 0, 50, 100, 250 and 500 μM neutral red. All incubations were carried out at least in triplicates.In situ application of neutral red
In triplicate, dissolved neutral red was added to coal associated groundwater wells in NSW, Australia to a final concentration of 250 μM. The wells open onto a 3 m thick non-gassy subbituminous coal seam 80 m below and contain 450 L of static groundwater (pH 5.5, 16 °C, 265 mg L−1 sulfate) with a 1000 L headspace and approximately 2 m2 of exposed coal surface area. Headspace gas was sampled via gas taps integrated into the gas tight wellhead using a gas-tight syringe.Methane analysis by gas chromatography
Methane concentrations were monitored monthly in 100 μl headspace samples by gas chromatography using a Shimadzu GC-2010 with flame ionization detection (GC-FID) and a GasPro PLOT column (60 m × 0.32 mm, Agilent Technologies, Australia).28Total cell counts, FISH and microscopic detection of Neutral Red Crystals (NRCs)
Culture slurries (1 mL) were taken monthly, fixed by the addition of glutaric dialdehyde (0.2 μm filtered, 2% final concentration) and stored at 4 °C in the dark. Fluorescence in situ hybridisation (FISH) was carried out using the general probe for Archaea Arch915 and the specific probe MSMX860 targeting the order Methanosarcinales.29,30 Samples (10 μL) were transferred to a microscope slide treated with a mounting medium (9.6% polyvinylalcohol 4-88 [moviol, Sigma Aldrich], 24% glycerol). Cell staining using SYBR Green I (Life Technologies), and cell counting was performed using a BX51WI epifluorescence microscope (Olympus).31X-ray crystallography
The initial structure the NRCs could be determined using a laboratory X-ray source and later using the MX2 beamline at the Australian Synchrotron to improve diffraction intensities and resolution. For X-ray crystallography at the laboratory source, single crystals were selected using a polarizing microscope (Leica M165Z), mounted on a MicroMount (MiTeGen, USA) that consists of a thin polymer tip with a wicking aperture. The X-ray diffraction measurements were carried out on a Bruker kappa-II CCD diffractometer at 150 K using an IμS Incoatec Microfocus Source with a Mo-Kα radiation (λ = 0.710723 Å). The single crystal, mounted on the goniometer using cryo loops for intensity measurements, was coated with paraffin oil and quickly transferred to the cold stream using an Oxford Cryo stream attachment. Symmetry related absorption corrections using the program SADABS were applied and the data were corrected for Lorentz and polarisation effects using Bruker APEX2 software. For X-ray diffraction measurements at the Synchrotron beam line MX2, a single crystal was mounted on the goniometer using cryo loop for diffraction measurements, coated with paraffin oil and then quickly transferred to the cold stream using cryo stream attachment. Si-111 monochromated synchrotron X-ray radiation (λ = 0.71023 Å) at 100(2) K was used to collect the data output and was corrected for the Lorentz and polarization effects by XDS software.32 The structure was solved subsequently to both analyses by direct-methods and the full-matrix least-squares refinement was carried out using SHELXL.33 The non-hydrogen atoms were refined anisotropically. The molecular graphic was generated using mercury.34DNA extraction and pyrotag sequencing
DNA was extracted from 2 mL of culture slurry after 0, 5 and 10 months of incubation using a modified phenol–chloroform extraction method.35 Deviations from this protocol were a change in the pH of the extraction buffer to 11. Subsequently, the DNA was precipitated using polyethylene glycol 6000 (Sigma-Aldrich) and the DNA pellet was washed once with 70% ethanol and was dissolved in 50 μL elution buffer (Quiagen). DNA was checked by standard agarose gel electrophoresis and was quantified fluorometrically using RiboGreen (Qubit Assay Kit, Invitrogen) according to the manufacturer's instructions. Extracted DNA was immediately used as target for 454 pyrosequening using a 454 GS FLX Titanium sequencer (University of Western Sydney (UWS), Penrith, Australia) and universal primers 926F (5′-AAA CTY AAA KGA ATT GRC GG-3′) and 1392R (5′-ACG GGC GGT GTG TRC-3′) targeting bacteria and archaea.36 Resulting sequences were checked and trimmed for quality and sequences shorter than 250 base pairs were discarded using MOTHUR. Chimeras were checked using the Uchime algorithm36 and remaining sequences were classified using the Ribosomal Database Project37 with 50% confidence interval and a screening for OTUs with total read abundances > 1 for all combined samples.Cyclic voltammetry measurements
A single chamber electrochemical cell containing three electrodes was used to characterize and compare the electro-activity of NRC modified electrodes by cyclic voltammetry (CV). The reference electrode (Ag/AgCl electrode in 1 M KCL, CH Instruments, Inc.), counter electrode (platinum wire, CH Instruments, Inc.) and working electrode (carbon felt, 30 mm × 30 mm × 5 mm, ALS Technologies, Australia) were connected to a potentiostat (EC-Lab software, Bio-Logic Instruments). The cell sealed with silicone rubber septa contained 250 mL of anaerobic Black-sea media as a negative control. The CV analysis of dissolved neutral red was carried out immediately after the addition of neutral red to a final concentration of 250 μM before crystal formation commenced. The CV measurement of NRCs was achieved by adding 250 μM neutral red to media and incubating for 3 weeks to allow complete crystal formation on the surface of the working electrode. A standard CV measurement was performed with a potential range of −1.0 V to +1.0 V (vs. Ag/AgCl) and back at a scan rate of 10 mV s−1 for 3 cycles. The current was recorded at each potential voltage point and the CV graphs were obtained by plotting the current against the cycling voltage from the last cycle.Conductive Atomic-Force Microscopy (c-AFM)
A scanning probe microscopy system (AIST-NT SmartSPM) was used to determine the topography of the NRCs and the conductive-AFM was used to measure NRC resistance. A conductive platinum coated tip (14 N m−1) was used for imaging and I–V curve analysis. The applied force through the tip during c-AFM measurements was 80 nN. The current and voltage characteristics were obtained by sweeping a bias range from −5 to +5 V (100 points, 2 passes and 30 ms per point). The NRCs were transferred onto an insulating glass plate and subsequently half-coated with gold. In order to measure conductivity, silver paste was used to laterally cover the whole sample composite from the bottom of the glass to the top of the gold surface. The I–V curve measurements were taken on the bare side of the NRC in steps of a few microns away from the gold-coated NRC end. The total resistance consists of the contact resistance between the tip and the sample and the lateral resistance that occurs from the current flow through the NRC.Neutral red membrane-bound electron transport
Membrane fractions of Methanosarcina mazei DSM 7222 were prepared as reported previously.5 Enzyme assays were conducted in N2 flushed rubber-stoppered glass cuvettes containing 1 mL potassium phosphate buffer (40 mM KH2PO4/K2HPO4, pH 7.0, 5 mM dithiothreitol, 1 μg mL−1 resazurin), 20 μM neutral red and 80 μg membrane protein. For hydrogenase activity measurement, the headspace was exchanged for a hydrogen atmosphere. For heterodisulfide reductase activity measurement, neutral red was first reduced with 15 nmol Ti(III)citrate, followed by addition of 20 nmol heterodisulfide. Enzyme activity measurements were followed at 530 nm (ε = 16.53 mM−1 cm−1) on a V-550 UV/Vis spectrophotometer (Jasco, Germany), 1 Unit of activity was defined as 1 μmol neutral red reduced/oxidized per minute.Acknowledgements
M. M. was supported by a Future Fellowship FT100100078 from the Australian Research Council and an August Wilhelm Scheer Visiting Professorship from the Technical University of Munich. S. B. and X. L. and project costs were supported by the Australian Research Council Linkage Project LP100100128, Discovery Project DP140100621 and industry partner Biogas Energy Pty Ltd. Y. M. O. was supported by a scholarship from industry partner Micronovo Pty Ltd.References
- M. Madsen, J. B. Holm-Nielsen and K. H. Esbensen, Renewable Sustainable Energy Rev., 2011, 15, 3141–3155 CrossRef CAS.
- R. Chandra, H. Takeuchi and T. Hasegawa, Renewable Sustainable Energy Rev., 2012, 16, 1462–1476 CrossRef CAS.
- A. E. Rotaru, P. M. Shrestha, F. Liu, M. Shrestha, D. Shrestha, M. Embree, K. Zengler, C. Wardman, K. P. Nevin and D. R. Lovley, Energy Environ. Sci., 2014, 7, 408–415 CAS.
- D. Strapoc, M. Mastalerz, K. Dawson, J. Macalady, A. V. Callaghan, B. Wawrik, C. Turich and M. Ashby, Annu. Rev. Earth Planet. Sci., 2011, 39, 617–656 CrossRef CAS.
- C. Welte and and U. Deppenmeier, Methods Enzymol., 2011, 494, 257–280 Search PubMed.
- C. Welte and U. Deppenmeier, Biochim. Biophys. Acta, 2014, 1837, 1130–1147 CrossRef CAS PubMed.
- P. J. Weimer and J. G. Zeikus, Arch. Microbiol., 1978, 119, 175–182 CrossRef CAS PubMed.
- H. T. Dinh, J. Kuever, M. Mussmann, A. W. Hassel, M. Stratmann and F. Widdel, Nature, 2004, 427, 829–832 CrossRef CAS PubMed.
- S. Kato, K. Hashimoto and K. Watanabe, Environ. Microbiol., 2012, 14, 1646–1654 CrossRef CAS PubMed.
- F. Liu, A. E. Rotaru, P. M. Shrestha, N. S. Malvankar, K. P. Nevin and D. R. Lovley, Energy Environ. Sci., 2012, 5, 8982–8989 CAS.
- Y. D. Scherson, G. F. Wells, S. G. Woo, J. Lee, J. Park, B. J. Cantwell and C. S. Criddle, Energy Environ. Sci., 2012, 6, 241–248 Search PubMed.
- S. Cheng, D. Xing, D. F. Call and B. E. Logan, Environ. Sci. Technol., 2009, 43, 3953–3958 CrossRef CAS PubMed.
- D. H. Park, M. Laivenieks, M. V. Guettler, M. K. Jain and J. G. Zeikus, Appl. Environ. Microbiol., 1999, 65, 2912–2917 CAS.
- K. Watanabe, M. Manefield, M. Lee and A. Kouzuma, Curr. Opin. Biotechnol., 2009, 20, 633–641 CrossRef CAS PubMed.
- F. Kastury, A. Juhasz, S. Beckmann and M. Manefield, Ecotoxicol. Environ. Saf., 2015, 122, 186–192 CrossRef CAS PubMed.
- L. Cheng, T. L. Qiu, X. B. Yin, X. L. Wu, G. Q. Hu, Y. Deng and H. Zhang, Int. J. Syst. Evol. Microbiol., 2007, 57, 2964–2969 CrossRef CAS PubMed.
- H. Penning and R. Conrad, Appl. Environ. Microbiol., 2006, 72, 178–184 CrossRef CAS PubMed.
- D. R. Lovley and M. J. Klug, Appl. Environ. Microbiol., 1983, 45, 187–192 CAS.
- O. J. Hao, J. M. Chen, L. Huang and R. L. Buglass, Crit. Rev. Environ. Sci. Technol., 1996, 26, 155–187 CrossRef CAS.
- W. Buckel and R. K. Thauer, Biochim. Biophys. Acta, 2013, 1827, 94–113 CrossRef CAS PubMed.
- M. Tietze, A. Beuchle, I. Lamla, N. Orth, M. Dehler, G. Greiner and U. Beifuss, ChemBioChem, 2003, 4, 333–335 CrossRef CAS PubMed.
- J. Brodersen, S. Baeumer, H. J. Abken, G. Gottschalk and U. Deppenmeier, Eur. J. Biochem., 1999, 259, 218–224 CrossRef CAS PubMed.
- D. H. Park and J. G. Zeikus, J. Bacteriol., 1999, 181, 2403–2410 CAS.
- A. A. Nixon, S. Berchmans and V. Yegnaraman, Bull. Electrochem., 1998, 14, 309–314 Search PubMed.
- M. W. Clark and M. E. Perkins, J. Am. Chem. Soc., 1932, 54, 1228–1248 CrossRef.
- S. Beckmann, M. Y. Oo, M. Lee and M. Manefield, Enhanced biogas production, NewSouth Innovations Pty Limited, Australia, 2014 Search PubMed.
- F. Widdel and F. Bak, in The Prokaryotes IV, ed. A. Balows, et al., Springer-Verlag, New York, 1992, vol. 183, pp. 3352–3378 Search PubMed.
- M. Lee, A. Low, O. Zemb, J. Koenig, A. Michaelsen and M. Manefield, Environ. Microbiol., 2012, 14, 883–894 CrossRef CAS PubMed.
- M. Krueger, S. Beckmann, B. Engelen, T. Thielemann, B. Cramer, A. Schippers and H. Cypionka, Geomicrobiol. J., 2008, 25, 315–321 CrossRef.
- V. J. Orphan, L. L. Jahnke, T. Embaye, K. A. Turk, A. Pernthaler, R. E. Summons and D. J. Des Marais, Geobiology, 2008, 6, 376–393 CrossRef CAS PubMed.
- M. Lunau, A. Lemke, K. Walther, W. Martens-Habbena and S. Meinhard, Environ. Microbiol., 2005, 7, 961–968 CrossRef PubMed.
- W. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. Van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466–470 CrossRef CAS.
- T. Lueders, M. Manefield and M. W. Friedrich, Environ. Microbiol., 2004, 6, 73–78 CrossRef CAS PubMed.
- T. Matsuki, K. Watanabe, J. Fujimoto, Y. Miyamoto, T. Takada, K. Matsumoto, H. Oyaizu and R. Tanaka, Appl. Environ. Microbiol., 2002, 68, 5445–5451 CrossRef CAS PubMed.
- P. D. Schloss, S. L. Westcott, T. Ryabin, J. R. Hall, M. Hartmann, E. B. Hollister, R. A. Lesniewski, B. B. Oakley, D. H. Parks, C. J. Robinson, J. W. Sahl, B. Stres, G. G. Thallinger, D. J. Van Horn and C. F. Weber, Appl. Environ. Microbiol., 2009, 75, 7537–7541 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1020368. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ee03085d |
| ‡ Present address: Radboud University Nijmegen, IWWR, Department of Microbiology, Heyendaalseweg 135, 6525 AJ, Nijmegen, The Netherlands. |
| § Present address: University of Southern Denmark, Department of Biology, Campusvej 55, 5230, Odense, Denmark. |
| This journal is © The Royal Society of Chemistry 2016 |