
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nonaqueous redox-flow batteries: organic solvents, supporting electrolytes, and redox pairs†
Ke
Gong
a,
Qianrong
Fang
a,
Shuang
Gu
*a,
Sam Fong Yau
Li
b and
Yushan
Yan
*a
aDepartment of Chemical and Biomolecular Engineering, Center for Catalytic Science and Technology, University of Delaware, Newark, DE 19716, USA. E-mail: shuang.gu@wichita.edu; yanys@udel.edu
bDepartment of Chemistry, National University of Singapore, Singapore 117543, Singapore
First published on 17th August 2015
Abstract
As members of the redox-flow battery (RFB) family, nonaqueous RFBs can offer a wide range of working temperature, high cell voltage, and potentially high energy density. These key features make nonaqueous RFBs an important complement of aqueous RFBs, broadening the spectrum of RFB applications. The development of nonaqueous RFBs is still at its early research stage and great challenges remain to be addressed before their successful use for practical applications. As such, it is essential to understand the major components in order to advance the nonaqueous RFB technology. In this perspective, three key major components of nonaqueous RFBs: organic solvents, supporting electrolytes, and redox pairs are selectively focused and discussed, with emphasis on providing an overview of those components and on highlighting the relationship between structure and properties. Urgent challenges are also discussed. To advance nonaqueous RFBs, the understanding of both components and systems is critically needed and it calls for inter-disciplinary collaborations across expertise including electrochemistry, organic chemistry, physical chemistry, cell design, and system engineering. In order to demonstrate the key features of nonaqueous RFBs, herein we also present an example of designing a 4.5 V ultrahigh-voltage nonaqueous RFB by combining a BP/BP˙− redox pair and an OFN˙+/OFN redox pair.
1. Introduction
The first redox-flow battery (RFB) was invented by Thaller in 1974.1 Unlike traditional rechargeable batteries, the energy-carrying redox pairs of RFBs are liberated from the solid electrodes into liquid electrolytes. The decoupling between energy storage and power delivery provides unprecedented design flexibility and scalability.2,3 Significant efforts and progress have been made in developing efficient and economical RFB systems in the last two decades,4–22 mostly driven by the need for addressing the intermittency of the fast growing renewable energy like wind and solar.The majority of RFBs are based on aqueous solutions as electrolytes, and aqueous RFBs have demonstrated high cell performance, low battery cost, and excellent system reliability. Similar to aqueous solutions, organic solvents can also be used to prepare electrolytes for RFBs. Since the first concept of nonaqueous RFBs was proposed by Singh in 1984,23 many types of nonaqueous RFBs have been invented and studied, which clearly validate the feasibility of using organic solvents for RFBs. Milestone examples for nonaqueous RFBs constructed with metal-based redox systems include: Ru(acac)3, Ru(bpy)3, or Fe(bpy)3 by Matsuda et al. in 1988;24 a series of U-ligand based redox pairs by Yamamura et al. in 2002;25 V(acac)3 by Thompson et al. in 200926 and Mn(acac)3 in 2011;27 Cr(acac)3 by Sleightholme et al. in 2010;28 Ni(bpy)3 by K. J. Kim and Y. J. Kim et al. in 2011;29 Co(acacen) by Li et al. in 2012;30 polyoxometalate by Anderson et al. in 201331 and V(mnt)3 in 2014.32
Regarding nonaqueous RFBs that are constructed by metal-free redox systems (or all-organic nonaqueous RFBs), Chakrabarti et al. reported rubrene based nonaqueous RFBs in 2007;33 Liu et al. reported 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO˙, “˙” stands for the radical, hereinafter) and N-methylphthalimide (NMPI) based nonaqueous RFBs in 2011;34 Jansen et al. introduced 2,5-di-tert-butyl-1,4-bis(2-methoxyethoxy)benzene (DBBB) and a variety of quinoxaline based nonaqueous RFBs in 2012;16 and Wang, Xu, and co-workers introduced anthraquinone redox systems into nonaqueous RFBs in 2012.18
In addition to the conventional all-soluble nonaqueous RFBs, the territory of nonaqueous RFBs is also extended by utilizing a solid metal (e.g., Li) as the negative electrode,18,35 or deploying a suspension electrode–electrolyte.36,37 Furthermore, the combination of the nonaqueous electrolyte and aqueous one opens up an important research direction, exemplified by the recent breakthroughs.14,15,38–49
Compared with aqueous RFBs, nonaqueous RFBs can offer a wide range of working temperature, high cell voltage, and potentially high energy density, thanks to the nature of organic solvents. As members of the RFB family, nonaqueous RFBs, especially those with the ability to work at low temperatures, are an important complement of aqueous RFBs, broadening the spectrum of RFB applications.
In spite of increasing interest received worldwide, the development of nonaqueous RFBs is still at its early research stage and great challenges remain to be addressed before their successful use for practical applications. As such, it is essential to understand the major components in order for us to advance nonaqueous RFB technologies. Cell types and structures of current nonaqueous RFBs have been summarized in an excellent review paper by Wang et al. in 2013.12 Membranes as well as cell performances of nonaqueous RFBs have been discussed in a comprehensive review paper by Moon et al., in the same year 2013.21 A comprehensive cost analysis comparing nonaqueous and aqueous RFBs has been presented by Darling, Gallagher, and their colleagues in their outstanding paper published in 2014.50 Note that lithium-ion batteries are also a class of competing nonaqueous batteries, and recent advances have been achieved and summarized in a recent book.51 In this perspective, the key features of nonaqueous RFBs are highlighted; and three key components of nonaqueous RFBs: organic solvents, supporting electrolytes, and redox pairs, are selectively focused and discussed, with emphasis on providing an overview of those components and on highlighting the relationship between structure and properties. In addition, two key urgent challenges are also discussed. At the end we also present an example of designing a 4.5 V ultrahigh-voltage nonaqueous RFB by combining a BP/BP˙− redox pair and an OFN˙+/OFN redox pair.
2. Features of nonaqueous RFBs
2.1. Wide working temperature range
The air temperature of about a half of the Earth's land can drop below 0 °C in winter (e.g., January at the northern hemisphere, 1959–1997, Fig. 1, from Global Climate Animations), and the lowest temperature recorded on the Earth – which was measured by the Russian Vostok Station on July 21, 1983 – was −89.2 °C in Antarctica. Low temperature could pose a significant challenge to the traditional aqueous RFBs since water solvent-based electrolytes tend to freeze at sufficiently low temperatures. By contrast, many organic solvents have low freezing points and appropriate boiling points. For example, tetrahydrofuran (THF) and 1-propanol have a freezing point of −108 °C and −126 °C, respectively, and these solvent based electrolytes would never freeze spontaneously on the Earth even in the coldest areas. There are many other solvents with different freezing points and distinctive properties available for nonaqueous RFBs (Table 1). | ||
| Fig. 1 Mean air temperature in January on the Earth's surface (1959–1997). Source of original modified image: Climate Lab Section of the Environmental Change Research Group, Department of Geography, University of Oregon – Global Climate Animations: Digital Library for Earth System Education (Reviewed Collection), Permission to use this image has been generously granted from Prof. J. J. Shinker. | ||
| Solvent |
T
f![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (°C) a (°C) |
T
bb (°C) |
d (g cm−3) | μ (mPa s) |
ε
re (—) |
LD50f (goral kgrat−1) |
p*g (kPa) |
E
redh (V vs. SHE) |
E
oxii (V vs. SHE) |
ECWj (V) |
|---|---|---|---|---|---|---|---|---|---|---|
| a T f: freezing point of pure solvent and data from ref. 52. b T b: boiling point of pure solvent and data from ref. 52. c d: density and data from ref. 52. d μ: viscosity and data from ref. 52. e ε r: relative permittivity and data from ref. 52. f LD50: median lethal dose (50%), data from Material Safety Data Sheets of Sigma-Aldrich. g p*: saturated vapour pressure at room temperature and data from ref. 52. h E red: limiting reduction potential (0.65 M Et4NBF4, 25 °C, glassy carbon, 5 mV s−1, and 1 mA cm−2 as threshold) and data from ref. 59. The potential is converted by SCE = 0.24 V vs. SHE. i E oxi: limiting oxidation potential (0.65 M Et4NBF4, 25 °C, glassy carbon, 5 mV s−1, and 1 mA cm−2 as threshold) and data from ref. 59. The potential is converted by SCE = 0.24 V vs. SHE. j ECW: electrochemical window (ECW = Eoxi − Ered). | ||||||||||
| N,N-Dimethylacetamide (DMAc) | −20 | 166 | 0.94 | 0.93 | 37.8 | 5.68 | 9.77 | |||
| N-Methyl-2-pyrrolidone (NMP) | −24 | 204 | 1.03 | 1.70 | 32.2 | 3.91 | 0.84 | |||
| Nitromethane (NM) | −29 | 101 | 1.13 | 0.61 | 36.7 | 0.94 | 4.88 | −1.0 | 2.9 | 3.9 |
| γ-Valerolactone (GVL) | −31 | 208 | 1.05 | 2.00 | 42.0 | 8.80 | 0.027 | −2.8 | 5.4 | 8.2 |
| Methoxyacetonitrile (MAN) | −35 | 120 | 0.96 | 0.70 | 36.0 | 0.98 | 2.50 | −2.5 | 3.2 | 5.7 |
| γ-Butyrolactone (GBL) | −43 | 204 | 1.13 | 1.73 | 39.1 | 1.54 | 0.43 | −2.8 | 5.4 | 8.2 |
| Acetonitrile (AN) | −44 | 82 | 0.79 | 0.34 | 35.9 | 6.69 | 11.81 | −2.6 | 3.5 | 6.1 |
| Trimethyl phosphate (TMP) | −46 | 197 | 1.07 | 2.20 | 21.0 | 0.84 | 0.13 | −2.7 | 3.7 | 6.4 |
| Propylene carbonate (PC) | −49 | 242 | 1.20 | 2.53 | 64.9 | 5.00 | 0.017 | −2.8 | 3.8 | 6.6 |
| 1,2-Butylene carbonate (BC) | −53 | 240 | 1.14 | 3.20 | 53.0 | 5.00 | 0.0056 | −2.8 | 4.4 | 7.2 |
| 3-Methoxypropionitrile (MPN) | −57 | 165 | 0.94 | 1.10 | 36.0 | 4.39 | 0.28 | −2.5 | 3.3 | 5.8 |
| N,N-Dimethylformamide (DMF) | −60 | 153 | 0.94 | 0.92 | 36.7 | 2.80 | 0.49 | |||
| Diglyme | −64 | 160 | 0.94 | 0.99 | 7.23 | 5.40 | 0.45 | |||
| 1,2-Dimethoxyethane (DME) | −69 | 85 | 0.86 | 0.46 | 7.20 | 5.37 | 6.38 | |||
| 4-Methyl-2-pentanone | −84 | 117 | 0.80 | 0.55 | 13.1 | 2.08 | 2.50 | |||
| Ethyl acetate (EA) | −84 | 77 | 0.89 | 0.43 | 6.02 | 5.62 | 12.57 | |||
| 2-Propanol | −88 | 82 | 0.78 | 2.04 | 19.9 | 5.05 | 5.76 | |||
| Nitroethane (NE) | −90 | 115 | 1.05 | 0.68 | 28.0 | 1.10 | 2.08 | −1.1 | 3.2 | 4.5 |
| Toluene | −95 | 111 | 0.86 | 0.55 | 2.38 | 5.58 | 3.79 | |||
| Hexane | −95 | 69 | 0.65 | 0.29 | 1.88 | 25.00 | 20.12 | |||
| Acetone | −95 | 56 | 0.78 | 0.30 | 20.6 | 5.80 | 30.72 | |||
| Dichloromethane (DCM) | −95 | 40 | 1.32 | 0.39 | 8.93 | 2.00 | 57.99 | |||
| Methanol (MeOH) | −98 | 65 | 0.79 | 0.55 | 32.7 | 1.98 | 16.89 | |||
| Tetrahydrofuran (THF) | −108 | 66 | 0.89 | 0.46 | 7.58 | 2.45 | 21.55 | |||
| Ethanol (EtOH) | −115 | 78 | 0.78 | 1.08 | 24.6 | 10.47 | 7.85 | |||
| 1-Propanol | −126 | 97 | 0.80 | 1.94 | 20.5 | 8.04 | 2.79 | |||
2.2. High cell voltage
Water solvent has an electrochemical window (ECW) of 1.23 V (25 °C, 100 kPa) and such a narrow ECW fundamentally limits the cell voltage of aqueous RFBs and the choice of RFB chemistries. By contrast, many organic solvents can offer an ECW of over 5 V. Examples include acetonitrile (AN) and γ-valerolactone (GVL) which have an ECW of 6.1 V and 8.2 V, respectively (Table 1). The wide range of ECWs allows for the use of redox pairs with very low redox potentials for the negative electrolyte and very high redox potentials for the positive electrolyte at the same time, offering high cell voltages without the concern of solvent breakdown. For example, in AN solvent with 0.5 M Et4NBF4 as the supporting electrolyte, the V(acac)3 based nonaqueous RFB a formal cell voltage of 2.2 V,26 and the Cr(acac)3 based counterpart does a formal cell voltage of 3.4 V.28 Since higher cell voltage leads proportionally to higher energy density and power density, nonaqueous RFBs in principle have the possibility to achieve high specific energy and/or high power density.2.3. Potentially high energy density
The solubility of redox compounds in aqueous electrolytes is generally low (around 1 M), except for some special redox pairs such as polybromide and polyiodide. The low solubility, together with low cell voltage, is the key cause for the generally low specific energy of aqueous RFBs, compared with other rechargeable battery technologies. The increase of solubility of redox compounds in aqueous electrolytes has been recognized as a major hurdle, largely because the water solvent substantially dictates the solubility. By contrast, there are many types of organic solvents available to work with redox compounds. Every solvent has a distinctive ability to dissolve certain compounds, and hence it is possible to find some solvents with sufficiently high solubility to construct RFBs with high energy density.3. Organic solvents
3.1. Freezing/boiling temperatures
Dissolving solutes usually depresses the freezing point and elevates the boiling point of a solvent, but the suppression and elevation are generally limited. For example, water and ethanol have a cryoscopic constant (Kf) of 1.86 and 1.99 K kgsolvent molsolute−1, respectively; and they have an ebullioscopic constant (Kb) of 0.51 and 1.19 K kgsolvent molsolute−1, respectively (data from http://www.vaxasoftware.com). These small cryoscopic constants (and ebullioscopic constants) indicate that the unit concentration of a solute can suppress (and elevate) only up to a few degrees for the freezing point (and boiling point) of the solvent, by the following equations (eqn (1) and (2)):| ΔTf = Kf·m·n | (1) |
| ΔTb = Kb·m·n | (2) |
Organic solvents can offer a very wide range of working temperatures, and there are a large number of solvents that have been used for electrochemical applications. The comprehensive book entitled “Electrochemistry in Nonaqueous Solutions” written by Izutsu is a must read for choosing nonaqueous solvents and understanding nonaqueous electrochemistry.52Table 1 lists some common organic solvents as possible choices for nonaqueous RFBs. Only organic solvents with a freezing point of −20 °C or below and a boiling point of 40 °C or above are included. Note that 40 °C is the highest average temperature in summer on the Earth surface (e.g., July at the northern hemisphere, 1959–1997). There are also other considerations (saturated vapour pressure, toxicity, and electrochemical window) for inclusion in Table 1.
3.2. Electrochemical window
Under the standard conditions (25 °C and 100 kPa), water solvent has well-defined ECWs with reversible reduction potentials of water (i.e., hydrogen evolution) of −0.83 and 0 V vs. SHE at pH = 14 and 0, respectively; and reversible oxidation potentials of water (i.e., oxygen evolution) of 0.40 and 1.23 V vs. SHE at pH = 14 and 0, respectively. However, the reversible reduction and oxidation potentials of organic solvents are neither practically useful nor easily obtainable. Instead, empirical methods are often used to define the ECWs of organic solvents and different criteria have been used for the evaluation of limiting reduction potentials (Ered) and limiting oxidation potentials (Eoxi). The Ered and Eoxi data of some organic solvents are from Ue's excellent work with a threshold current density of 1 mA cm−2 on a glassy carbon electrode at 25 °C (0.65 M Et4NFB4 as the supporting electrolyte and 5 mV s−1 as the scan rate, Table 1). Note that caution is needed to compare the ECWs among organic solvents because different criteria and different test conditions (such as electrode materials, supporting electrolytes, and the cyclic voltammetry method) may substantially impact both Ered and Eoxi. Regardless of the evaluation methods used, the oxidation is generally much higher for those organic solvents (2.9–5.4 V vs. SHE) than for water solvent (0.40–1.23 V vs. SHE depending on pH); at the same time the reduction potential is lower for the former than for the latter (Table 1). Electrochemically, organic solvents are much more stable than water solvent.3.3. Viscosity
The viscosity of organic solvents plays an important role in determining the ionic conductivity for organic solutions that contain supporting electrolytes. In general, lower solvent viscosity leads to higher ionic conductivity for a given supporting ion, governed by Stokes' law (eqn (3))| Λm = (z2·F2/π·NA)/(k·μ·r) | (3) |
Since the ionic conductivity of electrolytes controls the cell resistance and thus the voltage efficiency of RFBs, the viscosity of the organic solvent deserves careful consideration in designing nonaqueous RFBs. Table 2 shows the limiting ionic conductivity of some common supporting ions in four typical organic solvents with distinctive viscosities. Clearly, the lower the viscosity, the higher the ionic conductivity. In particular, AN and dichloromethane (DCM) solvents have a very low viscosity of 0.34 and 0.39 mPa s, respectively, and they usually offer very high limiting molar conductivity for supporting ions. In fact, AN has been the most often used organic solvent for nonaqueous electrochemistry studies and nonaqueous RFB applications as well. Water solvent is not an exception for Stokes' law. Water has a moderate viscosity of 0.89 mPa s, and thus water offers a moderate limiting molar conductivity for supporting ions. In addition to the impact on the limiting molar conductivity of supporting ions, the viscosity of organic solvents determines the pumping cost: higher viscosity brings higher pumping cost at a given flow rate of electrolyte.
| Supporting ion | r (nm) |
Λ
mb (S cm2 mol−1) |
E
redc (V vs. SHE) |
E
oxid (V vs. SHE) |
|||||
|---|---|---|---|---|---|---|---|---|---|
| PC (μ: 2.53 mPa s) | GBL (μ: 1.73 mPa s) | H2O (μ: 0.89 mPa s) | THF (μ: 0.46 mPa s) | AN (μ: 0.34 mPa s) | |||||
| a r: ionic radius and data from ref. 106. b Λ m: limiting molar conductivity at 25 °C, data in solvents PC and GBL from ref. 106, solvents EtOH and AN from ref. 52. c E red: limiting reduction potential, data from ref. 59 (BF4− as the supporting anion, 0.65 M, 25 °C, glassy carbon, 5 mV s−1, and 1 mA cm−2 as the threshold). The potential is converted by SCE = 0.24 V vs. SHE. Caution is needed for using the potential conversion. d E oxi: limiting oxidation potential, data from ref. 107 (Et4N+ as the supporting cation, 0.65 M, 25 °C, glassy carbon, 5 mV s−1, and 1 mA cm−2 as the threshold). The potential is converted by Li+/Li = −3.00 V vs. SHE. | |||||||||
| Anion | BF4− | 0.229 | 20.43 | 30.77 | 75.193 | 108.594 | 3.6 | ||
| ClO4− | 0.237 | 18.93 | 28.45 | 67.36 | 88.995 | 103.6 | 3.1 | ||
| PF6− | 0.254 | 17.86 | 26.70 | 65.593 | 77.596 | 102.897 | 3.8 | ||
| AsF6− | 0.260 | 17.58 | 25.92 | 32.498 | 100.197 | 3.8 | |||
| CF3SO3− | 0.270 | 16.89 | 24.93 | 96.3 | 3.0 | ||||
| (CF3SO2)2N− | 0.325 | 14.40 | 20.55 | 32.299 | 83.7299 | 3.3 | |||
| C4F9SO3− | 0.339 | 13.03 | 18.66 | 3.3 | |||||
| BPh4− | 0.419 | 8.52 | 11.67 | 19.8100 | 58.02 | 1.0 | |||
| Cation | Li+ | 0.076 | 8.43 | 13.99 | 38.68 | 69.9797 | −3.0 | ||
| Me4N+ | 0.283 | 14.50 | 21.52 | 44.9 | 94.52 | −2.9 | |||
| Et4N+ | 0.343 | 13.50 | 19.32 | 32.7 | 85.19 | −2.8 | |||
| Pr4N+ | 0.381 | 10.47101 | 23.45102 | 70.20103 | −2.8 | ||||
| Bu4N+ | 0.415 | 9.09 | 14.03 | 19.5 | 43.896 | 61.63 | −2.8 | ||
| Am4N+ | 0.467101 | 8.05101 | 17.13102 | 55.81104 | |||||
| Hex4N+ | 0.469105 | 6.14105 | 50.58104 | −2.9 | |||||
3.4. Relative permittivity
Another very important parameter to consider for an organic solvent is its relative permittivity (εr, or dielectric constant). The relative permittivity can impact not only the solubility of supporting electrolytes but also the dissociation constant of the supporting electrolyte when dissolved. Both solubility and dissociation constant can significantly influence the practical ionic conductivity of the organic solvent with the supporting electrolyte.The general trend is that higher relative permittivity of an organic solvent leads to higher solubility for supporting electrolytes (Table 3). Apparently, organic solvents with large relative permittivity (e.g., εr: 35.9 and 36.7 for AN and DMF, respectively) have higher solubility for supporting electrolytes than those with small relative permittivity (e.g., εr: 7.58 and 7.20 for THF and DME, respectively). PC, BC, and GVL have very high relative permittivity (εr: 64.9, 53.0, and 42.0, respectively) and thus they are expected to offer even higher solubility for supporting electrolytes. By contrast, the organic solvents with very low relative permittivity such as hexane and toluene (εr: 1.88 and 2.38, respectively) have very limited solubility for common supporting electrolytes. Even though they have very low viscosity (εr: 0.29 and 0.55 mPa s, respectively), their solutions have low ionic conductivity, limiting their use as nonaqueous solvents for electrochemical applications. In general, supporting electrolytes have lower solubility in organic solvents than in water since water has a much higher relative permittivity (78.39). For example, NaCl has a solubility of 5.4 M in water, which is substantially higher than those of organic supporting electrolytes in organic solvents. Note that other parameters (such as dipole moment, and acidity & basicity) also have substantial impacts on solubility and caution is needed in predicting solubility in organic solvents.
| Supporting electrolyte | S (mol L−1) |
σ
1Mc (mS cm−1) |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| DMF (εr: 36.7) | AN (εr: 35.9) | THF (εr: 7.58) | DME (εr: 7.20) | DMF (εr: 36.7; μ: 0.92 mPa s) | AN (εr: 35.9; μ: 0.34 mPa s) | THF (εr: 7.58; μ: 0.46 mPa s) | DME (εr: 7.20; μ: 0.46 mPa s) | ||
| a All data in this table are from ref. 52, unless otherwise noted. b S: solubility of the supporting electrolyte. c σ 1M: conductivity of the solution with 1 M supporting electrolyte. | |||||||||
| Bu4N+ salt | Bu4NBF4 | 2.34 | 2.21 | 2.02 | 1.70 | 14.5 | 32.3 | 2.7 | 4.4 |
| Bu4NClO4 | 2.29 | 2.05 | 1.48 | 1.10 | 12.059 | 27.059 | 2.7 | 3.2 | |
| Bu4NCF3SO3 | 2.25 | 2.50 | 2.35 | — | 10.9 | 23.3 | 3.3 | — | |
| Et4N+ salt | Et4NBF4 | 1.24 | 1.69 | <0.01 | <0.01 | 26.3 | 55.5 | — | — |
| Et4NClO4 | 1.00 | 1.13 | <0.01 | <0.01 | 24.059 | 50.059 | — | — | |
| Et4NCF3SO3 | 2.58 | 3.10 | 0.08 | — | 20.8 | 41.7 | — | — | |
Besides impacting the solubility of supporting electrolytes, the relative permittivity also dictates the dissociation constant (Kd) for supporting electrolytes. The relationship between Kd and εr can be described by the Denison–Ramsey equation (eqn (4))53
| −ln(Kd) = qe2/(ε0·εr·L·kB·T) | (4) |
Note that L could be far larger than the sum of radii of two ions of a salt, e.g., an L value of 16.4 nm was observed for Bu4NPF6 in fitting a group of organic solvents in a recent study54 and such a large L value is about 25 times that the sum of radii of Bu4N+ and PF6− (Table 2). The physical meaning of L has not been well understood so far.
Basically, the Denison–Ramsey equation shows the linear relationship between the natural logarithms of Kd of a given supporting electrolyte and the reciprocal of the relative permittivity of the solvent. Larger relative permittivity of organic solvent leads to larger dissociation constant for a given supporting electrolyte. For example, the dissociation constant of Bu4NClO4 drastically increases from 10−5.57 in THF (εr: 7.58) to 10−0.45 in PC (εr: 64.9) (Table 4). The dissociation constant of those supporting electrolytes in organic solvents is significantly lower than that in water. For example, NaCl has a large dissociation constant of 100.74 in water.55 Note that organic supporting electrolytes also have a similarly large dissociation constant (e.g., Kd: 100.78 for Bu4NI)56 in water, but their solubility is remarkably low, due to the strong hydrophobicity of hydrocarbon groups. The sharp comparison suggests that the small dissociation constant of supporting electrolytes in organic solvents is the root cause for the low conductivity of their solutions. This highlights the need for the development of high-relative permittivity organic solvents for electrochemical applications with respect to improving the conductivity for supporting electrolytes.
| Supporting electrolyte | log[Kd/(mol L−1)]a | |||||
|---|---|---|---|---|---|---|
| PC (εr: 64.9) | GBL (εr: 39.1) | AN (εr: 35.9) | DCM (εr: 8.93) | THF (εr: 7.58) | ||
| a K d: dissociation constant of the supporting electrolyte. The Kd data are converted from data of the association constant (Ka) by the equation Kd = 1/Ka. Ka data for solvents PC and GBL are from ref. 106. | ||||||
| Bu4N+ salt | Bu4NBF4 | −1.08108 | ||||
| Bu4NClO4 | −0.45 | −0.94 | −1.5896 | −4.5396 | −5.7196 | |
| Bu4NPF6 | −1.5596 | −3.1954 | −5.5796 | |||
| Bu4BPh4 | −0.38 | −0.80 | −1.04109 | −2.7154 | −3.6854 | |
| Et4N+ salt | Et4NBF4 | −0.48 | −1.00 | −4.69110 | ||
| Et4NClO4 | −0.49 | −0.98 | −4.66111 | |||
| Et4NPF6 | −0.30 | −0.89 | −4.61110 | |||
| Et4NCF3SO3 | −0.36 | −0.98 | ||||
| Et4N(CF3SO2)2N | −0.26 | −0.68 | −4.50110 | |||
3.5. Toxicity and other considerations
The toxicity of organic solvents is also an important consideration. The median lethal dose (LD50) is used to compare the toxicity of different organic solvents. In general, an LD50 value of 0.5–5.0 goral kgrat−1 is considered to be slightly toxic. Note that the organic solvents with LD50 values lower than 0.5 goral kgrat−1 are not included in Table 1, because of the strong toxic concerns. One should use low or less toxic solvents at all possible circumstances. In addition to LD50, other toxicity parameters should also be considered in selecting organic solvents.The saturated vapour pressure (p*) is also important in evaluating the toxicity of organic solvents. The lower the p* value is, the less the concern is over possible solvent intake when handling. Most of organic solvents are flammable and a low p* can also help reduce the fire risk.
Similar to viscosity, the density of the organic solvent affects the pumping cost. The density is also related to the specific energy: lower density leads to higher specific energy at a given cell voltage and capacity.
Organic solvents can be used in their pure form, but can also be used as mixtures for which possible combinations are almost unlimited. The engineering of these mixed solvents may provide unique properties that are not available from pure solvents for nonaqueous RFB applications as the solvent mixing has been practiced successfully in lithium-based rechargeable batteries.57
4. Supporting electrolytes
4.1. Overview
In general, pure solvents have extremely low ionic conductivity (Λs stands for the ionic conductivity of pure solvent). Pure water has a Λs of 6 × 10−8 S cm−1, and pure organic solvents also have extremely low ionic conductivities. For instance, pure PC and AN solvents have a Λs of 1 × 10−8 and 6 × 10−10 S cm−1, respectively.52 As such, a supporting electrolyte is necessary for organic solvents to provide sufficient ionic conductivity.A supporting electrolyte consists of the supporting cation(s) and the supporting anion(s), both of which contribute ionic conductivity to their solutions. Each supporting ion has a certain ability to offer ionic conductivity, evidenced by its distinctive limiting molar conductivity in a given solvent (Table 2). Following Stokes' law, a smaller ionic radius of supporting ions leads to higher limiting molar conductivity. Note that Stokes' law applies to most of the common supporting cations and anions, but it does not hold for small alkali metal cations (e.g., Li+) and halogen anions in terms of their ionic radii. The limiting molar conductivity of metal alkali cations and halogen anions is smaller than that predicted by Stokes' law, which is generally rationalized by the strong interaction between those ions and organic solvents. In addition, halogen anions are less electrochemically stable than the supporting anions shown in Table 2, and thus they are rarely used in nonaqueous electrochemistry applications.
4.2. Supporting anions
Tetrafluoroborate (BF4−), perchlorate (ClO4−), and hexafluorophosphate (PF6−) are the most often used supporting anions, since they have the smallest ionic radii (r: 0.229, 0.237, and 0.254 nm, respectively) and thus the highest limiting ionic conductivities (e.g., Λm: 108.5, 103.6, and 102.8 S cm2 mol−1, respectively, in AN, Table 2). The trend is the same for other solvents but the limiting molar conductivity is smaller due to the large solvent viscosity. Special consideration should be given to the use of ClO4− supporting anions, because most perchlorate salts are of explosion concern when heated or shocked; and thus the safer BF4− and PF6− anions are strongly recommended. For benchmark, the Cl− supporting anion in water has a comparable limiting molar conductivity (Λm: 76.35 S cm2 mol−1 in water). Note that some aqueous RFBs may use OH− as the working ion that has the highest limiting molar conductivity (Λm: 198.0 S cm2 mol−1 in water) among all known anions.The limiting oxidation potentials (Eoxi) of those common supporting anions overlap with those of some common organic solvents (e.g., Eoxi: 2.9–5.4 V vs. SHE, Table 1). In particular, the limiting oxidation potentials of PF6−, AsF6−, and BF4− are among the highest, e.g., Eoxi: 3.6–3.8 V vs. SHE in PC (Table 2). It should be noted that AsF6− salts are highly toxic and PF6− salts are strongly recommended as supporting anions when possible. In general, supporting anions lose electrons to form radicals when the electrode potential is higher than their limiting oxidation potentials and the radicals may further react with solvents. As such, the oxidation of supporting ions is irreversible (similar to solvent breakdown). The electrochemical window of a supporting electrolyte-containing organic solvent solution is determined by either supporting ions or solvents, whichever is limiting. The interaction between supporting ions and organic solvents may exist and the practical electrochemical window for their solution may differ.
4.3. Supporting cations
Tetraalkylammoniums are commonly used supporting cations, with Et4N+ and Bu4N+ being the two most popular cations. Similar to anions, a smaller ionic radius of supporting cations leads to higher limiting molar conductivity. In particular, Me4N+, Et4N+, and Pr4N+ cations have the smallest ionic radii (r: 0.283, 0.343, and 0.381 nm, respectively), and thus the highest limiting molar conductivities (e.g., Λm: 94.52, 85.19, and 70.20 S cm2 mol−1, respectively, in AN, Table 2). The molar limiting conductivity of supporting cations is lower than that of supporting anions due to their larger ionic radius; and both supporting cations and supporting anions have a similar molar limiting conductivity when they have close ionic radii (e.g., Λm: 61.63 vs. 58.02 S cm2 mol−1 for Bu4N+ of 0.415 nm vs. BPh4− of 0.419 nm, Table 2). Metal cations are also used as supporting cations sometimes since they have simple cation structure and good reductive stability. Although the Li+ cation has the smallest ionic radius (r: 0.076 nm), its molar limiting conductivity is only close to that of the Pr4N+ cation that has a much larger ionic radius (r: 0.381 nm). For benchmark in water, Na+ has a limiting molar conductivity of 50.10 S cm2 mol−1. Note that some aqueous RFBs (e.g., aqueous all-vanadium RFBs) can use H+ as the working ion that has the highest limiting molar conductivity: (Λm: 349.81 S cm2 mol−1) among all known cations (and anions as well).Unlike the supporting anions whose oxidative limiting potentials overlap with those of organic solvents, tetraalkylammonium supporting cations have a slightly lower limiting reduction potential (Ered) than organic solvents: e.g., Ered: from −2.8 V to −2.9 V vs. SHE in PC. Note that the limiting reduction potentials of tetraalkylammonium cations are very close to that of Li+ (Ered: −3.0 V vs. SHE in PC), suggesting excellent reductive stability. Opposite to supporting anions that can lose electrons, tetraalkylammonium cations can gain electrons to form radicals when the electrode potential is lower than their limiting reductive potentials. Combining supporting anions and supporting cations, supporting electrolytes overall can offer a much wide range of electrochemical windows, such as 6.6 V (Bu4NPF6 in PC).
Compared with lower tetraalkylammonium cations (such as Me4N+ and Et4N+), higher counterparts (e.g., Pr4N+ and Bu4N+) have higher solubility especially in organic solvents that have a low relative permittivity (e.g., THF and DME). For example, Et4NBF4 has a solubility of less than 0.01 M in both THF and DME, but the solubility of Bu4BF4 is up to 2.02 and 1.70 M in THF and DME, respectively. For extremely low-relative permittivity solvents, like benzene, Am4N+ (tetraamylammonium) and Hex4N+ (tetrahexylammonium) are needed to have a decent solubility as supporting electrolytes.
Owing to the similar cationic structure and close ionic radius, tetraalkylphosphonium cations are similar to tetraalkylammonium cations in many ways including conductivity and stability. For example, Bu4P+ and Bu4N+ have a similar limiting molar conductivity (Λm: 64.8658 and 61.63 S cm2 mol−1, respectively, in AN) and the same limiting reductive potential (Ered: −2.8 V vs. SHE in PC).59
4.4. Concentration and competition
In organic solvents, different solutes are found to compete with each other. For instance, the solubility of Et4NBF4 is strongly influenced by the concentration of the V(acac)3 redox compound in AN solvent.60 The higher the concentration of the V(acac)3 redox compound, the lower the solubility of Et4NBF4: the solubility of Et4NBF4 in AN decreases from 1.60 M without V(acac)3, to 1.10 M with 0.2 M V(acac)3, and further to 0.35 M with 0.5 M V(acac)3. This phenomenon can be well explained by the theory of partial molar volume for solutes. The effect of competing solubility must be considered when choosing the concentration of supporting electrolytes.Due to the strong interaction among ions at high concentration, the molar conductivity of supporting ions decreases with increasing concentration of supporting electrolytes (typically, via a cubic root of the relationship).61 On the other hand, increasing concentration of the supporting electrolyte will increase dissociated free supporting ions (depending on its Kd). The two competing trends on conductivity can lead to a critical concentration of the supporting electrolyte (if solubility is allowed) that reaches the highest ionic conductivity. For example, LiClO4 was observed to have a critical concentration of around 0.7 M in PC at 25 °C, giving the highest ionic conductivity of 54 mS cm−1.62 Such a small critical concentration is caused by the strong interaction between Li+ and solvent. For less-interacting ammonium cations, the critical concentration of the supporting electrolytes is much higher than Li+ salts. For example, MeEt3NBF4 was shown to have a critical concentration higher than 2 M in the same PC solvent.59 In the same work, Et4NBF4 showed a solubility of 1 M in PC, a value that has not reached the critical concentration yet.
5. Redox pairs
5.1. Overview
Two redox pairs are needed to construct an RFB cell, and the one with the lower redox potential serves as the negative redox pair and the other with the higher redox potential acts as the positive redox pair. When charging or discharging, electrical energy is stored into or released from the combination of both redox pairs via reductive–oxidative flipping of each pair simultaneously. The choice of redox pairs is crucial as it not only determines the cell voltage and electrode kinetics but also dictates the electrolyte cost and cell durability. There are two groups of redox pairs in nonaqueous RFBs: metal-based redox pairs and metal-free redox pairs.5.2. Metal-based redox pairs
The use of metal-based redox pairs in nonaqueous RFBs is a natural extension of their success in aqueous RFBs. Unlike the simple metal ions in aqueous RFBs, metal–organic ligand coordination complexes are used to construct metal-based redox pairs in nonaqueous RFBs, largely because of the need for improving its solubility in organic solvents. Simple metal salts have very limited solubility in most organic solvents, but metal–organic ligand complexes are reasonably soluble in many organic solvents.A metal–ligand complex is composed of a metal centre and several chelating ligands. The metals used to construct nonaqueous RFB pairs include Ru,24,63–65 Fe,24,29,63,64,66 U,25,67–72 V,26,32,60,73–75 Cr,28 Ni,29,66 Mn,27 and Co.30 Among those metals, V becomes increasingly attractive because it has good redox reversibility and moderate material cost. Ru also shows good redox reversibility but its cost is prohibitive. For example, the retail price of Ru(acac)3 is over 30 times higher than that of V(acac)3, i.e., $72.4 per g vs. $1.96 per g, from Sigma-Aldrich (with the same ligand and the same purity of 97%). With the aim of reusing the massive amount of depleted and recovered radioactive elements from the nuclear industry, Yamanura et al. has pioneered the use of U as redox pairs for nonaqueous RFBs. Fe, Cr, Mn, and Co are inexpensive and have the potential to drastically lower the materials cost for nonaqueous RFBs. However, these metals have worse redox reversibility than Ru and V.
Based on the atoms that are directly linked to the metal centre, the organic ligands used to construct redox pairs can be categorized into several groups: double-oxygen bidentate ligands, double-nitrogen bidentate ligands, double-sulfur bidentate ligands, and hybrid-atom (oxygen and nitrogen) bidentate ligands.
Double-oxygen bidentate ligands include “acac” (acetylacetone),25–28,60,63–65,69,72–75 “hfa” (hexafluroacetylacetone),25 “tfa” (1,1,1-trifluoroacetylacetone), “fod” (hexaflurobutanoylpivaloylmethane),25 “pta” (pivaloyltrifluoroacetone),25 “ba” (benzoylacetone), “dpm” (dipivaloylmethane),25 “btk” [m-bis(2,4-dioxo-1-pentyl)benzene],67,68,71,72 “etk” (8-oxo-2,4,12,14-acetylacetone),67,68,72 and “acacen” [bis(acetylacetone)ethylenediamine].30 Among those organic ligands, “acac” is the most frequently used one to construct metal–organic coordination complexes as redox pairs for nonaqueous RFBs, since it is one of the simplest bidentate ligands that form strong coordination bonds with many transition metals.
The double-nitrogen bidentate ligand used includes “bpy” (2,2′-bipyridine),24,29,63,64,66 and the double-sulfur bidentate ligand used includes “mnt” (maleonitriledithiolene);32 hybrid-atom bidentate ligand includes “tmma” (N,N,N′,N′-tetramethylmalonamide)70 where both the oxygen atom and the nitrogen atom serve as each dentate of the bidentate. In addition, there are monodentate ligands such as (single-oxygen) “dmso” (dimethyl sulfoxide) and (single-oxygen) “dmf” (dimethylformamide).25
For oxygen-dentate ligands (such as acac), the metal in metal–ligand complexes offers electron transfer via the change of its oxidation number upon redox reaction, in which ligands are redox-innocent. For nitrogen- (such as bpy) or sulphur-dentate ligands (such as mmt), both metals and ligands can provide electron transfer, in which ligands are redox-non-innocent. Combining different metals (with various oxidation numbers) and different ligands (with various substituents), metal–ligand complex redox pairs offer a wide range of redox potentials, shown in Table 5. Note that, the redox potential can be influenced substantially by both the organic solvent and the supporting electrolyte.76
| Redox paira |
φ′b (V vs. SHE) |
k
0c (cm s−1) |
Test conditiond |
|---|---|---|---|
| a Full names of the ligand abbreviations: “bpy” stands for 2,2′-bipyridine, “acac” for acetylacetone, “mnt” for maleonitriledithiolene, “dmso” for dimethyl sulfoxide, “hfa” for hexafluroacetylacetone, “tfa” for 1,1,1-trifluoroacetylacetone, “tmma” for N,N,N′,N′-tetramethylmalonamide, “fod” for hexaflurobutanoylpivaloylmethane, “pta” for pivaloyltrifluoroacetone, “ba” for benzoylacetone, “dpm” for dipivaloylmethane, “btk” for m-bis(2,4-dioxo-1-pentyl)benzene, “etk” for 8-oxo-2,4,12,14-acetylacetone, and “acacen” for bis(acetylacetone)ethylenediamine. b φ′: formal redox potential. Potential was converted to the SHE scale by relationships: Ag+/Ag = 0.54 V vs. SHE, SCE = 0.24 V vs. SHE, Li+/Li = −3.00 V vs. SHE, and Fc+/Fc = 0.69 V vs. SHE.76 Caution is needed for using the potential conversion. c k 0: standard rate constant of redox reaction. d Test conditions are AN as the solvent, 0.1 M Et4NBF4 as the supporting electrolyte, and GC as the electrode, unless otherwise noted. e i 0: exchange current density. | |||
| [Ru(bpy)3]3+/[Ru(bpy)3]2+ | 1.5 (from Ag+/Ag)24 | 3.4 × 10−3 (from i0e)112 | |
| [Fe(bpy)3]3+/[Fe(bpy)3]2+ | 1.3 (from SCE)112 | 1.3 × 10−2 (from i0)112 | |
| [Ru(acac)3]+/Ru(acac)3 | 1.2 (from SCE)112 | 4.6 × 10−2 (from i0)112 | |
| [Mn(acac)3]+/Mn(acac)3 | 1.2 (from Ag+/Ag)27 | 0.5 M Et4NBF4 | |
| [V(acac)3]+/V(acac)3 | 1.0 (from Ag+/Ag)26 | 6.5 × 10−4 (DMSO, 0.05 M Et4NPF6)74 | |
| [Cr(acac)3]+/Cr(acac)3 | 1.0 (from Ag+/Ag)28 | 0.5 M Et4NBF4 | |
| [V(mnt)3]−/[V(mnt)3]2− | 0.9 (from Fc+/Fc)32 | 0.1 M Bu4NPF6 | |
| [Co(acacen)]+/Co(acacen) | 0.3 (from Ag+/Ag)30 | 0.1 M Et4NPF6 | |
| Mn(acac)3/[Mn(acac)3]− | 0.1 (from Ag+/Ag)27 | 0.5 M Et4NBF4 | |
| [V(mnt)3]2−/[V(mnt)3]3− | −0.2 (from Fc+/Fc)32 | 0.1 M Bu4NPF6 | |
| [UO2(dmso)5]−/[UO2(dmso)5]2− | −0.3 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| [UO2(hfa)2]−/[UO2(hfa)2]2− | −0.3 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| [UO2(tfa)2]−/[UO2(tfa)2]2− | −0.3 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| [U(tmma)4]4+/[U(tmma)4]3+ | −0.4 (from Fc+/Fc)70 | 4.8 × 10−770 |
DMF, 0.1 Bu4NBPh4, Pt |
| Ru(acac)3/[Ru(acac)3]− | −0.5 (from SCE)112 | ||
| [UO2(fod)2]−/[UO2(fod)2]2− | −0.5 (from Fc+/Fc)25 | DMF, 0.1 M Bu4NClO4, Pt | |
| [UO2(pta)2]−/[UO2(pta)2]2− | −0.5 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| [SiV3W9O40]7−/[SiV3W9O40]10− | −0.5 (from Li+/Li)31 | PC, 0.5 M Bu4NOTf, GC | |
| [UO2(ba)2]−/[UO2(ba)2]2− | −0.7 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| UO2(acac)2/[UO2(acac)2]− | −0.8 (from Fc+/Fc)71 | 7.7 × 10−371 |
DMSO, 0.1 M Bu4NPF6, GC |
| [UO2(dpm)2]−/[UO2(dpm)2]2− | −0.8 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| UO2(btk)/[UO2(btk)]− | −0.8 (from Fc+/Fc)71 | 1.03 × 10−371 |
DMSO, 0.1 M Bu4NPF6, GC |
| UO2(etk)/[UO2(etk)]− | −0.8 (from Fc+/Fc)72 | 1.1 × 10−272 |
DMSO, 0.1 M Bu4NClO4, GC |
| [Ru(bpy)3]2+/[Ru(bpy)3]+ | −1.1 (from Ag+/Ag)24 | 2.0 × 10−1 (DMF, 0.2 M Bu4NClO4, Pt)113 | |
| [Fe(bpy)3]2+/[Fe(bpy)3]+ | −1.1 (from SCE)112 | 1.6 × 10−1 (DMF, 0.2 M Bu4NClO4, Pt)113 | |
| V(acac)3/[V(acac)3]− | −1.2 (from Ag+/Ag)26 | 8.7 × 10−4 (from i0)73 | 0.5 M Et4NBF4 |
| [Ni(bpy)3]2+/Ni(bpy)3 | −1.2 (from Ag+/Ag)66 | PC, 0.05 M Et4NBF4 | |
| U(pta)4/[U(pta)4]−(or U(pta)3) | −1.3 (from Fc+/Fc)25 | DMSO, 0.1 M Bu4NClO4, Pt | |
| [V(mnt)3]3−/[V(mnt)3]4− | −1.4 (from Fc+/Fc)32 | 0.1 M Bu4NPF6 | |
| Co(acacen)/[Co(acacen)]− | −1.7 (from Ag+/Ag)30 | 0.1 M Et4NPF6 | |
| U(btk)2/[U(btk)2]− | −1.8 (from Fc+/Fc)72 | 8.8 × 10−372 |
DMF, 0.1 M Bu4NClO4 |
| U(acac)4/[U(acac)4]−(or U(acac)3) | −1.8 (from Fc+/Fc)72 | 1.7 × 10−272 |
DMF, 0.1 M Bu4NClO4 |
| U(etk)2/[U(etk)2]− | −1.8 (from Fc+/Fc)72 | 1.5 × 10−272 |
DMF, 0.1 M Bu4NClO4 |
| Cr(acac)3/[Cr(acac)3]− | −1.8 (from Ag+/Ag)28 | 0.5 M Et4NBF4 | |
| [SiV3W9O40]10−/[SiV3W9O40]13− | −2.2 (from Li+/Li)31 | PC, 0.5 M Bu4NOTf | |
The properties of metal–ligand coordination complexes are the result of the interactions between metals and ligands. As such it is difficult to compare one group of ligands to the others. Staying within the same group of ligands, the impact of the ligand however can be revealed. For example, the increase of the basicity of the oxygen-dentate ligand (via introducing electron-withdrawing substituents) can lead to the negative shift for the redox potential of U–oxygen-dentate ligand complexes: an increase of eight units of pKa value for basicity of ligands leads to a negative shift of roughly 500 mV for redox potential of U(VI)/U(V) complex redox pairs. It is expected that the rate constant of redox reaction will also be impacted, but the correlation has not been elucidated yet.
Generally speaking, the redox kinetics of metal–ligand complex redox pairs in nonaqueous solvents is more facile than that of simple metal ion-based redox pairs in the aqueous system. As seen in Table 5, the standard rate constant is generally within the level of 10−3 to 10−1 cm s−1 for most of the metal–ligand complex redox pairs, which is statistically one to two orders of magnitude higher than those of aqueous metal ion-based redox pairs (a wide range of 10−6 to 10−2 cm s−1).8 This can be rationalized by the fact that the electron transfer in metal–ligand complexes does not involve the change of coordinating groups.77,78 Facile redox kinetics can lower the electrode overpotential, which is useful for achieving high voltage efficiency.
Besides the metal–ligand complexes, polyoxometalates are an emerging class of redox compounds to serve as metal-based redox pairs in nonaqueous RFBs.31 Polyoxometalates are polyatomic ions consisting of three or more transition metal oxyanions linked together by shared oxygen atoms to form a large, closed 3-dimensional framework. Two polyoxometalate redox pairs were introduced recently by Anderson et al. for both aqueous and nonaqueous RFBs: [SiV3W9O40]10−/[SiV3W9O40]13− with metal W being the electroactive element and [SiV3W9O40]7−/[SiV3W9O40]10− with metal V being the electroactive element.31 Both pairs offer up to three electrons of transfer upon redox reaction. With organic cations such as Bu4N+, these polyoxometalates are soluble in many organic solvents. Considering many structures available, polyoxometalates may be a promising class of metal-based redox systems to be explored for advancing nonaqueous RFBs.
5.3. Metal-free redox pairs
Redox pairs can also be constructed by metal-free organic redox compounds for nonaqueous RFBs (Table 6). Unlike the metal-based redox pairs, the electron transfer involves the formation of stable radicals. By gaining or losing electrons, neutral organic molecules can form radical anions or radical cations, respectively. In turn, the electron transfer between radical ions associated with the neutral molecule lead to certain redox potentials. For example, the first proposed metal-free redox pairs for nonaqueous RFBs:33 one pair constructed by neutral rubrene molecules and rubrene radical anions (i.e., rubrene/rubrene˙− with a redox potential of −1.9 V vs. SHE) and the other constructed by neutral rubrene molecules and rubrene radical cations (i.e., rubrene˙+/rubrene with a redox potential of 1.4 V vs. SHE).| Redox paira |
φ′b (V vs. SHE) |
k
0c (cm s−1) |
Test conditionsd |
|---|---|---|---|
| a Full names of the abbreviations: DBBB: 2,5-di-tert-butyl-1,4-bis(2-methoxyethoxy)benzene; TEMPO: 2,2,6,6-tetramethyl-1-piperidinyloxy; NMPI: N-methylphthalimide; 15D3GAQ: 1,5-bis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)anthracene-9,10-dione. b φ′: formal redox potential. Potential was converted to the SHE scale by relationships: Ag+/Ag = 0.54 V vs. SHE, and Li+/Li = −3.00 V vs. SHE. Caution is needed for using the potential conversion. c k 0: standard rate constant of redox reaction. d Test conditions are PC as the solvent, 0.2 M LiPF6 as the supporting electrolyte, and Pt as the electrode, unless otherwise noted. | |||
| Rubrene˙+/rubrene | 1.4 (from Ag wire)33 | AN:Tol, 0.05 TBAP, GC | |
| DBBB˙+/DBBB | 1.0 (from Li+/Li)16 | 1.0 × 10−2 (estimated) | |
| TEMPO+/TEMPO˙ | 0.9 (from Ag wire)34 | 1.0 × 10−1 (0.1 TBABF4, Pt)114 | AN, 1 M NaClO4, GC |
| Quinoxaline/quinoxaline˙− | 0.1 (from Li+/Li)16 | ||
| DPh-quinoxaline/DPh-quinoxaline˙− | 0.0 (from Li+/Li)16 | ||
| Me-quinoxaline/Me-quinoxaline˙− | −0.1 (from Li+/Li)16 | ||
| DMe-quinoxaline/DMe-quinoxaline˙− | −0.2 (from Li+/Li)16 | ||
| TMe-quinoxaline/TMe-quinoxaline˙− | −0.2 (from Li+/Li)16 | ||
| DPh-quinoxaline˙−/DPh-quinoxaline˙2− | −0.3 (from Li+/Li)16 | ||
| Quinoxaline˙−/quinoxaline˙2− | −0.4 (from Li+/Li)16 | ||
| Me-quinoxaline˙−/Me-quinoxaline˙2− | −0.4 (from Li+/Li)16 | ||
| 15D3GAQ˙−/15D3GAQ2− | −0.5 (from Li+/Li)18 | PC, 1 M LiPF6, GF | |
| DMe-quinoxaline˙−/DMe-quinoxaline˙2− | −0.5 (from Li+/Li)16 | ||
| TMe-quinoxaline˙−/TMe-quinoxaline˙2− | −0.5 (from Li+/Li)16 | ||
| 15D3GAQ/15D3GAQ˙− | −0.8 (from Li+/Li)18 | 1 M LiPF6, GF | |
| NMPI/NMPI˙− | −0.8 (from Ag wire)34 | 4.6 × 10−2 (0.1 TBABF4, Pt)115 | AN, 1 M NaClO4, GC |
| Rubrene/rubrene˙− | −0.9 (from Ag wire)33 | AN:Tol, 0.05 TBAP, GC | |
Most radicals are extremely reactive and thus short-lived, but there are many radicals that are relatively stable and sometimes persistent, and thus they could be used to construct metal-free redox pairs. From the structural point of view, the stabilization of radicals can be through electronic resonance, steric crowding, and/or dimer formation.79
Besides rubrene, other metal-free redox compounds include 2,2,6,6-tetramethyl-1-piperidinyloxyl (TEMPO˙, note this radical is neutral),34N-methylphthalimide (NMPI),34 1,5-bis(2-(2-(2-methoxyethoxy)ethoxy)ethoxy)anthracene-9,10-dione (15D3GAQ),18 2,5-di-tert-butyl-1,4-bis(2-methoxyethoxy)benzene (DBBB),16 and a quinoxaline family16 (Table 6). The TEMPO˙ neutral radical is a classic persistent radical molecule, whose stability is provided by both steric crowding from four methyl groups and electronic resonance. The NMPI˙+ cationic radical is stabilized through the electronic resonance with its electron-deficient benzene ring.34 The 15D3GAQ˙− anionic radical is likely stabilized by the electronic resonance across two benzene rings.80 The DBBB˙+ cationic radical is largely stabilized via the electronic resonance from the para-methoxy-benzene ring as an extended conjugation system (steric crowding is also provided by two t-butyl substituents).81 With different substituents, a group of quinoxaline redox compounds have been introduced and those quinoxaline anionic radicals are likely stabilized by their electronic resonance.
Considering the typical energy-storing time in RFBs being a few hours to up to a few days, the stability of radical-involving metal-free redox pairs may be sufficient. For example, up to 30 charge–discharge cycles have been demonstrated by a nonaqueous RFB based on a quinoxaline redox pair and a DBBB based redox pair, without substantially compromising either charge or discharge capacity.16 Such cyclability suggests a great feasibility of using radical redox compound for nonaqueous RFBs.
The substituent of redox compounds has impacts not only on the redox potential but also on the redox activity for redox pairs. For example, electron-donating substituents are shown to lower the redox potential but enhance the redox activity of the quinoxaline/quinoxaline˙− redox pair.16 Note that the influence of substituents is expected to be different from anionic radicals and cationic radicals. Meanwhile, the substituent has a significant impact on the solubility of redox compounds. For instance, with two substituents of 2-(2-(2-methoxyethoxy)ethoxy)ethoxy, 15D3GAQ has a solubility five times higher than that of the substituent-free anthraquinone (Aq) (more than 0.25 M for 15D3GAQ vs. less than 0.05 M for Aq, in solvent PC).18
Based on various redox radicals, metal-free redox pairs offer a wide range of redox potentials and a large rate constant, similar to metal-based redox pairs (Table 6). Like metal-based redox pairs, the electron transfer does not involve any bond formation or breakage, and hence the redox kinetics is very facile. Unlike the metal-based redox compounds whose solubility is generally limited (mostly less than 1 M), some metal-free redox compounds can offer very high solubility, with the example being substituent-free quinoxaline which has a solubility of 7 M in PC.16
Considering the huge number of stable radicals and the versatile modifications by substituents, a great deal of possible choices of organic compounds exists for constructing metal-free redox pairs for nonaqueous RFBs.
6. Urgent challenges
6.1. High internal resistance and low cell performance
A low internal resistance for an RFB cell is necessary to operate at high current density and to deliver high power density. However, the state-of-the-art nonaqueous RFBs have significantly high internal resistance, for which the low ionic conductivity of both the electrolyte and the membrane is primarily responsible.The ionic conductivity is substantially lower for organic electrolytes than for aqueous solutions. For example, 1 M Et4NBF4 in AN has an ionic conductivity of 55.5 mS cm−1 (Table 3), which is only 65%, 27%, and 14% that of 1 M NaCl (85.76 mS cm−1), 1 M KOH (209 mS cm−1), and 1 M H2SO4 solution (394.5 mS cm−1), respectively.82 Such a low ionic conductivity of 55.5 mS cm−1 is equivalent to a resistance of 1.8 Ω cm2 per 1 mm thickness of electrolyte and further a voltage loss of 180 mV per 100 mA cm−2 current density.
The ionic conductivity of commercial ion-exchange membranes is also significantly lower in nonaqueous solutions than in aqueous solutions. For example, the ionic conductivity of typical commercial anion-exchange membranes (AEMs) in nonaqueous solution is around 0.2–0.5 mS cm−1, e.g., 0.16 mS cm−1 for the Neosepta AHA membrane from Tokuyama Co. in 0.1 M Et4NBF4-containing AN solution,83 and 0.48 mS cm−1 for the FAP4 membrane obtained from Fuma-Tech in 1 M Et4NBF4-containing PC solution.84 By contrast, the ionic conductivity of commercial AEMs in water is around 15 mS cm−1 for Cl− and 40 mS cm−1 for OH−; and the ionic conductivity of commercial cation-exchange membranes in water is around 20 mS cm−1 for Na+ and 100 mS cm−1 for H+. A conductivity level of 0.5 mS cm−1 will lead to a membrane resistance of 2.0 Ω cm2 per 10 μm thickness of membrane and further a voltage loss of 200 mV per 10 μm thickness of membrane at 100 mA cm−2 of current density.
As a result, the overall resistance of current nonaqueous RFBs is prohibitively high, ranging from a few tens to a few hundreds of Ω cm2, which drastically limits the discharge current density and discharge power density to up to only a few mA cm−2 and only a few mW cm−2, respectively. Such a low current density and power density are two to three orders of magnitude lower than those of the state-of-the-art aqueous RFBs. Achieving low internal resistance and high cell performance is the most urgent need for developing nonaqueous RFBs with possible practical applications.
It is critical to lower the electrolyte resistance through advanced electrode design.8 Reducing the electrode thickness will directly lower electrolyte resistance inside the electrode. Implementing flow-through electrode and engineering of electrode structure can also help, via increasing the effective surface area and improving electrolyte transport. It is possible to design high-performance electrodes by taking advantage of the great success of electrode engineering in aqueous RFBs.
It is also crucial to lower membrane resistance in nonaqueous solutions through improving ionic conductivity and reducing membrane thickness, yet without compromising ionic selectivity and other important requirements such as mechanic strength and chemical stability. It must be pointed out that the current commercial IEMs are not designed for nonaqueous electrochemical applications, and therefore there is substantial room to improve membrane performance via preparing membranes specifically tailored for nonaqueous RFB applications. In our previous studies, multiple ion-exchange membrane configurations were developed which allowed flexible choices of redox pairs with mixed ion charges and different electrolytes to construct RFBs (Fig. 2).22
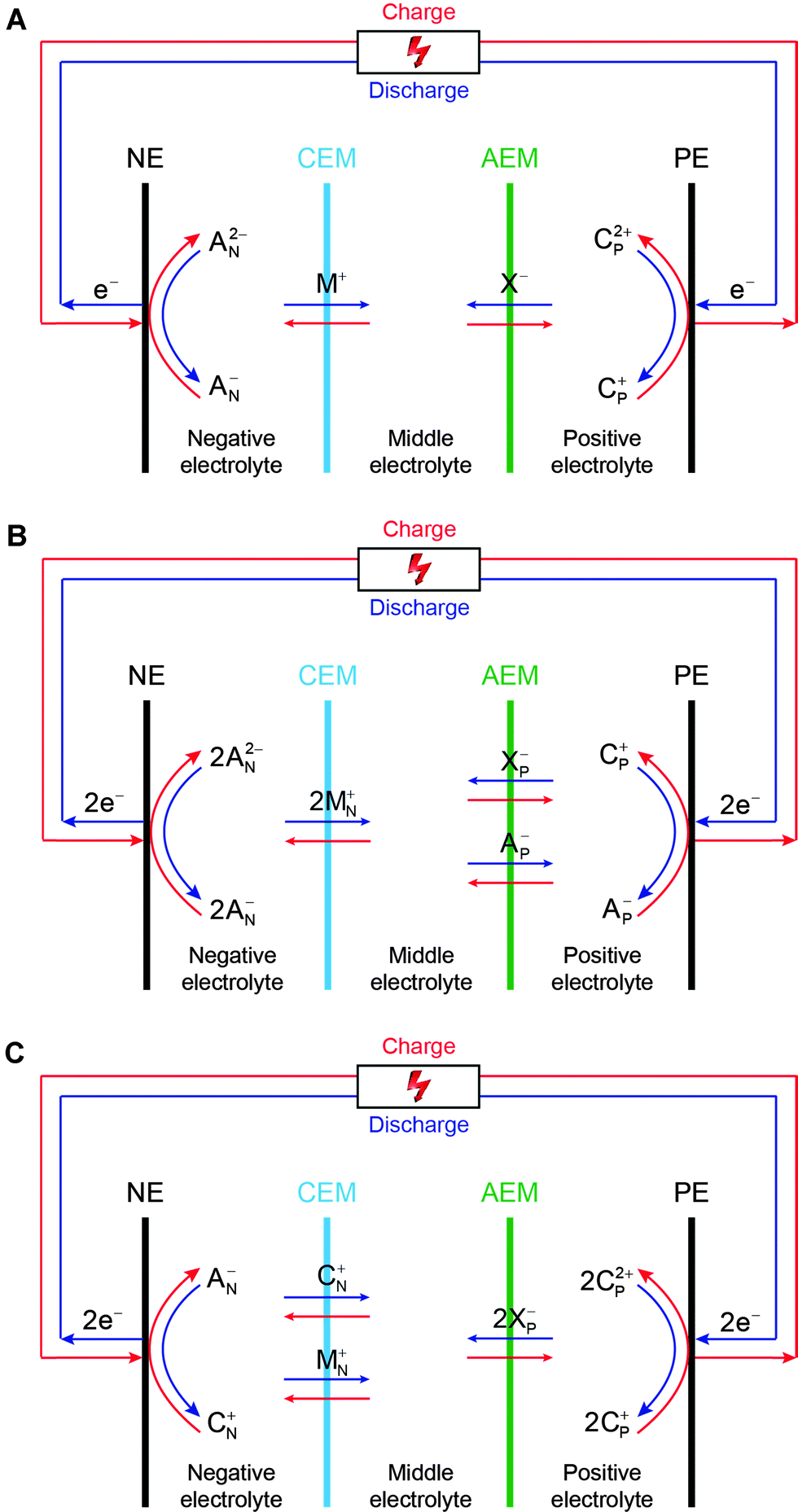 | ||
| Fig. 2 Working principles of the double-IEM RFB cell configuration. NE and PE represent the negative electrode and positive electrode, respectively. CEM and AEM stand for the cation-exchange membrane and anion-exchange membrane, respectively. (A) Combination of an anion–anion (negative) redox pair (AN−/AN2−) and a cation–cation (positive) redox pair (CP2+/CP+). (B) Combination of an anion–anion (negative) redox pair (AN−/AN2−) and an anion–cation hybrid (positive) redox pair (CP+/Ap−). (C) Combination of an anion–cation hybrid (negative) redox pair (CN+/AN−) and a cation–cation (positive) redox pair (CP2+/Cp+). M+, X−, MN+, and XP− are balancing ions. Note that the general working principles are, for the sake of simplicity, based on the assumptions that cations with more positive charge have a higher oxidation number than those with less positive charge, and anions with more negative charge have a lower oxidation number than those with less negative charge. When ions that do not follow those assumptions are used, the working principles are still applicable with minor alterations. (Reproduced with permission from ref. 22.) | ||
6.2. Sensitive redox system and limited cell durability
As for applications in renewable electricity storage, RFBs are required to have good cyclability and durability, and nonaqueous RFBs are not an exception. However, sufficient robustness of existing redox systems has not been demonstrated, toward which the stability of redox pairs and resilience of redox systems are the central focus.Both electrochemical stability and chemical stability of redox pairs are the prerequisite for durable nonaqueous RFBs. Nonaqueous redox pairs possess high rate constants, suggesting good electrochemical reversibility. However, their chemical stability can be of concern. For metal-based redox pairs, the organic ligands can be detached under certain circumstances. For metal-free redox pairs, the concern is their long-term chemical stability especially when nonaqueous RFBs are in the charged state.
It has been shown that oxygen and water from ambient atmosphere can also have substantial impacts on nonaqueous RFBs.85 Oxygen can passivate the electrode through reaction with both electrode materials and water deactivate redox pairs by forming oxo–metal complexes.
Designing and developing robust and resilient redox systems are also an urgent challenge for nonaqueous RFBs to be able to offer sufficient cyclability and durability for practical applications.
7. An example: designing a 4.5 V nonaqueous RFB
7.1. The idea and working principles
As discussed in Section 2, nonaqueous RFBs may offer a wide range of working temperatures, high cell voltage, and potentially high energy density. Herein we present an example of designing an ultrahigh-voltage (4.52 V) nonaqueous RFB with a high theoretical energy density (278 W h L−1) and a wide range of working temperatures (202 °C). Such an ultrahigh-voltage nonaqueous RFB is constructed by two redox pairs (one with ultra-negative redox potential in the negative electrolyte, and the other with ultra-positive redox potential in the positive electrolyte), and one Li+-conducting ceramic membrane between the negative electrolyte and the positive electrolyte (Fig. 3).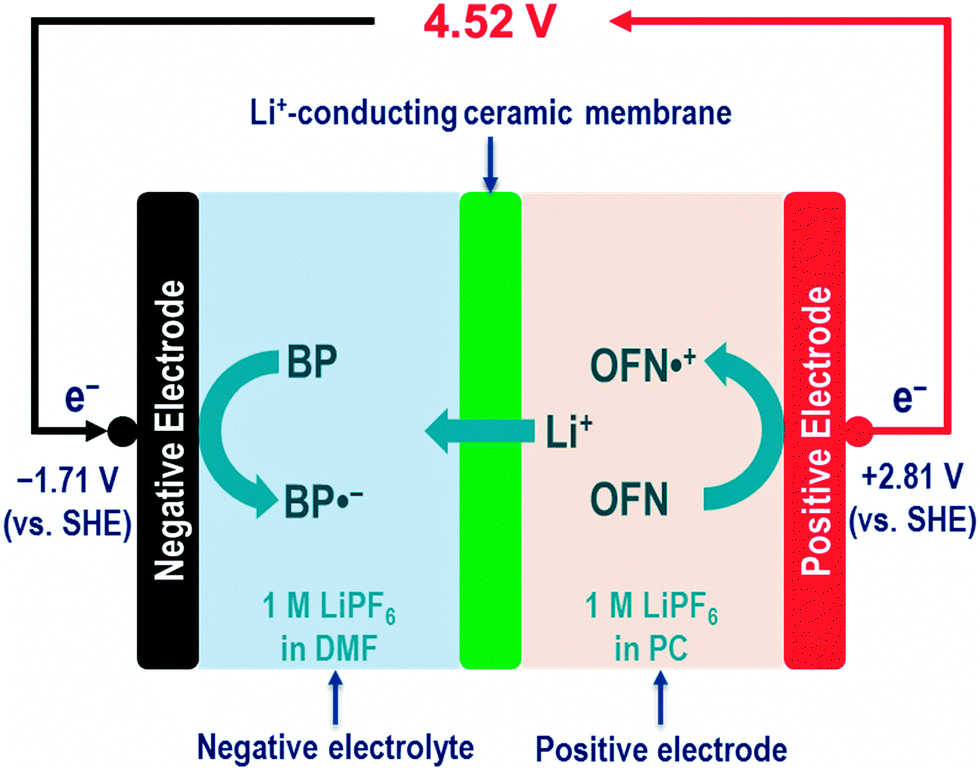 | ||
| Fig. 3 The BP–OFN nonaqueous RFB concept and its working principles. The negative electrolyte containing the BP/BP˙− redox pair and the positive electrolyte containing the OFN˙+/OFN redox pair are separated by a Li+-conducting ceramic membrane (e.g., LiSICON). 1 M LiPF6 is used as an example of the supporting electrolyte. When the cell is being charged, BP molecules are reduced to form BP˙− radical anions in the negative electrolyte (i.e., BP + e− = BP˙−), and OFN molecules are oxidized to form OFN˙+ radical cations in the positive electrolyte (i.e., OFN = OFN˙+ + e−). Meanwhile, Li+ ions pass through the Li+-conducting ceramic membrane from the positive electrolyte to the negative electrolyte. The discharging process is in reverse. | ||
It has long been known that some aromatic hydrocarbons are able to form stable radical anions, creating reversible redox pairs with very negative redox potentials.86–88 In particular, the biphenyl (BP) molecule can be reversibly reduced to form the biphenyl radical anion (BP˙−), with a very negative redox potential, e.g., a half-wave reduction potential of −2.70 V vs. SCE (or −2.46 V vs. SHE) observed for the BP/BP˙− redox pair in AN solvent. In addition, the comprehensive studies of both the equilibrium constant of the reaction (BP + Na = BP˙−Na+) and the dissociation constant of BP˙−Na+ salt suggest good redox reversibility and sufficient radical stability.87
On the other hand, a number of redox shuttle molecules have been recently explored with the main purpose of protecting lithium-ion batteries from being overcharged.89,90 Those redox shuttle compounds can form stable radical cations, leading to very positive redox potentials. In particular, the octafluoronaphthalene (OFN) molecule can be reversibly oxidized to form the octafluoronaphthalene radical cation (OFN˙+), with a very positive redox potential, e.g., 4.85 V vs. Li+/Li (or 1.85 V vs. SHE) measured for the OFN˙+/OFN redox pair in a mixed carbonate solvent. In addition to the very high redox potential, 70 charging–discharging cycles were demonstrated for the OFN˙+/OFN redox pair in an experimental lithium-ion battery, which clearly demonstrated the stability of the OFN˙+ radical cation.91
Herein our idea is to combine the BP/BP˙− redox pair and the OFN˙+/OFN redox pair in RFBs, which may promise to achieve unprecedentedly high cell voltage. DMF and PC are carefully chosen as the organic solvent for the two pairs, respectively, with criteria of both good electrochemical reversibility and high solubility.
7.2. Electrochemistry
The electrochemistry of both redox pairs was checked by performing cyclic voltammetry (CV) on a micro-Pt disk working electrode in their corresponding nonaqueous solvents, and Pt wire and Ag wire were used as the counter electrode and the reference electrode, respectively.Fig. 4A shows CV curves of 0.1 M BP in DMF solvent (0.1 M Bu4NClO4 as the supporting electrolyte, argon atmosphere). Regardless of the scan rate, every CV curve of BP shows a cathodic CV peak (i.e., BP reduction: BP + e− = BP˙−, around −2.3 V vs. Ag wire) and an anodic one (i.e., BP˙− oxidation: BP˙− = BP + e−, around −2.2 V vs. Ag wire), which leads to the formal redox potential of −2.25 V vs. Ag wire for the BP/BP˙− redox pair (or −1.71 V vs. SHE assuming the potential of Ag wire to be 0.54 V vs. SHE). Such a redox potential is 0.8+ V more negative than that of rubrene/rubrene˙− (−0.9 V vs. SHE, Table 6).
 | ||
| Fig. 4 Electrochemistry and solubility of the BP/BP˙− redox pair and the OFN˙+/OFN redox pair. (A) CV curves of the BP/BP˙− redox pair, test conditions: DMF solvent, 0.1 M BP, 0.1 M Bu4NClO4 as the supporting electrolyte; the Pt micro-working electrode (0.20 mm in diameter), and Pt wire and Ag wire as the counter electrode and the reference electrode, respectively. (B) Cathodic peak current density against the square root of scan rate for the BP/BP˙− redox pair. (C) Solubility measurement of the BP molecule in DMF solvent by NMR spectroscopy. (D) CV curves of the OFN˙+/OFN redox pair, test conditions: PC solvent, 0.1 M OFN, 0.1 M Bu4NClO4 as supporting electrolyte; the Pt micro-working electrode (0.20 mm in diameter), and Pt wire and Ag wire as the counter electrode and the reference electrode, respectively. (E) Anodic peak current density against the square root of scan rate for the OFN˙+/OFN redox pair. (F) Solubility measurement of the OFN molecule in PC solvent by NMR spectroscopy. | ||
Both the cathodic peak current and anodic one increased with the scan rate, and the diffusion coefficient of BP was obtained by using the Randles–Sevcik equation for reversible systems. Fig. 4B shows the fitting of the cathodic peak current against the square root of thr scan rate, giving a diffusion coefficient of 1.1 × 10−5 cm2 s−1 for BP. Furthermore, the standard rate constant of the BP/BP˙− redox pair was also estimated by using the Nicholson method.92 Table S1 (ESI†) shows the standard rate constant for each CV curve and the standard rate constant averages 4.8 × 10−3 cm s−1 for the BP/BP˙− redox pair. Although this value is lower than those of some reported metal-free redox pairs in Table 6, overall the kinetics is very facile. The obtained formal potential, diffusion coefficient, and standard rate constant of the BP/BP˙− redox pair are summarized in Table 7.
| Redox pair |
φ′a (V vs. SHE) |
D (cm2 s−1) |
k
0c (cm s−1) |
S (mol L−1) |
|---|---|---|---|---|
| Test conditions for the BP/BP˙− redox pair: DMF, 0.1 M BP, 0.1 M Bu4NClO4, Pt micro-working electrode (0.20 mm in diameter), Pt wire and Ag wire as the counter electrode and the reference electrode, respectively. Test conditions for the OFN˙+/OFN redox pair: PC, 0.1 M OFN, 0.1 M Bu4NClO4, all electrodes used were the same as the case of the BP/BP˙− redox pair.a φ′: formal redox potential [φ′ = (φpa+ φpc)/2, where φpa and φpc are the potential of anodic peak and the potential of cathodic peak, respectively.] The potential was converted to the SHE scale by the relationship: Ag wire = 0.54 V vs. SHE.b D: diffusion coefficient of the neutral compound, i.e., BP molecules in DMF and OFN molecules in PC, obtained by using the Randles–Sevcik equation.c k0: standard rate constant of redox reaction, obtained by adopting the Nicholson method (Tables S1 and S2, ESI).d S: solubility of the neutral command, i.e., BP molecules in DMF and OFN molecules in PC, measured by employing the NMR spectroscopy method (Fig. S1 and S2, ESI). | ||||
| BP/BP˙− | −1.71 | 1.1 × 10−5 | 4.8 × 10−3 | 9.7 |
| OFN˙+/OFN | 2.81 | 4.2 × 10−8 | 1.7 × 10−3 | 3.0 |
Fig. 4D shows CV curves of 0.1 M OFN in PC (0.1 M Bu4NClO4 as the supporting electrolyte, argon atmosphere). Similarly, every CV curve shows an anodic CV peak (i.e., OFN oxidation: OFN = OFN˙+ + e−, around 2.3 V vs. Ag wire) and a cathodic one (i.e., OFN˙+ reduction: OFN˙+ + e− = OFN, around 2.2 V vs. Ag wire). The two CV peaks defines the formal redox potential as 2.27 V vs. Ag wire (or 2.81 V vs. SHE assuming the potential of Ag wire to be 0.54 V vs. SHE) for the OFN˙+/OFN redox pair. Such a redox potential is 1.4+ V more positive than that of the rubrene˙+/rubrene redox pair (1.4 V vs. SHE, Table 6). Note that combining both the BP/BP˙ redox pair and the OFN˙+/OFN redox pair in an RFB cell may lead to a standard cell voltage of 4.52 V (Fig. 3), which is the highest among all reported nonaqueous RFBs.
Using the same Randles–Sevcik equation for reversible systems, the anodic CV peak current against the square root of the scan rate is plotted (Fig. 4E), giving a diffusion coefficient of OFN of 4.2 × 10−8 cm2 s−1. Note that such a diffusion coefficient of OFN in PC is much lower than that of BP in DMF (1.1 × 10−5 cm2 s−1), and this difference can be in part explained by the stronger solvent viscosity (2.53 vs. 0.92 mPa s for PC vs. DMF, Table 1) and the larger molecular weight (272.09 vs. 154.21 g mol−1 for OFN vs. BP). The standard rate constant of the OFN˙+/OFN redox pair was also obtained by using the same Nicholson method, and Table S2 (ESI†) shows the standard rate constant of each CV curve. The average standard rate constant is 1.7 × 10−3 cm s−1 for the OFN˙+/OFN redox pair. Despite the big difference in the diffusion coefficient of the neutral molecule, the standard rate constant of the OFN˙+/OFN redox pair is relatively close to that of the BP/BP˙− redox pair (4.8 × 10−3 cm s−1). Such a high level of the standard rate constant (>10−3 cm s−1) suggests that both pairs are sufficient for RFB applications.
7.3. Solubility and temperature
The NMR spectroscopy method was used to determine the solubility of redox compounds in organic solvents (1H NMR for BP, Fig. S1, ESI;† and 13C NMR for OFN, Fig. S2, ESI†). Fig. 4C shows the NMR-detected BP concentration in DMF solution against the apparent concentration of added BP. Before saturation, the detected BP concentration linearly increased with the added BP in the solution, and the detected BP levelled off when the added BP is high enough. Since only truly soluble species in solution can be detected by NMR spectroscopy, the observed saturated concentration is the solubility of BP in DMF: 9.7 mol L−1. Similarly, Fig. 4F shows the detected OFN concentration in PC solution against the apparent concentration of the added OFN. The observed solubility of OFN in PC was 3.0 mol L−1. Both solubility values are listed in Table 7.Such high solubility values may lead to a theoretical capacity density of 61.4 A h L−1 for the BP–OFN nonaqueous RFB. Note that this theoretical capacity density is solely based on the neutral redox compounds and pure solvents, not accounting for the supporting electrolytes or the possible solubility changes of their radical ions. Considering the ultrahigh cell voltage of 4.52 V, such a high capacity density may further lead to an unprecedented theoretical energy density of 278 W h L−1.
Under ambient pressure, DMF has a freezing point of −60 °C and a boiling point of 153 °C, and PC has a freezing point of −49 °C and a boiling point of 242 °C. Without considering the effects of freezing-point suppression and boiling-point elevation (discussed in Section 3.1), the BP–OFN nonaqueous RFB may operate at the working temperature between −49 °C and 153 °C, or as wide as 202 °C.
The full-cell design and assembly, utilizing both the BP/BP˙− redox pair and the OFN˙+/OFN redox pair, is currently under investigation and the results will be published in the due course.
8. Concluding remarks
Here we highlight the key features of nonaqueous RFBs and focus on their major components: organic solvents, supporting electrolytes, and redox pairs. As members of the RFB family, nonaqueous RFBs, especially those with the ability to work at low temperatures, are an important complement to aqueous RFBs, extending the range of RFB applications. Achieving low internal resistance and designing robust redox systems represent the two urgent needs for developing viable nonaqueous RFBs with high cell performance and good durability. To advance nonaqueous RFBs, it is important to understand both components and the system; and the understanding can be best achieved by collaborations across disciplines including electrochemistry, organic chemistry, physical chemistry, cell design, and system engineering. In order to demonstrate key features of nonaqueous RFBs, we also present an example of designing a 4.5 V ultrahigh-voltage nonaqueous RFB by combining the BP/BP˙− redox pair and the OFN˙+/OFN redox pair.Acknowledgements
This work was supported in part by the Department of Energy of U.S. through ARPA-E program (DE-AR0000346).Notes and references
- L. H. Thaller, The 9th Intersociety Energy Conversion Engineering Conference Proceedings, 1974, 924–928.
- Z. G. Yang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577–3613 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- W. A. Braff, M. Z. Bazant and C. R. Buie, Nat. Commun., 2013, 4, 2346 Search PubMed.
- B. Huskinson, M. P. Marshak, C. Suh, S. Er, M. R. Gerhardt, C. J. Galvin, X. Chen, A. Aspuru-Guzik, R. G. Gordon and M. J. Aziz, Nature, 2014, 505, 195–198 CrossRef CAS PubMed.
- C. P. de Leon, A. Frias-Ferrer, J. Gonzalez-Garcia, D. A. Szanto and F. C. Walsh, J. Power Sources, 2006, 160, 716–732 CrossRef PubMed.
- M. Skyllas-Kazacos, M. H. Chakrabarti, S. A. Hajimolana, F. S. Mjalli and M. Saleem, J. Electrochem. Soc., 2011, 158, R55–R79 CrossRef CAS PubMed.
- A. Z. Weber, M. M. Mench, J. P. Meyers, P. N. Ross, J. T. Gostick and Q. H. Liu, J. Appl. Electrochem., 2011, 41, 1137–1164 CrossRef CAS.
- X. F. Li, H. M. Zhang, Z. S. Mai, H. Z. Zhang and I. Vankelecom, Energy Environ. Sci., 2011, 4, 1147–1160 CAS.
- B. Schwenzer, J. L. Zhang, S. Kim, L. Y. Li, J. Liu and Z. G. Yang, ChemSusChem, 2011, 4, 1388–1406 CrossRef CAS PubMed.
- P. Leung, X. H. Li, C. P. de Leon, L. Berlouis, C. T. J. Low and F. C. Walsh, RSC Adv., 2012, 2, 10125–10156 RSC.
- W. Wang, Q. T. Luo, B. Li, X. L. Wei, L. Y. Li and Z. G. Yang, Adv. Funct. Mater., 2013, 23, 970–986 CrossRef CAS PubMed.
- R. Ferrigno, A. D. Stroock, T. D. Clark, M. Mayer and G. M. Whitesides, J. Am. Chem. Soc., 2002, 124, 12930–12931 CrossRef CAS PubMed.
- Y. H. Lu, J. B. Goodenough and Y. Kim, J. Am. Chem. Soc., 2011, 133, 5756–5759 CrossRef CAS PubMed.
- Y. R. Wang, Y. G. Wang and H. S. Zhou, ChemSusChem, 2011, 4, 1087–1090 CrossRef CAS PubMed.
- F. R. Brushett, J. T. Vaughey and A. N. Jansen, Adv. Energy Mater., 2012, 2, 1390–1396 CrossRef CAS PubMed.
- K. J. Kim, M. S. Park, J. H. Kim, U. Hwang, N. J. Lee, G. Jeong and Y. J. Kim, Chem. Commun., 2012, 48, 5455–5457 RSC.
- W. Wang, W. Xu, L. Cosimbescu, D. W. Choi, L. Y. Li and Z. G. Yang, Chem. Commun., 2012, 48, 6669–6671 RSC.
- Q. T. Luo, L. Y. Li, W. Wang, Z. M. Nie, X. L. Wei, B. Li, B. W. Chen, Z. G. Yang and V. Sprenkle, ChemSusChem, 2013, 6, 268–274 CrossRef CAS PubMed.
- J. Liu, J. G. Zhang, Z. G. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Y. Li, J. Z. Hu, C. M. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. L. Li, Y. Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929–946 CrossRef CAS PubMed.
- S. H. Shin, S. H. Yun and S. H. Moon, RSC Adv., 2013, 3, 9095–9116 RSC.
- S. Gu, K. Gong, E. Z. Yan and Y. S. Yan, Energy Environ. Sci., 2014, 7, 2986–2998 CAS.
- P. Singh, J. Power Sources, 1984, 11, 135–142 CrossRef CAS.
- Y. Matsuda, K. Tanaka, M. Okada, Y. Takasu, M. Morita and T. Matsumurainoue, J. Appl. Electrochem., 1988, 18, 909–914 CrossRef CAS.
- T. Yamamura, Y. Shiokawa, H. Yamana and H. Moriyama, Electrochim. Acta, 2002, 48, 43–50 CrossRef CAS.
- Q. H. Liu, A. E. S. Sleightholme, A. A. Shinkle, Y. D. Li and L. T. Thompson, Electrochem. Commun., 2009, 11, 2312–2315 CrossRef CAS PubMed.
- A. E. S. Sleightholme, A. A. Shinkle, Q. H. Liu, Y. D. Li, C. W. Monroe and L. T. Thompson, J. Power Sources, 2011, 196, 5742–5745 CrossRef CAS PubMed.
- Q. H. Liu, A. A. Shinkle, Y. D. Li, C. W. Monroe, L. T. Thompson and A. E. S. Sleightholme, Electrochem. Commun., 2010, 12, 1634–1637 CrossRef CAS PubMed.
- J. H. Kim, K. J. Kim, M. S. Park, N. J. Lee, U. Hwang, H. Kim and Y. J. Kim, Electrochem. Commun., 2011, 13, 997–1000 CrossRef CAS PubMed.
- D. P. Zhang, H. J. Lan and Y. D. Li, J. Power Sources, 2012, 217, 199–203 CrossRef CAS PubMed.
- H. D. Pratt, N. S. Hudak, X. K. Fang and T. M. Anderson, J. Power Sources, 2013, 236, 259–264 CrossRef CAS PubMed.
- P. J. Cappillino, H. D. Pratt, N. S. Hudak, N. C. Tomson, T. M. Anderson and M. R. Anstey, Adv. Energy Mater., 2014, 4, 1300566 Search PubMed.
- M. H. Chakrabarti, R. A. W. Dryfe and E. P. L. Roberts, J. Chem. Soc. Pak., 2007, 29, 294–300 CAS.
- Z. Li, S. Li, S. Q. Liu, K. L. Huang, D. Fang, F. C. Wang and S. Peng, Electrochem. Solid-State Lett., 2011, 14, A171–A173 CrossRef CAS.
- S. Hamelet, T. Tzedakis, J. B. Leriche, S. Sailler, D. Larcher, P. L. Taberna, P. Simon and J. M. Tarascona, J. Electrochem. Soc., 2012, 159, A1360–A1367 CrossRef CAS PubMed.
- M. Duduta, B. Ho, V. C. Wood, P. Limthongkul, V. E. Brunini, W. C. Carter and Y. M. Chiang, Adv. Energy Mater., 2011, 1, 511–516 CrossRef CAS PubMed.
- F. Y. Fan, W. H. Woodford, Z. Li, N. Baram, K. C. Smith, A. Helal, G. H. McKinley, W. C. Carter and Y. M. Chiang, Nano Lett., 2014, 14, 2210–2218 CrossRef CAS PubMed.
- Y. H. Lu and J. B. Goodenough, J. Mater. Chem., 2011, 21, 10113–10117 RSC.
- Y. R. Wang, P. He and H. S. Zhou, Adv. Energy Mater., 2012, 2, 770–779 CrossRef CAS PubMed.
- N. M. Asl, S. S. Cheah, J. Salim and Y. Kim, RSC Adv., 2012, 2, 6094–6100 RSC.
- Y. Zhao, L. N. Wang and H. R. Byon, Nat. Commun., 2013, 4, 1896 CrossRef PubMed.
- J. Chun, M. Chung, J. Lee and Y. Kim, Phys. Chem. Chem. Phys., 2013, 15, 7036–7040 RSC.
- Y. Y. Hou, X. J. Wang, Y. S. Zhu, C. L. Hu, Z. Chang, Y. P. Wu and R. Holze, J. Mater. Chem. A, 2013, 1, 14713–14718 CAS.
- S. L. Chou, Y. X. Wang, J. T. Xu, J. Z. Wang, H. K. Liu and S. X. Dou, Electrochem. Commun., 2013, 31, 35–38 CrossRef CAS PubMed.
- Y. Zhao and H. R. Byon, Adv. Energy Mater., 2013, 3, 1630–1635 CrossRef CAS.
- Y. Zhao, M. Hong, N. B. Mercier, G. H. Yu, H. C. Choi and H. R. Byon, Nano Lett., 2014, 14, 1085–1092 CrossRef CAS PubMed.
- J. K. Kim, W. Yang, J. Salim, C. Ma, C. W. Sun, J. Q. Li and Y. Kim, J. Electrochem. Soc., 2014, 161, A285–A289 CrossRef CAS PubMed.
- P. Liu, Y. L. Cao, G. R. Li, X. P. Gao, X. P. Ai and H. X. Yang, ChemSusChem, 2013, 6, 802–806 CrossRef CAS PubMed.
- N. F. Yan, G. R. Li and X. P. Gao, J. Electrochem. Soc., 2014, 161, A736–A741 CrossRef CAS PubMed.
- R. M. Darling, K. G. Gallagher, J. A. Kowalski, S. Ha and F. R. Brushett, Energy Environ. Sci., 2014, 7, 3459–3477 CAS.
- R. Matthé and U. Eberle, in Lithium-Ion Batteries: Advances and Applications, ed. G. Pistoia, Elsevier, 1st edn, 2014, ch. 8, pp. 151–176 Search PubMed.
- K. Izutsu, Electrochemistry in Nonaqueous Solutions, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2009 Search PubMed.
- J. T. Denison and J. B. Ramsey, J. Am. Chem. Soc., 1955, 77, 2615–2621 CrossRef CAS.
- N. G. Tsierkezos and A. I. Philippopoulos, Fluid Phase Equilib., 2009, 277, 20–28 CrossRef CAS PubMed.
- F. J. Millero, J. Phys. Chem., 1970, 74, 356–362 CrossRef CAS.
- G. Petrella, M. Castagnolo, A. Sacco and A. Degiglio, J. Solution Chem., 1976, 5, 621–629 CrossRef CAS.
- K. Xu, Chem. Rev., 2004, 104, 4303–4417 CrossRef CAS.
- D. Ekka and M. N. Roy, J. Phys. Chem. B, 2012, 116, 11687–11694 CrossRef CAS PubMed.
- M. Ue, K. Ida and S. Mori, J. Electrochem. Soc., 1994, 141, 2989–2996 CrossRef CAS PubMed.
- A. A. Shinkle, T. J. Pomaville, A. E. S. Sleightholme, L. T. Thompson and C. W. Monroe, J. Power Sources, 2014, 248, 1299–1305 CrossRef CAS PubMed.
- A. Chagnes, B. Carre, P. Willmann and D. Lemordant, Electrochim. Acta, 2001, 46, 1783–1791 CrossRef CAS.
- J. Barthel, H. J. Gores, R. Neueder and A. Schmid, Pure Appl. Chem., 1999, 71, 1705–1715 CrossRef CAS.
- M. H. Chakrabarti, R. A. W. Dryfe and E. P. L. Roberts, Electrochim. Acta, 2007, 52, 2189–2195 CrossRef CAS PubMed.
- M. H. Chakrabarti, E. P. L. Roberts and M. Saleem, Int. J. Green Energy, 2010, 7, 445–460 CrossRef CAS PubMed.
- M. H. Chakrabarti, E. P. L. Roberts, C. Bae and M. Saleem, Energy Convers. Manage., 2011, 52, 2501–2508 CrossRef CAS PubMed.
- J. Mun, M. J. Lee, J. W. Park, D. J. Oh, D. Y. Lee and S. G. Doo, Electrochem. Solid-State Lett., 2012, 15, A80–A82 CrossRef CAS PubMed.
- T. Yamamura, K. Shirasaki, Y. Shiokawa, Y. Nakamura and S. Y. Kim, J. Alloys Compd., 2004, 374, 349–353 CrossRef CAS PubMed.
- Q. Sun, Q. Wang, Y. Shiokawa and Y. Kawazoe, Chem. Phys. Lett., 2005, 415, 243–245 CrossRef CAS PubMed.
- K. Shirasaki, T. Yamamura, T. Herai and Y. Shiokawa, J. Alloys Compd., 2006, 418, 217–221 CrossRef CAS PubMed.
- T. Yamamura, K. Shirasaki, D. X. Li and Y. Shiokawa, J. Alloys Compd., 2006, 418, 139–144 CrossRef CAS PubMed.
- K. Shirasaki, T. Yamamura and Y. Shiokawa, J. Alloys Compd., 2006, 408, 1296–1301 CrossRef.
- T. Yamamura, K. Shirasaki, H. Sato, Y. Nakamura, H. Tomiyasu, I. Satoh and Y. Shiokawa, J. Phys. Chem. C, 2007, 111, 18812–18820 CAS.
- A. A. Shinkle, A. E. S. Sleightholme, L. T. Thompson and C. W. Monroe, J. Appl. Electrochem., 2011, 41, 1191–1199 CrossRef CAS.
- T. Herr, J. Noack, P. Fischer and J. Tubke, Electrochim. Acta, 2013, 113, 127–133 CrossRef CAS PubMed.
- D. P. Zhang, Q. H. Liu, X. S. Shi and Y. D. Li, J. Power Sources, 2012, 203, 201–205 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- D. H. Evans, Chem. Rev., 1990, 90, 739–751 CrossRef CAS.
- D. H. Evans, Chem. Rev., 2008, 108, 2113–2144 CrossRef CAS PubMed.
- A. R. Forrester and R. H. Thomson, Nature, 1964, 203, 74–75 CrossRef CAS.
- Z. P. Song, H. Zhan and Y. H. Zhou, Chem. Commun., 2009, 448–450 RSC.
- L. Zhang, Z. C. Zhang, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2012, 5, 8204–8207 CAS.
- V. M. M. Lobo and J. L. Quaresma, Handbook of Electrolyte Solutions, Elsevier, Amsterdam, Netherland, 1989 Search PubMed.
- S. Maurya, S. H. Shin, K. W. Sung and S. H. Moon, J. Power Sources, 2014, 255, 325–334 CrossRef CAS PubMed.
- D. H. Kim, S. J. Seo, M. J. Lee, J. S. Park, S. H. Moon, Y. S. Kang, Y. W. Choi and M. S. Kang, J. Membr. Sci., 2014, 454, 44–50 CrossRef CAS.
- A. A. Shinkle, A. E. S. Sleightholme, L. D. Griffith, L. T. Thompson and C. W. Monroe, J. Power Sources, 2012, 206, 490–496 CrossRef CAS PubMed.
- S. Wawzonek and D. Wearring, J. Am. Chem. Soc., 1959, 81, 2067–2069 CrossRef CAS.
- N. L. Holy, Chem. Rev., 1974, 74, 243–277 CrossRef CAS.
- A. C. Aten, C. Buthker and G. J. Hoijtink, Trans. Faraday Soc., 1959, 55, 324–330 RSC.
- C. Buhrmester, J. Chen, L. Moshurchak, J. W. Jiang, R. L. Wang and J. R. Dahn, J. Electrochem. Soc., 2005, 152, A2390–A2399 CrossRef CAS PubMed.
- R. L. Wang and J. R. Dahn, J. Electrochem. Soc., 2006, 153, A1922–A1928 CrossRef CAS PubMed.
- J. H. Chen, L. M. He and R. L. Wang, J. Electrochem. Soc., 2012, 159, A1636–A1645 CrossRef CAS PubMed.
- R. S. Nicholso, Anal. Chem., 1965, 37, 1351–1355 CrossRef.
- H. Y. Wang, J. J. Wang, S. L. Zhang, Y. C. Pei and K. L. Zhuo, ChemPhysChem, 2009, 10, 2516–2523 CrossRef CAS PubMed.
- W. Libus, B. Chachulski and L. Fraczyk, J. Solution Chem., 1980, 9, 355–369 CrossRef CAS.
- O. N. Kalugin, V. G. Panchenko, A. P. Dolgareva, A. G. Nikolaichuk and I. N. V'yunnik, Russ. J. Phys. Chem. A, 2008, 82, 1480–1483 CrossRef CAS.
- R. J. LeSuer, C. Buttolph and W. E. Geiger, Anal. Chem., 2004, 76, 6395–6401 CrossRef CAS PubMed.
- F. Croce, A. DAprano, C. Nanjundiah, V. R. Koch, C. W. Walker and M. Salomon, J. Electrochem. Soc., 1996, 143, 154–159 CrossRef CAS PubMed.
- A. Apelblat, J. Mol. Liq., 2010, 156, 89–94 CrossRef CAS PubMed.
- M. Salomon, J. Solution Chem., 1993, 22, 715–725 CrossRef CAS.
- B. Das and D. K. Hazra, J. Solution Chem., 1998, 27, 1021–1031 CrossRef CAS.
- J. F. Reardon, Electrochim. Acta, 1987, 32, 1595–1600 CrossRef CAS.
- H. M. Daggett, E. J. Bair and C. A. Kraus, J. Am. Chem. Soc., 1951, 73, 799–803 CrossRef CAS.
- M. Salomon and E. J. Plichta, Electrochim. Acta, 1984, 29, 731–735 CrossRef CAS.
- B. S. Krumgalz, J. Chem. Soc., Faraday Trans. 1, 1983, 79, 571–587 RSC.
- Y. Marcus, Ion properties, Marcel Dekker, Inc., New York, 1997 Search PubMed.
- M. Ue, J. Electrochem. Soc., 1994, 141, 3336–3342 CrossRef CAS PubMed.
- M. Ue, M. Takeda, M. Takehara and S. Mori, J. Electrochem. Soc., 1997, 144, 2684–2688 CrossRef CAS PubMed.
- I. Banik and M. N. Roy, J. Chem. Thermodyn., 2013, 63, 52–59 CrossRef CAS PubMed.
- D. S. Gill, M. S. Chauhan and M. B. Sekhri, J. Chem. Soc., Faraday Trans. 1, 1982, 78, 3461–3466 RSC.
- S. Katsuta, K. Imai, Y. Kudo, Y. Takeda, H. Seki and M. Nakakoshi, J. Chem. Eng. Data, 2008, 53, 1528–1532 CrossRef CAS.
- I. Svorstol, H. Hoiland and J. Songstad, Acta Chem. Scand., Ser. B, 1984, 38, 885–893 CrossRef.
- M. Morita, Y. Tanaka, K. Tanaka, Y. Matsuda and T. Matsumurainoue, Bull. Chem. Soc. Jpn., 1988, 61, 2711–2714 CrossRef CAS.
- T. Saji, T. Yamada and S. Aoyagui, Bull. Chem. Soc. Jpn., 1975, 48, 1641–1642 CrossRef CAS.
- T. Suga, Y. J. Pu, K. Oyaizu and H. Nishide, Bull. Chem. Soc. Jpn., 2004, 77, 2203–2204 CrossRef CAS.
- K. Oyaizu, A. Hatemata, W. Choi and H. Nishide, J. Mater. Chem., 2010, 20, 5404–5410 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee02341f |
| This journal is © The Royal Society of Chemistry 2015 |