
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Does it have to be carbon? Metal anodes in microbial fuel cells and related bioelectrochemical systems†
André
Baudler‡
a,
Igor
Schmidt‡
a,
Markus
Langner
b,
Andreas
Greiner
*b and
Uwe
Schröder
*a
aInstitute of Environmental and Sustainable Chemistry, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: uwe.schroeder@tu-braunschweig.de; Fax: +49 5313918424; Tel: +49 5313918425
bChair of Macromolecular Chemistry II, Universität Bayreuth, Universitätsstrasse 30, 95440 Bayreuth, Germany. E-mail: greiner@uni-bayreuth.de
First published on 27th May 2015
Abstract
Copper and silver are antimicrobial metals, on whose surface bacteria do not grow. As our paper demonstrates, this commonly reported antimicrobial property does not apply to electrochemically active, electrode respiring bacteria. These bacteria readily colonize the surface of these metals, forming a highly active biofilm. Average anodic current densities of 1.1 mA cm−2 (silver) and 1.5 mA cm−2 (copper) are achieved – data that are comparable to that of the benchmark material, graphite (1.0 mA cm−2). Beside the above metals, nickel, cobalt, titanium and stainless steel (SUS 304) were systematically studied towards their suitability as anode materials for microbial fuel cells and related bioelectrochemical systems. The bioelectrochemical data are put in relation to physical data of the materials (specific conductivity, standard potential) and to basic economic considerations. It is concluded that especially copper represents a highly promising anode material, suitable for application in high-performance bioelectrochemical systems.
Broader contextMicrobial fuel cells and related bioelectrochemical systems (BESs) have developed impressively over the past decade. In order to take this technology from (fundamental) research to application, the costs and the performance of these systems need to be further optimized. Carbon is generally considered as the anode material of choice, since it is biocompatible, chemically and microbially stable and it can be produced at comparatively low costs from biological and chemical polymer precursors via carbonization. But is it really suitable for large scale application, where its low conductivity represents a severe drawback? Here we demonstrate that copper is a promising alternative anode material. Its high conductivity allows minimising the amount of electrode material and thus the material costs, and contrary to its usual antimicrobial behaviour, high-performing electrochemically active biofilms grow readily on this metal. |
1 Introduction
Carbon in its different electroconductive modifications – particularly graphite – may be seen as the most versatile electrode material for electrochemical systems. In bioelectrochemical systems (BES)1 such as microbial fuel cells it is considered as the material of choice, since it is biocompatible, chemically and microbially stable and it can be produced at comparatively low costs from biological and chemical polymer precursors via carbonization.2 The use of three-dimensional polymer templates allows the preparation of highly efficient 3D graphite electrodes for high-performance bioelectrochemical systems.3–5 Further, many bioelectrochemical processes like the oxidation and reduction of outer membrane cytochromes at carbon is electrochemically reversible, i.e., it proceeds at high rates.6Yet, despite these positive properties, carbon possesses a major disadvantage: its electric conductivity lies two to three orders of magnitude below that of most metals. For example, whereas copper has a specific conductivity of 58 × 106 S m−1,7 the conductivity of polycrystalline graphite lies only in the region of 3 × 104–1 × 105 S m−1 (see Table 2). Conversely, the specific electrical resistivity of graphite exceeds that of metals by up to three orders of magnitude. In an electrochemical system like a (microbial) fuel cell, any increasing electrode resistivity leads to a decreasing cell voltage and thus to decreasing power. This power loss may be low for small, lab based systems, but in an up-scaled system the effect may result in the complete collapse of the electrochemical performance.
It thus seems consequent to consider metals as electrode materials for microbial BES. For abiotic BES cathodes this has already been realized, e.g., in the form of nickel cathodes in microbial electrolysis cells.8,9 For BES anodes, however, examples are scarce. Despite the variety of metals in the periodic table of elements only a very limited number may appear suitable as anode material. The metal (or alloy) should be electrochemically inert in the operational potential window of the bioelectrochemical system, which means that it should be either electrochemically noble or can become electrochemically passivated. Gold and platinum belong to the first group of metals. These noble metals are frequently used as electrodes in fundamental BES research,10–13 since their defined metal surface allows a high degree of electrochemical reversibility. Their high price, however, would not allow them to be used in large technical systems. At the other end of the scale, inexpensive base metals (or their alloys) can potentially be used as anode material, provided that a compact oxide layer (passivation layer) protects the metal from further oxidation. An example is stainless steel, which has been proposed as anode material for microbial fuel cells.14 A disadvantage of such electrodes is the additional resistance caused by the passivating oxide layer, which causes an often strong irreversibility of the electron transfer.15
Between both extremes ( and
and  ), there are metals that a microbiologist would most likely not consider as an electrode material for a bioelectrochemical system: silver and copper. These metals are known to be natural antimicrobial materials on which “no live microorganisms were generally recovered ... after prolonged incubation”.16 The antimicrobial effect is often referred to as oligodynamic effect.17 It is based on the antimicrobial action of traces of metal ions liberated from the metal surface upon oxidation. Yet, with standard potentials of
), there are metals that a microbiologist would most likely not consider as an electrode material for a bioelectrochemical system: silver and copper. These metals are known to be natural antimicrobial materials on which “no live microorganisms were generally recovered ... after prolonged incubation”.16 The antimicrobial effect is often referred to as oligodynamic effect.17 It is based on the antimicrobial action of traces of metal ions liberated from the metal surface upon oxidation. Yet, with standard potentials of  and
and  they are comparatively noble and they are extremely good electric conductors. Thus, the conductivity of copper is around 900 times better than that of polycrystalline graphite, which would allow to decrease the internal resistance of microbial BES significantly and to reduce the amount of required electrode material considerably.
they are comparatively noble and they are extremely good electric conductors. Thus, the conductivity of copper is around 900 times better than that of polycrystalline graphite, which would allow to decrease the internal resistance of microbial BES significantly and to reduce the amount of required electrode material considerably.
A number of studies indicate that electrochemically active bacteria, especially of Geobacteraceae, are tolerant against the oligodynamic effect of heavy metal ions. Thus, it has been shown that Geobacter dominated biofilms are not affected by the presence of heavy metal ions.18 Moreover, these electroactive bacteria are able to colonize the copper19,20 and silver21,22 surfaces.
This study is dedicated to a thorough investigation of copper, gold, silver, stainless steel, nickel, cobalt, and titanium as anode materials for microbial bioelectrochemical systems. Systematic biotic and abiotic tests were performed to characterize the electrochemical and bioelectrochemical behaviour of these metals. The biofilm growth was characterized using confocal laser scanning microscopy (CLSM). The bioelectrochemical data were combined with a rudimental economic analysis to assess the suitability of the individual metals as anode material in microbial bioelectrochemical systems.
2 Methods
2.1 Electrochemical setup and conditions
The chemicals used in this study were purchased from Sigma Aldrich or Roth and were of analytical grade. The electrochemical and bioelectrochemical measurements were carried out in half cell setups under potentiostatic control (VMP3 or MPG 2, BioLogic, France and PGSTAT302, Metrohm Autolab B. V., The Netherlands). Round-bottom flasks (250 mL) were used as electrochemical cells, containing a working electrode (Section 2.2), a counter electrode (graphite rod, CP Graphite GmbH, Germany) and a Ag/AgCl (sat. KCl, 0.197 V vs. SHE) reference electrode (Sensortechnik Meinsberg GmbH, Germany). All potentials in this article refer to this reference electrode. For an easier handling we did not use electrochemical cells, in which the counter electrode is separated from the working electrode via an ion exchange membrane. Yet, we regularly performed comparative measurements between divided cells and one-chamber cells, to exclude the impact of an electrochemical recycling of redox equivalents at the counter electrode.2.2 Working electrode preparation
The majority of bioelectrochemical experiments in this study were performed using wires or sheets of pure metals or stainless steel (SUS304), purchased from Chempur, Germany in a purity ≥99.9%. Additionally, metal plated graphite (MPG) electrodes were used to verify bioelectrochemical data achieved with the monolithic metal electrodes and for biofilm characterization via confocal laser scanning microscopy. The preparation of the MPG electrodes is described in the ESI.†There were no significant differences in the electrochemical behaviour of the pure metal electrodes and the MPG electrodes. The same applies to the cultivation and the performance of the electrochemically active biofilms on these different electrode types. Averaged current density data comprise the data of both, solid electrodes and MPG electrodes.
All electrodes had a geometric surface area of 1.5 cm2. Electrical connections were isolated with heat shrinking tubes. Prior to the electrochemical measurements the electrodes were rinsed with isopropyl ethanol to remove organic residue and were sonicated in deionised water for 30 minutes (Emmi 12HC, EMAG AG, Germany).
2.3 Biofilm cultivation
All bioelectrochemical experiments were performed under strictly anaerobic conditions, at a temperature of 35 °C. The electrochemically active biofilms were cultivated in a standard growth medium using acetate (10 mM) as the carbon source. The growth medium contained NH4Cl (0.31 g L−1), KCl (0.13 g L−1), NaH2PO4·H2O (2.69 g L−1), Na2HPO4 (4.33 g L−1), trace metal (12.5 mL L−1) and vitamin (12.5 mL L−1) solutions.23 The cultivation media were purged with nitrogen for at least 20 minutes to ensure anaerobic conditions before use.The study is based on the use of secondary biofilms, i.e., biofilms cultivated from preselected, primary electrochemically active biofilms. In the first step, primary wastewater (Wastewater treatment plant Steinhof, Braunschweig, Germany) was used as inoculum for the cultivation of primary biofilms. These biofilms, which were cultivated on graphite rods (CP Graphite GmbH, Germany), were scratched off with a sterile spatula into a falcon tube filled with 5 mL buffer solution and were dispersed with a vortex mixer (Vortex Genie 2, Scientific Industries, USA) for 2 minutes. Afterwards these suspensions were used as inoculum to cultivate secondary biofilms on the studied metal electrodes. A detailed description of the procedure is provided in ref. 24 and 25.
For biofilm cultivation, a constant potential was applied depending on the anticipated oxidation stability of the anode materials: carbon, gold, silver were polarised at 0.2 V, whereas the biofilms on copper, steel, nickel and titanium were cultivated at a potential of −0.2 V. As illustrated exemplarily in Fig. S2 (ESI†), this variation of the applied electrode potential did not have any significant impact on the biofilm cultivation and electrocatalytic biofilm performance.
The biofilm growth was monitored by measuring the bioelectrocatalytic current density of the acetate oxidation (eqn (1)). The experiments were carried out in batch mode for at least 20 days of operation, replenishing the entire substrate solution after complete substrate consumption (indicated by low current). Addition of inoculum was limited to the first cycle of operation.
All bioelectrochemical measurements were carried out in triplicates. The determination of the biofilm thickness was carried out in duplicates using two different methods (Section 2.4).
2.4 Optical biofilm characterization
In order to determine the thickness of the electrochemically active biofilms, a straightforward CLSM (Confocal laser scanning microscopy) reflection method was used. This method, which is a standard method in materials science26,27 and has also been applied in biofilm research,28 relies on the measurement of the laser reflectance at interfaces, at which changes of the refractive index occur. Since the electrochemically active biofilms are very dense (in terms of structure and optical density), the reflection of the upper cell layer can be easily determined in relation to the electrode surface. Using this method, no staining is involved; the measurement is straightforward and the biofilms can in principle be used for further bioelectrochemical experiments.For the measurement, established biofilm anodes were taken out of the reactor and the biofilm was scratched off from half of the electrode in one movement using a scalpel to gain a clean cut edge that allows determining the biofilm thickness. The prepared electrode was transferred into a small container, which was carefully filled up with deionised water until the electrode was well covered. After that, a z-stack of the electrode surface over the total height of the biofilm was performed (see Fig. 1 for illustration). For this measurement, a Leica “TCS SPE” microscope (Leica Microsystems, Germany) with a 25× water immersion objective lens was used. The wavelength of the laser was 532 nm (laser output 1.301 mW, Gain 614 V). The thickness of the biofilms was examined using “Leica Map 7.0” software (Digital Surf, France).
 | ||
| Fig. 1 Confocal laser scanning microscopy image (reflectance data) of an anodic electrochemically active biofilm cultivated on a gold electrode. The biofilms were cultivated for three batch cycles, at a temperature of 35 °C, 10 mM acetate, 0.2 V (vs. Ag/AgCl). | ||
2.5 Chemical trace analysis
The amount of metal ions released from the anodes into the electrolyte/buffer solutions during the bioelectrochemical measurements was determined using ICP-OES (ICP-OES Vista MPX, Varian, Germany). In all tested cases, in which the concentration of the respected metal ions was determined from the effluent of semi-batch-experiment, the metal concentration was below the detection limit of the ICP-OES. This means that the corrosion level was negligible.3 Results and discussion
3.1 Bioelectrochemical measurements
The most common and most intensively studied type of microbial biofilm electrode is that of the anodic, acetate based, mixed culture biofilm cultivated on carbon (graphite) e.g.ref. 2, 29 and 30. Although generally cultivated from natural bacterial inoculi (wastewater, soil, sludge, etc.31,32), very often a preselection of the bacterial culture is applied in order to improve the electrocatalytic performance of the biofilm electrodes.24,33,34Fig. 2 (main figure) depicts a typical example of the cultivation of an electrochemically active biofilm on a graphite electrode under semi-batch conditions and at a fixed electrode potential. The current flow resulting from the bioelectrocatalytic acetate oxidation (eqn (1)) was monitored.
| CH3COO− + 2H2O → 2CO2 + 7H+ + 8e− | (1) |
 | ||
| Fig. 2 Main figure: exemplary cultivation and resulting bioelectrocatalytic current generation of a primary, acetate based electrochemically active biofilm at polycrystalline graphite in a semi-batch experiment. The biofilm was cultivated in a half-cell setup under potentiostatic control. The electrode potential was 0.2 V (vs. Ag/AgCl). Inset figure (a) cylic voltammogram recorded under turnover conditions (depicted by the red dot indexed “a” in the main figure). Inset figure (b) cylic voltammogram recorded under non-turnover conditions (depicted by the red dot indexed “b” in the main figure). An analogous experiment at a secondary biofilm is depicted in Fig. S3 (ESI†). | ||
Such biofilm electrodes deliver a typical maximum current density in the range of 0.5–1.5 mA cm−2. Our previous analyses of such acetate grown, electrochemically active biofilms showed a strong dominance of Geobacteraceae as the prevailing species in these biofilms.35,36 The voltammetric behaviour of such Geobacter dominated biofilms corresponds to the voltammetry illustrated in Fig. 2. The shape of the voltammograms and the depicted redox potentials are characteristic35,37,38 and strongly differ from that of, e.g., Shewanella based biofilms.39 For this reason we can assume that the electrochemically active biofilms in this study are highly Geobacter dominated.
Is it now possible to apply the above results and biofilm cultivation steps to other electrode materials like metals? Fig. 3 illustrates a respective biofilm growth and biofilm performance at a copper electrode. It shows that electrochemically active bacteria colonize its surface readily, forming a highly active biofilm.
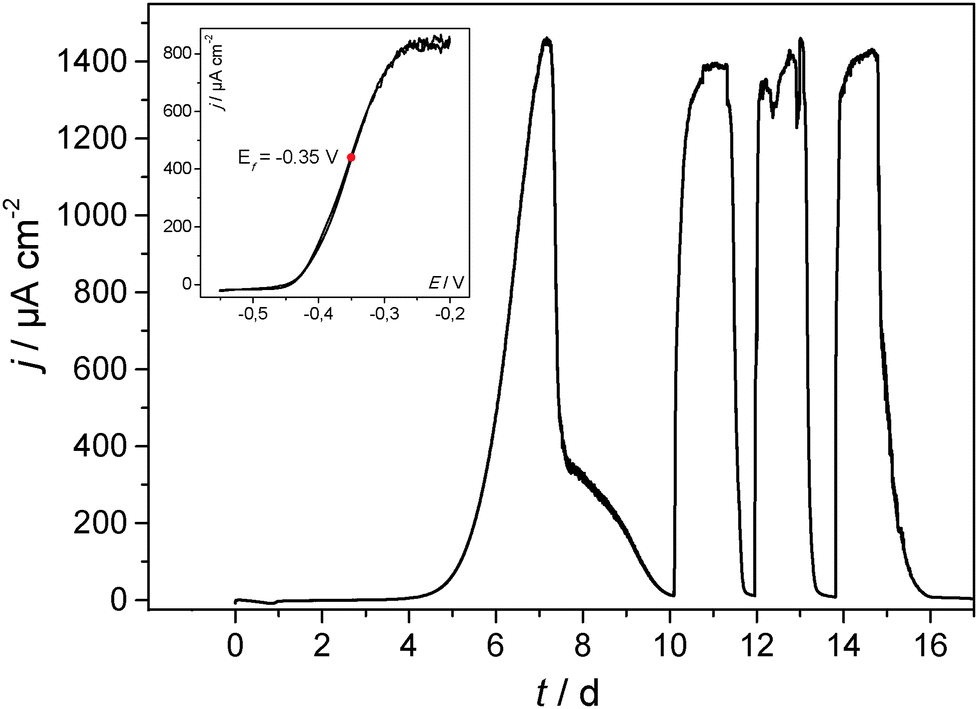 | ||
| Fig. 3 Main figure: cultivation and the resulting bioelectrocatalytic current generation of a secondary, acetate based electrochemically active biofilm at copper (copper sheet) in a model semi-batch experiment. The biofilm was cultivated in a half-cell setup under potentiostatic control. The electrode potential was −0.2 V (vs. Ag/AgCl). Inset: cyclic voltammogram recorded under turnover conditions. | ||
The voltammetry of the biofilm-copper electrode (Fig. 3 and Fig. S4, ESI†) is virtually identical with that of the carbon electrode (Fig. 2), illustrating that there are no specific (electrochemical) interactions between the bacteria and the respective electrode material, and apparently no remarkable differences in the biofilm composition. Voltammetric measurements in the sterile growth medium (depicted for copper in comparison to graphite in Fig. S5, ESI†) show that there is no redox signal/catalytic interaction of the pure metal with the components of the growth medium.
As illustrated by means of the optical images in Fig. 4, the formation of electrochemically active microbial biofilms takes place also on other metal surfaces, such as gold, silver or nickel. In all cases, a uniform and optically dense, reddish coloured biofilm is formed, supporting the above discussed dominance of Geobacter species in these biofilms.
 | ||
| Fig. 4 Digital photographs of the bare anode materials (MPG electrodes, left) and the corresponding biofilm electrodes (right). | ||
In order to systematically compare colonization and biofilm performance on metal electrodes in comparison to carbon, and in order to derive the principal suitability of the respective metals as anode material in bioelectrochemical systems, the following metals/alloys were studied in detail: gold, silver, copper, nickel, cobalt, titanium and stainless steel (SUS 304). The red columns in Fig. 4 summarize the mean maximum peak current densities (jmax) of secondary biofilms grown on these metals.
As Fig. 5 illustrates, the three noblest metals gold (1175 μA cm−2), silver (1119 μA cm−2) and copper (1515 μA cm−2) delivered a biofilm performance similar or even slightly higher than that of graphite (984 μA cm−2). Whereas for gold the result was expected, the biocompatibility of silver and copper is surprising, because of their frequent application as antimicrobial metals.
 | ||
| Fig. 5 Electrochemical performance of electrochemically active secondary biofilms cultivated on different metals and on polycrystalline graphite. Red columns: average maximum current densities achieved during semi-batch cultivation (see Fig. 1 and 2); blue columns: mean values of the slopes of the turnover cyclic voltammograms of the respective biofilm electrodes. The error bars indicate the standard deviations. Substrate: 10 mM acetate, temperature 35 °C. | ||
The oxides of the studied non-noble metals possess semiconducting properties40–43 and their thickness may vary between few nanometers to tens of nanometers.44,45 It may generally be assumed that – depending on thickness and conductivity – these passivating oxide layers play a major role as a barrier in the charge transfer process between the microorganism and the metals. As expected, the performance of the non-noble metals titanium, cobalt, nickel and stainless steel lies below that of graphite and of copper, silver and gold. Here, the highest performance was found for stainless steel (674 μA cm−2), followed by nickel (384 μA cm−2). Compared to the other electrode materials, the current densities at cobalt and titanium were negligible. In the case of titanium some biofilm formation was microscopically observed. Nevertheless, the electron transfer from biofilm to the electrode seems to be strongly hindered.
For a further evaluation of the electrocatalytic behaviour of the individual biofilm electrodes, the slopes of normalized turnover CVs (see inset figures in Fig. 2 and 3 for example voltammograms) were determined as a measure for the electrochemical reversibility of the electron transfer at the biofilm–electrode interface. A steep slope would stand for a Nernstian behaviour. This means that the electron transfer across the metal–biofilm interface is very fast (i.e., electrochemically reversible), and the redox state of the involved redox proteins follows the Nernst equation. Slower interfacial electron transfer kinetics (e.g., caused by an oxide layer on top of a metal surface) means that an additional overpotential needs to be applied in order to overcome the slower kinetics and to reach the desired redox states. This leads to decreasing slopes in the catalytic curves.14,46 For most of the metal samples, the slopes of the voltammograms corresponded to the current density achieved with these electrodes. An exception is nickel, which showed a rather high reversibility but comparatively low current density. The reason for this deviation may lie in specific interactions between the microbes and the electrode. For a deeper analysis of the role of these passivating layers, a kinetic, electrochemical impedance spectroscopy study is planned.
3.2 CLSM biofilm analysis
The thickness of the electrochemically active biofilms, as determined for the different electrode materials via CLSM analysis, was 117 ± 13 μm for graphite, 127 ± 11 μm for gold, 154 ± 10 μm for silver, 249 ± 21 μm for copper and 77 ± 9 μm for nickel. Fig. 6 correlates these thickness data with the bioelectrocatalytic current densities achieved at the electrode materials. | ||
| Fig. 6 Correlation of the electrocatalytic current density of electrochemically active secondary biofilms cultivated on different electrode materials and of the biofilm thickness as determined via CLSM analysis. | ||
Fig. 6 clearly shows that a higher current density corresponds to thicker biofilms. This finding – the correlation of current density and biofilm thickness or cell biomass is in agreement with literature e.g.ref. 47 and 48. It indicates the dominance of the bulk biofilm properties over the electrode–biofilm interface.
3.3 Electrochemical stability window
Except for gold, the redox potentials of all studied metals (see Table 1) lies below that of oxygen (at pH 7). Thus, for a potential application as anode materials in bioelectrochemical systems, the limits of the electrochemical stability of the respective metals have to be taken into account.| Electrode material | Specific electric resistivity [10−8 Ω m] | Standard oxidation potential vs. Ag/AgCl [V] |
|---|---|---|
| a Data from ref. 7. b Data from goodfellow. c Averaged value from different brands: “Properties and Characteristics of Graphite”, Entegris Inc. 2013. | ||
| Gold | 2.3a | 1.69 (Au/Au+) |
| Silver | 1.6a | 0.80 (Ag/Ag+) |
| Copper | 1.7a | 0.34 (Cu/Cu2+) |
| Nickel | 7.1a | −0.26 (Ni/Ni2+) |
| Cobalt | 5.6a | −0.28 (Co/Co2+) |
| S. Steel (SUS 304) | 71b | −0.41 (Fe/Fe2+) |
| Titanium | 39a | −1.37 (Ti/Ti3+) |
| Graphite, polycrystalline | 1590c | — |
As illustrated in Fig. 7, two major processes can be observed when metal electrodes are polarized towards positive potentials. Stainless steel and nickel are typical examples for passivated metals. Their oxidation layer protects the metal from becoming further oxidized. The dominant oxidation process at these metals is the oxygen evolution reaction. The reaction takes place at potentials above 1.2 V (vs. Ag/AgCl). The stability of the metals against oxidative dissolution requires the absence of certain ions, such as chloride, which are known to cause corrosion at stainless steel.
 | ||
| Fig. 7 Oxidative linear sweep voltammograms of selected metals (blank metal sheets), recorded in 0.1 M potassium nitrate solution. Scan rate 1 mV s−1. Additional linear sweep voltammograms of copper and stainless steel, recorded in filtrated, primary wastewater, are illustrated in Fig. S6 (ESI†). | ||
In the absence of ions that cause the precipitation of an insoluble, passivating salt layer (e.g., phosphate, carbonate), copper and silver undergo an oxidative dissolution. This reaction takes place at comparatively low potentials (Fig. 7). But would this impede the use of these metals as anode materials in bioelectrochemical systems? Fig. 8 puts the oxidative potential window of copper (measured in a nitrate solution) in relation to the bioelectrocatalytic acetate oxidation and to the oxygen reduction reaction at a platinum cathode.
 | ||
| Fig. 8 Linear sweep voltammogram of the copper oxidation (in a 0.1 M potassium nitrate solution) in relation to the bioelectrocatalytic acetate oxidation and the oxygen reduction at a platinum electrode at two different pH values. Scan rate 1 mV s−1. | ||
As Fig. 8 illustrates, the onset potential of the bioelectrocatalytic acetate oxidation is −0.4 V. The copper oxidation commences at an onset potential of +0.1 V, which provides an operative potential window of 0.5 V. In a microbial fuel cell, the oxidation power is delivered by an oxygen electrode. As shown in the figure, the oxygen reduction reaction at a platinum electrode has an onset potential of 0.1 V, which adjoins to the copper oxidation. Since the pH values of BES cathodes becomes generally alkaline during operation,49 the ORR potential shifts to more negative values (onset potential of 0 V at pH 9; Fig. 8), which lies clearly within the stability window of copper. Taking further into account the internal resistance of the electrochemical cells, MFC anodes are rarely polarized to potentials above 0 V (vs. Ag/AgCl). This means that copper (or silver) can be safely used well below their oxidation potential.
Zhu & Logan showed that copper corrosion prevents the formation of electrochemically active biofilms on copper anodes.50 Such an effect can occur especially during the BES start-up period, when biofilm formation still has to take place and the anode potential – due to the missing reduction power of a fully established microbial biofilm – may shift into the corrosion potential. Here, the copper ions are toxic for the planktonic cells in the bacterial inoculum, preventing biofilm formation. For this reason, care should be taken during the start-up of the bioelectrochemical system.
But what would happen when a copper–biofilm electrode would be polarized towards a potential at which copper is oxidized and copper ions are released from the metal into the biofilm? In a previous study, it was shown that electrochemically active biofilms are very robust in the presence of antibiotic compounds in the solution.18 The oxidation of copper may lead to the liberation of high concentrations of heavy metal ions at the metal–biofilm interface and thus damage the adjacent cell layers.51 Yet, despite the polarization of biofilm–copper electrode to a potential of 0.5 V (vs. Ag/AgCl), no remarkable impact on the bioelectrocatalytic activity of the biofilm was observed (data not shown). Different reasons for this resistance can be discussed. First of all, the presence of phosphate ions in the cultivation medium may lead to the formation of a passivating, insoluble layer of copper(II) phosphate, preventing the liberation of higher, cell-damaging copper ion concentrations. Similarly, biofilm inherent components, such as carbohydrates, proteins and nucleic acids52,53 may bind metal ions,54 thus also preventing damaging effects55 and producing passivating layers. A more detailed investigation is necessary to clarify these effects.
3.4 Economic and technologic considerations
An evaluation of the suitability of an electrode material must necessarily integrate economic considerations. Here, the major point of interest is the price of a respective electrode. For simplicity reasons, our analysis relies solely on the material price, based on world market prices. For a more detailed analysis, processing costs would have to be included.Table 2 summarizes the world market prices for selected electrode materials at the point of manuscript preparation. Gold and silver are not listed since it does not require a detailed analysis of the market to know that these noble metals can be excluded as electrode material for practical bioelectrochemical systems. For fundamental research, however, these metals will still be of great interest.
| Electrode material | Price per ton (US $) |
|---|---|
| a London metal exchange, 1 Feb. 2015, http://www.lme.com. b Global composite stainless steel price, Grade 304, MEPS international, Feb. 2015, http://www.meps.co.uk. c Averaged price, Northern graphite, Feb. 2015, http://www.northerngraphite.com. | |
| Copper | 5528a |
| S. Steel (SUS 304) | 2645b |
| Nickel | 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 898a 898a |
| Graphite (natural, large flakes) | 1450c |
Nickel showed a reasonable performance in the bioelectrochemical experiments, yet, as Table 2 shows, its high price (in combination with the relatively high ohmic resistivity – see Table 1) would not favour its use in bioelectrochemical systems. Following Table 2, graphite has the lowest price per ton electrode material followed by stainless steel and copper.
Would this order favour graphite and rule out copper as electrode material? Therefore, also the amount of material needed to achieve a certain performance has to be considered. Table 3 illustrates the example of a simplified cost estimation for an electrode with the size of 1 m2. A polycrystalline graphite electrode with a thickness of 10 mm and a specific conductivity of 6.3 × 104 S m−1 was used as the reference system. Using the gravimetric density of polycrystalline graphite the electrode weight and the deriving, mass based costs were calculated (in the case of polycrystalline graphite, the costs were estimated based on the price for natural graphite flakes).
| Electrode material | Specific conductivitya (S m−1) | Material density (g cm−3) | Electrode thicknessb (mm) | Electrode weightc | Electrode costsd (US $) |
|---|---|---|---|---|---|
| a The specific conductivity was calculated from as the reciprocal of the specific resistivity, Table 2. b The required electrode thickness was calculated from the specific conductivity of the metal (copper/stainless steel) in relation to that of graphite. c The electrode weight was calculated from the volume of the electrode (1 m2 × thickness) multiplied with the gravimetric density. d The electrode costs were calculated on the basis of the used weight and the commodity costs per ton (Table 2). | |||||
| Copper | 58 × 106 | 8.9 | 0.011 | 97 g | 0.53 |
| S. Steel (SUS 304) | 1.4 × 106 | 8.0 | 0.447 | 3.6 kg | 9.47 |
| Graphite, polycrystalline | 6.3 × 104 | 1.8 | 10 | 18 kg | 26.1 |
Based on the specific conductivity of copper and stainless steel, the necessary thickness of the respective metal electrodes to achieve the same conductivity as the graphite electrode, the deriving electrode weight and costs were calculated. In this calculation, mechanical issues like a minimum mechanical strength of the electrode were not considered.
Table 3 clearly shows that the electrode with the significantly lowest material costs (0.53 $) is the copper electrode, followed by stainless steel (10.53 $). The graphite electrode is the most expensive electrode (26 $). The key issue for the electrode costs is the factor 920 lower specific resistivity of copper in comparison to graphite, allowing to minimize the electrode thickness and thus the amount of electrode material.
Certainly, a copper electrode with a thickness of 11 μm (this is a typical thickness range of copper foils commercially used in lithium ion batteries) would require mechanical stabilization. This could be achieved by, e.g., by cell stacking. It has to be stated that even a 1 cm thick graphite electrode is far from being mechanically stable and needs to be stabilized as well.
4 Conclusion
In this study we have analysed the suitability of gold, silver, copper, nickel, cobalt, titanium and stainless steel as anode materials for microbial fuel cells and related bioelectrochemical systems and compared it to the current benchmark material – polycrystalline graphite. All materials except cobalt and titanium allowed the cultivation of well-performing electrochemically active biofilms. The highest performance was achieved at copper electrodes. We demonstrated that the oxidative dissolution potential of copper lies outside the potential range of a BES anode. Under the conditions of this study, no toxic effects of copper against the electrochemically active bacteria were observed. Whether the missing antimicrobial effect of copper and silver can be ascribed to a tolerance of the electrochemically active bacteria against these metals or simply to the comparatively negative redox potentials in the anaerobic environment, can so far not be answered.An exemplary price estimation showed that, due to the high conductivity of copper, electrodes can be fabricated at a minimum electrode thickness and thus at material costs that are considerably lower than those of graphite and also stainless steel.
As the essential finding of this study, copper can clearly be proposed as a high performance electrode material for microbial bioelectrochemical systems.
Acknowledgements
The authors gratefully thank the Deutsche Forschungsgemeinschaft (DFG) for support (DFG grant SCHR 753/10-1). Special thanks to Alexander Fröhlich (research group of Prof. Dr. Krull, Institute for Biochemical Engineering, TU Braunschweig) for performing the biofilm staining experiments.References
- U. Schroeder, F. Harnisch and L. T. Angenent, Energy Environ. Sci., 2015, 8, 513–519 CAS.
- M. Zhou, M. Chi, J. Luo, H. He and T. Jin, J. Power Sources, 2011, 196, 4427–4435 CrossRef CAS PubMed.
- S. Chen, G. He, A. A. Carmona-Martinez, S. Agarwal, A. Greiner, H. Hou and U. Schröder, Electrochem. Commun., 2011, 13, 1026–1029 CrossRef CAS PubMed.
- S. Chen, G. He, Q. Liu, F. Harnisch, Y. Zhou, Y. Chen, M. Hanif, S. Wang, X. Peng, H. Hou and U. Schröder, Energy Environ. Sci., 2012, 5, 9769 CAS.
- K. Guo, A. Prévoteau, S. A. Patil and K. Rabaey, Curr. Opin. Biotechnol., 2015, 33, 149–156 CrossRef CAS PubMed.
- H. Richter, K. P. Nevin, H. Jia, D. A. Lowy, D. R. Lovley and L. M. Tender, Energy Environ. Sci., 2009, 2, 506–516 CAS.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, Internet V., 2005 Search PubMed.
- P. A. Selembo, M. D. Merrill and B. E. Logan, J. Power Sources, 2009, 190, 271–278 CrossRef CAS PubMed.
- The Penn State Research Foundation, USA., 2010, 40pp., Cont.–in–part of U.S. Ser. No. 177,962.
- A. Kuzume, U. Zhumaev, J. Li, Y. Fu, M. Füeg, A. Esteve-Nuñez and T. Wandlowski, Electrochim. Acta, 2013, 112, 933–942 CrossRef CAS PubMed.
- R. M. Snider, S. M. Strycharz-Glaven, S. D. Tsoi, J. S. Erickson and L. M. Tender, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15467–15472 CrossRef CAS PubMed.
- D. Pocaznoi, B. Erable, M.-L. Delia and A. Bergel, Energy Environ. Sci., 2012, 5, 5287 CAS.
- H. Yi, K. P. Nevin, B.-C. Kim, A. E. Franks, A. Klimes, L. M. Tender and D. R. Lovley, Biosens. Bioelectron., 2009, 24, 3498–3503 CrossRef CAS PubMed.
- D. Pocaznoi, A. Calmet, L. Etcheverry, B. Erable and A. Bergel, Energy Environ. Sci., 2012, 5, 9645–9652 CAS.
- C. Dumas, A. Mollica, D. Féron, R. Basseguy, L. Etcheverry and A. Bergel, Bioresour. Technol., 2008, 99, 8887–8894 CrossRef CAS PubMed.
- G. Grass, C. Rensing and M. Solioz, Appl. Environ. Microbiol., 2011, 77, 1541–1547 CrossRef CAS PubMed.
- U. Krause, Pharm. Verfahrenstech. Heute, 1983, 1, 103–107 CAS.
- S. Patil, F. Harnisch and U. Schröder, ChemPhysChem, 2010, 11, 2834–2837 CrossRef CAS PubMed.
- E. Gamal, A. R. Mohamed, M. Bahgat and A. Dahshan, El-Minia Sci. Bull. Vol. Physic. Sect., 2013, 24, 1–12 Search PubMed.
- F. Kargi and S. Eker, J. Chem. Technol. Biotechnol., 2007, 82, 658–662 CrossRef CAS PubMed.
- D. Millo, F. Harnisch, S. A. Patil, H. K. Ly, U. Schröder and P. Hildebrandt, Angew. Chem., Int. Ed., 2011, 50, 2625–2627 CrossRef CAS PubMed.
- H. K. Ly, F. Harnisch, S. F. Hong, U. Schröder, P. Hildebrandt and D. Millo, ChemSusChem, 2013, 6, 487–492 CrossRef CAS PubMed.
- W. E. Balch, G. E. Fox, L. J. Magrum, C. R. Woese and R. S. Wolfe, Microbiol. Rev., 1979, 43, 260–296 CAS.
- Y. Liu, F. Harnisch, K. Fricke, R. Sietmann and U. Schröder, Biosens. Bioelectron., 2008, 24, 1012–1017 CrossRef PubMed.
- A. Baudler, S. Riedl and U. Schröder, Front. Energy Res., 2014, 2, 1–6 Search PubMed.
- T. R. Corle and G. S. Kino, Confocal Scanning Optical Microscopy and Related Imaging Systems, Academic Press, San Diego, 1996 Search PubMed.
- B. V. R. Tata and B. Raj, Bull. Mater. Sci., 1998, 21, 263–278 CrossRef CAS.
- S. R. Wood, J. Kirkham, P. D. Marsh, R. C. Shore, B. Mattress and C. Robinson, J. Dent. Res., 2000, 79, 21–27 CrossRef CAS PubMed.
- J. Wei, P. Liang and X. Huang, Bioresour. Technol., 2011, 102, 9335–9344 CrossRef CAS PubMed.
- F. Harnisch, C. Koch, S. A. Patil, T. Hübschmann, S. Müller and U. Schröder, Energy Environ. Sci., 2011, 4, 1265–1267 CAS.
- B. Cercado, N. Byrne, M. Bertrand, D. Pocaznoi, M. Rimboud, W. Achouak and A. Bergel, Bioresour. Technol., 2013, 134, 276–284 CrossRef CAS PubMed.
- S. F. Ketep, A. Bergel, M. Bertrand, W. Achouak and E. Fourest, Biochem. Eng. J., 2013, 73, 12–16 CrossRef CAS PubMed.
- K. Rabaey, G. Lissens, S. D. Siciliano and W. Verstraete, Biotechnol. Lett., 2003, 25, 1531–1535 CrossRef CAS.
- L. Huang and B. E. Logan, Appl. Microbiol. Biotechnol., 2008, 80, 655–664 CrossRef CAS PubMed.
- F. Harnisch, C. Koch, S. A. Patil, T. Hübschmann, S. Müller and U. Schröder, Energy Environ. Sci., 2011, 4, 1265 CAS.
- C. Koch, F. Harnisch, U. Schröder and S. Müller, Front. Microbiol., 2014, 5, 273 Search PubMed.
- K. Fricke, F. Harnisch and U. Schröder, Energy Environ. Sci., 2008, 1, 144 CAS.
- S. M. Strycharz-Glaven and L. M. Tender, ChemSusChem, 2012, 5, 1106–1118 CrossRef CAS PubMed.
- A. A. Carmona-Martinez, F. Harnisch, L. A. Fitzgerald, J. C. Biffinger, B. R. Ringeisen and U. Schröder, Bioelectrochemistry, 2011, 81, 74–80 CrossRef CAS PubMed.
- K. Hübner and G. Leonhardt, Phys. Status Solidi, 1975, 68, K175–K179 CrossRef PubMed.
- T. L. Barr, J. Phys. Chem., 1978, 82, 1801–1810 CrossRef CAS.
- M. Thieme, D. Scharnweber, L. Drechsler, C. Heiser, B. Adolphi and A. Weiss, J. Nucl. Mater., 1992, 189, 303–317 CrossRef CAS.
- D. J. Blackwood and L. M. Peter, Electrochim. Acta, 1989, 34, 1505–1511 CrossRef CAS.
- Y. Sul, C. B. Johansson, S. Petronis, A. Krozer, Y. Jeong, A. Wennerberg and T. Albrektsson, Biomaterials, 2002, 23, 491–501 CrossRef CAS.
- C.-C. Shih, C.-M. Shih, Y.-Y. Su, L. H. J. Su, M.-S. Chang and S.-J. Lin, Corros. Sci., 2004, 46, 427–441 CrossRef CAS.
- S. F. Ketep, A. Bergel, A. Calmet and B. Erable, Energy Environ. Sci., 2014, 7, 1633 CAS.
- S. Ishii, K. Watanabe, S. Yabuki, B. E. Logan and Y. Sekiguchi, Appl. Environ. Microbiol., 2008, 74, 7348–7355 CrossRef CAS PubMed.
- G. He, Y. Gu, S. He, U. Schröder, S. Chen and H. Hou, Bioresour. Technol., 2011, 102, 10763–10766 CrossRef CAS PubMed.
- F. Zhao, F. Harnisch, U. Schröder, F. Scholz, P. Bogdanoff and I. Herrmann, Environ. Sci. Technol., 2006, 40, 5191–5199 Search PubMed.
- X. Zhu and B. E. Logan, J. Chem. Technol. Biotechnol., 2014, 89, 471–474 CrossRef CAS PubMed.
- S. Chen, P. Wang and D. Zhang, Corros. Sci., 2014, 87, 407–415 CrossRef CAS PubMed.
- H.-C. Flemming and J. Wingender, Nat. Rev. Microbiol., 2010, 8, 623–633 CAS.
- J. Wingender, T. R. Neu and H. C. Flemming, Microbial extracellular polymeric substances, Springer, 1999 Search PubMed.
- G.-P. Sheng, J. Xu, H.-W. Luo, W.-W. Li, W.-H. Li, H.-Q. Yu, Z. Xie, S.-Q. Wei and F.-C. Hu, Water Res., 2013, 47, 607–614 CrossRef CAS PubMed.
- G. Bitton and V. Freihofer, Microb. Ecol., 1978, 25, 119–125 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ee00866b |
| ‡ Both authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |