
Direct observation of an inhomogeneous chlorine distribution in CH3NH3PbI3−xClx layers: surface depletion and interface enrichment†
Abstract
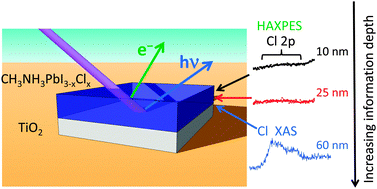
We have used hard X-ray photoelectron spectroscopy (HAXPES) at different photon energies and fluorescence yield X-ray absorption spectroscopy (FY-XAS) to non-destructively investigate CH3NH3PbI3−xClx perovskite thin films on compact TiO2. This combination of spectroscopic techniques allows the variation of information depth from the perovskite layer surface to the top-most part of the underlying compact TiO2 layer. We have taken advantage of this to understand the distribution of chlorine throughout the perovskite/TiO2 layer stack. No Cl is detected using HAXPES, indicating surface depletion of Cl and allowing us to place an upper limit on the amount of Cl in the perovskite layer: x < 0.07 and x < 0.40 to depths of ∼10 nm and ∼26 nm, respectively, beneath the perovskite film surface. Our FY-XAS results, however, demonstrate that there is a higher average concentration of Cl throughout the perovskite layer than at the surface (x > 0.40) consistent with both enhanced concentrations of Cl deep beneath the perovskite film surface and near the CH3NH3PbI3−xClx perovskite/TiO2 interface. The consequences of this distribution of Cl in the CH3NH3PbI3−xClx perovskite layer on device performance are discussed.
- This article is part of the themed collection: Celebrating the 150th anniversary of the German Chemical Society
 Please wait while we load your content...
Please wait while we load your content...

