
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electronic structures of cyclometalated palladium complexes in the higher oxidation states†
Bao N.
Nguyen‡
a,
Luis A.
Adrio
a,
Tim
Albrecht
a,
Andrew J. P.
White
a,
Mark A.
Newton
b,
Maarten
Nachtegaal
c,
Santiago J. A.
Figueroa§
 b and
King Kuok (Mimi)
Hii
*a
b and
King Kuok (Mimi)
Hii
*a
aDepartment of Chemistry, Imperial College London, Exhibition Road, South Kensington, London SW7 2AZ, UK. E-mail: mimi.hii@imperial.ac.uk
bEuropean Synchrotron Radiation Facility (ESRF), 6 rue Jules Horowitz, 38043, Grenoble, France
cPaul Scherer Institut, 5232 Villigen PSI, Switzerland
First published on 18th August 2015
Abstract
The electronic and redox properties of a series of cyclometalated Pd complexes with oxidation states of +2, +3 and +4 were examined using a range of currently available spectroscopic and electrochemical techniques. Changes in metal–ligand bond lengths were established by X-ray crystallography and correctly predicted by DFT calculations, from which the frontier orbitals and partial atomic charges can be obtained. X-ray absorption spectroscopy (XAS) revealed interesting XANES features that suggest a synergistic relationship between metal–ligand interactions. The electrochemical study of the Pd(II) dimer was found to contain two sequential oxidative potentials indicative of a weak metal–metal interaction.
Introduction
The direct activation of Ar–H bonds towards C–C and C–X bond forming reactions is of significant interest, as they have the potential to deliver greater step- and atom-economy than ‘classical’ cross-coupling reactions. Indeed, ‘C–H activation of aromatics without the need for haloaromatics’ was identified as one of ‘the most aspirational reactions’ and an ‘unmet need’, in a survey conducted by the ACS Green Chemistry Institute's Pharmaceutical Roundtable some years ago,1 and still remains as one of the most significant challenges for synthesis in the 21st century. Palladium catalysis plays a critical role within this arena: many reactions can be performed simply by using Pd(OAc)2 without extraneous ligands, in the presence of an appropriate oxidant.2–5 Often referred to as ‘oxidative coupling’ reactions, it is believed that the reactions involve palladacycle intermediates of higher oxidation states (+3 and/or +4).6–9 A classic example is the Pd-catalysed ortho-acetoxylation of 2-phenylpyridine (2-PhPy, Scheme 1) and its closely-related benzo[h]quinoline derivatives. The reaction proceeds in the presence of Pd(OAc)2 and iodosobenzene diacetate, PhI(OAc)2, which functions simultaneously as an oxidant and a source of the acetate. | ||
| Scheme 1 Pd-catalysed ortho-acetoxylation of 2-phenylpyridine. | ||
First reported by the group of Sanford,10 the reaction was further investigated by Ritter and co-workers.11 It is accepted that the reaction initiates with a chelated-assisted cyclometalation to form a stable diacetato-bridged palladacycle 1 (Scheme 2, step i), which undergoes oxidative addition with PhI(OAc)2 to form a diamagnetic bimetallic Pd(III) intermediate 2 (step ii), prior to reductive elimination (step iii) to afford the ortho-acetoxylated product. An earlier proposal favoured the formation of Pd(IV) catalytic intermediates. Although a discreet monomeric complex was never isolated directly from the catalytic mixture, the viability of the +4 oxidation state was provided by the reaction of PhI(OAc)2 with a bis-C,N-chelated complex (3) to give the octahedral Pd(IV) complex 4, which undergoes reductive elimination at 60 °C spontaneously to afford the expected product (Scheme 3).
 | ||
| Scheme 2 Proposed catalytic steps of the Pd-catalyzed acetoxylation of 2-PhPy. | ||
 | ||
| Scheme 3 Oxidative addition of PhI(OAc)2 to complex 3. | ||
These studies provided a series of palladacycles 1–4 with formal oxidation states of +2, +3 or +4, with only 2-PhPy and acetate as ligands.12 Thus far, characterisation of these complexes has been achieved by NMR spectroscopy, X-ray crystallography and, more recently, mass spectrometry.13 While these techniques provide useful structural information, they do not offer direct information on the electronic properties of these cyclometalated complexes; particularly, electronic (re)distribution that occurs upon the increase in oxidation state that may account for their subsequent reactivity. The aim of this work is to combine spectroscopic measurements with theoretical methods to afford direct comparisons and insights into the electronic and redox properties of these closely-related palladacycles. As the extended π-system in 2-phenyl pyridine can also be redox active, we wish to determine the charge distribution and the extent of ligand participation14 in these compounds.
Results and discussion
X-ray crystallography and DFT calculations
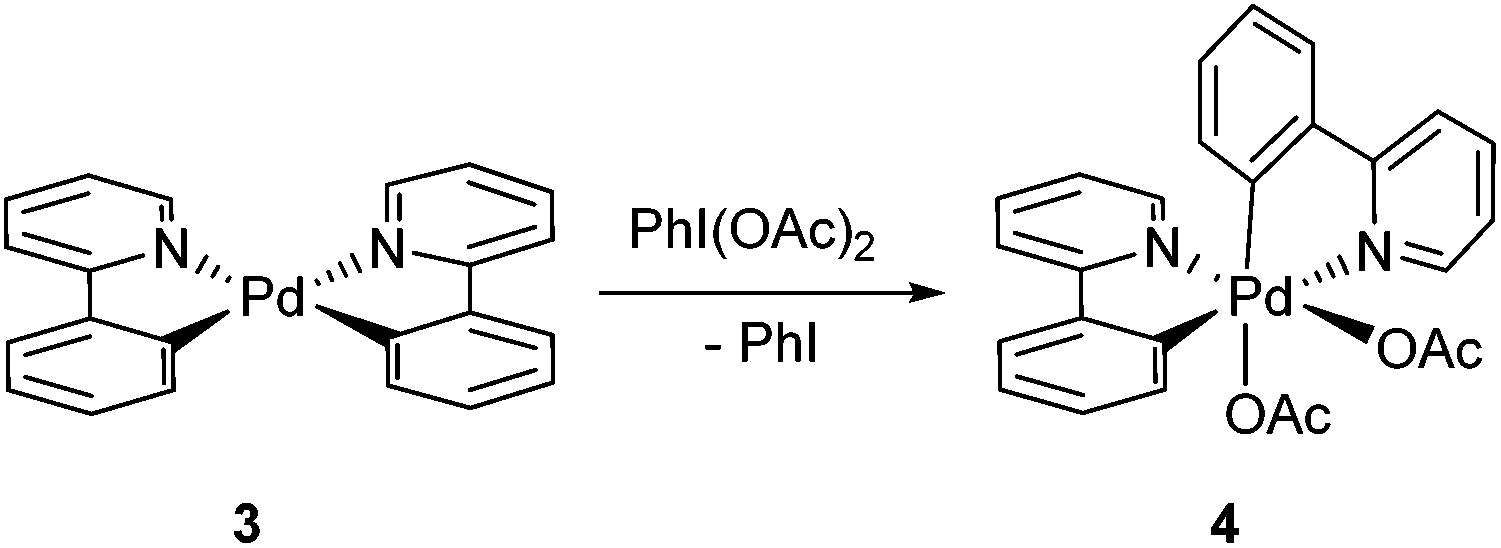
Solid-state structures for Pd(II) complexes 115 and 216 had been previously established by X-ray crystallography. In this study, crystals suitable for X-ray diffraction were also obtained for the bis-chelated complexes 3 and 4 (Fig. 1). The structure of 3 shows the expected twisted square planar coordination of two chelated 2-PhPy ligands to the palladium centre, the two [Pd,N,C] coordination planes being inclined to each other by ca. 24°. The coordination distances of the two ligands are very similar, with the Pd–N bond length being ca. 0.13 Å longer than its Pd–C counterpart in both instances. The X-ray crystal structure for complex 4 had been previously reported as a C2-symmetrical molecule (CCDC code: LUFQIN) with 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 disorder between the coordinated C and N atoms.21 In this work, however, we were able to obtain an ordered crystal structure showing an asymmetric, all-cis geometry, which is commensurate with the solution state structure observed by NMR spectroscopy (more extensive discussion on the structure determination is provided in the ESI†).
:50 disorder between the coordinated C and N atoms.21 In this work, however, we were able to obtain an ordered crystal structure showing an asymmetric, all-cis geometry, which is commensurate with the solution state structure observed by NMR spectroscopy (more extensive discussion on the structure determination is provided in the ESI†).
 | ||
| Fig. 1 Molecular structures of complexes 3 and 4. | ||
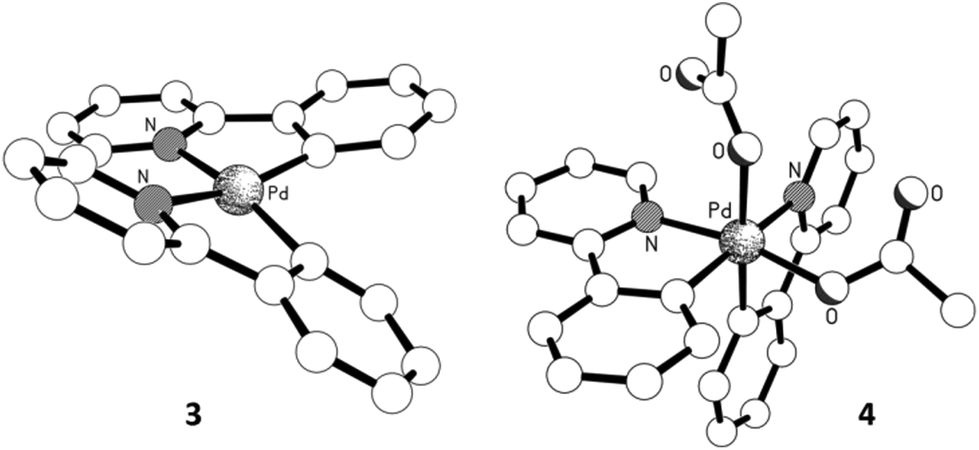
As the oxidation state of the Pd increases from +2 to +3, the primary coordination sphere of its oxidative adduct might be expected to contract due to increased charge. However, a comparison of the structural parameters of complexes 1 and 2 showed that this only appears to affect the mutually trans Pd–O and Pd–C bonds, where a shortened Pd–O bond was counteracted by a corresponding lengthening of the Pd–C bond (by ca. 0.02 Å). In contrast, the mutually opposite Pd–N and Pd–O bonds remained essentially the same (Fig. 2). The same trends were also observed in complexes 3 and 4. In the latter, the bond lengths in the primary coordination sphere of the highly asymmetric oxidative adduct are influenced by different trans effects; nevertheless, the equivalent Pd–N and Pd–C bonds (trans to C- and N-donor atoms, respectively) were found to be largely unaffected by the change in the formal oxidation state from +2 to +4. In terms of the covalent bonds, no statistically significant changes were observed in the bond lengths composing the chelate ring of the 2-PyPh ligand. Among the acetate ligands, the one positioned trans to the C(σ)-donor group in the Pd(IV) complex 4 is particularly notable by having practically identical C–O bond lengths of ca. 1.23 Å, while in all other instances the coordinated C–O bond is substantially longer.17
 | ||
| Fig. 2 Experimental bond lengths (Å) of the primary coordination sphere (solid state structure). Calculated bond lengths are given in red in parenthesis (dichloroethane solvent field applied). | ||
In order to rationalise these structural features, DFT calculations were performed, whereby the structures of all four complexes were optimised using the WB97XD functional18 with a mixed Def2TZVP basis set19 at Pd, 6-31G(d,p) basis set for all other atoms, and solvent correction (CPCM, dichloroethane). Taking into account the differences between solid and solution structures, the calculated bond lengths were in very good accord with the experimental values, with changes in the primary coordination spheres accurately predicted (Fig. 2), including the greater trans effects imposed by C- over N-donor atoms in the octahedral complex 4.
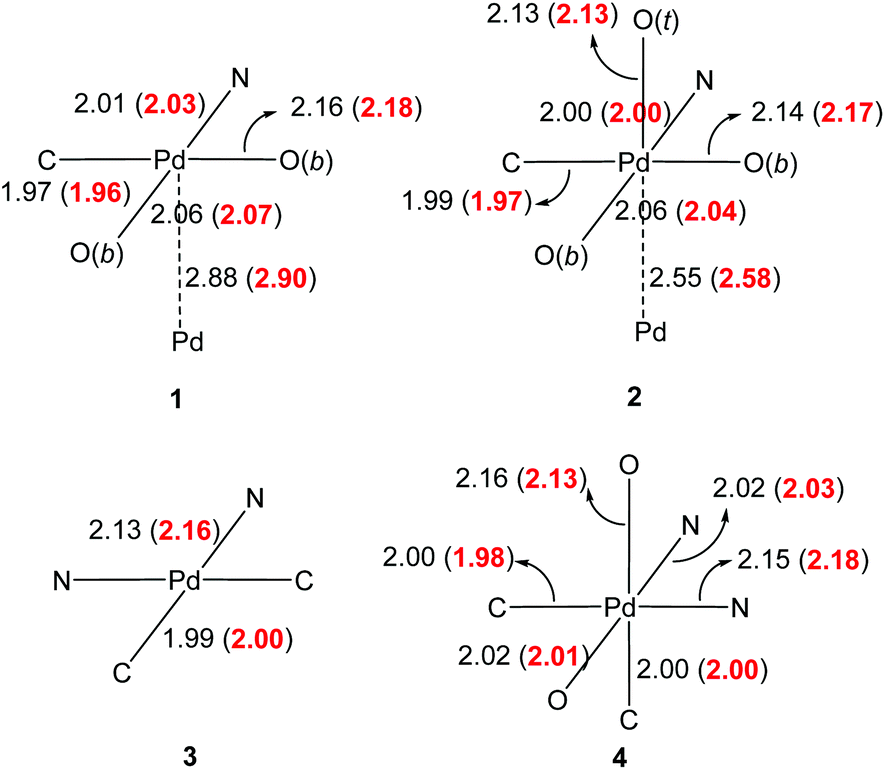
Good agreement in the predicted bond lengths allows us to analyse the frontier orbitals of these molecules (Fig. 3). In terms of dimeric complexes 1 and 2, it is interesting to note the change from antibonding and bonding interactions between the metal centres, thus supporting the development of M–M interactions during the oxidation process. With the exception of complex 1, there is substantial ligand participation by both 2-phenylpyridyl and acetate, which are likely to work synergistically to stabilize the oxidative adducts. The participation of the acetate ligand trans to the C(σ)-donor group in complex 4 is particularly notable. Natural bond order analysis revealed nearly identical C–O bonds, which accounts for the shorter bond lengths observed previously.
 | ||
| Fig. 3 HOMO's of compounds 1–4. | ||
The MO analysis was further supplemented by implementing Charge Model 5 (CM5), developed by Cramer and Truhlar20 to afford prediction of partial atomic charges that are essentially independent of the level of theory and basis set (Fig. 4). As expected, positive charges of the Pd metal centre increased by 50 and 100% upon oxidation to Pd(III) and Pd(IV), respectively. Interestingly, changes in the ligand charges are not only dependent on the donor atom, but also their relative positions. In the case of Pd(III) complex 2, the axial acetate carries a greater charge than its bridging counterpart. At the same time, only a slight reduction in the partial charge on the C(σ)-donor atom was observed (−0.14 to −0.12). In comparison, a more dramatic electronic redistribution occurred in the transformation of complex 3 to the Pd(IV) adduct 4. In this case, a significant reduction of C(σ)-donor atom charges was observed (from −0.16 to ca. −0.1, irrespective of the trans-donor atom). The greater charge separation between the reactive centres, combined with the orbital contributions obtained from the DFT study, supports the experimental observations – the greater propensity for 4 to undergo reductive elimination to generate a C–O bond, compared to 2.
 | ||
| Fig. 4 Partial atomic charges in complexes 1–4 obtained by CM5 analysis. | ||
In the next part of our study, we attempted to obtain further experimental verification for the above analysis, in particular, whether changes in the metal's electronic environment can be detected spectroscopically.
X-ray absorption studies
In a XAS measurement, an electron is excited from the metal's inner shell (in this case 1s, or ‘K-edge’) to its vacant valence orbitals (4d and 5p for Pd) or, when given more energy, to the continuum. This gives rise to an X-ray absorption spectrum corresponding to the local unoccupied density of states that can be directly correlated to the oxidation state. In the XAS spectrum of Pd, the dominant contribution arises from 1s → 5p transitions, giving rise to ‘p peaks’ (ΔL = 1, generally accepted as the K-edge). Other transitions into empty orbitals, i.e. 1s → 4d transitions (‘d peaks’) give rise to pre-edge features in the XAS spectra of non-centrosymmetric complexes, due to mixing of (partially occupied) 4d orbitals with 5p-orbitals.21Changes in the metal's oxidation state are expected to affect the positions of both the p- and d-peaks. For Pd complexes, an increase in the oxidation state of the metal can cause either an increase or decrease in the K-edge energy (p-peak) depending on whether the oxidation is metal- or ligand-based. For example, in a previous study of (diphosphine)Pd complexes, an increase in the oxidation state from Pd(0) to Pd(II) was reported to lower the K-edge energy.22 Conversely, an increase in the K-edge energy was observed in the oxidation of a cyclometalated pentaphenylferrocene-oxazoline Pd(II) complex to a paramagnetic Pd(III) species.23 In the present study, the relative positions of the d-peaks are also of interest,24 as they are expected to be more sensitive to ligand effects (metal–ligand interactions operate primarily through the d-orbitals).
XAS spectra of complexes 1 and 3 were recorded at the Pd K-edge (24.35 keV), initially in the solid state (pressed discs), and then as solutions in 1,2-dichloroethane (DCE). The oxidative adducts 2 and 4 are unstable in the solid state and have limited stability in solution25 at ambient temperature. For these compounds, PhI(OAc)2 was added to the Pd(II) precursors, to generate these complexes in situ.26 Due to the diluted sample, acquisition of multiple scans was required in order to improve the signal-to-noise ratio. The experiments were repeated with freshly prepared solution, such that spectra of sufficient quality can be extracted and inspected, before they were combined for analysis. The EXAFS regions of complexes 1–4 recorded as solution samples were compared with the structural parameters extracted from the X-ray diffraction studies. The goodness-of-fit of the primary coordination shell confirmed that the metal–ligand distances in the solid-state structures are maintained in solution (Fig. S4–S7, ESI†), although the poor resolution and the presence of multiple scatterers did not permit accurate determination of structural parameters beyond the primary coordination environment.
The expected increase in the absorption energy was observed in the XANES spectra for 3 → 4 (+2 → +4). However, the XANES regions for complexes 1 and 2 appeared to be identical (Fig. 5A). The XANES can be deconvoluted into p and d peaks by their transformation into negative second derivative plots (Fig. 5B), revealing three distinct features I–III.27 Feature I at lower-energy is assigned as the d-peak, while the more energetic features II and III are the p-peaks. In the case of complexes 1 and 2, the d peak remained unchanged, while the p peaks decreased and increased in energies, respectively. This suggests the presence of ligand contributions that can exert an opposite effect on the different energy states. In comparison, lowering in energy of the d-peak of complex 4 is accompanied by a rise in energy of both the p-peaks, relative to that of complex 3, which is expected for a metal-based oxidation process.
 | ||
| Fig. 5 A = normalised Pd–K-edge XAS of complexes 1–4; B = second-derivative XAS spectra of complexes 1–4; C = energetic positions of the features corresponding to d- and p-peaks. | ||
Electrochemistry
During the course of this work, the redox behaviour of the dinuclear complex 1 by cyclic voltammetry (CV) in acetonitrile (the reaction solvent – Scheme 1) was investigated. In a study by Bercaw and Green,28 two irreversible 1-electron oxidative potentials at +0.43 and +1.4 V (vs. Fc/Fc+) were recorded in acetonitrile, which were attributed to PdII–PdII → PdII–PdIII and PdII–PdIII → PdIII–PdIII couples, respectively. More recently, the electrochemical behaviour of complex 1 was re-examined by Dudkina et al.,29 where two oxidative potentials were similarly observed at +0.88 and +1.82 V (vs. SCE). However, these were reported to be 2-electron transitions, leading the authors to reassign these to PdII–PdII→PdIII–PdIII and PdIII–PdIV→PdIV–PdIV transitions, respectively.Comparing these two reports, the assignment by Dudkina et al. is more consistent with the relatively low reductive potential of PhI(OAc)2 (vide infra), which is able to oxidize complex 1 to 2 (PdII–PdII → PdIII–PdIII), and also used as an oxidant in the catalytic process. If this is the case, it was speculated that the two-electron transition observed at the lower potential could be the result of two successive 1-electron oxidations, due to M⋯M interactions.28,30 To pursue this further, the electrochemistry of complex 1 was revisited by duplicating the CV measurements under the reported conditions: using a glassy carbon (GC) electrode with [Bu4N][PF6] as an electrolyte in acetonitrile. At a scan rate of 1 V s−1, we were indeed able to resolve an additional, quasi-reversible transition at +0.28 V, in addition to the previously reported redox transition at ∼+0.5 V (vs. Fc/Fc+) (Fig. 6A). The two redox potentials were observed reproducibly using a Au(111) electrode, although the signal at higher potential appeared to be less prominent on the more reactive Au surface (S8, ESI†). The peak current ratio was found to depend strongly on the scan rate (Fig. 6B), changing from ∼0.7 at 1.0 V s−1 to about ∼0.15 at 0.1 V s−1, which may explain why it was not observed in earlier experiments (recorded at a scan rate of 0.1 V s−1). At the same time, the peak observed on the reversed scan at ca. +0.23 V also becomes more prominent at higher scan rates.
 | ||
| Fig. 6 A (top) CV of complex 1 recorded using a GC electrode (0.1 M [Bu4N]PF6 in CH3CN, 1 V s−1) in the low potential region (grey dotted line = background scan). B (bottom) at different scan rates. | ||
Hence, we have identified successive PdII–PdII → PdII–PdIII and PdII–PdIII → PdIII–PdIII transitions of complex 1 to occur in acetonitrile at redox potentials of +0.28 and ∼+0.5 V (vs. Fc/Fc+), respectively. The mixed-valent complex is highly unstable and can only be observed (as a reversible oxidation) at high scan rates. The revised oxidative potentials of complex 1 are commensurate with the use of PhI(OAc)2 as an effective 2-electron oxidant. It is known that in the presence of an acid, the hypervalent iodonium reagent dissociates in acetonitrile to form phenylacetoxyiodonium ions that undergo irreversible 2-electron reduction at −0.91 V (vs. SCE) to the corresponding iodobenzene and acetate ion (Scheme 4),31i.e. the pre-dissociation of PhI(OAc)2 generates nucleophilic acetate ions, as well as the active oxidant [PhI(OAc)]+ with the requisite over-potential to effect the transformation of 1 to 2, but not to PdIV.
 | ||
| Scheme 4 Redox chemistry of PhI(OAc)2. | ||
Conclusions
A number of spectroscopic and theoretical techniques have been applied to examine the electronic features of two 2-PhPy–Pd complexes, as they undergo oxidative addition reactions with PhI(OAc)2. Highly complementary information on the structure and reactivity of these complexes can be obtained from this study, offering a coherent picture of the electronic nature of these complexes: (1) a study of the solid-state structures of the complexes revealed some interesting features, with the ligands trans-effect being the overriding factor in M–L bond lengths, rather than changes in the formal oxidation states; (2) an appropriate method has been found for the DFT calculation, which correctly predicts the changes in M–L distances upon oxidative addition. The corresponding analysis of the frontier orbital gave insights into the participation of ligands in the stability of the oxidative adducts; (3) charge distributions in these metal complexes were revealed by CM5 calculations; (4) XAS studies revealed that the oxidation of a Pd(II) palladacycle 3 to Pd(IV) adduct 4 is predominantly a metal-based process (rise in K-edge), while an adjustment in the metal–ligand interaction was observed in the oxidative addition of a Pd(II) dimer to a Pd(III) dimer (opposite shifts in p-peaks); and (5) during the course of this work, the oxidative potentials for complex 1 in acetonitrile have been revised, commensurate with the use of PhI(OAc)2 as an oxidant. This work provides a useful benchmark for future studies of similar palladacycles with different ligands, while highlighting additional challenges involve further refinement of these experiments to afford more accurate predictions/quantification of electronic properties that are relevant to redox catalysis. The practical and theoretical challenges of determining the structure of discreet molecular complexes (exacerbated by their limited solubility) are also highlighted in this work. This will require further design of sample environments/acquisition and detection methods to achieve better resolution of EXAFS spectra.Acknowledgements
This work was supported by an EPSRC grant: ‘Elucidate and Separate’ (ELSEP, grant number: EP/G070172/1), with further contributions from the Pharmacat Consortium (AstraZeneca, GlaxoSmithKline and Pfizer). Travel to the SLS was supported by the European Commission's FP6 program (Strengthening the European Research Area, Research Infrastructures. No: RII3-CT-2004-506008). We thank Johnson Matthey PLC for an academic loan scheme, which provided Pd salts for this work. We are also grateful to ESRF (BM23) and SLS (SuperSAXS) for provision of beam time and access to their facilities.Notes and references
- D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC.
- T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed.
- K. M. Engle, T. S. Mei, M. Wasa and J. Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed.
- T. S. Mei, L. Kou, S. Ma, K. M. Engle and J. Q. Yu, Synthesis, 2012, 1778 CAS.
- S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- A. J. Canty, Dalton Trans., 2009, 10409 RSC.
- K. Muniz, Angew. Chem., Int. Ed., 2009, 48, 9412 CrossRef CAS PubMed.
- P. Sehnal, R. J. K. Taylor and I. J. S. Fairlamb, Chem. Rev., 2010, 110, 824 CrossRef CAS PubMed.
- K. J. Bonney and F. Schoenebeck, Chem. Soc. Rev., 2014, 43, 6609 RSC.
- (a) A. R. Dick, K. L. Hull and M. S. Sanford, J. Am. Chem. Soc., 2004, 126, 2300 CrossRef CAS PubMed; (b) D. Kalyani and M. S. Sanford, Org. Lett., 2005, 7, 4149 CrossRef CAS PubMed.
- (a) D. C. Powers, M. A. L. Geibel, J. E. M. N. Klein and T. Ritter, J. Am. Chem. Soc., 2009, 131, 17050 CrossRef CAS PubMed; (b) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 419 CrossRef CAS PubMed; (c) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302 CrossRef CAS PubMed.
- Defined as the charge left on the metal after all ligands have been removed in their normal, close-shell configuration.
- L. Guo, Y. Xu, X. Wang, W. Liu and D. Lu, Organometallics, 2013, 32, 3780 CrossRef CAS.
- For example, see: (a) V. Lyaskovskyy and B. de Bruin, ACS Catal., 2012, 2, 270 CrossRef CAS; (b) V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228 CrossRef CAS PubMed.
- (a) M. Dincer, N. Ozdemir, M. E. Gunay and B. Cetinkaya, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, M381 CAS; (b) H. Y. Thu, W. Y. Yu and C. M. Che, J. Am. Chem. Soc., 2006, 128, 9048 CrossRef CAS PubMed.
- J. M. Racowski, A. R. Dick and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 10974 CrossRef CAS PubMed.
- Experimental bond lengths for unbound acetate ions are 1.21 and 1.36 Å for C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O, respectively, see: M. Ibrahim and E. Koglin, Acta Chim. Slov., 2004, 51, 453 CAS.
O and C–O, respectively, see: M. Ibrahim and E. Koglin, Acta Chim. Slov., 2004, 51, 453 CAS. - J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- A. V. Marenich, S. V. Jerome, C. J. Cramer and D. G. Truhlar, J. Chem. Theory Comput., 2012, 8, 527 CrossRef CAS.
- K. Getty, M. U. Delgado-Jaime and P. Kennepohl, Inorg. Chim. Acta, 2008, 361, 1059 CrossRef CAS PubMed.
- M. Tromp, J. A. van Bokhoven, M. Q. Slagt, R. J. M. K. Gebbink, G. van Koten, D. E. Ramaker and D. C. Koningsberger, J. Am. Chem. Soc., 2004, 126, 4090 CrossRef CAS PubMed.
- S. H. Eitel, M. Bauer, D. Schweinfurth, N. Deibel, B. Sarkar, H. Kelm, H. J. Kruger, W. Frey and R. Peters, J. Am. Chem. Soc., 2012, 134, 4683 CrossRef CAS PubMed.
- In theory, better resolved d-peaks can be obtained from XAS recorded at the Pd L3-edge. But it is not possible to acquire such low-energy spectra (ca. 3 keV) in this experiment as complexes 2 and 4 can only be generated in solution.
- Under these conditions, complexes 2 and 4 undergo slow decomposition over 3 h, where noticeable degradation of the spectra can be observed. The limited operational window prevents the application of other XAS techniques with long acquisition times, e.g. those based on florescence emissions.
- The generation of the oxidative adduct under the conditions of XAS experiments were established by off-line NMR studies (see the ESI†).
- In principle, better resolution of the absorption edges may be obtained through high energy resolution fluorescence detection (HERFD) XAS. However, this is currently not feasible at such high energies (Pd K-edge 24.35 keV) at either SLS or ESRF, where these experiments were conducted.
- J. E. Bercaw, A. C. Durrell, H. B. Gray, J. C. Green, N. Hazari, J. A. Labinger and J. R. Winkler, Inorg. Chem., 2010, 49, 1801 CrossRef CAS PubMed.
- Y. B. Dudkina, D. Y. Mikhaylov, T. V. Gryaznova, A. I. Tufatullin, O. N. Kataeva, D. A. Vicic and Y. H. Budnikova, Organometallics, 2013, 32, 4785 CrossRef CAS.
- I. O. Koshevoy, P. Lahuerta, M. Sanau, M. A. Ubeda and A. Domenech, Dalton Trans., 2006, 5536 RSC.
- (a) M. Giffard, G. Mabon, E. Leclair, N. Mercier, M. Allain, A. Gorgues, P. Molinié, O. Neilands, P. Krief and V. Khodorkovsky, J. Am. Chem. Soc., 2001, 123, 3852 CrossRef CAS; (b) G. Kokkinidis, M. Papadopoulou and A. Varvoglis, Electrochim. Acta, 1989, 34, 133 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental data, X-ray crystallographic analysis, details of EXAFS fitting, electrochemical data, and theoretical calculations. CCDC 905536 and 948332. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt02104a |
| ‡ Current affiliation: School of Chemistry, University of Leeds, Woodhouse Lane, Leeds LS2 9JT, UK. |
| § Current affiliation: Laboratório Nacional de Luz Síncrotron (LNLS), Caixa Postal 6192, CEP, 13083-970, Campinas – SP, Brazil. |
| This journal is © The Royal Society of Chemistry 2015 |