DOI:
10.1039/C5DT00435G
(Paper)
Dalton Trans., 2015,
44, 7258-7267
Solid state and solution studies of lithium tris(n-butyl)magnesiates stabilised by Lewis donors†‡
Received
30th January 2015
, Accepted 9th March 2015
First published on 9th March 2015
Abstract
Several Lewis base adducts of the synthetically important lithium tris(n-butyl)magnesiate LiMg(nBu)3 have been prepared and structurally characterised. The complexes were prepared by a co-complexation approach i.e., by combining the monometallic nBuLi and nBu2Mg reagents in hydrocarbon solution before adding a molar equivalent of a donor molecule (a bidentate amine, tridentate amine or cyclic ether). The lithium magnesiates all adopt variants of the “Weiss motif” structure, i.e., contacted ion pair dimers with a linear arrangement and metals connected by butyl anions, where tetrahedral magnesium ions are in the central positions and the lithiums occupy the outer region, solvated by a neutral Lewis donor [(donor)Li(μ-nBu)2Mg(μ-nBu)2Mg(μ-nBu)2Li(donor)]. When TMPDA, PMDETA or (R,R)-TMCDA [TMPDA = N,N,N′N′-tetramethylpropanediamine; PMDETA = N,N,N′,N′′,N′′-pentamethyldiethylenetriamine; and (R,R)-TMCDA = (R,R)-N,N,N′,N′-tetramethylcyclohexane-1,2-diamine], are employed, dimeric tetranuclear lithium magnesiates are produced. Due to the tridentate nature of the ligand, the PMDETA-containing structure (2) has an unusual ‘open’-motif. When TMEDA (TMEDA = N,N,N′,N′-tetramethylethylenediamine) is employed, a n-butoxide-containing complex [(TMEDA)Li(μ-nBu)(μ-OnBu)Mg2(nBu)2(μ-nBu)(μ-OnBu)Li(donor)] has been serendipitously prepared and adopts a ladder conformation which is commonly observed in lithium amide chemistry. This complex has also been prepared using a rational methodology. When 1,4-dioxane is employed, the donor stitches together a polymeric array of tetranuclear dimeric units (6). The hydrocarbon solution structures of the compounds have been characterised by 1H, 7Li, 13C NMR spectroscopy; 2 has been studied by variable temperature and DOSY NMR.
Introduction
Despite the first organolithium compounds being reported almost 100 years ago,1 they are without doubt, still one of the most widely utilised classes of reagents in modern synthetic chemistry. This is primarily due to their versatility and their ease of use. The metallation reaction (i.e., transforming a relatively inert C–H bond to a more reactive, more useful C–Metal bond) is fundamental to many synthetic procedures and is commonly achieved by reacting an organic substrate with an organolithium reagent. The resultant organometallic compound can then be electrophilically quenched, to furnish new organic molecules.2 However, lithium organometallics often promote side reactions as a result of their high polarity/reactivity (including reacting with solvents such as THF and diethylether) and they often require cryogenic temperatures to encourage selectivity. Alternative methods have been established using magnesium reagents, which generally offer a better functional group tolerance; however, their drawbacks include they generally have low solubility in common solvents and due to their sluggish reactivity there is often a need for an excess of the base to be used to give adequate yields.3 As energy efficiency is becoming a key driver in modern synthesis,4 it is important to develop protocols where chemistry can be performed at temperatures close to ambient. Over the past couple of decades, a new category of bimetallic alkali metal magnesiates, which often exhibit reactivities that cannot be replicated by the monometallic agents on their own, has been investigated.5 In many cases, they can be used under mild reaction conditions and can display unusual deprotonation regioselectivities.6 Magnesiates have recently received a high degree of attention from the synthetic chemist in a large part due to the work of Knochel.7 His group has discovered that simply adding one equivalent of lithium chloride to a conventional Grignard or Hauser (amidomagnesium halide) reagent can have a marked difference in selectivity and reactivity. These lithium magnesiate reagents have been coined as ‘turbo Grignard’8 and ‘turbo-Hauser’9 [with general formulae RMgCl·LiCl and (NR2)MgCl·LiCl respectively]. The alkyl magnesiate RMgCl·LiCl is a useful reagent for inducing halogen–magnesium exchange in unactivated functionalised aryl and heteroaryl bromides in high yields, working at non-cryogenic temperatures with good group tolerance.10 The amido containing (TMP)MgCl·LiCl (where TMP is 2,2,6,6-tetramethylpiperidide) is an efficient base for the hydrogen–magnesium exchange of aromatics and heteroaromatics substrates.11 Homoleptic alkyllithium magnesiates, which depending on their stoichiometry can be grouped as (lower-order) triorganomagnesiates LiMgR3 or (higher-order) tetraorganomagnesiates Li2MgR4, have also been established as useful synthetic reagents.12 Both, homoleptic and heteroleptic alkyllithium magnesiates have been broadly employed in organic synthesis, so much so that LiMgnBu3 and LiMgiPrnBu2 are commercially available as mixtures in hexane–diethyl ether. In 1951, Wittig employed a lithium magnesiate, [LiMgPh3]13 – prepared by combining phenyllithium with diphenylmagnesium – in addition reactions across ketones. Since this first report, lithium magnesiates has been established as useful reagents for nucleophilic additions.13,14 They have also been found to be excellent bases for the deprotonation of aromatic substrates15 such as fluoroaromatics,15d furans15g or pyridine carboxamides.15h The most common reagent employed in these reactions is [LiMgnBu3]. This complex has also been shown to have high activity in halogen–magnesium exchange reactions. Oshima treated dibromomethylsilanes with the magnesiate to prepare α-silylalkyl-magnesium compounds,16 and Iida and Mase used it to carry out selective-monosubstitution of dibromoarenes.17 Many other magnesiate systems have been employed, establishing lithium magnesiates as important reagents for halogen–magnesium exchange reactions.16–18 Surprisingly, despite these extensive applications just a few examples of ‘simple’ homoleptic lithium magnesiates have been structurally characterised to date.19 The structure of the pioneering Wittig's lithium magnesiate was elucidated by Weiss by adding TMEDA to the magnesiate mixture.20 With respect to the lower order species, Weiss isolated and crystallised two different dimeric forms, firstly the contacted ion pair [(TMEDA·Li(μ-Ph)2Mg(μ-Ph)]2 and the solvent separated ion pair [(TMEDA)2Li]+[Ph2Mg(μ-Ph)]−. He also reported several other homoleptic organolithiums by using methyl,21 benzyl,22 or alkynyl22 ligands. More recently, Power,23 Hevia24 and Westerhausen25 have made further contributions by incorporating the 2,4,6-iPr3C6H2, CH2Si(CH3)3 (methyl(trimethylsilyl)) and C4H6 (butane-1,4-diide) anion respectively. In most cases, the lithium magnesiates adopt what has become known as the “Weiss-motif” with a tetrahedrally-disposed magnesium in the central positions and the alkali-metal occupying the outer region, solvated by neutral Lewis donors. With the exception of one compound (5), only homoleptic alkyl (or aryl) lithium magnesiates have been considered in this work but this is not a prerequisite as magnesiates also exist with combinations of different functional groups (heteroleptic) including oxide26 or amido groups.27
Results and discussion
Syntheses
By combining (co-complexing) the monometallic reagents nBuLi and nBu2Mg in hexane solution, a white suspension of the homoleptic mixture [LiMgnBu3] was produced. On the addition of one molar equivalent of the corresponding Lewis base the suspension is solubilised. For 1, one equivalent of TMPDA (N,N,N′N′-tetramethylpropanediamine) was added to [LiMgnBu3] in hexane affording a colourless solution. Crystallisation at −35 °C resulted in colourless crystals of dimeric [(TMPDA)·Li(μ-nBu)2Mg(μ-nBu)]2 (1) in 67% yield. In contrast, addition of PMDETA (N,N,N′,N′′,N′′-pentamethyldiethylenetriamine) in an equimolar ratio results in the formation of a yellow oil in the bulk hexane. Concentration of the hexane solution and storage at −35 °C resulted in colourless crystals of [(PMDETA)·Li(μ-nBu)(nBu)Mg(μ-nBu)]2 (2) in 35% yield. When the chiral auxiliary ligand (R,R)-TMCDA [(R,R)-N,N,N′,N′-tetramethylcyclohexane-1,2-diamine] was added to an equimolar mixture of n-butyllithium and di-n-butylmagnesium in hexane, a white suspension was produced which on gently heating it dissolves in the reaction medium. Storage at −35 °C for 36 hours deposited dimeric crystalline 3, [{(R,R)-TMCDA}·Li(μ-nBu)2Mg(μ-nBu)]2, in high yield (82%). Compound 5, [(TMEDA)·Li(μ-nBu)(μ-OnBu)Mg(nBu)]2, can be synthesised by the equimolar reaction of lithium-n-butoxide (synthesised in situ by reacting n-butyllithium with n-butanol) with an equimolar quantity of di-n-butylmagnesium and TMEDA (N,N,N′,N′-tetramethylethylenediamine) (33%). The synthesis of the homoleptic n-butyllithium magnesiate stabilised by TMEDA produced crystalline material (4) in 59%. Disappointingly, and despite repeated attempts at the analysis, the crystals degraded before the data collection was completed (at 123 K). Due to the synthetic importance of the bimetallic mixture [LiMgnBu3] in ethereal solution,14d,15d–i we decided to study the role of oxygen donors such as THF or diethylether. [LiMgnBu3] was dissolved in hexane and the Lewis base was added in stoichiometric and substoichiometric amounts, and also in excess; however, no crystalline material was obtained. Next, it was decided to focus on the bidentate donor 1,4-dioxane by adding one molar equivalent of the cyclic ether to an equimolar mixture of n-butyllithium and di-n-butylmagnesium in benzene. Precipitation of a white solid suggested the formation of a new adduct. Toluene was added to the resultant white solid, which redissolved the complex on strong heating. Cooling the solution slowly to ambient temperature afforded a crop of colourless needles of polymeric [(dioxane)·LiMgnBu3]∞, 6 (47%).
X-ray crystallography
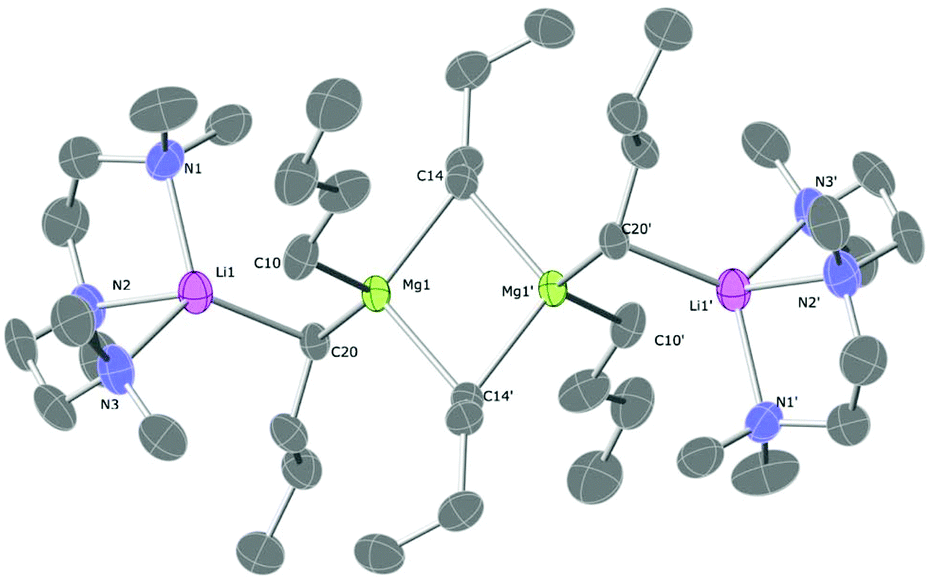
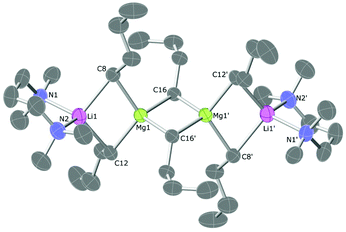
The TMPDA-solvated lithium magnesiate 1 crystallises in the space group Pbca (Fig. 1). 1 was found to be a dimer with the classical ‘Weiss-type’ motif (Scheme 1) in which six butyl anions bridge the four metals (two Li and two Mg atoms), giving rise to three four-membered rings (i.e., two Li–C–Mg–C rings and one Mg–C–Mg–C ring). The former rings are fused to the latter at different Mg atoms. Each Mg atom is bound solely to (four) C atoms, and each Li atom binds to two C atoms and their coordination sphere is completed with the ancillary ligand forming a six-membered Li–N–C–C–C–N ring. The Li–C distances range from 2.348(4) to 2.303(4) Å and the Mg–C distances from 2.205(6) to 2.298(2)Å are as expected for such species. A search in the Cambridge Structural Database28 reveals that only four lithium complexes stabilised by TMPDA have been crystallographically characterised, bearing anionic groups such as phenylacetylene,29 isocyanate,30 boratabenzene31 or benzyl.32 Li–N distances of 1, [2.085(4) to 2.092(4) Å], are similar to those found for other lithium compounds stabilised by chelating TMPDA. Diamines have proven being a key additive in the field of alkali metal-mediated magnesiation, dramatically modifying both structural pattern and reactivity,33 so different nitrogen-donors were employed in this work (Scheme 2). Apart from lithium magnesiates, tridentate ligand PMDETA has been used to chelate lithium zincates like PMDETA·LiZn(HMDS)Me2,34 PMDETA·LiZnMe3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 35 or PMDETA·LiZn(CH2SiMe3).36 The employment of the tridentate ligand PMDETA causes the lithium magnesiate to crystallise as dimeric 2 whereby the lithium atoms bind in the usual way to the ancillary ligand crystallising in space group P21/n (Fig. 2). The same methodology was used by Hevia et al. to produce [(PMDETA)LiMg(CH2SiMe3)3].24n-Butyl's less sterically demanding nature in comparison to methyl(trimethylsilyl) presumably results in two key differences between the structures. While the lithium magnesiate containing methyl(trimethylsilyl) groups was found to be a monomer, compound 2 can be described as an ‘open dimeric’ motif, in which the lithium and magnesium atoms are bridged by only one butyl group so the angle Li–C20–Mg1 is more open than in the closed structures, which contain Li–C–Mg–C four-membered rings. This angle is also affected by the nature of the anionic group, being 83.31° in the n-butyllithium magnesiate dimer and 149.4° for Hevia's monomer.24 Complex 2 resembles the dimer obtained by Weiss and co-workers when they employed the alkynyl ligand with TMEDA, although the structure found by them is again a closed dimer.22 Strohmann crystallographically characterized a Mg–C four-membered rings. This angle is also affected by the nBuLi adduct stabilised by PMDETA, where the tetranuclear dimer [(nBuLi)2PMDETA]2 is produced.37 This dimer is structurally related to compound 2 as both can be interpreted as a dimer with two outer PMDETA·nBuLi units linked to a dimeric core of (nBuLi)2 for Strohmann's complex or (nBu2Mg)2 for 2. Strohmann's lithium dimer presents longer Li–C interatomic distances for the outer unit [2.498(6) Å] than compound 2 [2.271(5) Å] suggesting a stronger interaction between the outer PMDETA·nBuLi and the central ring in 2. The C–Mg distance [2.237(3) Å] is within the range of typical bond lengths in organomagnesiates. Next, a chiral bidentate donor was employed and produced the complex [{(R,R)-TMCDA}·Li(μ-nBu)2Mg(μ-nBu)]2 (3) which crystallises in space group P21 (Fig. 3). (R,R)-TMCDA solvates the triorganomagnesiate leading to a contacted ion pair dimer where the atoms in molecule are not related by any symmetry operations. Several alkali-metal magnesium amide complex stabilised by chiral donors33c,38 [namely (−)-sparteine and (R,R)-TMCDA] have been characterised, but to the best of our knowledge, this is the first example of a crystallographically elucidated chiral tris(alkyl)lithium magnesiate. When TMEDA was employed, crystals of 4 could be isolated; however, they could not be analysed by X-ray crystallography, even when low temperatures were employed (123 K). The crystals were found to degrade in the X-ray beam, presumably due to them undergoing a phase change. Although no solid-state structural data for 4 could be obtained, a hydrocarbon solution of the complex was studied by NMR spectroscopy (vida infra). In one of the attempts to analyse 4, data was successfully collected and the structure was found to be [(TMEDA)·Li(μ-nBu)(μ-OnBu)Mg(nBu)]2 (5). It is a heteroleptic dimer that contains both n-butyl and n-butoxide groups. In light of this serendipitous discovery, a rational synthetic procedure for the complex was employed (vida supra) involving the co-complexation of lithium-n-butoxide and di-n-butylmagnesium. Compound 5, which crystallises in space group P21/c, is a dimer of [(TMEDA)·Li(OnBu)MgnBu2] units which bind through a Mg–O interaction exhibiting ladder-like features39 (Fig. 4). Each unit is composed of a four-membered (C–Mg–O–Li) ring. Lithium is linked to magnesium via one butyl and one butoxide group and it completes its coordination sphere coordinating to TMEDA in order to have tetrahedral (sum of angles 657.44°) coordination.
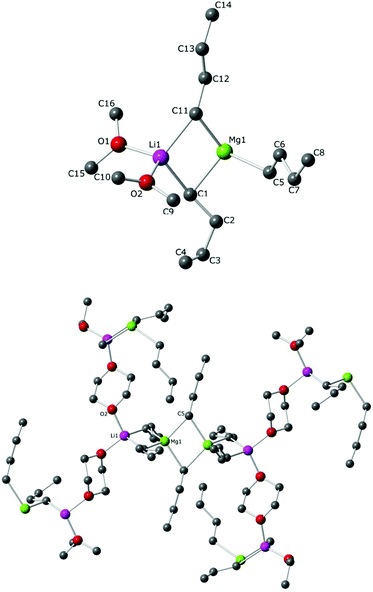
35 or PMDETA·LiZn(CH2SiMe3).36 The employment of the tridentate ligand PMDETA causes the lithium magnesiate to crystallise as dimeric 2 whereby the lithium atoms bind in the usual way to the ancillary ligand crystallising in space group P21/n (Fig. 2). The same methodology was used by Hevia et al. to produce [(PMDETA)LiMg(CH2SiMe3)3].24n-Butyl's less sterically demanding nature in comparison to methyl(trimethylsilyl) presumably results in two key differences between the structures. While the lithium magnesiate containing methyl(trimethylsilyl) groups was found to be a monomer, compound 2 can be described as an ‘open dimeric’ motif, in which the lithium and magnesium atoms are bridged by only one butyl group so the angle Li–C20–Mg1 is more open than in the closed structures, which contain Li–C–Mg–C four-membered rings. This angle is also affected by the nature of the anionic group, being 83.31° in the n-butyllithium magnesiate dimer and 149.4° for Hevia's monomer.24 Complex 2 resembles the dimer obtained by Weiss and co-workers when they employed the alkynyl ligand with TMEDA, although the structure found by them is again a closed dimer.22 Strohmann crystallographically characterized a Mg–C four-membered rings. This angle is also affected by the nBuLi adduct stabilised by PMDETA, where the tetranuclear dimer [(nBuLi)2PMDETA]2 is produced.37 This dimer is structurally related to compound 2 as both can be interpreted as a dimer with two outer PMDETA·nBuLi units linked to a dimeric core of (nBuLi)2 for Strohmann's complex or (nBu2Mg)2 for 2. Strohmann's lithium dimer presents longer Li–C interatomic distances for the outer unit [2.498(6) Å] than compound 2 [2.271(5) Å] suggesting a stronger interaction between the outer PMDETA·nBuLi and the central ring in 2. The C–Mg distance [2.237(3) Å] is within the range of typical bond lengths in organomagnesiates. Next, a chiral bidentate donor was employed and produced the complex [{(R,R)-TMCDA}·Li(μ-nBu)2Mg(μ-nBu)]2 (3) which crystallises in space group P21 (Fig. 3). (R,R)-TMCDA solvates the triorganomagnesiate leading to a contacted ion pair dimer where the atoms in molecule are not related by any symmetry operations. Several alkali-metal magnesium amide complex stabilised by chiral donors33c,38 [namely (−)-sparteine and (R,R)-TMCDA] have been characterised, but to the best of our knowledge, this is the first example of a crystallographically elucidated chiral tris(alkyl)lithium magnesiate. When TMEDA was employed, crystals of 4 could be isolated; however, they could not be analysed by X-ray crystallography, even when low temperatures were employed (123 K). The crystals were found to degrade in the X-ray beam, presumably due to them undergoing a phase change. Although no solid-state structural data for 4 could be obtained, a hydrocarbon solution of the complex was studied by NMR spectroscopy (vida infra). In one of the attempts to analyse 4, data was successfully collected and the structure was found to be [(TMEDA)·Li(μ-nBu)(μ-OnBu)Mg(nBu)]2 (5). It is a heteroleptic dimer that contains both n-butyl and n-butoxide groups. In light of this serendipitous discovery, a rational synthetic procedure for the complex was employed (vida supra) involving the co-complexation of lithium-n-butoxide and di-n-butylmagnesium. Compound 5, which crystallises in space group P21/c, is a dimer of [(TMEDA)·Li(OnBu)MgnBu2] units which bind through a Mg–O interaction exhibiting ladder-like features39 (Fig. 4). Each unit is composed of a four-membered (C–Mg–O–Li) ring. Lithium is linked to magnesium via one butyl and one butoxide group and it completes its coordination sphere coordinating to TMEDA in order to have tetrahedral (sum of angles 657.44°) coordination.
 |
| | Fig. 1 Molecular structure of 1. Ellipsoids are displayed at 30% probability and all hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Li1–N1, 2.092(4); Li–N2, 2.085(4); Li–C8, 2.348(4); Li1–C12, 2.303(4); Mg1–C8, 2.205(6); Mg1–C12, 2.225(2); Mg1–C16 2.298(2); N1–Li1–N2, 99.8(2); N1–Li1–C8, 106.9(2); N1–Li1–C12, 120.4(2); N2–Li1–C8, 107.4(2); N2–Li1–C12, 117.1(2); C8–Li1–C12, 104.5(1); C8–Mg1–C12, 112.2(1); C8–Mg1–C16, 104.1(1); C8–Mg1–C16′, 112.6(1); C12–Mg1–C16, 110.1(1); Li1–C8–Mg1, 70.5(1); Li1–C12–Mg1, 71.0(1). | |
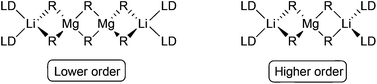 |
| | Scheme 1 Weiss-motifs of a lower order [(donor)·LiMgR3)]2 (left) and a higher order lithium magnesiate [(donor)2 Li2MgR4], (right). LD = Lewis donor. | |
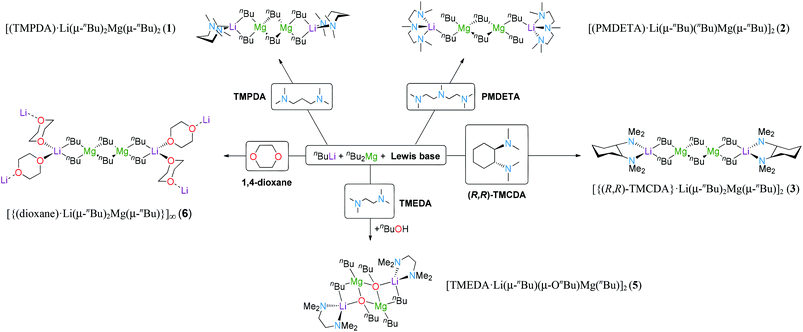 |
| | Scheme 2 Synthesis of homoleptic n-butyllithium magnesiates. | |
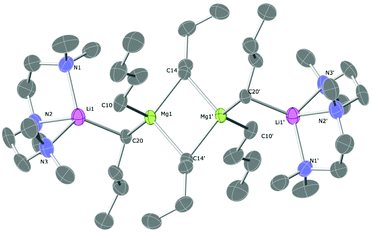 |
| | Fig. 2 Molecular structure of 2. Ellipsoids are displayed at 30% probability and all hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Li–N1, 2.178(5); Li–N2, 2.273(5); Li–N3, 2.161(5); Li1–C20, 2.271(5); Li1⋯C10, 3.161(1); Mg1–C10, 2.186(3); Mg1–C20, 2.237(3); Mg1–C14, 2.321(3); N1–Li–N2, 82.3(2); N1–Li–N3, 120.0(2); N2–Li1–N3, 83.3(2); N1–Li1–C20, 117.3(2); N2–Li1–C20, 108.1(2); N3–Li1–C20, 122.7(2); C10–Mg1–C14, 107.4(1); C10–Mg1–C20, 121.2(1); C14–Mg1–C20, 105.5(1). | |
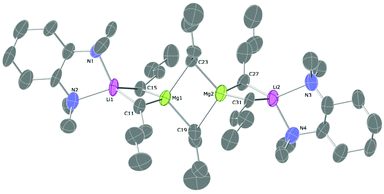 |
| | Fig. 3 Molecular structure of 3. Ellipsoids are displayed at 30% probability and all hydrogen atoms are omitted for clarity. | |
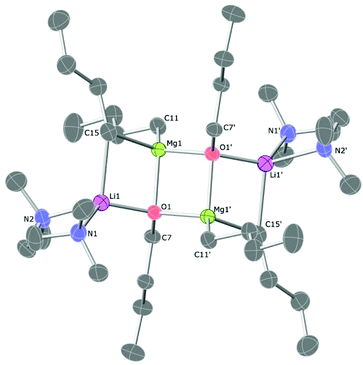 |
| | Fig. 4 Molecular structure of 5. Ellipsoids are displayed at 30% probability and all hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angles (°): Li1–N1, 2.114(3); Li1–N2, 2.191(3); Li1–C15, 2.343(3); Li1–O1, 1.946(3); Mg1–C11, 2.156(2); Mg1–C15, 2.211(2); Mg1–O1, 2.044(1); N1–Li1–N2, 86.5(1); N1–Li1–C15, 111.3(1); N1–Li–O1, 125.5(1); N2–Li1–O1, 123.7(1); N2–Li1–C15, 112.4(1); O1–Li1–C15, 98.1(1); C11–Mg1–C15, 118.3(1); C11–Mg1–O1, 115.5(1); C15–Mg1–O1, 99.0(1). | |
Magnesium ions are linked to two butyl groups (one bridge and one terminal). A very close precedent can be found in the sodium (or potassium) magnesium alkoxides crystallographically characterised by Mulvey40 (Fig. 5). The core of these examples and 5 is a O–Mg–O–Mg ring and the alkali metal is bonded to one oxygen atom and one molecule of TMEDA. While the tetrahedrally-disposed lithium atom is bridged to magnesium by one n-butyl chain in 5, sodium and potassium atoms are penta-coordinated as they bond to two butyl groups to form a closed structure. Since 1998, this motif has been known as an inverse crown compound.27c,41 All the n-butyl anions are bridging in the inverse crown molecules whereas two of these groups adopt terminal binding positions in 5. The difference in the steric nature of t-butoxide versus n-butyl may be an explanation for this structural variation; however, it seems more likely that it is due to an alkali metal effect as it could be envisaged that bulkier alkoxide groups (albeit not at the α-position of the ligand) would more likely generate open structures. The cyclic ether 1,4-dioxane has been previously used to solvate different lithium ‘ate’ complexes.24,42 When one equivalent is added to the unsolvated LiMg(nBu)3, it was possible to crystallise polymeric 6, which crystallises in space group C2/c (Fig. 6). Lithium magnesiate 6 comprises dimeric [(dioxane)2LiMgnBu3]2 units where two dioxane molecules bridge with lithium acting as a bridge between different dimers undergoing a three-dimensional polymeric structure. Unfortunately, a large amount of unresolved disorder adversely affects the precision of the n-butyl groups, dioxane and benzene molecules (present as crystallisation solvent in the unit cell) and therefore precludes discussion of any geometrical parameters, although its connectivity is unequivocal.
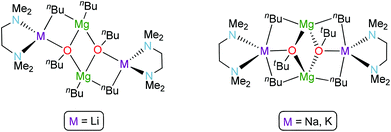 |
| | Fig. 5 ChemDraw representations for [(TMEDA)LiMg(nBu)2(OnBu)]2 (5) (left) and Mulvey's alkali metal alkoxide–magnesium bis(alkyl) dimers (inverse crowns) [(TMEDA)MMg(nBu)2(OtBu)]2, M = Na, K (right). | |
 |
| | Fig. 6 Molecular structure of 6, showing asymmetric unit; (top) and polymer propagation (bottom). All hydrogen atoms are omitted for clarity. | |
NMR spectroscopic studies
Complementing their solid-state characterization, lithium magnesiates 1–6 have also been examined using multinuclear (1H, 7Li and 13C) NMR spectroscopy. In order to maintain the Lewis base coordination in solution, the solvent of choice was D12-cyclohexane for all the complexes, except for 6, which required D8-toluene and an elevated acquisition temperature (353 K) because of its low solubility. A comparison of the resonances in the 1H and 13C NMR spectra belonging to the ancillary ligands of 1–6 with those observed for the free Lewis bases suggests that the donors remain coordinated to the metal centres in solution (see ESI‡ for full details). As a solid state structure for ‘[(TMEDA)LiMg(nBu)3]2′ 4 was frustratingly not forthcoming, we examined hydrocarbon solutions of this compound. The 1H NMR spectrum shows two singlets at 2.33 and 2.39 ppm while the signals for free TMEDA in cyc-C6D12 appear at 2.30 and 2.14 ppm. 1H NMR spectra showed that both TMEDA and nBu resonances were present (in an approximate 1:3 ratio) which is in agreement with a low order lithium magnesiate. Turning to 7Li NMR resonances, the resonance for n-butyllithium is shifted upfield from 2.40 ppm to 1.01 ppm when it is co-complexed with the ‘low polarity’ di-n-butylmagnesium (Table 1). The further shift of this resonance after the addition of the donor also highlights that the Lewis base remains coordinated to the lithium in solution. Tridentate ligand PMDETA causes the most shielded resonance, appearing at 0.77 ppm for 2. Although the structure parameters for 1–6 show the presence of at least two chemically distinct n-butyl chains, the NMR spectroscopic studies at 300 K show only one set of signals for the alkyl groups in hydrocarbon solution is present. This fact suggests that rapid exchange of the alkyl positions occurs in solution, causing the signals for the n-butyl chains to become equivalent. In all cases (see ESI‡ for full details) the resonance for the CH2–M resonance is broad. As a case study, we choose to probe further a D8-toluene solution of 2 using a variable-temperature experiment in an attempt to decipher the three different n-butyl groups (one terminal, one bridging Li and Mg centres, and one bridging two Mg atoms) which exist in the solid state structure (N.B., cyc-C6D12 is solid below 268 K).The temperature was lowered to 240 K and 1H NMR spectroscopic measurements were obtained every 10 K (Fig. 7). At the lowest temperature, the CH2–M resonance decoalesces into two signals of an approximate integration of 1:2, not the three resonances that we expected. This seems to point to two possible scenarios. Firstly, in arene, the dimer stays intact but the terminal n-butyl group now adopts a bridging position (or is in rapid exchange between bridging and terminal with respect to the NMR timescale); or secondly, the dimer dissociates to a monomer in D8-toluene and the resonances therefore correspond to two bridging and one terminal butyl chain (Scheme 3). In order to find out whether 2 retains its dimeric structure in solution, we performed 1H NMR diffusion-ordered nuclear magnetic resonance spectroscopy (1H-DOSY) to gain insight into its aggregation in D8-toluene. DOSY measurements are useful to estimate molecular weights of species in solution,43 as species with higher molecular weight diffuse slower that those with lower molecular weight. A relationship can be established between coefficient diffusion and molecular weight, which are inversely proportional. In our study we chose 1,2,3,4-tetraphenylnaphthalene (TPhN), 1-phenylnaphthalene (PhN), and tetramethylsilane (TMS) as internal standards, as they exhibit good solubility in toluene with minimal overlapping of signals and are inert to magnesiates.
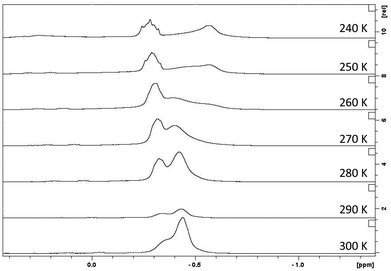 |
| | Fig. 7 Variable-temperature 1H NMR experiment of 2 in D8-toluene at 300 K. | |
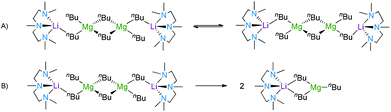 |
| | Scheme 3 Representation of two different equilibrium processes of 2 in solution. | |
Table 1 Chemical shifts in the 7Li spectrum in cyc-C6D12 at 300 K
| Complex |
n
BuLi |
LiMgnBu3 |
1
|
2
|
3
|
4
|
5
|
|
7Li (ppm) |
2.40 |
1.01 |
0.79 |
0.77 |
1.04 |
0.87 |
0.99 |
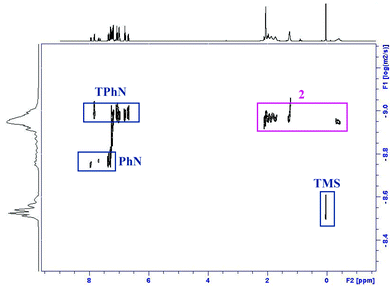
The 1H-DOSY NMR spectrum of a mixture of 2 with these standards in D8-toluene at 300 K showed only one set of signals for the lithium magnesiate confirming that there is only one species in solution and PMDETA remains attached to the lithium magnesiate in arene solvent (Fig. 8). A correlation between logD (average diffusion value, D, 7.2 × 10−10 m2 s−1) and logFW (FW = molecular weight) can be established (calibration curve obtained from the internal standards, logD = −0.698logFW − 7.358; r2 = 0.995). This correlation gave an experimental molecular weight which was estimated to be 370 g mol−1, which differs in a 1.23% from the molecular weight for the monomer [(PMDETA)LiMgnBu3]. Thus, analysis of this data suggests that the dimeric constitution of 2 in solid state is not retained in solution.
 |
| | Fig. 8
1H DOSY NMR spectrum of complex 2 in D8-toluene solution at 300 Kin the presence of inert standards 1,2,3,4-tetraphenylnaphthalene (TPhN), 1-phenylnaphthalene (PhN) and tetramethylsilane (TMS). | |
Conclusions
A systematic study of a series of n-butyllithium magnesiates in solid state and solution has been performed by employing neutral bidentate, tridentate and chiral Lewis bases. It is possible to rationally synthesise homoleptic dimers which adopt the ‘Weiss motif’ in solid state. Solution studies have shown that the Lewis bases remain attached to the lithium magnesiate species in aliphatic and arene solvents; however, there is evidence that the aggregation state can be lowered, even in the presence of potentially π-donating arene molecules. In future studies, we will look to utilise these compounds in organic transformations and compare the results with the previously published data.
Experimental
General methods
n-Hexane and toluene were distilled from sodium/benzophenone, 1,4-dioxane, TMEDA, TMPDA and PMDETA were distilled from CaH2. (R,R)-TMCDA was synthesised according to the standard procedure.44n-Butyllithium was freshly standardised by titration against menthol–1,10-phenanthroline in THF at 0 °C nBu2Mg was standardised with iodine in presence of an excess of LiCl, in THF at 0 °C. All synthetic work was carried out under argon by using standard Schlenk techniques. 1H, 13C NMR and 7Li NMR spectra were recorded on a Bruker DPX 400 MHz spectrometer. All 13C NMR spectra were proton decoupled. 1H-DOSY experiments were performed in an NMR tube that was charged with 20 mg of 2, TPhN (15 mg), PhN (13 μL), TMS (19 μL), and D8-toluene up to 0.7 mL of solution, before NMR analysis. Data for X-ray crystal structure determinations were measured with Oxford Diffraction Xcalibur and Gemini A diffractometers using graphite monochromated radiation. All structures were refined using all unique reflections and against F2 to convergence using SHELXL-97.45 Disordered butyl groups in 1, 2 and 6 were modelled over two sites with appropriate restraints and constraints applied. High degrees of motion were also observed for the dioxane and benzene solvents present in structure 6. The sample used for structure 3 was twinned by a 180° rotation about 001. The raw data was processed to give an hklf 5 formatted reflection file. Refinement with this data gave a much improved model, but still required restraints to be added to the displacement parameters of atoms Li2, N3, N4, C36 and C41. Selected parameters are reported in Table 1 of ESI‡ and full details are available in cif format, see CCDC 1046063 to CCDC 1046067 contain the supplementary crystallographic data for this paper. Elemental analyses were attempted, but due to the extreme air sensitivity of the complexes variable results were obtained.
Synthesis of [TMPDA·Li(μ-nBu)2Mg(nBu)]2 (1)
n
BuLi (0.25 mL, 4 mmol of 1.6 M solution in hexanes) was dissolved in 15 mL of dried hexane in an oven-dried Schlenk. 4 mmol of nBu2Mg (4 mL of 1 M solution in heptanes) were added, turning the solution to slightly cloudy. After stirring for 30 minutes, 4 mmol of TMPDA (0.68 mL) were added. Gently heating gave a colourless solution. All volatiles were removed under vacuum and 10 mL of pentane were added. The solution was transferred to a freezer (−80 °C) affording colourless crystals after 24 hours. (0.89 g, 67%).
1H NMR (400.13 MHz, 300 K, cyc-C6D12): δ −0.80 to −0.42 (12H, br, M–CH2CH2CH2CH3, nBu), 0.85 (18H, t, 3JHH = 7.2 MHz, M–CH2CH2CH2CH3, nBu), 1.22–1.32 (12H, m, M–CH2CH2CH2CH3, nBu), 1.50–1.58 (12H, m, M–CH2CH2CH2CH3, nBu), 1.66 (4H, br, N–CH2CH2CH2–N, TMPDA), 2.25 (24H, s, N–CH3, TMPDA), 2.45–2.47 (8H, t, 3JHH = 4.8 Hz, N–CH2, TMPDA). 13C{1H} NMR (100.62 MHz, 300 K, cyc-C6D12): δ 9.99 (M–CH2CH2CH2CH3, nBu), 13.26 (M–CH2CH2CH2CH3, nBu), 23.34 (N–CH2CH2CH2–N, TMPDA), 31.74 (M–CH2CH2CH2CH3, nBu), 32.09, (M–CH2CH2CH2CH3, nBu) 45.97 (N–CH3, TMPDA), 60.82 (N–CH2CH2CH2–N, TMPDA). 7Li NMR (155.50 MHz, 300 K, cyc-C6D12): δ 0.79.
Synthesis of [PMDETA·Li(μ-nBu)Mg(nBu)(μ-nBu)]2 (2)
n
BuLi (2.5 mL, 4 mmol of 1.6 M solution in hexanes) was dissolved in 15 mL of dried hexane in an oven-dried Schlenk tube. 4 mmol of nBu2Mg (4 mL of 1 M solution in heptanes) were added and solution turned slightly cloudy. After stirring for 30 minutes, 4 mmol of PMDETA (0.84 mL) were added. Solution is gently heated to give a colourless solution with a yellow oil at the bottom of the Schlenk tube. It was and stored at −30 °C, affording colourless crystals after 1 day. (0.53 g, 35%).
1H NMR (400.13 MHz, 300 K, cyc-C6D12): δ −0.82 to −0.62 (12H, br, M–CH2CH2CH2CH3, nBu), 0.86 (18H, t, 3JHH = 7.2 MHz, M–CH2CH2CH2CH3, nBu), 1.23–1.30 (12H, m, M–CH2CH2CH2CH3, nBu), 1.50–1.56 (m, 12H, M–CH2CH2CH2CH3, nBu), 2.29 (30H, s, N–CH3 PMDETA), 2.44–2.53 (16H, br, N–CH2, PMDETA). The relatively low solubility of 2 over prolonged periods of time in cyc-C6D12 solutions precluded the collection of its 13C{1H} spectrum. 7Li NMR (155.50 MHz, 300 K, cyc-C6D12): δ 0.77.
Synthesis of [(R,R)-TMCDA·Li(μ-nBu)2Mg(nBu)]2 (3)
n
BuLi (2.5 mL, 4 mmol of 1.6 M solution in hexanes) was dissolved in 15 mL of dried hexane in an oven-dried Schlenk. 4 mmol of nBu2Mg (4 mL of 1 M solution in heptanes) were added and solution turned slightly cloudy. After stirring for 30 minutes, 4 mmol of (R,R)-TMCDA (0.76 mL) were added. The mixture was gently heated to yield a transparent colourless solution. The solution was transferred to a freezer operating at −30 °C, affording colourless crystals after 24 hours. (1.22 g, 82%).
1H NMR (400.13 MHz, 300 K, cyc-C6D12): δ −0.73 to −0.71 (12H, br, M–CH2CH2CH2CH3, nBu), 0.86 (18H, t, 3JHH = 7.2 MHz, M–CH2CH2CH2CH3, nBu), 1.07–1.13 (4H, m, γ-CH2, (R,R)-TMCDA), 1.15–1.20 (4H, m, β-CH2, (R,R)-TMCDA), 1.22–1.31 (12H, m, M–CH2CH2CH2CH3, nBu), 1.50–1.56 (12H, m, M–CH2CH2CH2CH3, nBu), 1.78–1.80 (4H, m, γ′-CH2, (R,R)-TMCDA), 1.90–1.93 (4H, m, β′-CH2, (R,R)-TMCDA), 2.18 (12H, s, N–CH3, (R,R)-TMCDA), 2.39 (14H, s, N–CH3 and α-CH, (R,R)-TMCDA). 13C{1H} NMR (100.62 MHz, 300 K, cyc-C6D12): δ 10.31 (M–CH2CH2CH2CH3, nBu), 13.32 (M–CH2CH2CH2CH3, nBu), 21.81 (γ-CH2, (R,R)-TMCDA), 25.04 (β-CH2, (R,R)-TMCDA), 31.56 (M–CH2CH2CH2CH3, nBu), 32.22 (M–CH2CH2CH2CH3, nBu), 37.06 (N–CH3, (R,R)-TMCDA), 44.63 (N–CH3, (R,R)-TMCDA), 64.07 (α-CH, (R,R)-TMCDA). 7Li NMR (155.50 MHz, 300 K, cyc-C6D12): δ 1.04.
Synthesis of [TMEDA·LiMgnBu3]x (4)
n
BuLi (2.5 mL, 4 mmol of 1.6 M solution in hexanes) was dissolved in 15 mL of dried hexane in an oven-dried Schlenk. 4 mmols of nBu2Mg (4 mL of 1 M solution in heptanes) were added and solution turned slightly cloudy. After stirring for 30 minutes, 4 mmol of TMEDA (0.60 mL) were added. The mixture was gently heated to yield a transparent colourless solution. The solution was transferred to a freezer operating at −35 °C, affording colourless crystals after 48 hours. (0.75 g, 59%).
1H NMR (400.13 MHz, 300 K, cyc-C6D12): δ −0.75 to −0.31 (6H, br, M–CH2CH2CH2CH3, nBu), 0.84 (9H, t, 3JHH = 7.3 MHz, M–CH2CH2CH2CH3, nBu), 1.21–1.30 (6H, m, M–CH2CH2CH2CH3, nBu), 1.46–1.54 (6H, m, M–CH2CH2CH2CH3, nBu), 2.28 (12H, s, N–CH3, TMEDA), 2.34 (4H, s, N–CH2, TMEDA). 13C{1H} NMR (100.62 MHz, 300 K, cyc-C6D12): δ 10.81 (M–CH2CH2CH2CH3, nBu), 14.23 (M–CH2CH2CH2CH3, nBu), 32.39 (N–CH2CH2CH2–N, TMPDA), 32.99 (M–CH2CH2CH2CH3, nBu), 46.74, (N–CH3, TMEDA) 57.93 (N–CH2, TMEDA). 7Li NMR (155.50 MHz, 300 K, cyc-C6D12): δ 0.84.
Synthesis of [TMEDA·Li(μ-nBu)(μ-OnBu)Mg(nBu)]2 (5)
n
BuLi (3.35 mL, 5 mmol of 1.6 M solution in hexanes) was dissolved in 15 mL of dried hexane in an oven-dried Schlenk tube. 5 mmol of nBuOH were added (0.45 mL) at 0 °C and white solid immediately precipitates. 5 mmol of nBu2Mg (5 mL of 1 M solution in heptanes) were added and solution turned slightly cloudy. After stirring for 30 minutes, 5 mmol of TMEDA (0.75 mL) were added. Suspension is stirred for 2 hours and filtrated over celite with a filter stick. Solution is concentrated and stored at −35 °C, affording a crop of crystals after 24 hours (0.44 g, 33%).
1H NMR (400.13 MHz, 300 K, cyc-C6D12): δ −0.94 to −0.46 (8H, br, M–CH2CH2CH2CH3, nBu), 0.84–0.93 (18H, m, M–(O)–CH2CH2CH2CH3, nBu and OnBu), 1.26 (12H, br, M–(O)–CH2CH2CH2CH3, nBu and OnBu), 1.53 (12H, br, M–(O)–CH2CH2CH2CH3, nBu and OnBu), 2.25 (24H, s, N–CH3, TMEDA), 2.35 (8H, m, N–CH2, TMEDA), 3.63 (8H, br, M–O– CH2CH2CH2CH3, OnBu), The relatively low solubility of 2 over prolonged periods of time in cyc-C6D12 solutions precluded the collection of its 13C{1H} spectrum. 7Li NMR (155.50 MHz, 300 K, cyc-C6D12): δ 0.99.
Synthesis of [{(dioxane)·Li(μ-nBu)2Mg(nBu)}2]∞ (6)
n
BuLi (0.125 mL, 2 mmol of 1.6 M solution in hexanes) was dissolved in 5 mL of dried hexane in an oven-dried Schlenk. 2 mmol of nBu2Mg (2 mL of 1 M solution in heptanes) were added, turning the solution to slightly cloudy. After stirring for 30 minutes, 2 mmol of 1,4-dioxane (0.17 mL) were added. All the volatiles are removed under vacuum and the white precipitated is suspended in 5 mL of dried benzene. Addition of 2 mL of dried toluene and vigorously heating affords in a colourless solution. The flask containing the colourless solution was placed in a Dewar flask of hot water and allowed to cool slowly to room temperature affording colourless needles. (0.27 g, 47%).
1H NMR (400.13 MHz, 343 K, D8-toluene): δ −0.31 (12H, t, 3JHH MHz M–CH2CH2CH2CH3, nBu), 1.09 (18H, t, 3JHH = 7.2 MHz, M–CH2CH2CH2CH3, nBu), 1.52–1.61 (12H, m, M–CH2CH2CH2CH3, nBu), 1.71–1.79 (12H, m, M–CH2CH2CH2CH3, nBu), 3.39 (16H, s, dioxane). 13C{1H} NMR (100.62 MHz, 343 K, D8-toluene): δ 9.66 (M–CH2CH2CH2CH3, nBu), 14.35 (M–CH2CH2CH2CH3, nBu), 23.34 (N–CH2CH2CH2–N, TMPDA), 32.10 (M–CH2CH2CH2CH3, nBu), 67.59, (M–CH2CH2CH2CH3, nBu) 137.79 (dioxane). 7Li NMR (155.50 MHz, 343 K, D8-toluene): δ 0.67.
Acknowledgements
We gratefully acknowledge the support of the EPSRC (J001872/1 and L001497/1) for the award of a Career Acceleration Fellowship to CTOH.
Notes and references
- W. Schlenk and E. Bergmann, Justus Liebigs Ann. Chem., 1928, 463, 98 CrossRef CAS.
-
(a) P. Beak and V. Snieckus, Acc. Chem. Res., 1982, 15, 306 CrossRef CAS;
(b) V. Snieckus, Chem. Rev., 1990, 90, 879 CrossRef CAS;
(c) T. K. Macklin and V. Snieckus, Org. Lett., 2005, 7, 2519 CrossRef CAS PubMed.
-
(a) R. W. Hoffmann, Chem. Soc. Rev., 2003, 32, 225 RSC;
(b) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed.
-
(a) L. S. Bennie, W. J. Kerr, M. Middleditch and A. J. B. Watson, Chem. Commun., 2011, 47, 2264 RSC;
(b) J. Francos, S. Zaragoza-Calero and C. T. O'Hara, Dalton Trans., 2014, 43, 1408 RSC.
- R. E. Mulvey, Organometallics, 2006, 25, 1060 CrossRef CAS.
-
(a) A. J. Martínez-Martínez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O'Hara, Chem. Sci., 2014, 5, 771 RSC;
(b) A. J. Martínez-Martínez, A. R. Kennedy, R. E. Mulvey and C. T. O'Hara, Science, 2014, 346, 834 CrossRef PubMed.
-
P. Knochel, A. Gavryushin and K. Brade, in The Chemistry of Organomagnesium Compounds, ed. Z. Rappoport and I. Marek, John Wiley and Sons, Chichester, 2008, p. 511 Search PubMed.
- A. Krasovskiy and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 3333 CrossRef CAS PubMed.
-
(a) P. García-Álvarez, D. V. Graham, E. Hevia, A. R. Kennedy, J. Klett, R. E. Mulvey, C. T. O'Hara and S. Weatherstone, Angew. Chem., Int. Ed., 2008, 47, 8079 CrossRef PubMed;
(b) D. R. Armstrong, P. García-Álvarez, A. R. Kennedy, R. E. Mulvey and J. A. Parkinson, Angew. Chem., Int. Ed., 2010, 49, 3185 CrossRef CAS PubMed.
-
(a) H. Ila, O. Baron, A. J. Wagner and P. Knochel, Chem. Commun., 2006, 583 RSC;
(b) N. M. Barl, V. Werner, C. Sämann and P. Knochel, Heterocycles, 2014, 88, 827 CrossRef CAS PubMed.
-
(a) A. Krasovskiy, V. Krasovskaya and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 2958 CrossRef CAS PubMed;
(b) W. Lin, O. Baron and P. Knochel, Org. Lett., 2006, 8, 5674 Search PubMed;
(c) S. H. Wunderlich, C. J. Rohbogner, A. Unsinn and P. Knochel, Org. Process Res. Dev., 2010, 14, 339 CrossRef CAS;
(d) B. Haag, M. Mosrin, H. Ila, V. Malakhov and P. Knochel, Angew. Chem., Int. Ed., 2011, 50, 9794 CrossRef CAS PubMed.
- F. Mongin and A. Harrison-Marchand, Chem. Rev., 2013, 113, 7563 CrossRef CAS PubMed.
- G. Wittig, F. J. Meyer and G. Lange, Justus Liebigs Ann. Chem., 1951, 571, 167 CrossRef CAS.
-
(a) E. C. Ashby, L.-C. Chao and J. Laemmle, J. Org. Chem., 1974, 39, 3258 CrossRef CAS;
(b) H. G. Richey and B. A. King, J. Am. Chem. Soc., 1982, 104, 4672 CrossRef CAS;
(c) H. G. Richey and J. Farkas, Organometallics, 1990, 9, 1778 CrossRef CAS;
(d) M. Hatano, T. Matsumura and K. Ishihara, Org. Lett., 2005, 7, 573 CrossRef CAS PubMed;
(e) J. G. Sośnicki, Tetrahedron Lett., 2006, 47, 6809 CrossRef PubMed.
-
(a) M. Yasuda, M. Ide, Y. Matsumoto and M. Nakata, Synlett, 1997, 899 CrossRef CAS PubMed;
(b) M. Ide, M. Yasuda and M. Nakata, Synlett, 1998, 936 CrossRef CAS PubMed;
(c) H. Awad, F. Mongin, F. Trécourt, G. Quéguiner and F. Marsais, Tetrahedron Lett., 2004, 45, 7873 CrossRef CAS PubMed;
(d) H. Awad, F. Mongin, F. Trécourt, G. Quéguiner, F. Marsais, F. Blanco, B. Abarca and R. Ballesteros, Tetrahedron Lett., 2004, 45, 6697 CrossRef CAS PubMed;
(e) O. Bayh, H. Awad, F. Mongin, C. Hoarau, L. Bischoff, F. Trécourt, G. Quéguiner, F. Marsais, F. Blanco, B. Abarca and R. Ballesteros, J. Org. Chem., 2005, 70, 5190 CrossRef CAS PubMed;
(f) O. Bayh, H. Awad, F. Mongin, C. Hoarau, F. Trécourt, G. Quéguiner, F. Marsais, F. Blanco, B. Abarca and R. Ballesteros, Tetrahedron, 2005, 61, 4779 CrossRef CAS PubMed;
(g) F. Mongin, A. Bucher, J. P. Bazureau, O. Bayh, H. Awad and F. Trécourt, Tetrahedron Lett., 2005, 46, 7989 CrossRef CAS PubMed;
(h) H. Hawad, O. Bayh, C. Hoarau, F. Trécourt, G. Quéguiner and F. Marsais, Tetrahedron, 2008, 64, 3236 CrossRef CAS PubMed;
(i) G. Bentabed-Ababsa, F. Blanco, A. Derdour, F. Mongin, F. Trécourt, G. Quéguiner, R. Ballesteros and B. Abarca, J. Org. Chem., 2009, 74, 163 CrossRef CAS PubMed.
- J. Kondo, A. Inoue, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2001, 40, 2085 CrossRef CAS.
- T. Iida, T. Wada, K. Tomimoto and T. Mase, Tetrahedron Lett., 2001, 42, 4841 CrossRef CAS.
-
(a) K. Kitagawa, A. Inoue, H. Shinokubo and K. Oshima, Angew. Chem., Int. Ed., 2000, 39, 2481 CrossRef CAS;
(b) A. Inoue, K. Kitagawa, H. Shinokubo and K. Oshima, J. Org. Chem., 2001, 66, 4333 CrossRef CAS PubMed;
(c) A. Inoue, J. Kondo, H. Shinokubo and K. Oshima, Chem. – Eur. J., 2002, 8, 1730 CrossRef CAS;
(d) S. Dumouchel, F. Mongin, F. Trécourt and G. Quéguiner, Tetrahedron Lett., 2003, 44, 2033 CrossRef CAS;
(e) S. Dumouchel, F. Mongin, F. Trécourt and G. Quéguiner, Tetrahedron Lett., 2003, 44, 3877 CrossRef CAS;
(f) S. Dumouchel, F. Mongin, F. Trécourt and G. Quéguiner, Tetrahedron, 2003, 59, 8629 CrossRef CAS PubMed;
(g) K. Fukuhara, Y. Takayama and F. Sato, J. Am. Chem. Soc., 2003, 125, 6884 CrossRef CAS PubMed;
(h) S. Ito, T. Kubo, N. Morita, Y. Matsui, T. Watanabe, A. Ohta, K. Fujimori, T. Murafuji, Y. Sugihara and A. Tajiri, Tetrahedron Lett., 2004, 45, 2891 CrossRef CAS PubMed;
(i) F. D. Therkelsen, M. Rottländer, N. Thorup and E. B. Pedersen, Org. Lett., 2004, 6, 1991 CrossRef CAS PubMed;
(j) T. Tsuji, T. Nakamura, H. Yorimitsu, H. Shinokubo and K. Oshima, Tetrahedron, 2004, 60, 973 CrossRef CAS PubMed;
(k) J. Xu, N. Jain and Z. Sui, Tetrahedron Lett., 2004, 45, 6399 CrossRef CAS PubMed;
(l) F. Buron, N. Plé, A. Turck and F. Marsais, Synlett, 2006, 1586 CAS;
(m) S. Kii, A. Akao, T. Iida, T. Mase and N. Yasuda, Tetrahedron Lett., 2006, 47, 1877 CrossRef CAS PubMed;
(n) T. Shoji, S. Ito, K. Toyota, T. Iwamoto, M. Yasunami and N. Morita, Eur. J. Org. Chem., 2009, 4307 CrossRef CAS;
(o) D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Chem. Rev., 2013, 114, 1207 CrossRef PubMed.
- A. Harrison-Marchand and F. Mongin, Chem. Rev., 2013, 113, 7470 CrossRef CAS PubMed.
- D. Thoennes and E. Weiss, Chem. Ber., 1978, 111, 3726 CrossRef CAS.
- T. Greiser, J. Kopf, D. Thoennes and E. Weiss, Chem. Ber., 1981, 114, 209 CrossRef CAS.
- B. Schubert and E. Weiss, Chem. Ber., 1984, 117, 366 CrossRef CAS.
- K. M. Waggoner and P. P. Power, Organometallics, 1992, 11, 3209 CrossRef CAS.
- S. E. Baillie, W. Clegg, P. García-Álvarez, E. Hevia, A. R. Kennedy, J. Klett and L. Russo, Organometallics, 2012, 31, 5131 CrossRef CAS.
- R. Fischer, R. Suxdorf, H. Görls and M. Westerhausen, Organometallics, 2012, 31, 7579 CrossRef CAS.
-
(a) K. W. Henderson, R. E. Mulvey, F. B. M. Reinhard, W. Clegg and L. Horsburgh, J. Am. Chem. Soc., 1994, 116, 10777 CrossRef CAS;
(b) M. Motevalli, D. Shah, S. A. A. Shah and A. C. Sullivan, J. Chem. Soc., Chem. Commun., 1994, 2427 RSC;
(c) F. Antolini, P. B. Hitchcock, M. F. Lappert and X.-H. Wei, Organometallics, 2003, 22, 2505 CrossRef CAS;
(d) M.-L. Hsueh, B.-T. Ko, T. Athar, C.-C. Lin, T.-M. Wu and S.-F. Hsu, Organometallics, 2006, 25, 4144 CrossRef CAS;
(e) S. Heitz, J.-D. Epping, Y. Aksu and M. Driess, Chem. Mater., 2010, 22, 4563 CrossRef CAS;
(f) P. J. Calderone, D. Banerjee, L. A. Borkowski and J. B. Parise, Inorg. Chem. Commun., 2011, 14, 741 CrossRef CAS PubMed;
(g) K. Wurst and M. R. Buchmeiser, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, m1106 CAS;
(h) C. Gallegos, V. Tabernero, F. M. García-Valle, M. E. G. Mosquera, T. Cuenca and J. Cano, Organometallics, 2013, 32, 6624 CrossRef CAS.
-
(a) W. Clegg, K. W. Henderson, R. E. Mulvey and P. A. O'Neil, J. Chem. Soc., Chem. Commun., 1993, 969 RSC;
(b) W. Clegg, K. W. Henderson, R. E. Mulvey and P. A. O'Neil, J. Chem. Soc., Chem. Commun., 1994, 769 RSC;
(c) A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, J. Am. Chem. Soc., 1998, 120, 7816 CrossRef CAS;
(d) J. K. Brask, T. Chivers, M. Parvez and G. P. A. Yap, Inorg. Chem., 1999, 38, 3594 CrossRef CAS;
(e) L. Barr, A. R. Kennedy, J. G. MacLellan, J. H. Moir, R. E. Mulvey and P. J. A. Rodger, Chem. Commun., 2000, 1757 RSC;
(f) G. C. Forbes, A. R. Kennedy, R. E. Mulvey, P. J. A. Rodger and R. B. Rowlings, J. Chem. Soc., Dalton Trans., 2001, 1477 RSC;
(g) T. Chivers, D. J. Eisler, C. Fedorchuk, G. Schatte, H. M. Tuononen and R. T. Boeré, Inorg. Chem., 2006, 45, 2119 CrossRef CAS PubMed;
(h) P. C. Andrikopoulos, D. R. Armstrong, A. R. Kennedy, R. E. Mulvey, C. T. O'Hara, R. B. Rowlings and S. Weatherstone, Inorg. Chim. Acta, 2007, 360, 1370 CrossRef CAS PubMed.
-
(a) F. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380 CrossRef PubMed;
(b)
Cambridge Structural Database v5.36 (February 2015), Cambridge Crystallographic Data Centre, UK, 2015 Search PubMed.
- B. Schubert and E. Weiss, Chem. Ber., 1983, 116, 3212 CrossRef CAS.
- D. R. Armstrong, A. H. Khandelwal, P. R. Raithby, R. Snaith, D. Stalke and D. S. Wright, Inorg. Chem., 1993, 32, 2132 CrossRef CAS.
- G. E. Herberich, B. Schmidt, U. Englert and T. Wagner, Organometallics, 1993, 12, 2891 CrossRef CAS.
- A. Lennartson, J. Sundberg, T. Wiklund, G. Hilmersson and M. Håkansson, Eur. J. Inorg. Chem., 2010, 2010, 3029 CrossRef.
-
(a) R. E. Mulvey, Acc. Chem. Res., 2009, 42, 743 CrossRef CAS PubMed;
(b) S. E. Baillie, T. D. Bluemke, W. Clegg, A. R. Kennedy, J. Klett, L. Russo, M. de Tullio and E. Hevia, Chem. Commun., 2014, 50, 12859 RSC;
(c) J. Francos, B. J. Fleming, P. García-Álvarez, A. R. Kennedy, K. Reilly, G. M. Robertson, S. D. Robertson and C. T. O'Hara, Dalton Trans., 2014, 43, 14424 RSC.
- D. R. Armstrong, E. Herd, D. V. Graham, E. Hevia, A. R. Kennedy, W. Clegg and L. Russo, Dalton Trans., 2008, 1323 RSC.
- S. Merkel, D. Stern, J. Henn and D. Stalke, Angew. Chem., Int. Ed., 2009, 48, 6350 CrossRef CAS PubMed.
- D. R. Armstrong, H. S. Emerson, A. Hernan-Gomez, A. R. Kennedy and E. Hevia, Dalton Trans., 2014, 43, 14229 RSC.
- C. Strohmann and V. H. Gessner, Angew. Chem., Int. Ed., 2007, 46, 4566 CrossRef CAS PubMed.
-
(a) A. R. Kennedy and C. T. O'Hara, Dalton Trans., 2008, 4975 RSC;
(b) P. García-Álvarez, A. R. Kennedy, C. T. O'Hara, K. Reilly and G. M. Robertson, Dalton Trans., 2011, 40, 5332 RSC.
-
(a) R. E. Mulvey, Chem. Soc. Rev., 1991, 20, 167 RSC;
(b) R. E. Mulvey, Chem. Soc. Rev., 1998, 27, 339 RSC.
- N. D. R. Barnett, W. Clegg, A. R. Kennedy, R. E. Mulvey and S. Weatherstone, Chem. Commun., 2005, 375 RSC.
- A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, Angew. Chem., Int. Ed., 1998, 37, 3180 CrossRef CAS.
-
(a) W. Uhl, K. W. Klinkhammer, M. Layh and W. Massa, Chem. Ber., 1991, 124, 279 CrossRef CAS;
(b) A. Fürstner, R. Martin, H. Krause, G. Seidel, R. Goddard and C. W. Lehmann, J. Am. Chem. Soc., 2008, 130, 8773 CrossRef PubMed.
-
(a) D. Li, I. Keresztes, R. Hopson and P. G. Williard, Acc. Chem. Res., 2008, 42, 270 CrossRef PubMed;
(b) A. Macchioni, G. Ciancaleoni, C. Zuccaccia and D. Zuccaccia, Chem. Soc. Rev., 2008, 37, 479 RSC;
(c) C. Su, R. Hopson and P. G. Williard, Eur. J. Inorg. Chem., 2013, 2013, 4136 CrossRef CAS.
- J.-C. Kizirian, N. Cabello, L. Pinchard, J.-C. Caille and A. Alexakis, Tetrahedron, 2005, 61, 8939 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnotes |
| † In memory of Professor Ken Wade, a truly inspirational man. |
| ‡ Electronic supplementary information (ESI) available: NMR spectra for 1–6. CCDC 1046063–104067. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00435g |
|
| This journal is © The Royal Society of Chemistry 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a