
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
New donor–acceptor conjugates based on a trifluorenylporphyrin linked to a redox–switchable ruthenium unit†‡
Areej
Merhi
ab,
Xu
Zhang
ab,
Dandan
Yao
ab,
Samuel
Drouet
a,
Olivier
Mongin
a,
Frédéric
Paul
a,
J. A. Gareth
Williams
c,
Mark A.
Fox
 *c and
Christine O.
Paul-Roth
*ab
*c and
Christine O.
Paul-Roth
*ab
aInstitut des Sciences Chimiques de Rennes, ISCR-UMR CNRS 6226, Université de Rennes 1, 35042 Rennes Cedex, France. E-mail: christine.paul@univ-rennes1.fr; christine.paul@insa-rennes.fr
bInstitut National des Sciences Appliquées, INSA-ISCR, 35043 Rennes Cedex and Université Européenne de Bretagne, France
cDepartment of Chemistry, Durham University, Durham, DH1 3LE, UK. E-mail: m.a.fox@durham.ac.uk
First published on 17th April 2015
Abstract
Reactions of the 16-electron ruthenium complex [Ru(dppe)2Cl][PF6] with metal-free and zinc ethynylphenyltrifluorenylporphyrins 1 and 2 respectively, gave the new dyads 3 and 4 with ethynylruthenium group as a potential electron donor and the porphyrin as a potential electron acceptor. The redox properties of the porphyrins 1–4 were investigated by cyclic voltammetry and UV spectroelectrochemistry (SEC), which reveal that the monocation and monoanion of metal-free porphyrin 1 are stable under these conditions whereas the formation of the corresponding radical cation or anion of the zinc porphyrin 2 was accompanied by partial decomplexation of the zinc ion. Oxidations of the dyads 3 and 4 gave stable radical cations as probed using IR, NIR and UV SEC methods. These cations show similar NIR and IR bands to those reported for the known 17-electron [Ru(dppe)2(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh)Cl]+ radical cation. Remarkably, the dyad 3 has four stable redox states +2/+1/0/−1 where the second oxidation and first reduction processes take place at the porphyrin unit. Simulated absorption spectra on 1–4 at optimised geometries obtained by TD-DFT computations with the CAM-B3LYP functional are shown to be in very good agreement with the observed UV absorption spectra of 1–4. The spectra of 1–4 and their oxidised and reduced species were interpreted with the aid of the TD-DFT data. Fluorescence measurements reveal that the dyads 3 and 4 are only weakly emitting compared to 1 and 2, indicative of quenching of the porphyrinic singlet excited state by the ruthenium centre.
CPh)Cl]+ radical cation. Remarkably, the dyad 3 has four stable redox states +2/+1/0/−1 where the second oxidation and first reduction processes take place at the porphyrin unit. Simulated absorption spectra on 1–4 at optimised geometries obtained by TD-DFT computations with the CAM-B3LYP functional are shown to be in very good agreement with the observed UV absorption spectra of 1–4. The spectra of 1–4 and their oxidised and reduced species were interpreted with the aid of the TD-DFT data. Fluorescence measurements reveal that the dyads 3 and 4 are only weakly emitting compared to 1 and 2, indicative of quenching of the porphyrinic singlet excited state by the ruthenium centre.
Introduction
The combination of electron-rich group 8 metal complex(es) around a porphyrin core within the same molecular assembly can lead to new organometallic chromophores with remarkable optical properties.1,2 Furthermore, when the peripheral metallic fragments are redox-active, the redox-activity of these assemblies might be exploited for switching their linear optical (LO) and nonlinear optical (NLO) properties by means of electron-transfer.3,4 For instance, we showed that tetra(ruthenium-chloride-ethynylphenyl)zinc(II) porphyrin (ZnTRuEP) presents particular cubic NLO properties which result from the association of the porphyrinic and Ru-based fragments (Chart 1).5–7 Akita and co-workers recently demonstrated that the fluorescence of a porphyrin linked to a CpM(P^P) unit via an acetylide, such as A and B, could be redox-switched.8 The neutral derivatives are essentially non-fluorescent due to an intramolecular redox quenching of the porphyrin excited singlet state by the organometallic substituent, while oxidation of the metal–alkynyl unit partially restores the red fluorescence characteristic of the Zn(II) porphyrin core. So far, only a handful of such organometallic conjugates have been reported8,9 despite their potential for use in molecular-based electronics or photonics.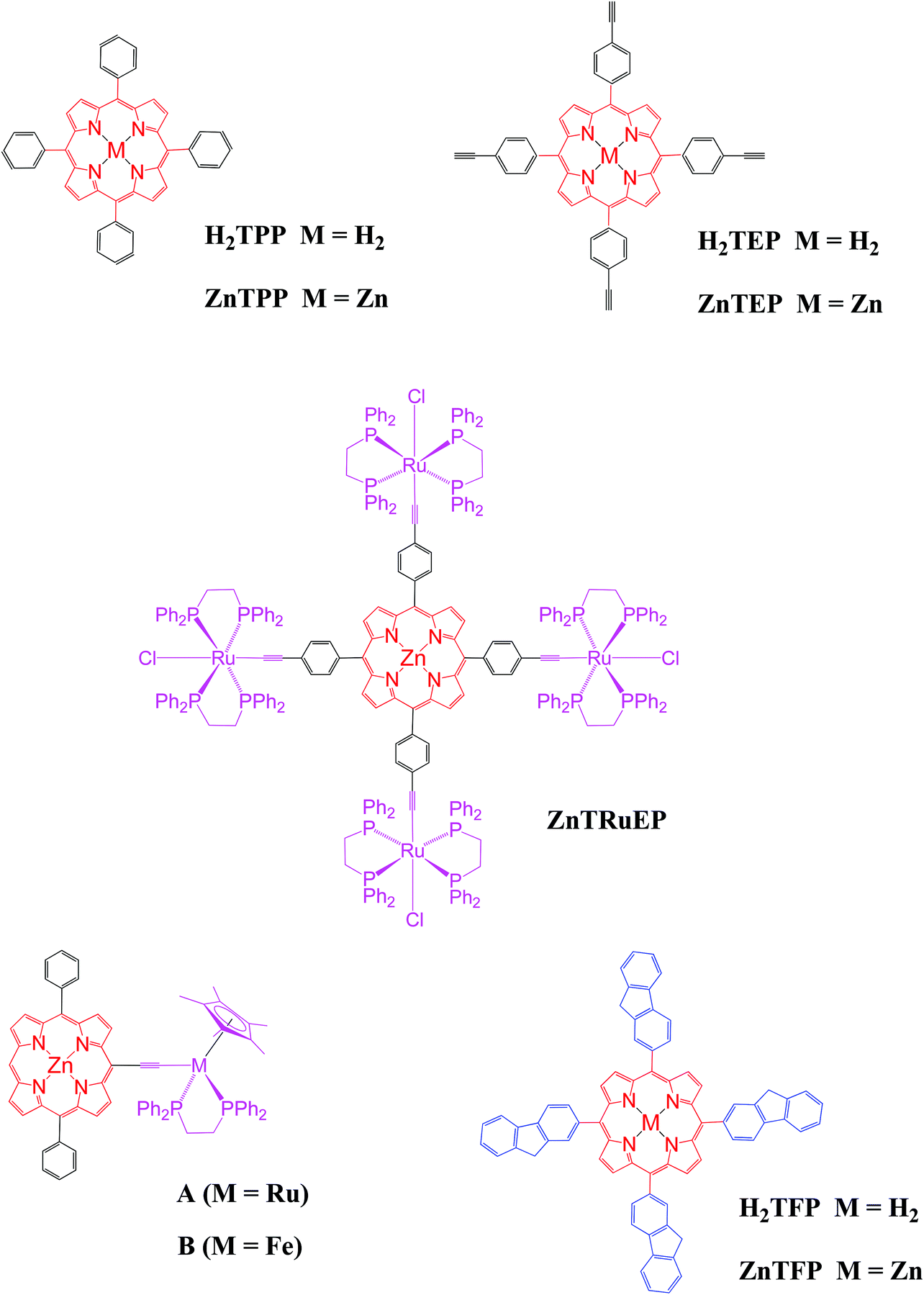 | ||
| Chart 1 Previously reported porphyrins relevant to the current study. | ||
While redox control of LO and NLO properties at the molecular level has ample precedence in the literature10 with metal acetylide derivatives,4,11 redox-control of the fluorescence with metal acetylide derivatives is much more rare in comparison.12 However, no fluorescence yields were quoted for the fluorescent (“on”) states in these examples apart from the dyads A and B where low quantum yields of ≤1.5% were measured.8 Although these examples clearly establish the proof of principle of fluorescence redox-switching for acetylide derivatives, more intense fluorescence in the “on” state is certainly desirable for any practical use compared to other systems developed so far.13
The low fluorescence yield reported in the case of A+ or B+ (“on” states) is not so surprising, given that the fluorescence of the ZnTPP core is intrinsically quite weak. In this respect, we reported the synthesis of zinc porphyrins possessing several fluorenyl arms (e.g.ZnTFP) and showed that the fluorescence of these derivatives was more intense than the corresponding phenyl (as opposed to fluorenyl) analogues (e.g.ZnTPP)14–16 eventually allowing their use as luminophores in OLEDs.5 In particular, a high fluorescence quantum yield (ΦF = 24%) was evidenced for the metal-free tetrafluorenylporphyrin macrocycle (H2TFP), demonstrating the good capacity of the fluorenyl units to enhance quantum yields by increasing the radiative deactivation process. Identifying new meso-tetraarylporphyrin conjugates resembling ZnTRuEP, A or B – but featuring fluorenyl groups instead of phenyl ones on the porphyrin core – may thus be an attractive approach to isolate new redox-switchable conjugates that would have more desirable fluorescence properties in the “on” (oxidised) state.
This work describes a first contribution toward this aim using the well-established17 Ru(dppe)2Cl metal–acetylide fragment as electrophore for redox-controlling the fluorescence of porphyrins with pendant fluorenyl arms. Thus, the syntheses and characterisation of the new dyads 3 and 4 from the recently synthesised18,19 ethynylphenyltrifluorenylporphyrins 1 and 2 respectively (Scheme 1) and the luminescence properties of these compounds are reported. The spectral properties of the four porphyrins 1–4 and their various oxidised and reduced species are also examined in detail by UV spectroelectrochemistry (SEC) and, for 3 and 4, by IR and NIR spectroelectrochemistry. These experimental studies are complemented by DFT computations to understand the nature of the observed absorption bands. The use of the new acetylide complexes 3 and 4 for redox-switching of their optical properties is briefly discussed.
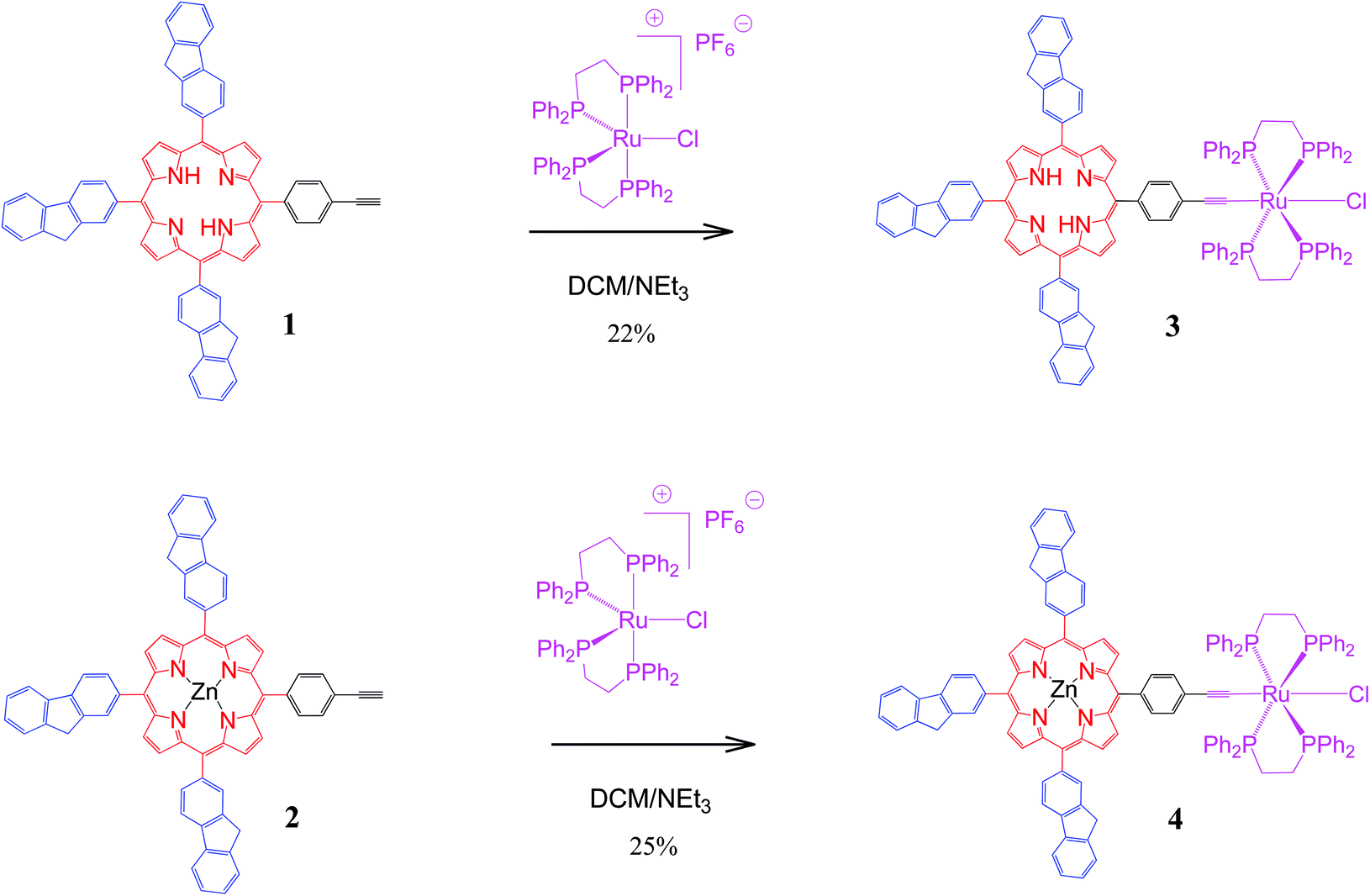 | ||
| Scheme 1 Syntheses of new dyads 3 and 4. | ||
Results and discussion
Syntheses and characterisation
The new dyad 3 was formed from the reaction of the ethynylphenyltrifluorenylporphyrin 1 with the 16-electron five-coordinate ruthenium complex [RuCl(dppe)2][PF6] in dichloromethane (DCM) followed by triethylamine (NEt3) (Scheme 1).20 The latter was used in excess to avoid an inseparable mixture of the starting material 1 and the desired product 3. The reaction was considered complete when the singlet peak of the terminal alkyne at 3.3 ppm disappeared on monitoring the mixture by 1H NMR spectroscopy prior to work-up, which gave a greenish-brown solid identified as 3. Dyad 4 was made from 2 using a similar procedure.The dyads 3 and 4 were characterised by multinuclear NMR spectroscopy, mass spectrometry and elemental analyses. 31P NMR spectra for 3 and 4 showed singlets at 49.7 and 50.0 ppm respectively, corresponding to four equivalent phosphorus atoms in the dppe ligands coordinated to the ruthenium group. The peaks in the 1H NMR spectrum of 4 can be identified by direct comparison with the 1H NMR spectra of 2 and a model compound21 RuCl(CCPh)(dppe)2 (Fig. 1). The spectrum of 4 contains a broad multiplet at 2.8 ppm corresponding to the eight CH2 dppe protons. Two peaks, a singlet at 4.20 ppm (2H), and another at 4.22 ppm (4H), characterise the CH2 protons of the fluorenyl units. The peaks between 7.0 and 7.4 ppm and the overlapped peak at 7.8 ppm are from the phenyl groups of the dppe ligands. Between 7.4 and 8.5 ppm, the seven peaks that correspond to the fluorene groups are identified along with two doublets at 7.04 and 7.98 ppm corresponding to the phenylene unit at the porphyrin (Fig. S1‡). In addition, the β pyrrolic protons are identified by the peaks at around 9 ppm. The 1H NMR spectrum of 3 differs from that of 4, with a singlet at −2.6 ppm characterizing the porphyrin NH protons. The 13C NMR spectra for 1–4 also show the expected peaks although many are overlapped – there are 43 unique carbons in 1 and 2 and 52 in 3 and 4 (Fig. S2‡).
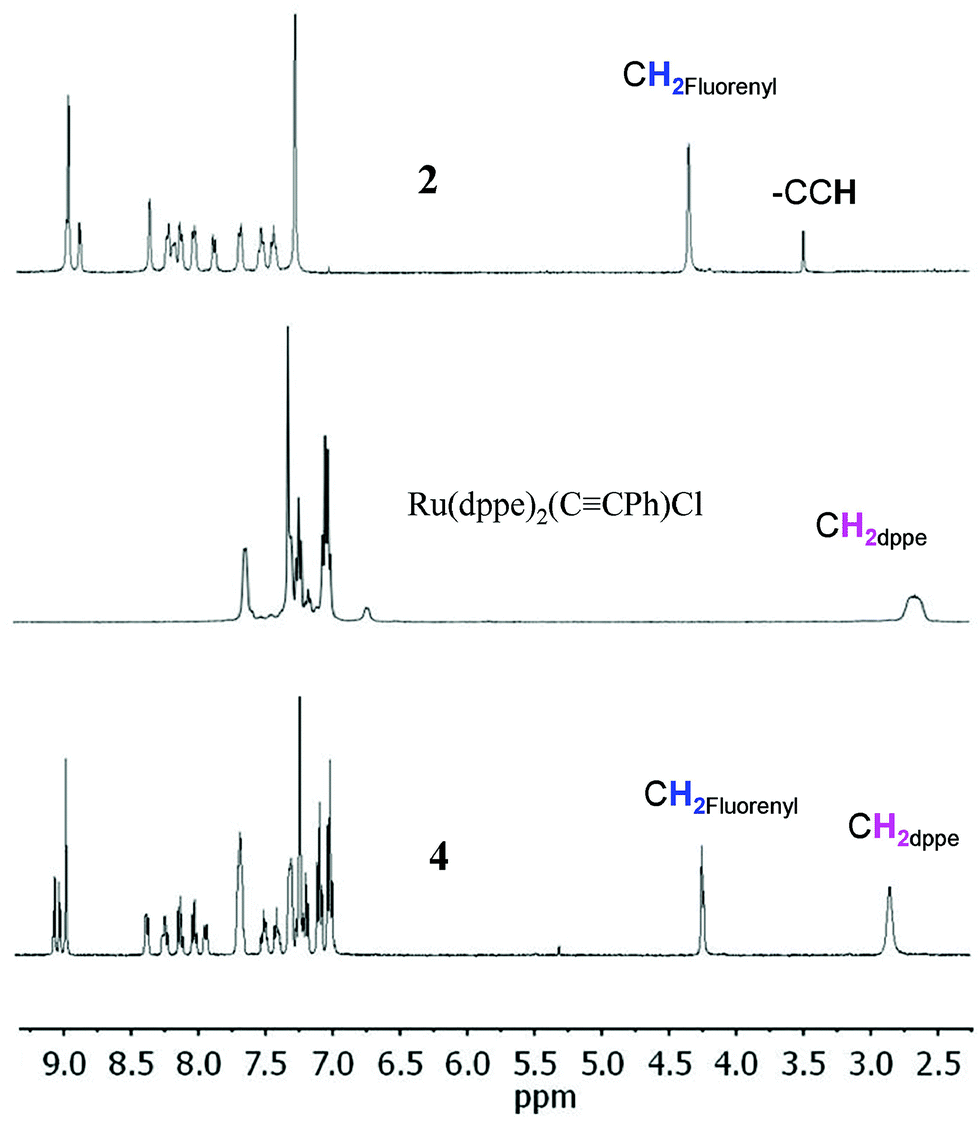 | ||
| Fig. 1
1H NMR spectra (400 MHz, in CDCl3) of zinc porphyrin 2, Ru(dppe)2(CCPh)Cl and the new dyad 4. | ||
Absorption and emission spectroscopy
The UV-visible absorption spectra were recorded at room temperature for 1–4 (Fig. 2 and Table 1). In all cases, the spectra exhibit an intense B-(Soret) band with a maximum absorption around 425 nm and several lower-energy Q-bands. Four characteristic Q-bands are observed for the free-base porphyrins 1 and 3 (two of them overlapped in 3), while only two bands are observed for the Zn(II) complexes 2 and 4, as is usual in metalloporphyrins. A characteristic blue shift is observed for all these bands upon metallation with zinc. In addition, a broad band centred around 275 nm is seen for 1–4, which corresponds to the absorption of the three fluorenyl arms of the porphyrin.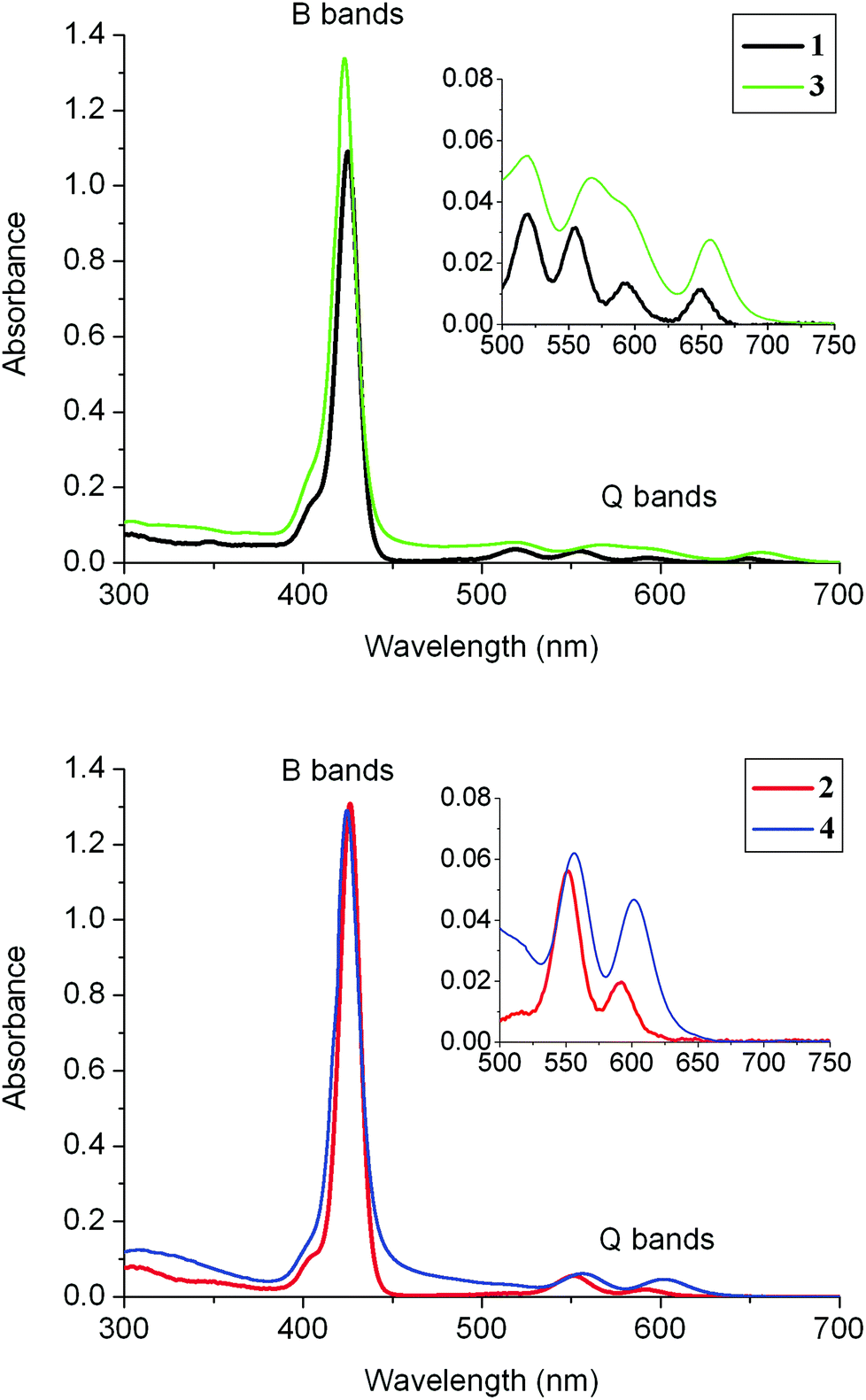 | ||
| Fig. 2 Absorption spectra (in DCM) of the new dyads 3 and 4 (10−5 M) at room temperature. The corresponding precursors 1 and 2 are included for comparison. | ||
| λ max /nm | λ max /nm | λ max/nm | λ em/nm | Φ f % | Ref. | |
|---|---|---|---|---|---|---|
| UV band | Soret band | Q bands | ||||
| a Wavelengths of the absorption maxima in the UV region (200–400 nm range). b Wavelengths of the absorption maxima in the Soret or B band region (400–450 nm range). c Fluorescence quantum yields using H2TPP in toluene as standard (ΦF = 0.12)22 following excitation into the Soret bands. | ||||||
| 1 | 275 | 425 | 518, 555, 594, 649 | 657, 700 | 21 | This work |
| 2 | 273 | 426 | 554, 595 | 607, 655 | 7 | This work |
| 3 | 263 | 423 | 520, 568, 590, 657 | 659, 718 | 1.54 | This work |
| 4 | 264 | 425 | 557, 602 | 606, 651 | 0.02 | This work |
| H2TPP | — | 417 | 513, 548, 589, 646 | 650, 714 | 12 | 22 |
| H2TFP | 272 | 426 | 519, 557, 593, 649 | 661, 725 | 24 | 23 |
| ZnTFP | 264 | 428 | 555, 601 | 608, 657 | 8.5 | 24 |
| ZnTEP | 302 | 424 | 552, 594 | — | — | 6 |
| ZnTPP | — | 421 | 556, 603 | 603, 650 | 3.3 | This work |
| ZnTRuEP | 327 | 418 | 452, 563, 615 | 601, 652 | 0.02 | This work |
The emission spectra of these compounds were recorded upon excitation of the Soret band (Fig. 3). Depending on their nature (free base or metallated porphyrin) they consist of several sub-bands assigned to a vibronic progression from the Q-state: for 3, the band near 720 nm assigned as Q(2,0) is very weak and appears often as an extended tail on the Q(1,0) band. The main fluorescence bands are blue-shifted upon metallation. For 3, the strongest emission band Q(0,0) is typically around 600 nm and the second peak, i.e. Q(1,0), appears at ∼650 nm. Compounds 1 and 2 without any Ru acetylide substituents have more intense fluorescence in solution than 3 and 4. Porphyrin 1 has a luminescence quantum yield of 21% which is higher than that of the reference H2TPP (12%) and comparable to that of H2TFP (24%). The Zn(II) porphyrin 2 has a quantum yield of 7%, again higher than that of the TPP analogue ZnTPP (3.3%) After substitution of 1 with the Ru complex to generate the dyad 3, the fluorescence quantum yield drops to 1.5%, comparable to the values reported for A+ and B+. Dyad 4 with the Zn(II) porphyrin is much more weakly emissive (Φ = 0.02%), similar to that determined for ZnTRuEP, A and B. It seems that the complexation of the porphyrin ligand by Zn(II) facilitates the non-radiative deactivation processes. The quenching of the porphyrin emission by the ruthenium unit is apparently less efficient for the free base porphyrins than for the zinc porphyrins. The emission spectra of 3 and 4 at 77 K are shown in Fig. S3.‡
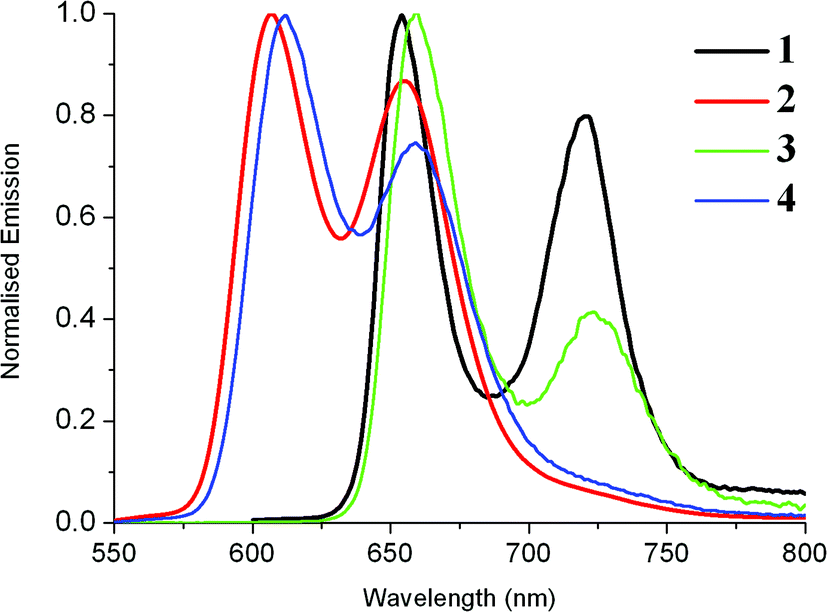 | ||
| Fig. 3 Emission spectra (in DCM) of the new dyads 3 (10−5 M) and 4 (10−6 M) at room temperature and comparison with emissions of 1 and 2. | ||
Electrochemistry
Cyclic voltammetry (CV) data of tetraarylporphyrins generally show two oxidation and two reduction waves corresponding to oxidation and reduction at the porphyrin unit. These waves are present in the CV measurements carried out here for 1 and 2 where the first oxidation and reduction waves are reversible (Fig. 4 and Table 2). Table 2 also lists reported CV values for related porphyrins (Chart 1) where zinc porphyrins are easier to oxidise by ∼0.1 V and more difficult to reduce by ∼0.2 V compared to the metal-free porphyrin analogues. The compounds 1 and 2 at 2.18 V and 2.26 V respectively have the smallest oxidation–reduction potential differences of the porphyrins listed in Table 2. Irreversible oxidation waves at even more positive potentials are also found in the porphyrins listed, which correspond to oxidations at the fluorenyl groups in 1 and 2. | ||
| Fig. 4 Cyclic voltammograms for 1–4 in DCM in the presence of 0.1 M [Bu4N][PF6] as the supporting electrolyte. The current is scaled at 5 μA between each tick mark. | ||
| E 1ox | E 2ox | E 3ox | E 1red | E 2red | E 1ox–E1red | Ref. | |
|---|---|---|---|---|---|---|---|
| a Values in italics are anodic potentials of irreversible waves. b Values in italics are cathodic potentials of irreversible waves. c Oxidation at the ruthenium unit. | |||||||
| 1 | 0.53 | 0.82 | 1.43 | −1.65 | −1.98 | 2.18 | This work |
| 2 | 0.39 | 0.66 | 1.35 | −1.87 | −2.27 | 2.26 | This work |
| 3 | −0.02c | 0.48 | 0.81, 0.94 | −1.70 | −2.15 | 1.68 | This work |
| 4 | 0.01c | 0.32 | 0.50, 0.63 | −1.80 | −2.19 | 1.81 | This work |
| H2TPP | 0.53 | 0.87 | 1.95 | −1.78 | −2.12 | 2.31 | 25 |
| ZnTPP | 0.46 | 0.72 | — | −1.90 | −2.28 | 2.36 | 25 |
| H2TFP | 0.58 | 0.90 | 1.50 | −1.78 | −2.12 | 2.36 | 26 |
| ZnTFP | 0.41 | 0.71 | 1.40 | — | 26 | ||
| Ru(dppe)2(CCPh)Cl |
0.01c | 0.89 | — | 27 | |||
| H2TEP | 0.58 | 0.90 | 1.05, 1.36 | −1.62 | −1.95 | 2.20 | 28 |
| ZnTEP | 0.46 | 0.72 | — | −1.90 | −2.32 | 2.36 | 28 |
| ZnTRuEP | 0.03c | 0.41 | 0.70 | — | 6 | ||
The CV data for the ruthenium–porphyrin complexes, 3 and 4, reveal reversible oxidation waves at −0.02 and 0.01 V respectively due to oxidations at the ruthenium ethynyl units at similar potentials to the ferrocenium/ferrocene couple.29,30 For comparison, Ru(dppe)2 (CCPh)Cl has an oxidation potential of 0.01 V27 which shows that the porphyrin moieties have very small effects on the potentials corresponding to the ruthenium ethynyl moieties, as found for ZnTRuEP.7 The presence of the ruthenium redox centres results in the oxidation waves associated with the porphyrin units being shifted to more positive potentials by ∼0.06 V in 3 and 4. This trend follows for the reduction wave potential of 4 but does not follow for 3 where its reduction wave potential is more negative by 0.05 V. Unlike their porphyrin precursors 1 and 2, the second oxidation and reduction waves at the porphyrin units in 3 and 4 are irreversible. There is an irreversible oxidation wave at 0.89 V27 for Ru(dppe)2(CCPh)Cl which suggests that such irreversible waves are expected in 3 and 4 in the same potential range as the second oxidation waves at the porphyrin units. The observed two irreversible oxidation waves for 3 and 4 (Table 2 and Fig. S4‡) support this.
Spectroelectrochemistry
Spectroelectrochemistry (SEC) measurements were carried out on the four porphyrins 1–4, to obtain the absorption spectra and establish the stabilities of their oxidised and reduced species. Absorption measurements of the oxidised and reduced species for all porphyrins are listed in Table 3. Apart from our published study,7 there is only one study on the parent porphyrins using a thin-layer SEC cell and two spectroscopic studies on porphyrin radical anions generated electrochemically. The reported data from these studies are shown in Table 3 for comparison.| Porphyrin | n | λ max/nm | λ max/nm | λ max/nm | λ max/nm |
|---|---|---|---|---|---|
| UV band | “Soret bands” | “Q bands” | NIR band | ||
| a Bulk electrolysis, [Et4N][ClO4], DMF, ref. 31. b Ref. 32. c Bulk electrolysis, [Pr4N][ClO4], THF, ref. 33. | |||||
| [1]n | 0 | 262 (65), 304 (39) | 424 (527) | 519 (27), 556 (20), 596 (8), 649 (9) | |
| +1 | 261 (58), 322 (36) | 464 (341) | 632 (sh, 15), 690 (76) | ||
| “+2” | 384 (70) | 440 (sh, 106), 485 (126) | 670 (158) | ||
| −1 | 257 (59), 305 (45) | 425 (238), 457 (sh, 43) | 519 (20), 557 (16), 601 (12), 656 (13), 704 (5) | 791 (5), 878 (6), 942 (sh, 4) | |
| [2]n | 0 | 427 (479) | 554 (22), 614 (14) | ||
| “+1” | 425 (140), 460 (94) | 689 (22) | 874 (12) | ||
| “−1” | 318 (70) | 437 (224) | 570 (40), 614 (46), 722 (8) | 835 (15) | |
| [3]n | 0 | 261 (133) | 423 (478) | ||
| +1 | 262 (117) | 424 (404) | 520 (20), 559 (18), 594 (9), 650 (7) | 840 (4), 1234 (3) | |
| +2 | 460 (195) | 705 (57) | |||
| −1 | 260 (153) | 411 (94), 466 (sh, 40) | 517 (sh, 20), 561 (18), 606 (17), 657 (9), 722 (8) | 839 (sh, 3) | |
| [4]n | 0 | 424 (480) | 553 (27), 597 (15) | ||
| +1 | 426 (435) | 553 (26), 593 (13) | 817 (5), 1302 (2) | ||
| “+2” | 425 (238), 454 (75) | 551 (20), 693 (19) | 880 (7) | ||
| “−1” | 261 (153), 312 (77) | 437 (473) | 572 (32), 616 (34) | 793 (7) | |
| [H2TPP]n | 0a | 418 (466) | 513 (18), 548 (8), 590 (5), 647 (4) | ||
| +1b | 417 (sh, 245), 436 (368) | 660 (49) | |||
| −1a | 405 (85), 430 (sh, 80), 448 (89) | 625 (9), 683 (13), 705 (sh, 12) | 765 (9), 873 (9) | ||
| −2a | 415 (sh, 61), 438 (82) | 548 (20), 595 (23) | |||
| [ZnTPP]n | 0c | 419 (560) | 545 (21), 585 (3) | ||
| +1c | 365 (35) | 409 (190), 460 (33) | 515 (10), 560 (10), 610 (12) | 770 (10), 840 (3) | |
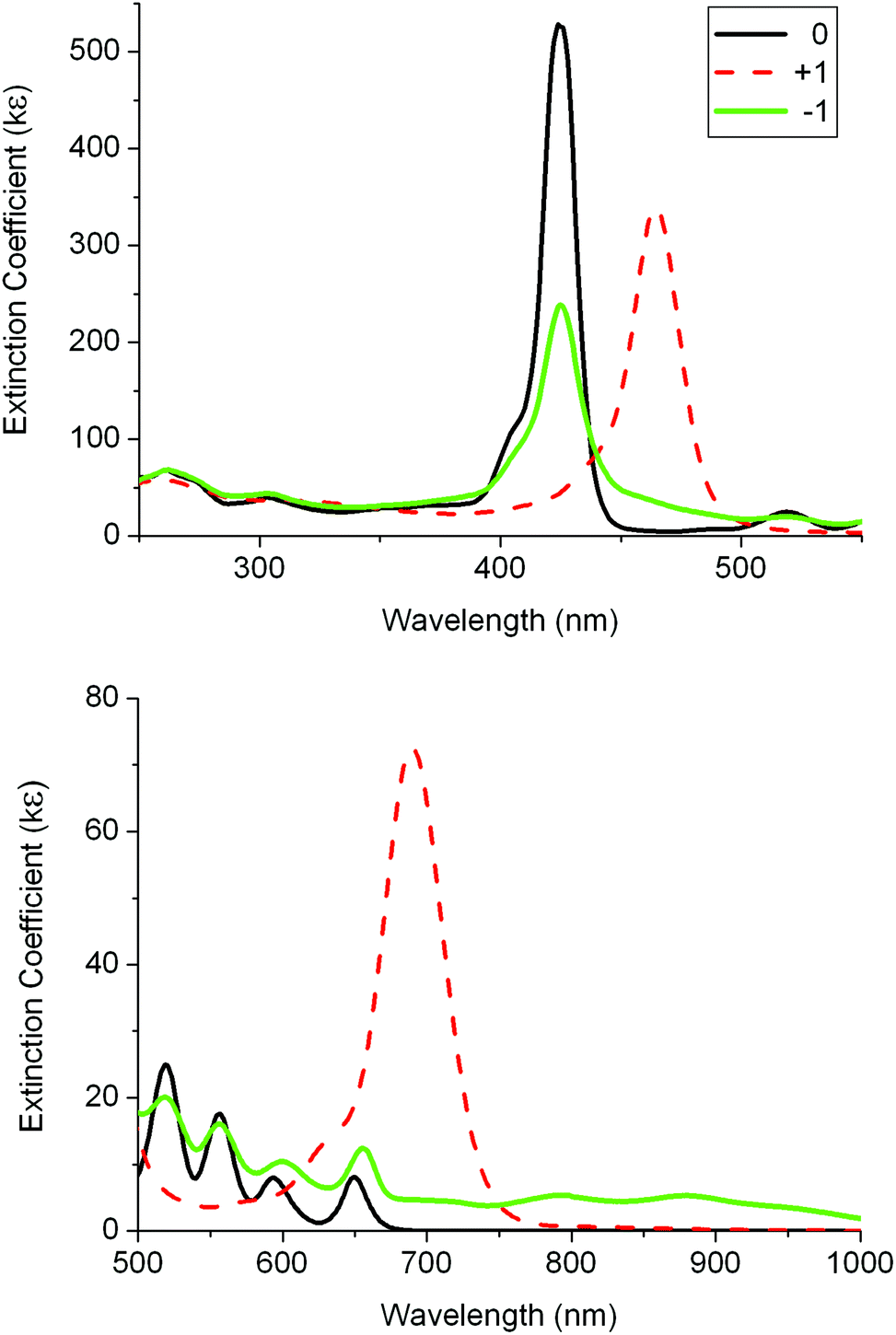 | ||
| Fig. 5 UV-visible absorption spectra for 1 and its oxidised and reduced species in 0.1 M [Bu4N][PF6]/DCM using a thin-layer spectroelectrochemistry (SEC) cell. | ||
A second SEC experiment was carried out on 1 in order to obtain the spectrum of the dication by two-electron oxidation. The two intense monocation bands at 464 and 690 nm were changed to two bands with smaller intensities at 440 and 485 nm and a broad, more intense band at 670 nm (Fig. S5‡). However, on back reduction the monocation spectrum was subtly different, displaying bands at 460 and 680 nm. This suggests that the dication is not stable under these thin-layer SEC cell conditions and the spectral data listed in Table 2 are thus labelled “2+”. Given the obvious similarities between the initial neutral species and the neutral species on back reduction, it is assumed here that the spectrum of pure dication would resemble “2+”.
SEC studies on 2 were less satisfactory. A spectrum of an oxidised species was obtained but on back-reduction the spectrum of neutral 2 was not fully restored. There were weak bands present at 520 and 650 nm on back reduction which imply that the Zn ion was replaced by protons during the process. A similar spectrum was found from back-oxidation on a second experiment investigating the reduction process of 2. The loss of the Zn ion in both processes suggests that zinc porphyrins with fluorenyl groups are susceptible to metal loss on oxidation and reduction processes. The spectra for the oxidised and reduced species “+1” and “−1” of 2 are listed in Table 3 and shown in Fig. S6.‡ While they are not robust species, some bands probably are from the expected monocation and monoanion of 2. The similarity of the “+1” bands to the reported data of the monocation species of ZnTPP supports this assumption.
Spectra were recorded for two oxidation species and a reduction species of 3 (Fig. 6 and Table 3). The original spectrum for the neutral species of 3 was recovered after recording these oxidised and reduced species. The first oxidation process showed little changes to the Soret and Q bands suggesting that the porphyrin unit is not involved in the oxidation process.7 The second oxidation process showed the same dramatic changes involving the Soret and Q bands as found for the first oxidation of 1 (Table 3 and Fig. S7‡). The spectrum on reduction of 3 has the Soret band intensity diminished and many weak bands between 450 and 850 nm. This resembles the spectrum for the reduced species of 1 and indicates that the porphyrin unit is reduced.
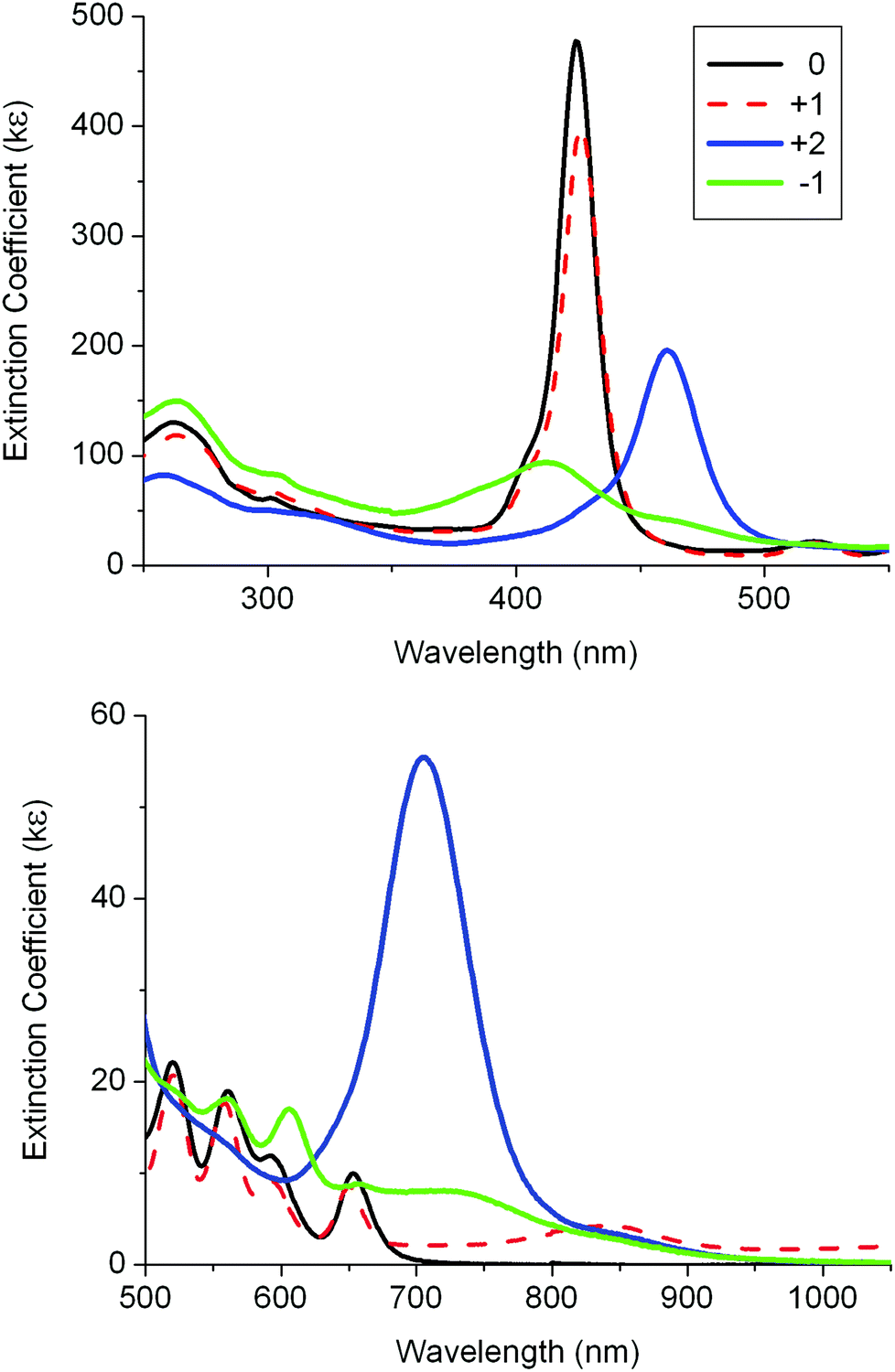 | ||
| Fig. 6 UV-visible absorption spectra for 3 and its oxidised and reduced species in 0.1 M [Bu4N][PF6]/DCM using a SEC cell. | ||
As in 3, the first oxidation process of 4 showed little change to the Soret and Q-bands (Table 3 and Fig. S8‡). On further oxidation, there are notable changes to the Q bands with new, weak, low-energy bands at 693 and 880 nm. However, back-reduction did not revert to the original spectrum corresponding to 4 as minor peaks at 520 and 650 nm were present. In fact, the back-reduction spectrum appears as a mixture of neutral 3 and 4. The loss of the zinc(II) metal presumably takes place during the second oxidation process. Reduction of 4 also resulted in some loss of the zinc on back oxidation. It is a very important detrimental process and one which needs to be overcome if fatigue-resistant redox-switchable systems are to be developed with such molecules. The spectra recorded for “+2” and “−1” are assumed to represent the spectra of the dication and monoanion of 4. There are obvious similarities between these spectra and the “+1” and “−1” spectra of the zinc precursor 2. More importantly, these spectra are different from those of the corresponding species of 1 and 3.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 8000 cm−1 (Fig. 7 and Table 4). These bands are characteristic of the arylethynylruthenium moiety on oxidation. For example, the spectrum of the monocation of Ru(dppe)2(CCPh)Cl shows two weak NIR bands at around 12000 and 9000 cm−1 (Fig. S9‡).29,30,34 Unlike the precursors 1 and 2 which contain very weak CC bands, the arylethynylruthenium complexes 3 and 4 contain strong CC bands at 2068 cm−1 and so IR spectroelectrochemistry experiments were also carried out on these ruthenium complexes. On oxidation, the CC bands corresponding to the neutral species disappeared and CC bands at 1910 cm−1 appeared along with the tails of the NIR bands (Fig. S10 and S11‡). The energy differences of 158 cm−1 for these CC bands on oxidation are typical of arylethynylruthenium complexes, with 165 cm−1 having been reported for Ru(dppe)2(CCPh)Cl.30,34 These NIR and IR data for the cations of 3 and 4 indicate that the porphyrin units have little influence on the spectral properties associated with the Ru units and may be considered as spectators.
000 and 8000 cm−1 (Fig. 7 and Table 4). These bands are characteristic of the arylethynylruthenium moiety on oxidation. For example, the spectrum of the monocation of Ru(dppe)2(CCPh)Cl shows two weak NIR bands at around 12000 and 9000 cm−1 (Fig. S9‡).29,30,34 Unlike the precursors 1 and 2 which contain very weak CC bands, the arylethynylruthenium complexes 3 and 4 contain strong CC bands at 2068 cm−1 and so IR spectroelectrochemistry experiments were also carried out on these ruthenium complexes. On oxidation, the CC bands corresponding to the neutral species disappeared and CC bands at 1910 cm−1 appeared along with the tails of the NIR bands (Fig. S10 and S11‡). The energy differences of 158 cm−1 for these CC bands on oxidation are typical of arylethynylruthenium complexes, with 165 cm−1 having been reported for Ru(dppe)2(CCPh)Cl.30,34 These NIR and IR data for the cations of 3 and 4 indicate that the porphyrin units have little influence on the spectral properties associated with the Ru units and may be considered as spectators.
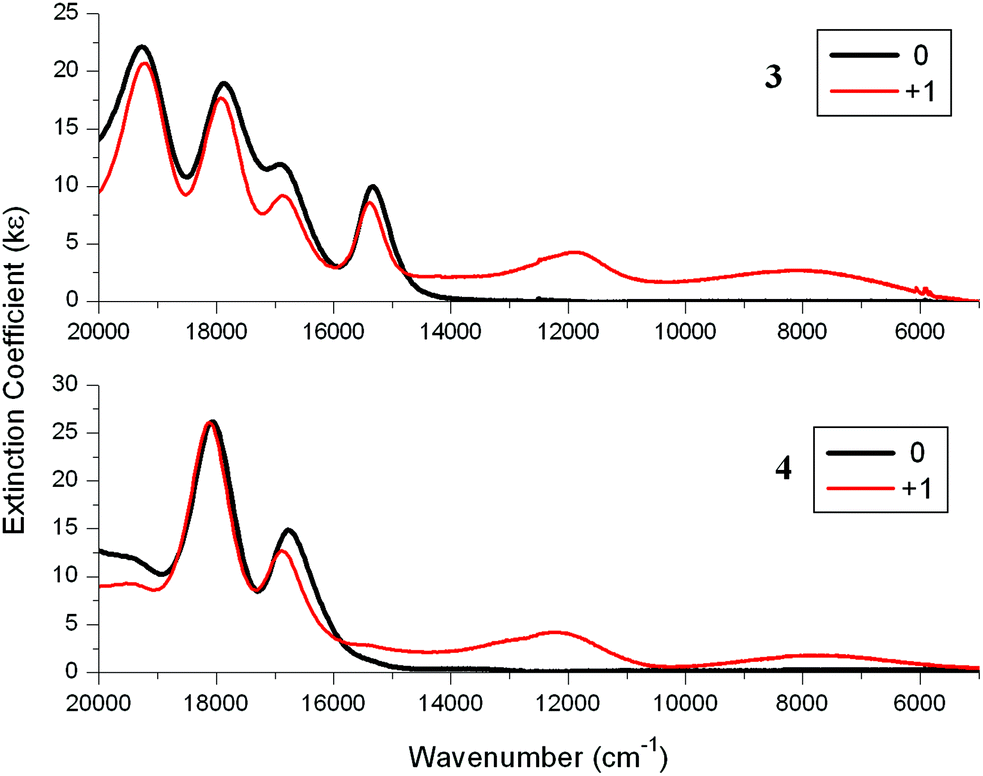 | ||
| Fig. 7 Near-IR (NIR) absorption spectra for 3 (top) and 4 (bottom) and their cations in 0.1 M [Bu4N][PF6]/DCM using a SEC cell. | ||
CPh)Cl, ZnTRuEP and their monocations obtained by spectroelectrochemistry (SEC) in 0.1 M [Bu4N][PF6]/DCM at 298 K
Computations
Geometry optimisations on 1–4 were carried out with the hybrid-DFT functional method (B3LYP) in order to aid interpretation of their observed spectroscopic data, with 1′–4′ denoted for DFT-optimised geometries to distinguish from the physical geometries. Metal-free porphyrins are difficult to model correctly for comparison between observed and calculated spectroscopic data as the two hydrogens at the nitrogens of the porphyrin unit are fluctional in solution. The most stable conformer 1′ involves the two hydrogens at opposite nitrogen atoms in the porphyrin centre. Three conformers of 1, where two hydrogens are at adjacent nitrogens, were also optimised and are 7.0, 7.1 and 7.2 kcal mol−1 less stable in energy than 1′. The predicted electronic structures and spectroscopic data from these three conformers are somewhat similar to that of 1′ so only the geometries of 1′ and, by implication, 3′ with hydrogens at opposite nitrogens are looked at in detail here.Electronic structure calculations reveal the expected frontier orbitals (HOMO and LUMO) located at the porphyrin units for 1′ and 2′ (Fig. 8). In 1′, the LUMO+1 and HOMO−1 orbitals are also at the porphyrin unit (Gouterman four-electron four-orbital model).35 While the LUMO and LUMO+1 orbitals are degenerate, the HOMO−1 is 0.4 eV lower in energy compared to HOMO (Table S1‡). The orbital make-up in 2′ is different due to an occupied orbital involving zinc so the ‘four orbitals’ in 1′ become five in 2′ (Table S2‡). This would explain the different Q-band characteristics in observed absorption spectra for metal-free and zinc porphyrins. Comparison of the HOMO and LUMO energies for 1′ and 2′ with the first oxidation reduction half-wave potentials in Table 1 shows the logical trends in the LUMO energies and the HOMO–LUMO energy differences. However the higher predicted HOMO energy in 1′ by 0.1 eV compared to 2′ does not support the fact that 2 is easier to oxidise by 0.14 V compared to 1. Calculated HOMO energies for metal-free porphyrins have been reported to be higher than for zinc porphyrins36 and it is assumed here that the zinc contribution to the stability of the HOMO energies is over-estimated in the computations.
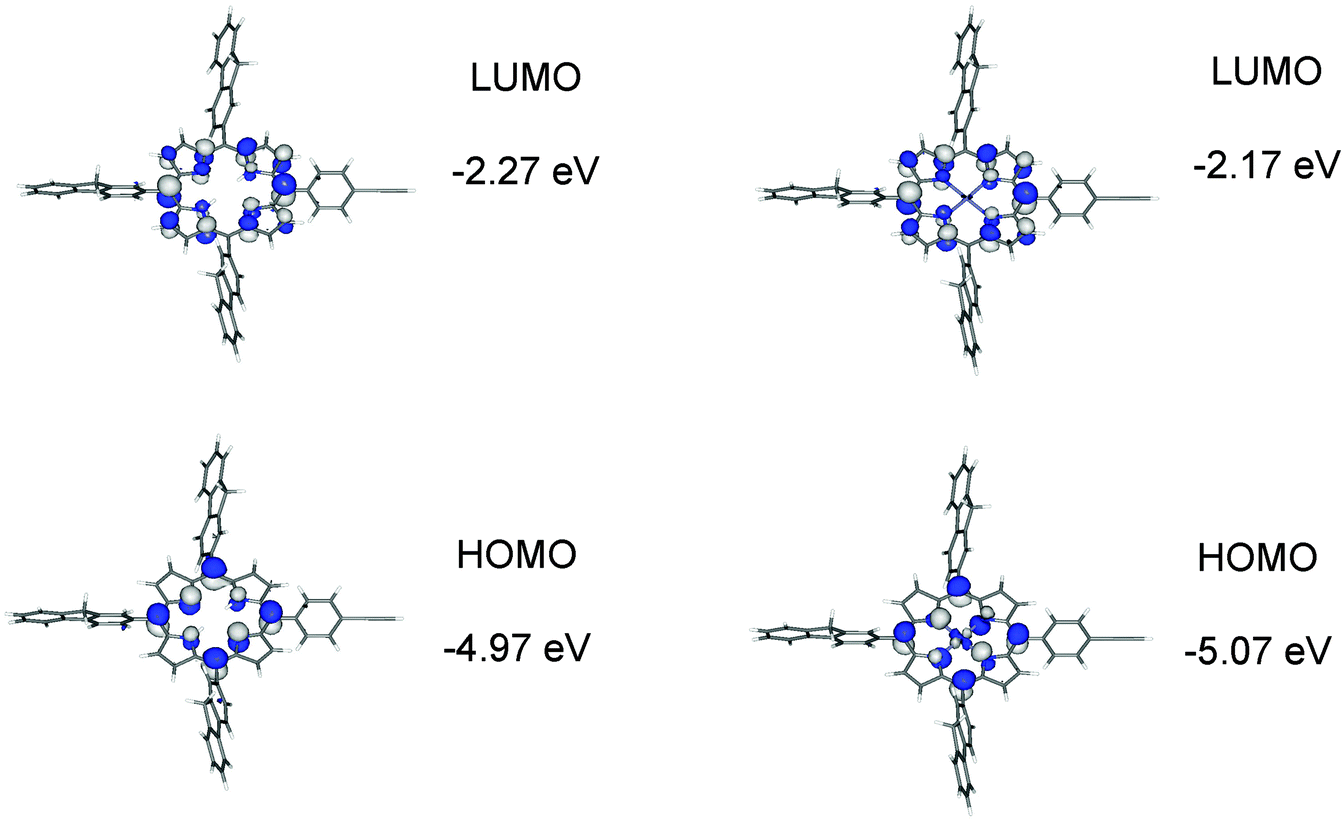 | ||
| Fig. 8 Frontier molecular orbitals for 1′ and 2′ plotted with a contour value of ±0.04 (e bohr−3)1/2. | ||
For the ruthenium dyad 3′, the HOMO, HOMO−1 and HOMO−2 with similar energies of −4.61, −4.81 and −4.91 eV are mainly at the ethynylruthenium unit whereas the degenerate LUMO and LUMO+1 are on the porphyrin unit (Fig. 9 and Table S3‡). While the LUMO is essentially located on the porphyrin only, the porphyrin contributes significantly (29%) to the HOMO which is quite surprising when the porphyrin group has little influence on the observed oxidation potential of 3 (−0.02 V) compared to 0.01 V for Ru(dppe)2(CCPh)Cl.
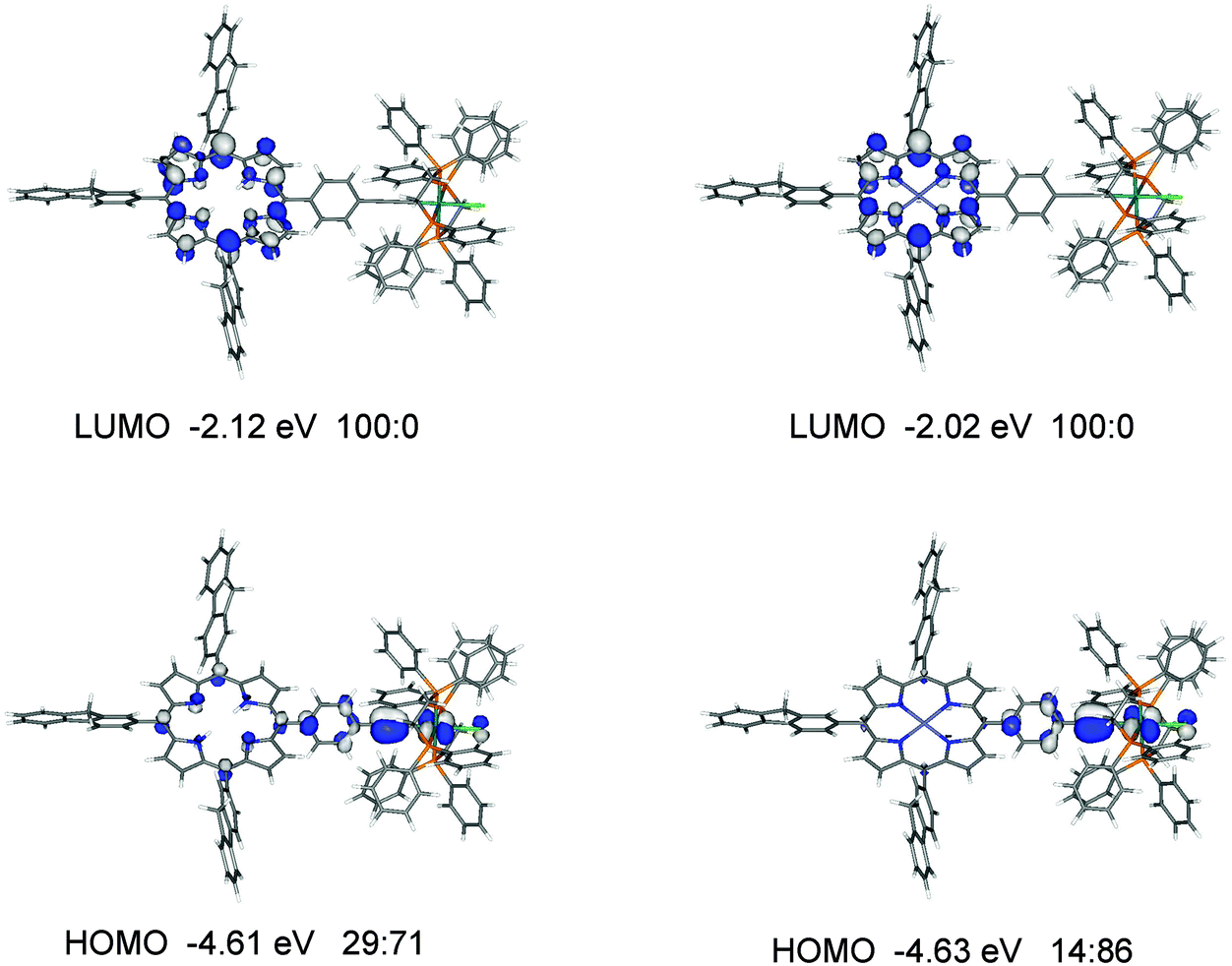 | ||
| Fig. 9 Frontier molecular orbitals for 3′ and 4′. The ratios listed correspond to % orbital contributions on the porphyrin and [(C6H4CC)Ru(dppe)2Cl] fragments. | ||
A similar orbital make up to 3′ is found for 4′ (Fig. 9 and Table S4‡) with one notable difference: the orbital with ruthenium character (HOMO−2) in 3′ is not present in the occupied orbitals of similar energies for 4′, presumably due to the contributions of the zinc atom to the orbitals in 4′. The HOMO in 4′ has considerably less porphyrin contribution (14%) than the HOMO in 3′ (29%) yet their energies are very similar at −4.63 and −4.61 eV respectively. The trend of the HOMO energies is in good agreement with the observed oxidation potentials (Table 2).
TD-DFT computations were carried out on all geometries 1′–4′ to simulate the absorption spectra and to aid assignments of the bands in the observed spectra of 1–4 and, by inference, assign the bands of the oxidised and reduced species. The CAM-B3LYP functional is used here instead of B3LYP as it is necessary to correctly model charge transfer over long distances37 for dyads like 3 and 4. One disadvantage with the predicted TD-DFT data is that the vibronic couplings typically observed in the Q-bands for porphyrins are not taken into account. While open-shell optimisations of these oxidised and reduced species can be carried out here, predicted open-shell TD-DFT data on porphyrin geometries are generally unreliable as shown elsewhere.38
Simulated absorption spectra on 1′–4′ from TD-DFT data are shown in Fig. 11. The observed strong Soret(B-) and the relatively weaker Q-bands are reproduced well computationally in 1′ and 2′ assuming that the computed Q-bands would be split by vibronic couplings. The predicted Soret bands at 410–431 nm are in good agreement with the observed maxima of 423–428 nm (Table 5). The computed lowest energy Q-bands are also in accord with observed lowest energy Q-bands as shown in Table 5. Computed extinction coefficients for these bands also reproduce similar values to those observed.
000 on the calculated oscillator strengths (f)
| B band | B band | Q band | Q band | |
|---|---|---|---|---|
| λ max/nm (ε) | λ max/nm (ε) | λ max/nm (ε) | λ max/nm (ε) | |
| Calculated | Observed | Calculated | Observed | |
| 1′ | 421 (488) | 425 (624) | 684 (6) | 649 (5) |
| 2′ | 410 (724) | 428 (316) | 607 (6) | 596 (3) |
| 3′ | 431 (484) | 423 (511) | 685 (9) | 657 (11) |
| 4′ | 420 (555) | 425 (524) | 609 (12) | 602 (20) |
The Soret or B-bands arise from allowed HOMO−1 → LUMO/LUMO+1 transitions where the π and π* orbitals largely overlap in the porphyrin unit in 1′ and 2′ (Fig. 11). The weaker Q-bands in 1′ result from two HOMO → LUMO/LUMO+1 transitions where the HOMO has significant N p-orbital character. The weaker Q-bands in 2′ are from the four HOMO/HOMO−2 → LUMO/LUMO+1 transitions where the HOMO and HOMO−2 have substantial zinc characters (Fig. 11).
In the case of the complexes 3′ and 4′, the simulated Soret and Q-bands are in agreement with observed absorption spectra for 3 and 4 (compare Fig. 10 with Fig. 2). As the Q-bands for 3′ resemble 1′ and 4′ with 2′, the orbital contributions from the arylethynylruthenium to these bands are very small (Fig. 10). Any bands resulting from transitions involving the ruthenium moiety and the porphyrin have low oscillator strengths – these are charge-transfers – relative to the local porphyrin–porphyrin transitions and thus have little impact on the overall spectral patterns for 3′ and 4′.
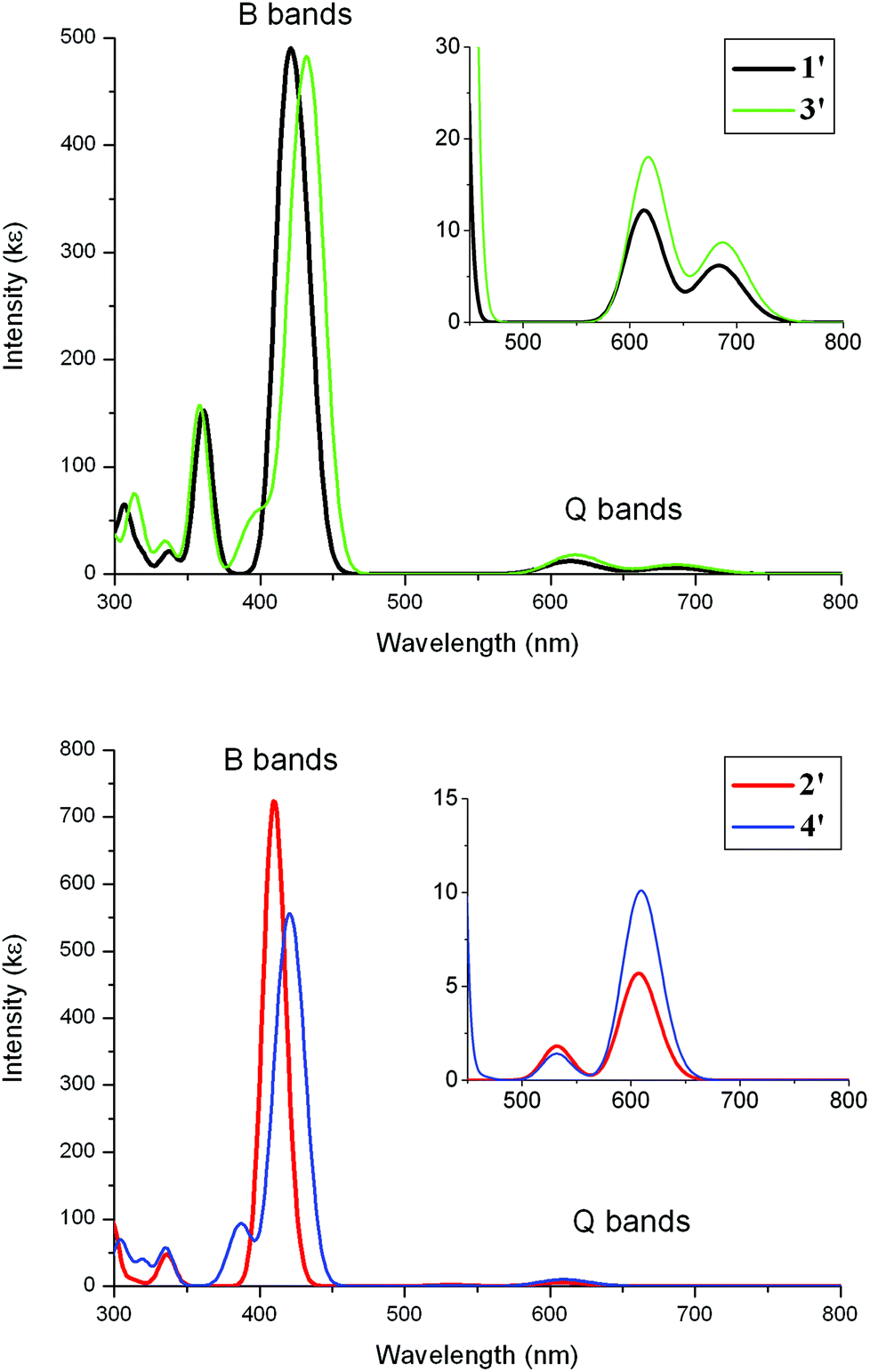 | ||
| Fig. 10 TD-DFT absorption spectra for 1′–4′. The bands were simulated using a half-height width of 0.08 eV. | ||
 | ||
| Fig. 11 Orbitals involved in the B-(Soret) and Q-bands for 2 based on TD-DFT data with computed wavelength and oscillator strength (f) listed for each transition. | ||
Oxidation of 1 to [1]+ would involve removal of an electron from the HOMO, forming a singly occupied molecular orbital SOMO, and thus result in significant changes in the energies of both HOMO (which would then become highest occupied single orbital, α-HOSO, and lowest unoccupied single orbital, β-LUSO, in open-shell computation data) and HOMO−1 (α-HOSO−1 and β-HOSO). Since the transitions corresponding to Soret bands and Q-bands involve HOMO and HOMO−1, dramatic changes in these bands are expected in the absorption spectra on oxidation of 1 to [1]+. These changes are observed experimentally, with the bands at 464 and 690 nm assigned to π(porphyrin) → π*(porphyrin) and π(porphyrin) → π*(N-character porphyrin) transitions, similar to those responsible for the Soret and Q-bands of 1. The 690 nm band lacks the vibronic couplings typically found in neutral porphyrins due to loss of orbital symmetries in the orbital make-up of the radical cation [1]+.
Reduction of 1 to [1]– adds an electron to the LUMO but this picture is complicated with the LUMO and LUMO+1 being degenerate in 1. When an electron is added, the LUMO becomes a SOMO (α-HOSO and β-LUSO). The observed spectrum of [1]– retains several bands observed in 1 which are the transitions involving the largely unchanged degenerate molecular orbital LUMO+1 i.e. responsible for the Soret and Q-bands. There are several unique bands in [1]– which arise from transitions involving the SOMO and all would involve porphyrin orbitals. Similar band assignments would apply to the spectra of the oxidised and reduced species of 2. The Q-bands in 2 are quite different to those in 1 due to the zinc contribution but the overall spectral changes on oxidation and reduction of 2 follow those of 1.
The first oxidations of 3 and 4 to [3]+ and [4]+ take place at the ethynylruthenium units where the HOMOs are located. As mentioned earlier, the charge transfer transitions involving the HOMO and HOMO−1 – which are essentially two ethynylruthenium d–π orbitals orthogonal to each other30,39 – give bands too weak to be observed above the strong Soret and the relatively weaker Q-bands from local porphyrin–porphyrin transitions in the absorption spectra of 3 and 4.
Removal of an electron from the HOMO in 3 would lead to a SOMO (α-HOSO and β-LUSO) on the ethynylruthenium moiety and thus a low-energy transition from an ethynylruthenium orbital (HOMO−1 in 3; α-HOSO−1 and β-HOSO in [3]+) into another ethynylruthenium orbital (SOMO; α-HOSO and β-LUSO) is expected. The weak NIR band of 8100 cm−1 observed for [3]+ is assigned to this formally β-HOSO → β-LUSO transition. A second weak NIR band of 11880 cm−1 is also the result of a transition involving the ethynylruthenium SOMO since similar NIR transitions are found in related ethynylruthenium cations (Table 4). No obvious changes are found for the Soret and Q-bands on oxidation of [3] to [3]+ which show that the transitions from the porphyrin molecular orbitals are not affected by the oxidation/reduction of the ethynylruthenium redox centre. The same conclusions apply for 4 and [4]+.
The observed IR v(CC) bands from 2068 cm−1 for the neutral species of the ruthenium complexes 3 and 4 to 1910 cm−1 on oxidation indicate weakening of the CC bonds on oxidation. The removal of an electron from the HOMO located on the ethynylruthenium unit (RuCC) in 3 (or 4) would be expected to significantly weaken the CC bond length (and strengthen the Ru–C bond in RuCC).39
On second oxidations, both 3 and 4 give absorption spectra like those of 1 and 2 on first oxidations, respectively, while 3 and 4 on first reductions give similar spectra to those of 1 and 2 on first reductions. The spectra from the first oxidations on the dyads 3 and 4 resemble overlap of the neutral porphyrins 1 and 2 respectively with a [Ru(dppe)2(CCPh)Cl]+ monocation. The spectroelectrochemical and computational results here thus show that the porphyrin and ethynylruthenium units in 3 and 4 can be viewed as largely independent redox centres.
The observed fluorescence quantum yields decrease when the ethynylruthenium units are attached to the corresponding porphyrins (Table 1). From our spectroelectrochemical and computational data on 3 and 4 here, a plausible explanation for the fluorescence yield decrease is the intramolecular electron transfer taking place after excitation of the complexes 3 and 4. The porphyrins 1 and 2 emit local fluorescence after light excitation (Fig. 12). The ruthenium complex 3 (or 4) is also initially excited at the porphyrin units but, while local porphyrin fluorescence of the complex is observed based on a similar emission spectrum (Fig. 3) to that of the corresponding porphyrin 1 (or 2), the major pathway is the intramolecular electron transfer to a diradical excited state. The diradical, which contains a negative charge at the porphyrin unit and a positive charge at the ruthenium centre, is then returned to the initial ground state via charge recombination. The latter process is non-radiative so the fluorescence quantum yields are decreased on going from 1 and 2 to 3 and 4 respectively.
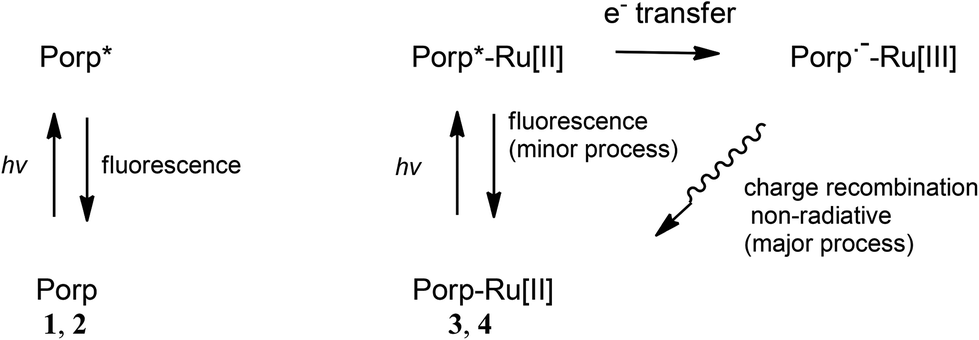 | ||
| Fig. 12 Proposed photophysical processes of the porphyrins 1 and 2 (Porp) and the porphyrin–ruthenium complexes 3 and 4 (Porp-Ru[II]). | ||
Conclusions
In summary, two donor–acceptor dyads (3 and 4) featuring the redox-active and electron-rich Ru(dppe)2Cl group as the donor site and a trifluorenylphenyl porphyrin as the acceptor site have been synthesised and characterised. The absorption and emission properties of these new compounds show that, while the precursors 1 and 2 are brighter fluorophores than H2TPP, both 3 and 4 are much poorer fluorophores than ZnTPP. Nevertheless, 3 has a higher fluorescence quantum yield than ZnTRuEP and other related chromophores containing Ru(II) and Fe(II) acetylide complexes at their periphery such as A or B (Chart 1), reflecting the desired effect of the 2-fluorenyl groups appended to the porphyrin ring on the fluorescence. Furthermore, compounds featuring free bases are much better luminophores than compounds made of Zn(II)-complexed porphyrins (ΦF(1) > ΦF(2) and ΦF(3) > ΦF(4)).Electrochemical (CV) and spectroelectrochemical studies on 1–4 show at least two kinetically stable redox states present. Compounds 1 and 3 featuring free base porphyrins have much better redox stabilities than compounds 2 and 4 with Zn(II)-complexed porphyrins. The nature of the first excited states of these derivatives in their mono-oxidised, di-oxidised and mono-reduced states are interpreted on the basis of computed electronic structures on optimised geometries of 1–4. For 3, there is the possibility for achieving a strong modulation of the cubic (and even quadratic) NLO properties at carefully selected wavelengths by reduction. In contrast, a much poorer modulation of the NLO properties is expected upon oxidation for 3 and 4 based on these studies. Controlling the luminescence of these derivatives, in particular for compounds 1 and 3 where three and four stable redox states respectively are accessible, by redox-switching appears promising. Current work on this aspect is underway.
Experimental section
General procedures
All reaction mixtures were performed under argon and were magnetically stirred. Solvents were distilled from appropriate drying agent prior to use, DCM from CaH2 and THF from sodium/benzophenone. Other solvents used were of HPLC grade. Commercially available reagents were used without further purification unless otherwise stated.1H NMR and 13C NMR in CDCl3 were recorded using Bruker 200 DPX, 300 DPX and 500 DPX spectrometers. The chemical shifts were referenced to 7.26 ppm for 1H, 77.2 ppm for 13C and external H3PO4 at 0.0 ppm for 31P. Peak assignments were performed by 2D NMR experiments where possible: COSY (Correlation Spectroscopy), HMBC (Heteronuclear Multiple Bond Correlation) and HMQC (Heteronuclear Multiple Quantum Coherence). UV spectra were recorded on a UVIKON XL spectrometer from Biotek instruments. PL emission spectra were recorded on a Photon Technology International (PTI) apparatus coupled on an 814 Photomultiplier Detection System, Lamp Power Supply 220B and MD-5020.
Steady-state fluorescence measurements were performed at room temperature on dilute solutions (ca. 10−6 M) using an Edinburgh Instruments (FLS 920) spectrometer working in photon-counting mode, equipped with a calibrated quantum counter for excitation correction. The dichloromethane solutions used were 10 times more concentrated for the zinc complex 4 (∼10−6 M) than for the free base 3 (∼10−5 M). Fluorescence quantum yields were measured using standard methods; H2TPP in DCM (Φf = 0.12 at λex = 417 nm) was used as a reference. The estimated uncertainty on the reported fluorescence quantum yields is ±10%.
Additional characterisation data; 1 FT-IR (KBr disc, cm−1): 3306 (w, NH stretch), 3272 (w, CC–H stretch), 3037 (s, aromatic CH stretch), 2920 (w, aliphatic CH stretch), 2104 (w, CC stretch); 13C{1H} NMR (400 MHz, CDCl3, δ in ppm): 143.9, 141.9, 141.7, 141.4, 140.8, 134.7, 133.7, 131.6 (CH in C6H4), 131.5, 130.8, 130.7 (CH in C6H4), 127.3, 125.5, 121.8, 121.0, 120.9, 120.5, 119.1, 118.2, 78.5 (CCH), 37.3 (fluorenyl CH2). 2 FT-IR (KBr disc, cm−1): 3294 (w, CC–H stretch), 3053 (s, aromatic CH stretch), 2920 (w, aliphatic CH stretch), 2108 (w, CC stretch); 13C{1H} NMR (400 MHz, CDCl3, δ in ppm): 150.7, 150.6, 150.0, 143.9, 143.7, 141.8, 141.4, 141.2, 137.6, 134.5, 133.6, 132.4, 132.3, 131.8 (CH in C6H4), 131.5, 130.6 (CH in C6H4), 127.2, 125.4, 122.0, 121.8, 121.5, 120.4, 120.12, 118.0, 78.3 (CCH), 37.3 (fluorenyl CH2).
C stretch).
C stretch).
Electrochemistry
Cyclic voltammograms were recorded with an Autolab PG-STAT 30 potentiostat at 20 °C from solutions of ca. 10−4 M analyte in dry DCM containing 0.1 M tetra-n-butylammonium hexafluorophosphate [Bu4N][PF6] at scan rate ν = 100 mV s−1 under a dry nitrogen atmosphere. The single-compartment three-electrode cell was equipped with platinum wire counter and reference electrodes and a glassy carbon working electrode. All redox potentials are reported with the decamethylferrocene/decamethylferrocenium (Cp*2Fe+/Cp*2Fe) redox couple used as an internal reference system at −0.53 V (ref. 27) vs. the usual ferrocene/ferrocenium (Cp2Fe+/Cp2Fe) redox couple at 0.0 V in DCM.Spectroelectrochemistry
Spectroelectrochemical (SEC) experiments were performed at room temperature in an airtight optically transparent thin-layer electrochemical (OTTLE) cell40 equipped with Pt minigrid working and counter electrodes (32 wires cm−1), Ag wire pseudo-reference electrode and CaF2 windows for a 200 μm path-length solvent compartment. DCM solutions containing 0.1 M [Bu4N][PF6] electrolyte were used in the cell for SEC experiments. The cell was fitted into the sample compartment of a Cary UV-Vis-IR spectrophotometer or a Nicolet Avatar 6700 FT-IR spectrometer. Bulk electrolysis was carried out using an Autolab PG-STAT 30 potentiostat.Computations
All computations were carried out with the Gaussian 09 package.41 The four model S0 geometries 1′–4′ with no symmetry constraints were fully optimised with the B3LYP functional42 using the 3-21G* basis set43 for all atoms. The 3-21G* basis set is used here as it has been shown to be suitable for ruthenium acetylides elsewhere.34,39,44 Frequency calculations on these geometries revealed no imaginary frequencies. Computed absorption data were obtained from TD-DFT45 calculations on S0 geometries using the CAM-B3LYP functional.46 As the CAM-B3LYP functional generally overestimates the transition energies, compared to that of experimental and B3LYP data, a scaling factor of 0.85 was applied.47 The MO diagrams were generated with the Gabedit package48 and the %MO contributions were obtained using the GaussSum software.49Acknowledgements
We thank “Région Bretagne” (ARED; A.M.), China Scholarship Council (CSC; D. Y. and X. Z.), MENRT (S.D.) and EPSRC (J.A.G.W. and M.A.F.) for funding. We also thank CNRS and Durham University for supporting the Rennes-Durham collaboration.References
- See for example: (a) P. Gautam, B. Dhokale, V. Shukla, C. P. Singh, K. S. Bindra and R. Misra, J. Photochem. Photobiol., A, 2012, 239, 24–27 CrossRef CAS PubMed; (b) V. N. Nemykin and R. G. Hadt, J. Phys. Chem. A, 2010, 114, 12062–12066 CrossRef CAS PubMed; (c) F. D'Souza, P. M. Smith, S. Gadde, A. L. McCarty, M. J. Kullman, M. E. Zandler, M. Itou, Y. Araki and O. Ito, J. Phys. Chem. B, 2004, 108, 11333–11343 CrossRef; (d) Q. Li, G. Mathur, G. Gowda, S. Surthi, Q. Zhao, L. Yu, J. S. Lindsey, D. F. Bocian and V. Misra, Adv. Mater., 2004, 16, 133–137 CrossRef CAS PubMed.
- S. di Bella, C. Dragonetti, M. Pizzotti, D. Roberto, F. Tessore and R. Ugo, Top. Organomet. Chem., 2010, 28, 1–55 CrossRef CAS.
- C. Bucher, C. H. Devillers, J.-C. Moutet, G. Royal and E. Saint-Aman, Coord. Chem. Rev., 2009, 253, 21–36 CrossRef CAS PubMed.
- G. Grelaud, M. P. Cifuentes, F. Paul and M. G. Humphrey, J. Organomet. Chem., 2014, 751, 181–200 CrossRef CAS PubMed.
- S. Drouet, A. Merhi, G. Grelaud, M. P. Cifuentes, M. G. Humphrey, K. Matczyszyn, M. Samoc, L. Toupet, C. O. Paul-Roth and F. Paul, New J. Chem., 2012, 36, 2192–2195 RSC.
- S. Drouet, A. Merhi, D. Yao, M. P. Cifuentes, M. G. Humphrey, M. Wielgus, J. Olesiak-Banska, K. Matczyszyn, M. Samoc, F. Paul and C. O. Paul-Roth, Tetrahedron, 2012, 68, 10351–10359 CrossRef CAS PubMed.
- A. Merhi, G. Grelaud, N. Ripoche, A. Barlow, M. P. Cifuentes, M. G. Humphrey, F. Paul and C. O. Paul-Roth, Polyhedron, 2015, 86, 64–70 CrossRef CAS PubMed.
- M. Murai, M. Sugimoto and M. Akita, Dalton Trans., 2013, 42, 16108–16120 RSC.
- J. Rochford, A. D. Rooney and M. T. Pryce, Inorg. Chem., 2007, 46, 7247–7249 CrossRef CAS PubMed.
- There are many NLO switching compounds reported in the literature (usually with ferrocenyl group as the redox-switching group): (a) K. L. Kott, D. A. Higgins, R. J. McMahon and M. Corn, J. Am. Chem. Soc., 1993, 115, 5342–5343 CrossRef CAS; (b) T. Kondo, S. Horiuchi, I. Yagi, S. Ye and K. Uosaki, J. Am. Chem. Soc., 1999, 121, 391–398 CrossRef CAS; (c) M. Malaun, Z. R. Reeves, R. L. Paul, J. C. Jeffery, J. A. MacCleverty, M. D. Ward, I. Asselberghs, K. Clays and A. Persoons, Chem. Commun., 2001, 49–50 RSC; (d) C. Sporer, I. Ratera, D. Ruiz-Molina, Y. Zhao, J. Vidal-Gancedo, K. Wurst, P. Jaitner, K. Clays, A. Persoons, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 5266–5268 CrossRef CAS PubMed; (e) L. Boubekeur-Lecaque, B. J. Coe, K. Clays, S. Foerier, T. Verbiest and I. Asselberghs, J. Am. Chem. Soc., 2008, 130, 3286–3287 CrossRef CAS PubMed; (f) W.-Y. Wang, N.-N. Ma, S.-L. Sun and Y.-Q. Qiu, Organometallics, 2014, 33, 3341–3352 CAS; (g) B. J. Coe, S. Houbrechts, I. Asselberghs and A. Persoons, Angew. Chem., Int. Ed., 1999, 38, 366–368 CrossRef CAS; (h) I. Asselberghs, K. Clays, A. Persoons, M. D. Ward and J. McCleverty, J. Mater. Chem., 2004, 14, 2831–2839 RSC; (i) I. Asselberghs, K. Clays, A. Persoons, M. D. Ward and J. McCleverty, J. Mater. Chem., 2004, 14, 2831–2839 RSC; (j) B. J. Coe, Acc. Chem. Res., 2006, 39, 383–393 CrossRef CAS PubMed.
- K. A. Green, M. P. Cifuentes, M. Samoc and M. G. Humphrey, Coord. Chem. Rev., 2011, 255, 2530–2541 CrossRef CAS PubMed.
- (a) K. M.-C. Wong, S. C.-F. Lam, C.-C. Ko, N. Zhu, V. W.-W. Yam, S. Roué, C. Lapinte, S. Fathallah, K. Costuas, S. Kahlal and J.-F. Halet, Inorg. Chem., 2003, 42, 7086–7097 CrossRef CAS PubMed; (b) L. Norel, E. Di Piazza, M. Feng, A. Vacher, X. He, T. Roisnel, O. Maury and S. Rigaut, Organometallics, 2014, 33, 4824–4835 CrossRef CAS; (c) E. Di Piazza, L. Norel, K. Costuas, A. Bourdolle, O. Maury and S. Rigaut, J. Am. Chem. Soc., 2011, 133, 6174–6775 CrossRef CAS PubMed; (d) M. Tropiano, N. L. Kilah, M. Morten, H. Rahman, J. J. Davis, P. D. Beer and S. Faulkner, J. Am. Chem. Soc., 2011, 133, 11847–11849 CrossRef CAS PubMed; (e) M. Yano, K. Matsuhira, M. Tatsumi, Y. Kashiwagi, M. Nakamoto, M. Oyama, K. Ohkubo, S. Fukuzumi, H. Misaki and H. Tsukube, Chem. Commun., 2012, 48, 4082–4084 RSC; (f) T. Sato and M. Higuchi, Chem. Commun., 2013, 49, 5256–5259 RSC.
- P. Audebert and F. Miomandre, Chem. Sci., 2013, 4, 575–584 RSC.
- C. Paul-Roth, J. A. G. Williams, J. Letessier and G. Simonneaux, Tetrahedron Lett., 2007, 48, 4317–4322 CrossRef CAS PubMed.
- C. O. Paul-Roth and G. Simonneaux, Tetrahedron Lett., 2006, 47, 3275–3278 CrossRef CAS PubMed.
- C. O. Paul-Roth and G. Simonneaux, C. R. Acad. Sci., Ser. IIb: Mec., Phys., Astron., 2006, 9, 1277–1286 CAS.
- For recent examples of Ru(dppe)2Cl-acetylides, see: (a) G. Grelaud, M. P. Cifuentes, T. Schwich, G. Argouarch, S. Petrie, R. Strange, F. Paul and M. G. Humphrey, Eur. J. Inorg. Chem., 2012, 1, 65–75 CrossRef PubMed; (b) P. J. West, T. Schwich, M. P. Cifuentes and M. G. Humphrey, J. Organomet. Chem., 2011, 696, 2886–2893 CrossRef CAS PubMed; (c) Y. Liu, C. M. Ndiaye, C. Lagrost, K. Costuas, S. Choua, P. Turek, L. Norel and S. Rigaut, Inorg. Chem., 2014, 53, 8172–8188 CrossRef CAS PubMed; (d) S. Marqués-González, M. Parthey, D. S. Yufit, J. A. K. Howard, M. Kaupp and P. J. Low, Organometallics, 2014, 33, 4947–4963 CrossRef; (e) M. Parthey, J. B. G. Gluyas, M. A. Fox, P. J. Low and M. Kaupp, Chem. – Eur. J., 2014, 20, 6895–6908 CrossRef CAS PubMed; (f) S. S. Chavan and B. G. Bharate, Inorg. Chim. Acta, 2013, 394, 598–604 CrossRef CAS PubMed; (g) R. Gatri, I. Ouerfelli, M. L. Efrit, F. Serein-Spirau, J.-P. Lère-Porte, P. Valvin, T. Roisnel, S. Bivaud, H. Akdas-King and J.-L. Fillaut, Organometallics, 2014, 33, 665–676 CrossRef CAS; (h) M. I. Bruce, M. L. Cole, K. Costuas, B. G. Ellis, K. A. Kramarczuk, C. Lapinte, B. K. Nicholson, G. J. Perkins, B. W. Skelton, A. H. White and N. N. Zaitseva, Z. Anorg. Allg. Chem., 2013, 639, 2216–2223 CrossRef CAS PubMed.
- A. Merhi, S. Drouet, N. Kerisit and C. O. Paul-Roth, Tetrahedron, 2013, 69, 7112–7124 CrossRef CAS PubMed.
- C. O. Paul-Roth, A. Merhi, D. Yao and O. Mongin, J. Photochem. Photobiol., A, 2014, 288, 23–33 CrossRef CAS PubMed.
- S. Guesmi, D. Touchard and P. H. Dixneuf, Chem. Commun., 1996, 2773–2774 RSC.
- D. Touchard, C. Morice, V. Cadierno, P. Haquette, L. Toupet and P. H. Dixneuf, J. Chem. Soc., Chem. Commun., 1994, 859–860 RSC.
- J. W. Owens, R. Smith, R. Robinson and M. Robins, Inorg. Chim. Acta, 1998, 279, 226–231 CrossRef CAS.
- S. Drouet and C. O. Paul-Roth, Tetrahedron, 2009, 65, 10693–10700 CrossRef CAS PubMed.
- S. Drouet, A. Merhi, G. Argouarch, F. Paul, O. Mongin, M. Blanchard-Desce and C. O. Paul-Roth, Tetrahedron, 2012, 68, 98–105 CrossRef CAS PubMed.
- C. Paul-Roth, J. Rault-Berthelot, G. Simonneaux, C. Poriel, M. Abdalilah and J. Letessier, J. Electroanal. Chem., 2006, 597, 19–27 CrossRef CAS PubMed.
- C. Paul-Roth, J. Rault-Berthelot, G. Simonneaux, J. Letessier and J.-F. Bergamini, J. Electroanal. Chem., 2007, 606, 103–116 CrossRef CAS PubMed.
- M. A. Fox, J. E. Harris, S. Heider, V. Pérez-Gregorio, M. E. Zakrzewska, J. D. Farmer, D. S. Yufit, J. A. K. Howard and P. J. Low, J. Organomet. Chem., 2009, 694, 2350–2358 CrossRef CAS PubMed.
- M. V. Sheridan, K. Lam and W. E. Geiger, Angew. Chem., Int. Ed., 2013, 52, 12897–12900 CrossRef CAS PubMed.
- C. E. Powell, M. P. Cifuentes, J. P. Morrall, R. Stranger, M. G. Humphrey, M. Samoc, B. Luther-Davies and G. A. Heath, J. Am. Chem. Soc., 2003, 125, 602–610 CrossRef CAS PubMed.
- N. Gauthier, N. Tchouar, F. Justaud, G. Argouarch, M. P. Cifuentes, L. Toupet, D. Touchard, J.-F. Halet, S. Rigaut, M. G. Humphrey, K. Costuas and F. Paul, Organometallics, 2009, 28, 2253–2266 CrossRef CAS.
- G. Peychal-Heiling and G. S. Wilson, Anal. Chem., 1971, 43, 550–556 CrossRef.
- C. Paliteiro and A. Sobral, Electrochim. Acta, 2005, 50, 2445–2451 CrossRef CAS PubMed.
- J. Fajer, D. C. Borg, A. Forman, D. Dolphin and R. H. Felton, J. Am. Chem. Soc., 1970, 92, 3451–3459 CrossRef CAS.
- M. A. Fox, J. D. Farmer, R. L. Roberts, M. G. Humphrey and P. J. Low, Organometallics, 2009, 28, 5266–5269 CrossRef CAS.
- M. Gouterman, J. Mol. Spectrosc., 1961, 6, 138–163 CrossRef CAS.
- C.-K. Tai, W.-H. Chuang and B.-C. Wang, J. Lumin., 2013, 142, 8–16 CrossRef CAS PubMed.
- L. Weber, D. Eickhoff, T. B. Marder, M. A. Fox, P. J. Low, A. D. Dwyer, D. J. Tozer, S. Schwedler, A. Brockhinke, H.-G. Stammler and B. Neumann, Chem. – Eur. J., 2012, 18, 1369–1382 CrossRef CAS PubMed.
- E. A. Alemán, J. M. Rocha, W. Wongwitwichote, L. A. G. Mora-Tovar and D. A. Modarelli, J. Phys. Chem. A, 2011, 115, 6456–6471 CrossRef PubMed.
- M. A. Fox, R. L. Roberts, W. M. Khairul, F. Hartl and P. J. Low, J. Organomet. Chem., 2007, 692, 3277–3290 CrossRef CAS PubMed.
- M. Krejcik, M. Danek and F. Hartl, J. Electroanal. Chem., 1991, 317, 179–187 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven Jr., J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
- (a) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS PubMed; (b) G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218 CrossRef CAS PubMed.
- (a) M.-C. Oerthel, D. S. Yufit, M. A. Fox, M. R. Bryce and P. J. Low, Organometallics, 2015 DOI:10.1021/om501186c; (b) M. A. Fox, B. Le Guennic, R. L. Roberts, D. A. Brue, D. S. Yufit, J. A. K. Howard, G. Manca, J.-F. Halet, F. Hartl and P. J. Low, J. Am. Chem. Soc., 2011, 133, 18433–18446 CrossRef CAS PubMed; (c) M. I. Bruce, A. Burgun, M. A. Fox, M. Jevric, P. J. Low, B. K. Nicholson, C. R. Parker, B. W. Skelton, A. H. White and N. N. Zaitseva, Organometallics, 2013, 32, 3286–3299 CrossRef CAS; (d) M. I. Bruce, M. A. Fox, P. J. Low, B. K. Nicholson, C. R. Parker, W. C. Patalinghug, B. W. Skelton and A. H. White, Organometallics, 2012, 31, 2639–2657 CrossRef CAS; (e) D. J. Armitt, M. I. Bruce, M. Gaudio, N. N. Zaitseva, B. W. Skelton, A. H. White, B. Le Guennic, J.-F. Halet, M. A. Fox, R. L. Roberts, F. Hartl and P. J. Low, Dalton Trans., 2008, 6763–6775 RSC; (f) M. A. Fox, R. L. Roberts, T. E. Baines, B. Le Guennic, J.-F. Halet, F. Hartl, D. S. Yufit, D. Albesa-Jové, J. A. K. Howard and P. J. Low, J. Am. Chem. Soc., 2008, 130, 3566–3578 CrossRef CAS PubMed; (g) J.-L. Xia, W. Y. Man, X. Zhu, C. Zhang, G.-J. Jin, P. A. Schauer, M. A. Fox, J. Yin, G.-A. Yu, P. J. Low and S. H. Liu, Organometallics, 2012, 31, 5321–5333 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- L. Weber, J. Halama, K. Hanke, L. Böhling, A. Brockhinke, H.-G. Stammler, B. Neumann and M. A. Fox, Dalton Trans., 2014, 43, 3347–3363 RSC.
- A. R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- N. M. O'Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
Footnotes |
| † In memory of Ken Wade – a superb chemist and a true gentleman. |
| ‡ Electronic supplementary information (ESI) available: NMR spectra for 1–4, emission spectra for 3 and 4 at 77 K, additional CV plots for 3, additional SEC spectra for 1–4, molecular orbital data and Cartesian coordinates for optimised geometries of 1′–4′. See DOI: 10.1039/c5dt00348b |
| This journal is © The Royal Society of Chemistry 2015 |