
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Why are the {Cu4N4} rings in copper(I) phosphinimide clusters [Cu{μ-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) PR3}]4 (R = NMe3 or Ph) planar?†‡
PR3}]4 (R = NMe3 or Ph) planar?†‡
Thomas P.
Robinson
a,
Richard D.
Price
a,
Matthew G.
Davidson
*a,
Mark A.
Fox
 b and
Andrew L.
Johnson
*a
b and
Andrew L.
Johnson
*a
aDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: M.G.Davidson@bath.ac.uk; A.L.Johnson@bath.ac.uk; Fax: +(44) (0)1225 386231; Tel: +(44) (0)1225 384467
bDepartment of Chemistry, University of Durham, Durham, DH1 3LE, UK
First published on 12th February 2015
Abstract
The copper phosphinimide complexes [Cu{μ-N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) PR3}]4 (1, R = NMe2 and 2, R = Ph) were obtained in good yields from the reactions of Cu[Mes] (Mes = mesityl, C6H2Me3-2,4,6) with the corresponding iminophosphoranes, HNPR3. The molecular structures of 1 and 2 reveal the presence of planar eight-membered {Cu4N4} rings which contrasts with the saddle-shaped {M4N4} rings found in related metal phosphinimide complexes. According to computations, there is negligible aromaticity in the planar {Cu4N4} rings in 1 and 2 and the saddle shape observed in related {M4N4} rings is due to steric factors.
PR3}]4 (1, R = NMe2 and 2, R = Ph) were obtained in good yields from the reactions of Cu[Mes] (Mes = mesityl, C6H2Me3-2,4,6) with the corresponding iminophosphoranes, HNPR3. The molecular structures of 1 and 2 reveal the presence of planar eight-membered {Cu4N4} rings which contrasts with the saddle-shaped {M4N4} rings found in related metal phosphinimide complexes. According to computations, there is negligible aromaticity in the planar {Cu4N4} rings in 1 and 2 and the saddle shape observed in related {M4N4} rings is due to steric factors.
Introduction
The significance of iminophosphoranes is well established in both organic synthesis1 and organometallic chemistry,2 with metal phosphinimide complexes (especially those of titanium and some rare earth elements) having been exploited in the development of highly efficient of ‘non-metallocene’ based catalysts,3 of the general form (R′3PN)2MRx and (Cp)MRx(NPR′3) (R′ = alkyl or aryl, R = alkyl). In comparison, exploitation of metal phosphinimide complexes in organic synthesis is predominantly limited to the use of lithium phosphinimide systems, which find utility in a number of areas including, as an [NH2−] synthon, in the preparation of non-ionic phosphazene bases, in dehydrocoupling of primary and secondary phosphines, in the synthesis of primary, secondary, cyclic or functional amines, as well as in the generation of heteroatomic linkages (P–N–P, P–N–As, P–N–S).1a,bThe chemistry of iminophosphoranes is intrinsically associated by an isolobal, isoelectronic and isoneutral relationship with phosphorus ylides and phosphine oxides. The PE bonding (E = CH2, NH and O) in these systems being viewed as a resonance hybrid between a double bonded neutral ‘ylene’ form and a zwitterionic ‘ylide’ form (Fig. 1).4
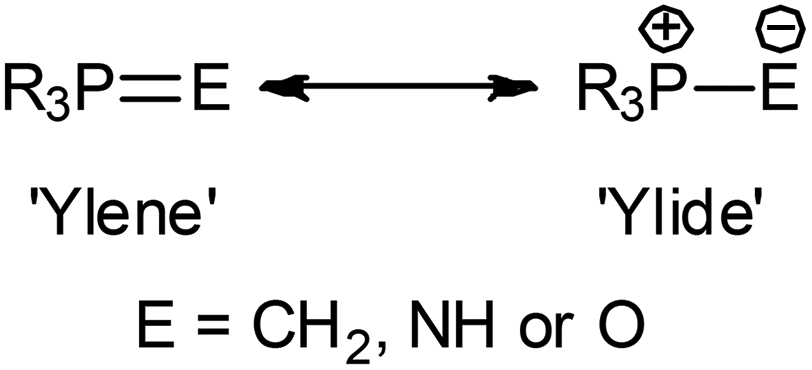 | ||
| Fig. 1 Hybrid resonance structures of R3PE. | ||
Given the developing utility of lithium phosphinamide complexes, it has been suggested that the preparation and development of potassium,5 magnesium,6 nickel, palladium and copper derivatives may lead to promising applications in organic synthesis.1a Indeed, the novel Co(I) and Ni(I) complexes [Co(μ2-NPtBu3)]4 and [Ni(μ2-NPtBu3)]4 have both been reported recently, along with their use as catalysts in the mild hydrogenation of alkenes and alkynes.7
Until now, the isolation and unambiguous characterisation of a neutral homoleptic N-Cu(I)-metallated iminophosphorane complexes has not, to our knowledge, been reported, although the related cationic systems, [Cu4(NHPEt3)4]4+,8 and the cubic [M12(NPEt3)8]4+ (M = Cu(I) or Ag(I)) clusters9 and [M3(μ-NPR3)(PR3)3]2+ (M = Ag(I) or Au(I); R = Me or Ph) systems10 have been described. Other structurally characterised phosphinimide complexes of copper are limited to the Cu(II)–acetate systems Cu(HNPPh3)2(OAc)2, [Cu2(HNPPh3)2(OAc)4]11 and [Cu4(NPMe3)3(OAc)5]12 and the mixed-valence species [Cu6Br6(NPMe3)4], [Cu6Cl7(NPMe3)4] and [Cu6Cl6(NPMe3)4]+.13 Continuing our ongoing research at Bath into the coordination chemistry of Group 11 metals with anionic nitrogen coordination ligands,14 we report here the syntheses and structural characterisations, by single crystal X-ray diffraction, of the copper(I) phosphinimide complexes [c-{Cu[μ-NP(NMe2)3]}4] (1) and [c-{Cu[μ-NPPh3]}4] (2).
Results and discussion
Syntheses and characterisation
Initial reactions to prepare Cu(I) phosphinimide complexes 1 and 2 focussed on the reaction of CuCl with either [LiNP(NMe2)3]15 or [LiNPPh3]16 in THF (Scheme 1). The [LiNP(NR2)3] complexes were made in situ from n-butyllithium and HNP(NR2)3. While successful, these reactions were low yielding (17–21%), therefore an alternative synthetic procedure utilising the reagent [Cu(Mes)] (Mes = C6H2Me3-2,4,6) was investigated.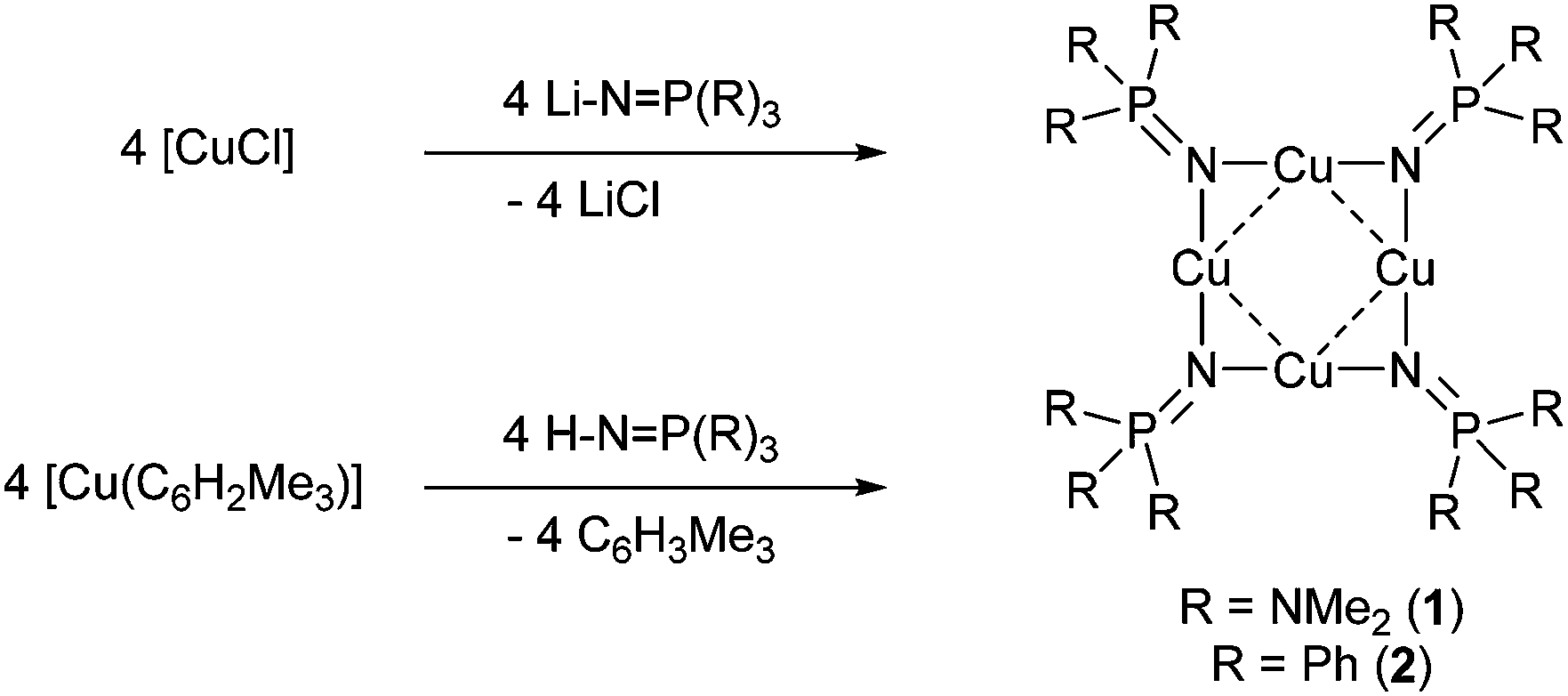 | ||
| Scheme 1 | ||
The reaction of [Cu(Mes)] with HNP(NMe2)3 in toluene (Scheme 1) at low temperature (−78 °C) produced an immediate reaction with the solution turning from pale yellow to colourless. Warming of the solution to ambient temperature followed by filtration, via cannula, and cooling gave a crop of pale yellow crystals (1) in 78% isolated yield. A similar reaction of HNPPh3 with [Cu(Mes)] followed by filtration and cooling afforded pale yellow crystalline material (2), in 70% isolated yield.
For both complexes 1 and 2, NMR spectroscopic data reveal the absence of resonances associated with phosphinamide hydrogen atoms.17 In the case of 1, the 1H NMR spectrum (in C6D6) shows resonances for the NMe2 moieties at δ = 2.72 ppm and a single resonance in the 31P NMR spectrum at δ = 32.9 ppm. Correspondingly, the 1H NMR spectrum of 2 (in CD2Cl2) shows the presence of the aromatic CH groups on the phosphinimide ligand and the 31P NMR spectrum shows a single resonance at δ = 15.9 ppm.
X-ray crystallography
Single-crystal X-ray diffraction studies were carried out on crystals of 1 and 2 to determine their solid-state structures. Complex 1 crystallises in the space group P21/n with the molecule sitting on a centre of symmetry such that only half of complex 1 is present in the asymmetric unit. Complex 2 crystallises in the space group P21/c and one molecule of the complex is present in the asymmetric unit cell (along with half of a disordered toluene molecule residing on a centre of crystallographic symmetry such that one toluene molecule is present for two molecules of 2). The molecular structures of complexes 1 and 2 are shown in Fig. 2 and selected structural parameters listed in Table 1. Complexes 1 and 2 are amongst only a relatively small number of known homoleptic planar, tetranuclear coinage metal(I) clusters singly bridged by monoanionic ligands, and represent the first examples of homoleptic Group 11 phosphinimide complexes. The planar core contrasts with other reported {M4N4} phosphinimide complexes where the {M4N4} cores are either cubic18 or saddled (approx. D2d symmetry, Fig. 3).7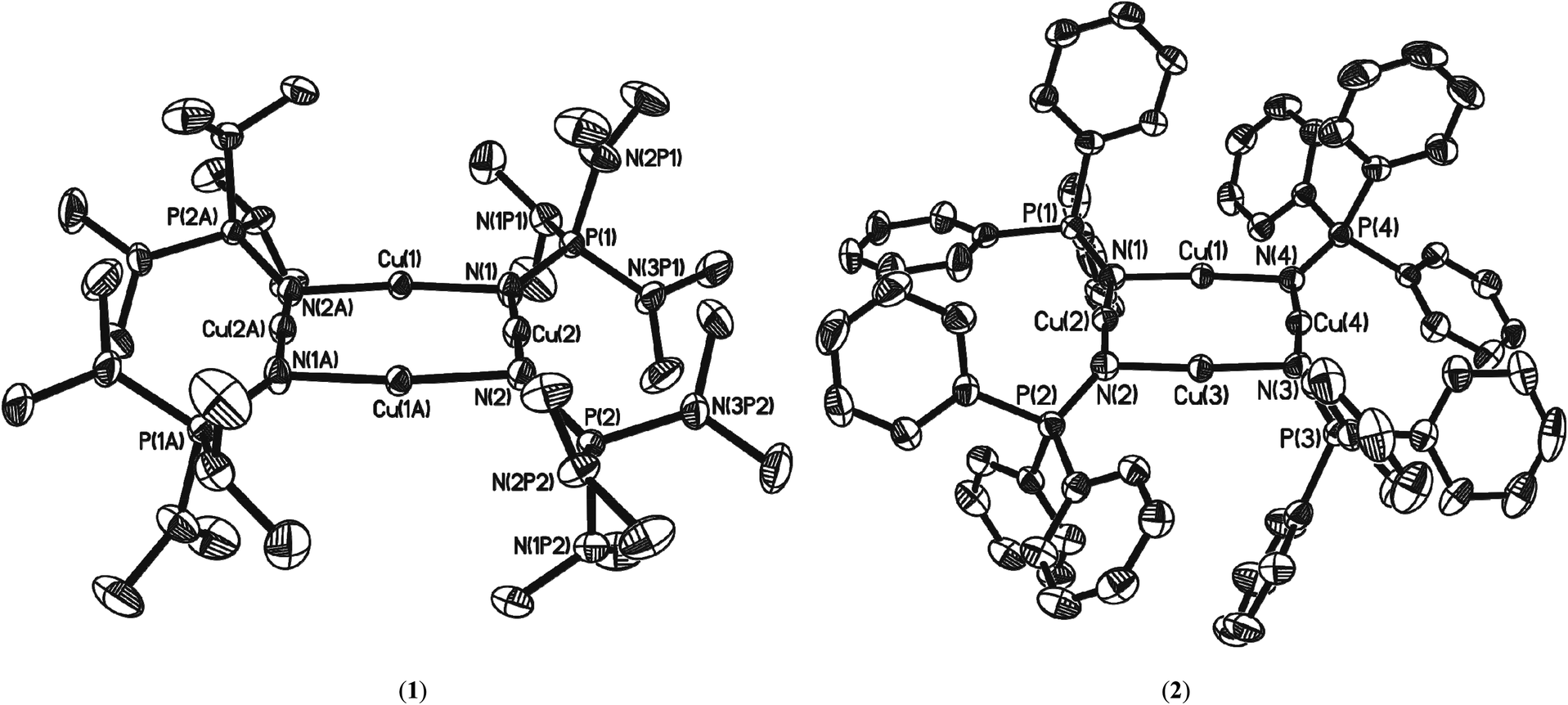 | ||
| Fig. 2 Molecular structures of the complexes 1 and 2 (50% probability ellipsoids). Hydrogen atoms in 1 and 2 have been omitted for clarity. Symmetry transformations used to generate equivalent atoms in 1: −X, −Y + 1, −Z + 1. | ||
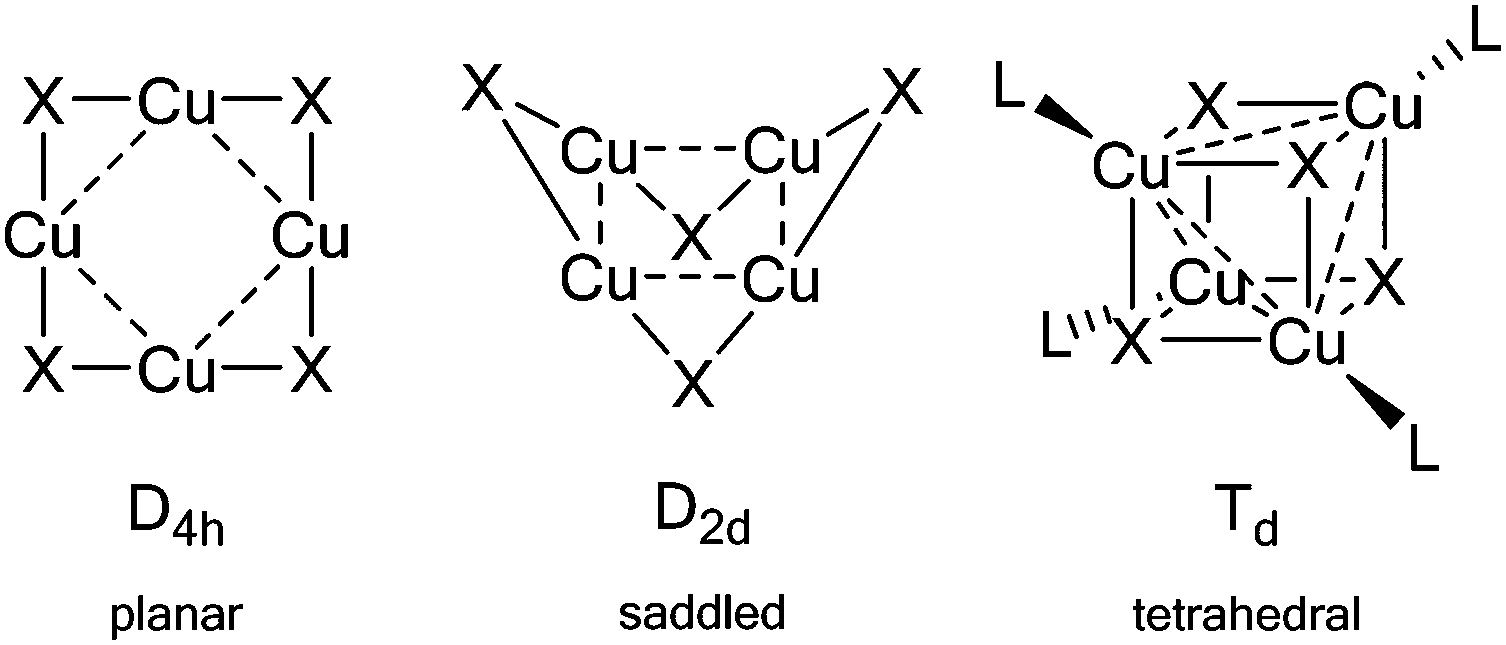 | ||
| Fig. 3 Geometries for Cu4X4 and Cu4X4L4 clusters. | ||
| 1 | 1 (calc) | 2 | 2 (calc) | |
|---|---|---|---|---|
| a Conformer A. b Conformer B (see Fig. 4). | ||||
| Cu(1)–Cu(2) | 2.7484(3) | 2.854 | 2.7479(7) | 2.830 |
| Cu(2)–Cu(1A/3) | 2.6556(3) | 2.760 | 2.6436(6) | 2.825 |
| Cu(3)–Cu(4) | 2.7508(7) | 2.830 | ||
| Cu(4)–Cu(1) | 2.6762(6) | 2.825 | ||
| Cu(1)–N(1) | 1.8454(17) | 1.860 | 1.861(4) | 1.860 |
| Cu(1)–N(2A/4) | 1.8550(17) | 1.863 | 1.855(3) | 1.864 |
| Cu(2)–N(1) | 1.8561(17) | 1.864 | 1.854(3) | 1.861 |
| Cu(2)–N(2/3) | 1.8576(17) | 1.869 | 1.861(3) | 1.864 |
| Cu(3)–N(2) | 1.848(3) | 1.861 | ||
| Cu(3)–N(3) | 1.861(3) | 1.864 | ||
| Cu(4)–N(3) | 1.850(3) | 1.860 | ||
| Cu(4)–N(4) | 1.846(3) | 1.864 | ||
| N(1)–P(1) | 1.5413(17) | 1.554 | 1.551(3) | 1.568 |
| N(2)–P(2) | 1.5480(17) | 1.558 | 1.559(4) | 1.568 |
| N(3)–P(3) | 1.557(3) | 1.569 | ||
| N(4)–P(4) | 1.555(4) | 1.568 | ||
| Cu(2)–Cu(1)–Cu(2A/4) | 85.518(10) | 89.73 | 91.30(2) | 90.018 |
| Cu(1)–Cu(2)–Cu(1A/3) | 92.482(10) | 90.27 | 88.74(2) | 89.956 |
| Cu(2)–Cu(3)–Cu(4) | 91.94(2) | 90.020 | ||
| Cu(3)–Cu(4)–Cu(1) | 88.02(2) | 89.961 | ||
| N(1)–Cu(1)–N(2A/4) | 172.91(8) | 171.66 | 176.11(15) | 170.14 |
| N(1)–Cu(2)–N(2) | 176.98(8) | 172.24 | 175.38(15) | 170.11 |
| N(2)–Cu(3)–N(3) | 176.65(16) | 170.17 | ||
| N(3)–Cu(4)–N(4) | 173.69(15) | 170.09 | ||
The structural element of interest in both 1 and 2 is the presence of a square-planar centro-symmetric eight-membered (CuN)4 ring with N–Cu–N angles close to 180° [N–Cu–N(Ave); (1) 174.95(8)°, (2) 175.46(15)°: Cu–N; (1) 1.854(2)Å, (2) 1.854(3)Å] which are comparable to those of other two-coordinate or quasi-two-coordinate Cu(I) complexes in a nitrogen coordination environment14a,d,19 and Cu–N–Cu angles close to 90° (av. 93.62(8)°). The planar {Cu4} cores of 1 and 2 (with approx. D4h symmetry, Fig. 3) have each Cu atom bonded to two doubly bridging phosphinimide ligands (μ2-NPR3) via the nitrogen atom creating a two-coordinate geometry about the copper atoms. While the average Cu⋯Cu distances [(1): 2.702(3)Å, (2): 2.705(6)Å] are shorter than the sum of the van der Waals radii of Cu (1.40 Å)20 and within the range for potential d10–d10 closed shell interactions as observed for unsupported Cu(I)–Cu(I) interactions, the Cu–Cu distances in 1 and 2 are at the longer end of the scale observed for ligand-supported cuprophilic interactions.14a
The average P–N bond lengths observed in both 1 and 2 [1: 1.545(2)Å; 2: 1.556(4)Å] are both marginally shorter than those found in the parent iminophosphorane systems (1.557(1) Å and 1.582(2) Å respectively)21 suggesting retention of similar P–N bond character to that the parent ligand with some electrostatic shortening. Pyramidalisation of the nitrogen atoms of the ligands is indicated by the sum of angles about each nitrogen atom [for 1 N1: ΣN = 355.33(10)°, N2: ΣN = 343.64(10)°; for 2 N1: ΣN = 351.9(2)°, N2: ΣN = 342.4(2)°, N3: ΣN = 341.4(2)°, N4: ΣN = 352.3(2)° ], such that the P–N vectors are at an angle to the {Cu4} planes in both 1 and 2 [For 1: P(1)–N(1)–X = 162.56(3)°; P(2)–N(2)–X = 147.58(3)°, For 2: P(1)–N(1)–X = 155.95(3)°; P(2)–N(2)–X = 146.32(3)°, P(3)–N(3)–X = 143.27(3)°; P(4)–N(4)–X = 157.85(3)° (where X is the midpoint between two Cu atoms)]. Similar bonding geometries have been reported previously for magnesium phosphinimide complexes and are proposed to originate from the ylidic character of the P–N bonding with a lone pair of electrons residing on the N atom in a predominantly p-type orbital (Fig. 1).6 The distortion of the ligands away from co-planarity with the {Cu4} cores result in a cis, trans, cis, trans (c,t,c t) orientation with respect to each Cu–Cu interaction around the {Cu4} ring (conformer A, Fig. 4).
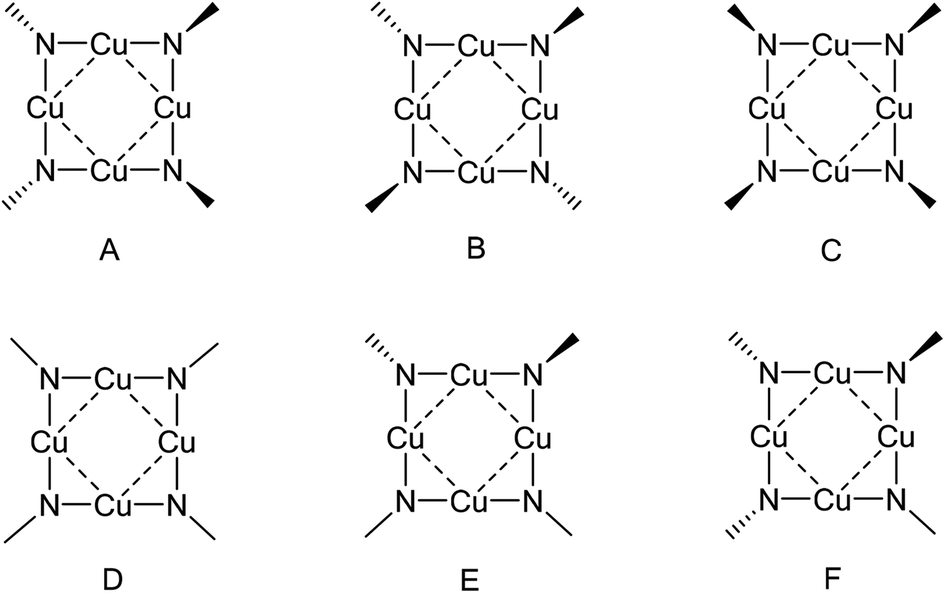 | ||
| Fig. 4 Conformers for {Cu4N4} complexes with N atoms in pyramidal and/or planar coordinations. | ||
In a more general context, the planar {Cu4N4} cores of 1 and 2 contrast to the saddle shaped geometries observed for other copper imido complexes (Fig. 5) such as [Cu(μ2-NCtBu2)]4 (saddle angles, θ = 95.2,94.1°),22 [Cu(μ2-NCtBuPh)]4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 (θ = 130.9,131.2°) and [Cu(μ2-NCPh2)]4, (θ = 141.9°)22 which contain {Cu4N4} rings with bridging imino ligands, in which each imino nitrogen atom has a planar coordination geometry at the nitrogen (conformer D, Fig. 4). The structurally related copper(I) amide complexes [Cu4(NR2)4] (NR2 = NMe2, NEt2, and N{c-(CH2)4}), also form tetrameric clusters with a central 8 membered {Cu4N4} core; while both [Cu4(NMe2)4] and [Cu4(N{c-(CH2)4})4]24 display planar geometries, the more sterically encumbered ethyl system [Cu4(NEt2)4] displays a saddle shaped geometry (θ = 141.87°).19c
23 (θ = 130.9,131.2°) and [Cu(μ2-NCPh2)]4, (θ = 141.9°)22 which contain {Cu4N4} rings with bridging imino ligands, in which each imino nitrogen atom has a planar coordination geometry at the nitrogen (conformer D, Fig. 4). The structurally related copper(I) amide complexes [Cu4(NR2)4] (NR2 = NMe2, NEt2, and N{c-(CH2)4}), also form tetrameric clusters with a central 8 membered {Cu4N4} core; while both [Cu4(NMe2)4] and [Cu4(N{c-(CH2)4})4]24 display planar geometries, the more sterically encumbered ethyl system [Cu4(NEt2)4] displays a saddle shaped geometry (θ = 141.87°).19c
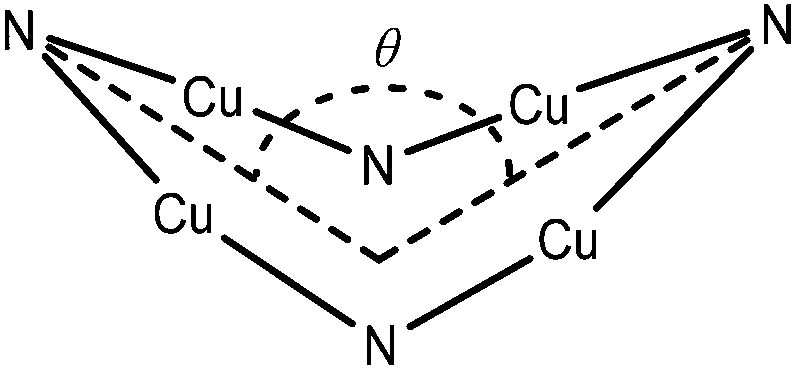 | ||
| Fig. 5 One saddle angle (θ) shown of two possible within the {Cu4N4} ring. | ||
A survey of the Cambridge Structural Database25 shows a number of complexes with {Cu4X4} cores (X = 1st row element, i.e. B, C, N or O as part of an anionic ligand) which can similarly be categorised as having either an approximate D4h or D2d core arrangement. Computational studies have attributed this preference for Group 11 transition metals tetramers to form clusters with D4h/D2d geometry to a significant electrostatic stabilisation and a dominant effect of the Pauli repulsion between metal atoms.26 This is in contrast to alkali metal tetramers, for which cubic geometries dominate and attractive electrostatic and orbital interaction terms compensate for large Pauli repulsion energies.26 This is supported by the fact that while tetrahedral/cubic {M4(NPR3)4} systems (M = Li, K, Cs and Rb)27 are known structurally, copper(I) based clusters with a central tetrahedral {Cu4X4} core are not known in the literature in the absence of ancillary groups coordinating to the metal centre i.e. [(L)CuX]4 systems (Td symmetry, Fig. 3).
It has been suggested that the steric demands of the anionic ligand play a dominant role in the solid state conformation of the cluster rather than a saddle-like geometry being indicative of strong metallophilic interactions.19a,c,d,24 However, the planarity of the {Cu4} rings in related clusters (and analogous Ag and Au systems) has also been attributed to a contribution from transition metal based σ-aromatic stabilisation resulting from a degree of cyclic electron conjugation within the cluster bonding (vide infra).28
In the cobalt and nickel phosphinimide complexes recently reported by Stryker et al.,7 and related to 1 and 2, saddled {Co4N4} (saddle angle, θ = 112.5°) and {Ni4N4} (θ = 117.7°) ring conformations are observed. The structures include two planar imido nitrogen atoms and two pyramidalised imido nitrogen atoms (Fig. 4, conformer E, for {Co4N4} ΣN = 359.6°, 347.5°; for {Ni4N4} ΣN = 359.7°, 347.4°). This geometry is suggested to result from repulsion between the bulky {NPtBu3} groups. However, it is worth noting that the different planar and pyramidal environments at the imido nitrogen have no significant influence on the corresponding bond lengths involving these imido nitrogens.7
Hybrid-DFT studies
In order to provide further insight as to whether the planarity of the {Cu4N4} ring present in the X-ray geometries of 1 and 2 is due to steric and/or electronic factors, geometry optimisations at the B3LYP/6-311G(d,p) level were carried out on 1 and 2. Using the molecular geometries obtained from single crystal X-ray diffraction experiments as starting geometries, a cis, trans, cis, trans- (c,t,c,t-) orientation (conformer A, Fig. 4) and planarity was retained for complex 1, but for complex 2 molecular rearrangement to a trans, trans, trans, trans- (t,t,t,t-) configuration (conformer B, Fig. 4) was observed upon optimisation with an average saddle angle of 159.4°. Selected parameters, for comparison between the experimental and computed geometries, are listed in Table 1 and reveal that bond lengths are consistently longer by 0.1 Å in the computed values giving some confidence in the accuracy of B3LYP/6-311G(d,p) for copper phosphinimides. Table 2 lists the sum of angles at the ring nitrogen atoms and the saddle angles for optimised geometries of Cu4(NR2)4 systems investigated here. The sum of angles at the ring nitrogen atoms are all similar at 350.4–350.8° for the optimised and rearranged geometry of 2 and are close to the sum of angles of 342.4–352.3° found for nitrogen atoms in the experimental data. The barrier between these two conformers, A and B, in 2 must be small reflecting little steric influence of the PPh3 groups.| R2 | Geometry | Rel. E. | Ring | Θ 1 | Θ 2 | ∑N1 | ∑N2 | ∑N3 | ∑N4 | NICS | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| H2 | Planar | 180.0 | 180.0 | 1.0 | |||||||
| Me2 | Planar | 179.1 | 179.1 | 0.0 | |||||||
| Me2 (Expt) | Planar | 180.0 | 180.0 | 24 | |||||||
| Et2 | 0.00 | Saddled | 132.8 | 132.7 | −1.7 | ||||||
| Et2 | 1.11 | Saddled | 144.6 | 144.6 | −2.0 | ||||||
| Et2 (Expt) | Saddled | 141.9 | 141.9 | 19c | |||||||
| H(PH3)+ | A | 1.20 | Planar | 180.0 | 180.0 | −0.7 | |||||
| H(PH3)+ | B | 0.00 | Saddled | 156.5 | 156.5 | −0.8 | |||||
| H(PEt3)+ (Expt) | B | Saddled | 125.8 | 125.8 | 8 | ||||||
| H(PH3)+ | C | 2.65 | Planar | 180.0 | 180.0 | −0.7 | |||||
| PH3 | A | 0.06 | Planar | 179.9 | 179.9 | 356.7 | 356.5 | 356.6 | 356.5 | 1.2 | |
| PH3 | B | 0.00 | Planar | 179.1 | 179.1 | 356.0 | 356.1 | 356.1 | 356.0 | 1.3 | |
| PH3 | C | 0.16 | Planar | 180.0 | 180.0 | 357.5 | 357.5 | 357.5 | 357.5 | 1.3 | |
| PH3 | D | 1.17 | Planar | 180.0 | 180.0 | 360.0 | 360.0 | 360.0 | 360.0 | 1.4 | |
| PMe3 | E | Saddled | 166.3 | 166.3 | 358.9 | 347.7 | 349.7 | 356.8 | 1.0 | ||
| PEt3 | F | Saddled | 151.1 | 149.9 | 359.9 | 347.9 | 331.6 | 350.9 | 0.1 | ||
| P(NMe2)3 | A | 3.06 | Planar | 180.0 | 180.0 | 356.6 | 346.3 | 356.5 | 346.3 | 0.2 | |
| P(NMe2)31 (Expt) | A | Planar | 180.0 | 180.0 | 355.3 | 343.6 | 355.3 | 343.6 | ibid | ||
| P(NMe2)3 | E | 0.00 | Saddled | 134.0 | 132.6 | 360.0 | 339.3 | 348.0 | 359.9 | −0.2 | |
| PPh3 | B | Saddled | 159.4 | 159.3 | 350.4 | 350.8 | 350.6 | 350.7 | 1.6 | ||
| PPh32 (Expt) | A | Planar | 179.0 | 179.0 | 351.9 | 342.4 | 341.4 | 352.3 | ibid | ||
| CH2 | D | Planar | 179.9 | 179.9 | 360.0 | 360.0 | 360.0 | 360.0 | 3.8 | ||
| CPh2 (Expt) | D | Saddled | 138.1 | 138.6 | 359.8 | 360.0 | 360.0 | 360.0 | 22 |
Geometry optimisation of complex 1 starting with a t,t,t,t-conformer (B, Fig. 4), however, gave a minimum with a {Cu4N4} ring containing a more acute saddle angle of θ = 132.6° compared to 159.4° for the optimised geometry of 2. There are two planar imido nitrogens (ΣN = 353.9°, 360.0°) and two pyramidal imido nitrogens (ΣN = 339.3°, 348.0°) resulting in conformer E (Fig. 4). This shows significant steric repulsion in accord with the higher Tolman cone angle for the bulky P(NMe2)3 groups compared to the PPh3 groups,29 thus resulting in a non-planar {Cu4N4} ring containing planar imido nitrogens (conformer E).
To our knowledge, there is only one comparable computational study30 on {Cu4N4} ring systems reported in the literature. The parent molecule Cu4(NH2)4 at BP86/cc-pVDZ-PP was identified as saddled not planar. Several {Cu4N4}-containing structures with tetrahedral nitrogen atoms, such as Cu4(NMe2)4, have been shown by X-ray crystallographic studies to be planar so the reported saddled form is surprising.
As B3LYP/6-311G(d,p) optimisations on the much more complex molecule 1 gave geometries in good agreement with experimental data (Table 1), B3LYP/6-311G(d,p) was used on simpler models with tetrahedral ring nitrogens to predict whether planar or saddled forms are in accord with experimental data. The results of Cu4(NR2)4 are summarised in Table 2 where R is H, Me and Et and the optimised molecular geometries ae shown in Fig. 6.
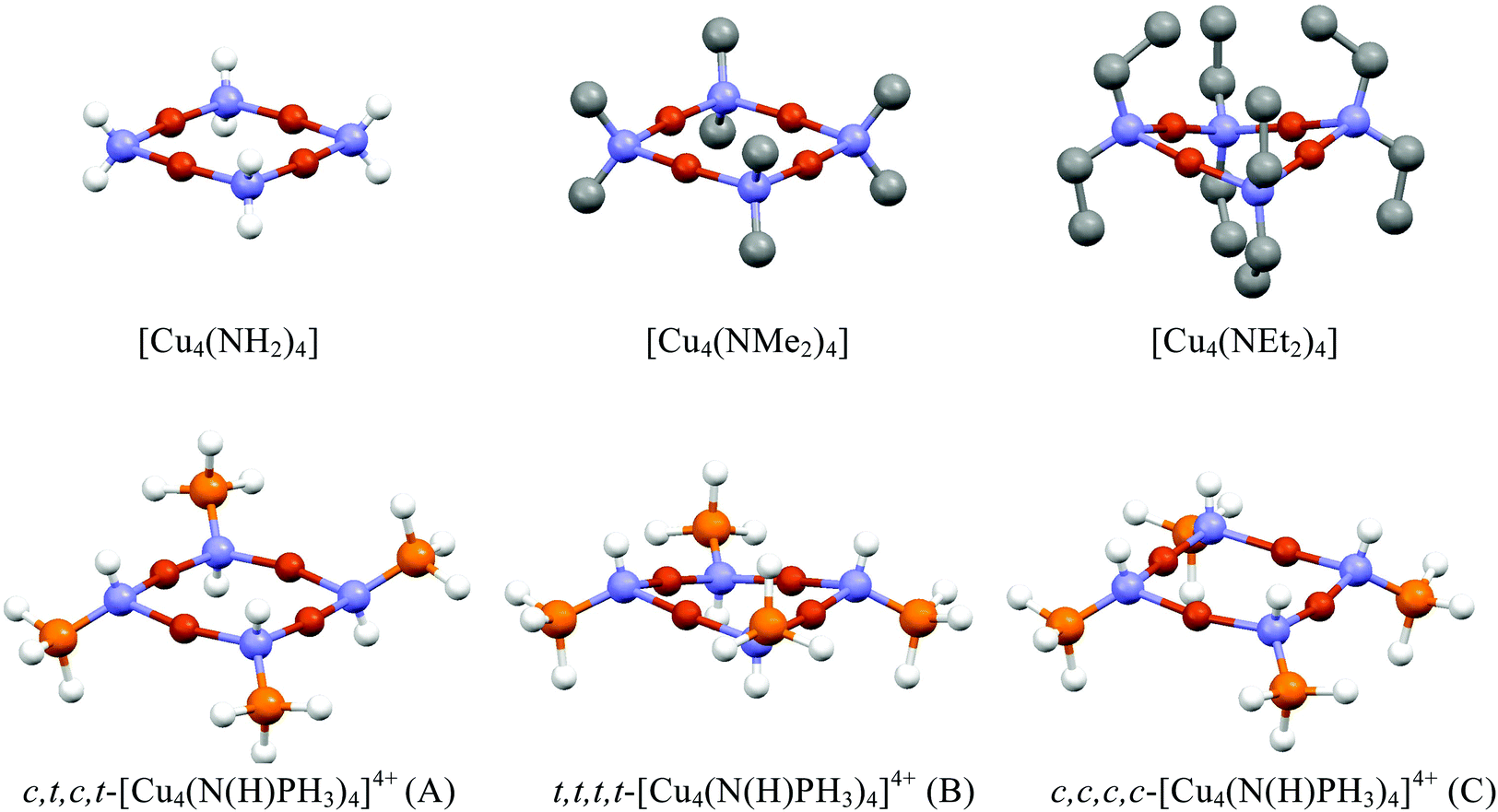 | ||
| Fig. 6 Diagrams showing the optimised molecular geometries of selected complexes containing 4-coordinate nitrogen ligands in {Cu4N4} clusters. In the case of [Cu4(NMe2)4] and [Cu4(NEt2)4] hydrogen atoms have been omitted for clarity. | ||
With B3LYP/6-311G(d,p), the parent molecule Cu4(NH2)4 is planar and attempts to locate the saddled form by starting with saddled geometries all resulted in the planar form. While this parent molecule has not been structurally determined experimentally, the methyl and ethyl analogues have been determined by X-ray crystallography. As already noted, the ethyl analogue Cu4(NEt2)4 is saddled while the methyl analogue Cu4(NEt2)4 is planar. Geometry optimisations of Cu4(NMe2)4 and Cu4(NEt2)4 only succeeded in locating planar and saddled minima respectively in total agreement with observed data. The presence of bulkier ethyl groups is clearly responsible for steric interactions between ligands leading to the saddled form being favoured over the planar form. The only structurally determined {Cu4N4} system from copper and iminophosphorane prior to our work is the tetracation [Cu4(NHPEt3)4]4+ which is found in the saddled form.8 The simpler model system [Cu4(NHPH3)4]4+ was looked at computationally to establish whether the saddled form can be attributed to the steric bulk of the ethyl groups or not. There are four possible conformers based on the positions of the PH3 and H at the nitrogens – three based on conformers A–C were looked at (see Fig. 6). Conformer B was found to be the most stable conformer and saddled whereas the other two are planar. This suggests that the sterics of the ethyl groups are not a determining factor in this case.
Since our experimental results concern {Cu4N4} systems with three-coordinate ring nitrogens (complexes 1 and 2), several systems containing three-coordinate ring nitrogens (Table 2), including the parent system [Cu4(NPH3)4], were looked at in detail (see Fig. 7). Optimised geometries of [Cu4(NPH3)4] based on conformers A, B, C and D were obtained with C and D requiring symmetry constraints to avoid rearrangements to the more stable forms A and B. All contained planar {Cu4N4} rings with near-planar nitrogen atoms for A, B and C. However, replacing hydrogens with methyl and ethyl groups gave optimised geometries with saddle angles of 166.3° (av) and 150.5° (av) respectively. Their planar forms could not be located from various starting planar geometries. It seems that even the less bulky PMe3 groups are responsible for steric interactions leading to saddled {Cu4N4} rings (Fig. 7). The planar forms observed experimentally for 1 and 2 seem to occur due to favourable packing of the PR3 groups leading to planar {Cu4N4} geometries.
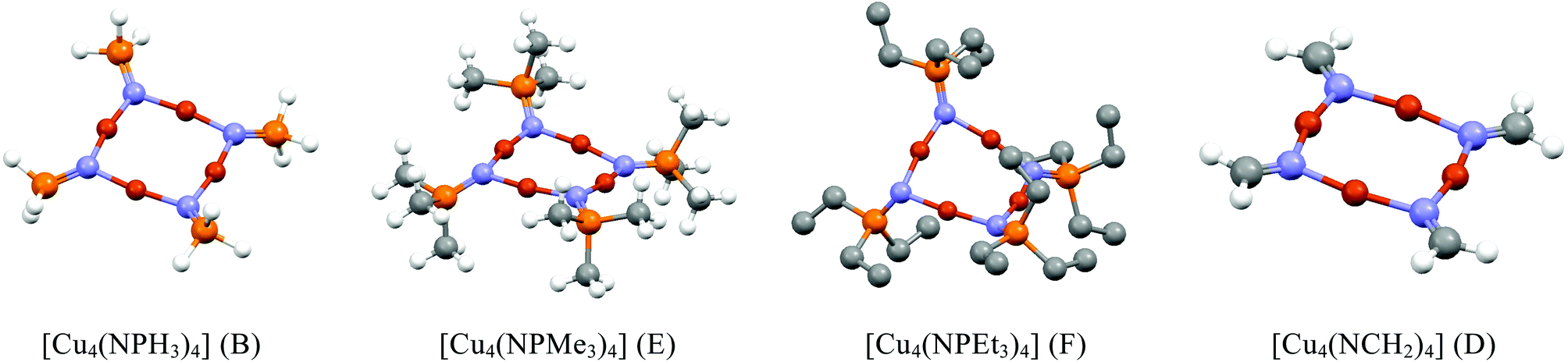 | ||
| Fig. 7 Diagrams showing the optimised molecular geometries of selected complexes containing 3-coordinate nitrogen ligands in {Cu4N4} clusters. In the case of [Cu4(NPEt3)4] hydrogen atoms have been omitted for clarity. | ||
The Cu4(NCR2)4 systems with {μ-NCR2} moieties resemble the Cu4(NPR3)4 systems in that the ring nitrogen atoms are three-coordinate. The parent Cu4(NCH2)4 is shown to be planar like Cu4(NPH3)4 at B3LYP/6-311G(d,p), but inevitably replacing the hydrogens with bulkier substituents will cause steric repulsions resulting in saddled forms as found experimentally.
As noted above, there have been theoretical studies on {Cu4} ring systems that suggest aromatic stabilisation resulting from cyclic electron conjugation within the planar ring.28 Here, the nucleus-independent chemical shift (NICS)31 calculations were carried out as a measure of (anti)aromaticity in 1, 2 and the related {Cu4N4} systems listed in Table 2. At the B3LYP/6-311G(d,p) level, benzene has a NICS value of −8.9 ppm and cyclobutadiene of 25.4 ppm which reflect aromatic and antiaromatic character respectively. The optimised geometries of 1 and 2 have values close to zero (0.2 and 1.6 ppm, respectively) indicative of have negligible aromaticity or antiaromaticity. The saddled form of 1 has a NICS value of −0.2 ppm which shows that the saddled form is slightly more aromatic than the planar form of 1 (0.2 ppm). Any degree of aromaticity as a result of the planarity in the {Cu4N4} ring is not supported here. While different functionals and basis sets have been used, the reported NICS value for the saddled Cu4(NH2)4 geometry is −1.7 ppm compared to 1.3 ppm here for the planar form i.e. again, the saddled form is more ‘aromatic’. Our computations suggest that the preference for planarity in the parent systems, where there are no steric effects from the ligand substituents, is very unlikely to be due to ring aromaticity based on the NICS data.
In conclusion, the planar geometries observed in the solid state structures of 1 and 2 arise from the ‘tuned’ steric demands of the phosphinimide ligands rather than on the basis of either strong Cu⋯Cu interactions and σ-bond delocalisation.32 Sterics are clearly important in determining the planarity of the {Cu4N4} ring in {Cu4N4} systems while according to computations here the planar forms are favoured in neutral parent {Cu4N4} systems.
Experimental section
General remarks
All manipulations were carried out under an atmosphere of dry dinitrogen or argon using standard Schlenk and glove-box techniques. Toluene and hexane were dried using an Innovative Technology Inc. solvent purification system (SPS) system and degassed under dinitrogen or argon prior to use. The starting materials, CuMes33 and HNPPh334 were prepared using literature procedures. HP(NMe2)3, was purchased from Sigma-Aldrich and used as received. NMR spectra were recorded at 298 K on Bruker Avance 500 and 400 MHz NMR spectrometers and referenced as follows for 1H and 13C{1H} spectra: benzene (1H, δ = 7.16 ppm; 13C, δ = 128.0 ppm) d2-dichloromethane (1H, δ = 5.32 ppm; 13C, δ = 53.84 ppm). 31P{1H} NMR chemical shifts were referenced to 85% H3PO4 (δ = 0.0 ppm). Elemental analyses were performed externally by the London Metropolitan University Microanalysis Service.
Syntheses of complexes
Single crystal X-ray crystallography
Experimental details relating to the single-crystal X-ray crystallographic studies are summarised in Table 3. For all structures, data were collected on a Nonius Kappa CCD diffractometer at 150(2) K using Mo-Kα radiation (λ = 0.71073 Å). Structure solution and refinements were performed using SHELX8635 and SHELX9736 software, respectively. Corrections for absorption were made in all cases. Data were processed using the Nonius Software.37 Structure solution,38 followed by full-matrix least squares refinement36b was performed using the WINGX-1.80 suite of programs throughout.39 For all complexes, hydrogen atoms were included at calculated positions. Crystals of the complex 2 were both small and weakly diffracting, with intensity loss at higher 2-theta angle. Hence a data completeness of >93.5% (max 2θ = 25.0 °) could not be met. The CCDC reference numbers for 1 and 2 are 955629 and 955630 respectively.| Compound | 1 | 2 |
|---|---|---|
| Chemical formula | C12H36Cu2N8P2 | C75.50H64Cu4N4P4 |
| Formula mass | 481.51 | 1405.35 |
| Crystal system | Monoclinic | Monoclinic |
| a/Å | 13.2320(1) | 9.1310(4) |
| b/Å | 13.9940(2) | 24.407(1) |
| c/Å | 13.3030(2) | 29.4590(7) |
| α/° | 90.00 | 90.00 |
| β/° | 117.240(2) | 98.382(2) |
| γ/° | 90.00 | 90.00 |
| Unit cell volume/Å3 | 2190.11(5) | 6495.1(4) |
| Temperature/K | 150(2) | 150(2) |
| Space group | P21/n | P21/c |
| No. of formula units per unit cell, Z | 4 | 4 |
| Absorption coefficient, μ/mm−1 | 2.100 | 1.438 |
| No. of reflections measured | 17527 |
24762 |
| No. of independent reflections | 4667 | 10711 |
| R int | 0.0475 | 0.0645 |
| Final R1 values (I > 2σ(I)) | 0.0289 | 0.0490 |
| Final wR (F2) values (I > 2σ(I)) | 0.0760 | 0.1073 |
| Final R1 values (all data) | 0.0329 | 0.0951 |
| Final wR (F2) values (all data) | 0.0793 | 0.1280 |
| Goodness-of-fit on F2 | 1.064 | 1.081 |
| Largest diff. peak and hole/e Å−3 | 0.702, −0.530 | 0.959, −0.436 |
| CCDC reference number | 955629 | 955630 |
Computational studies
Calculations were carried out using the Gaussian09 package.40 All starting geometries of 1, 2 and related systems were optimised without symmetry constraints at B3LYP/6-311G(d,p) level of theory.41 No imaginary frequencies were found from frequency calculations on these optimised geometries and indicate that the geometries are true minima. Symmetry constraints were however applied to conformers C (C4v) and D (C4h) of Cu4(NPH3)4. NICS values were obtained from dummy atoms placed in the centre of the {Cu4} rings using the GIAO42-NMR method at B3LYP/6-311G(d,p). Calculated 31 GIAO-NMR chemical shifts were obtained using the δ(31P) = 310.0 − σ(31P) scale while the 13C shifts were calculated using the δ(13C) = 182.5 − σ(31C) scale.References
- (a) H. J. Cristau, M. Taillefer and N. Rahier, J. Organomet. Chem., 2002, 646, 94–106 CrossRef CAS; (b) M. Taillefer, N. Rahier, E. Minta and H. J. Cristau, Phosphorus, Sulfur Silicon Relat. Elem., 2002, 177, 1847–1850 CrossRef CAS; (c) A. W. Johnson, Ylides and Imines of Phosphorus, Wiley, New York, Chichester, 1993 Search PubMed.
- (a) K. Dehnicke, M. Krieger and W. Massa, Coord. Chem. Rev., 1999, 182, 19–65 CrossRef; (b) K. Dehnicke and F. Weller, Coord. Chem. Rev., 1997, 158, 103–169 CrossRef CAS; (c) D. W. Stephan, Adv. Organomet. Chem., 2006, 54, 267–291 CrossRef CAS.
- (a) F. Guerin, J. C. Stewart, C. Beddie and D. W. Stephan, Organometallics, 2000, 19, 2994–3000 CrossRef CAS; (b) L. LePichon, D. W. Stephan, X. Gao and Q. Wang, Organometallics, 2002, 21, 1362–1366 CrossRef CAS; (c) K. Ma, W. E. Piers and M. Parvez, J. Am. Chem. Soc., 2006, 128, 3303–3312 CrossRef CAS PubMed; (d) D. W. Stephan, Organometallics, 2005, 24, 2548–2560 CrossRef CAS; (e) D. W. Stephan, F. Guerin, R. E. v. H. Spence, L. Koch, X. Gao, S. J. Brown, J. W. Swabey, Q. Wang, W. Xu, P. Zoricak and D. G. Harrison, Organometallics, 1999, 18, 2046–2048 CrossRef CAS; (f) D. W. Stephan, J. C. Stewart, F. Guerin, R. E. v. H. Spence, W. Xu and D. G. Harrison, Organometallics, 1999, 18, 1116–1118 CrossRef CAS; (g) N. Yue, E. Hollink, F. Guerin and D. W. Stephan, Organometallics, 2001, 20, 4424–4433 CrossRef CAS.
- (a) J. Koketsu, Y. Ninomiya, Y. Suzuki and N. Koga, Inorg. Chem., 1997, 36, 694–702 CrossRef CAS; (b) I. A. Koppel, R. Schwesinger, T. Breuer, P. Burk, K. Herodes, I. Koppel, I. Leito and M. Mishima, J. Phys. Chem. A, 2001, 105, 9575–9586 CrossRef CAS; (c) P. V. Sudhakar and K. Lammertsma, J. Am. Chem. Soc., 1991, 113, 1899–1906 CrossRef CAS.
- S. Chitsaz, B. Neumuller and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 9–10 CrossRef CAS.
- A. S. Batsanov, P. D. Bolton, R. C. B. Copley, M. G. Davidson, J. A. K. Howard, C. Lustig and R. D. Price, J. Organomet. Chem., 1998, 550, 445–448 CrossRef CAS.
- J. Camacho-Bunquin, M. J. Ferguson and J. M. Stryker, J. Am. Chem. Soc., 2013, 135, 5537–5540 CrossRef CAS PubMed.
- M. Krieger, S. Schlecht, K. Harms and K. Dehnicke, Z. Anorg. Allg. Chem., 1998, 624, 1565–1567 CrossRef CAS.
- U. Riese, N. Faza, W. Massa and K. Dehnicke, Angew. Chem., Int. Ed., 1999, 38, 528–531 CrossRef CAS.
- (a) A. Bauer, N. W. Mitzel, A. Schier, D. W. H. Rankin and H. Schmidbaur, Chem. Ber., 1997, 130, 323–328 CrossRef CAS; (b) G. Pivoriunas, C. Maichle-Mossmer, S. Schwarz and J. Strahle, Z. Anorg. Allg. Chem., 2005, 631, 1743–1745 CrossRef CAS.
- J. C. Stephens, M. A. Khan and R. P. Houser, Inorg. Chem., 2001, 40, 5064–5065 CrossRef CAS.
- R. M. Z. Kocker, J. Pebler, C. Friebel, K. Dehnicke and D. Fenske, Z. Anorg. Allg. Chem., 1995, 621, 1311–1317 CrossRef.
- (a) R. M. Z. Kocker, A. Behrendt, K. Dehnicke and D. Fenske, Z. Naturforsch., B: Chem. Sci., 1994, 49, 301–308 Search PubMed; (b) R. M. Z. Kocker, K. Dehnicke and D. Fenske, Z. Naturforsch., B: Chem. Sci., 1994, 49, 987–990 Search PubMed.
- (a) A. L. Johnson, A. M. Willcocks and S. P. Richards, Inorg. Chem., 2009, 48, 8613–8622 CrossRef CAS PubMed; (b) A. M. Willcocks, A. L. Johnson, P. R. Raithby, S. Schiffers and J. E. Warren, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2011, 67, M215–M217 CAS; (c) A. M. Willcocks, T. Pugh, J. A. Hamilton, A. L. Johnson, S. P. Richards and A. J. Kingsley, Dalton Trans., 2013, 42, 5554–5565 RSC; (d) A. M. Willcocks, T. P. Robinson, C. Roche, T. Pugh, S. P. Richards, A. J. Kingsley, J. P. Lowe and A. L. Johnson, Inorg. Chem., 2012, 51, 246–257 CrossRef CAS PubMed.
- K. Weber, K. Korn, M. Schulz, K. Korth and J. Sundermeyer, Z. Anorg. Allg. Chem., 1999, 625, 1315–1320 CrossRef CAS.
- (a) R. E. Cramer, F. Edelmann, A. L. Mori, S. Roth, J. W. Gilje, K. Tatsumi and A. Nakamura, Organometallics, 1988, 7, 841–849 CrossRef CAS; (b) H. Schmidbaur and G. Jonas, Chem. Ber., 1967, 100, 1120–1128 CrossRef CAS; (c) H. J. Cristau, L. Chiche, J. Kadoura and E. Torreilles, Tetrahedron Lett., 1988, 29, 3931–3934 CrossRef CAS.
- (a) A. Armstrong, L. H. Jones, J. D. Knight and R. D. Kelsey, Org. Lett., 2005, 7, 713–716 CrossRef CAS PubMed; (b) J. A. Reisz, E. B. Klorig, M. W. Wright and S. B. King, Org. Lett., 2009, 11, 2719–2721 CrossRef CAS PubMed.
- (a) M. Krieger, R. O. Gould, K. Harms, A. Greiner and K. Dehnicke, Z. Anorg. Allg. Chem., 2001, 627, 747–754 CrossRef CAS; (b) M. Krieger, R. O. Gould, K. Harms, S. Parsons and K. Dehnicke, Chem. Ber., 1996, 129, 1621–1625 CrossRef CAS; (c) M. Krieger, R. O. Gould, B. Neumuller, K. Harms and K. Dehnicke, Z. Anorg. Allg. Chem., 1998, 624, 1434–1442 CrossRef CAS; (d) U. Müller, O. Bock, H. Sippel, T. Grob, K. Dehnicke and A. Greiner, Z. Anorg. Allg. Chem., 2002, 628, 1703–1707 CrossRef; (e) B. Neumüller and K. Dehnicke, Z. Anorg. Allg. Chem., 2004, 630, 799–805 CrossRef; (f) U. Riese, N. Faza, W. Massa, K. Harms, T. Breyhan, P. Knochel, J. Ensling, V. Ksenofontov, P. Gutlich and K. Dehnicke, Z. Anorg. Allg. Chem., 1999, 625, 1494–1499 CrossRef CAS.
- (a) H. Chen, M. M. Olmstead, S. C. Shoner and P. P. Power, J. Chem. Soc., Dalton Trans., 1992, 451–457 RSC; (b) J. P. Coyle, W. H. Monillas, G. P. A. Yap and S. T. Barry, Inorg. Chem., 2008, 47, 683–689 CrossRef CAS PubMed; (c) H. Hope and P. P. Power, Inorg. Chem., 1984, 23, 936–937 CrossRef CAS; (d) A. M. James, R. K. Laxman, F. R. Fronczek and A. W. Maverick, Inorg. Chem., 1998, 37, 3785–3791 CrossRef CAS PubMed.
- A. Bondi, J. Phys. Chem., 1964, 68, 441–451 CrossRef CAS.
- (a) A. S. Batsanov, R. C. B. Copley, M. G. Davidson, M. A. Fox, T. G. Hibbert, J. A. K. Howard and K. Wade, J. Cluster Sci., 2006, 17, 119–137 CrossRef CAS PubMed; (b) M. Grun, K. Harms, R. M. Z. Kocker, K. Dehnicke and H. Goesmann, Z. Anorg. Allg. Chem., 1996, 622, 1091–1096 CrossRef; (c) N. W. Mitzel and C. Lustig, J. Chem. Soc., Dalton Trans., 1999, 3177–3183 RSC; (d) M. G. Davidson, A. E. Goeta, J. A. K. Howard, C. W. Lehmann, G. M. McIntyre and R. D. Price, J. Organomet. Chem., 1998, 550, 449–452 CrossRef CAS.
- R. A. D. Soriaga, S. Javed and D. M. Hoffman, J. Cluster Sci., 2010, 21, 567–575 CrossRef CAS PubMed.
- M. K. Davies, P. R. Raithby, M. A. Rennie, A. Steiner and D. S. Wright, J. Chem. Soc., Dalton Trans., 1995, 2707–2709 RSC.
- S. Gambarotta, M. Bracci, C. Floriani, A. Chiesivilla and C. Guastini, J. Chem. Soc., Dalton Trans., 1987, 1883–1888 RSC.
- F. H. Allen, O. Kennard, J. J. Galloy, O. Johnson and D. G. Watson, Chem. Struct. 2, 1993, 343–358 CAS.
- M. El-Hamdi, M. Sola, G. Frenking and J. Poater, J. Phys. Chem. A, 2013, 117, 8026–8034 CrossRef CAS PubMed.
- (a) S. Courtenay, P. R. Wei and D. W. Stephan, Can. J. Chem., 2003, 81, 1471–1476 CrossRef CAS; (b) T. Grob, S. Chitsaz, K. Harms and K. Dehnicke, Z. Anorg. Allg. Chem., 2002, 628, 473–479 CrossRef; (c) T. Grob, K. Harms and K. Dehnicke, Z. Anorg. Allg. Chem., 2000, 626, 1065–1072 CrossRef CAS.
- (a) A. C. Tsipis and C. A. Tsipis, J. Am. Chem. Soc., 2003, 125, 1136–1137 CrossRef CAS PubMed; (b) D. Y. Zubarev, B. B. Averkiev, H. J. Zhai, L. S. Wang and A. I. Boldyrev, Phys. Chem. Chem. Phys., 2008, 10, 257–267 RSC.
- (a) C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS; (b) S. Otto and A. Roodt, Inorg. Chim. Acta, 2004, 357, 1–10 CrossRef CAS.
- E. E. Karagiannis and C. A. Tsipis, Organometallics, 2010, 29, 847–859 CrossRef CAS.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- S. D. Bunge, J. A. Ocana, T. L. Cleland and J. L. Steele, Inorg. Chem., 2009, 48, 4619–4621 CrossRef CAS PubMed.
- E. M. Meyer, S. Gambarotta, C. Floriani, A. Chiesivilla and C. Guastini, Organometallics, 1989, 8, 1067–1079 CrossRef CAS.
- H. J. Cristau, J. Kadoura, L. Chiche and E. Torreilles, Bull. Soc. Chim. Fr., 1989, 515–520 CAS.
- G. M. Sheldrick, University of Göttingen, 1986.
- (a) G. M. Sheldrick, Acta Crystallogr., Sect. A: Fundam. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed; (b) G. M. Sheldrick, SHELXL97, Program for the Solution of Crystal Structures, University of Göttingen, Germany, 1997 Search PubMed.
- Z. Otwinowski and W. Minor, Macromol. Crystallogr., Part A, 1997, 276, 307–326 CrossRef CAS.
- A. Altomare, M. C. Burla, M. Camalli, G. L. Cascarano, C. Giacovazzo, A. Guagliardi, A. G. G. Moliterni, G. Polidori and R. Spagna, J. Appl. Crystallogr., 1999, 32, 115–119 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, 2009 Search PubMed.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed; (b) C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS; (c) G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090 CrossRef CAS PubMed.
- (a) R. Ditchfield, Mol. Phys., 1974, 27, 789–807 CrossRef CAS; (b) C. M. Rohlfing, L. C. Allen and R. Ditchfield, Chem. Phys., 1984, 87, 9–15 CrossRef CAS; (c) K. Wolinski, J. F. Hinton and P. Pulay, J. Am. Chem. Soc., 1990, 112, 8251–8260 CrossRef CAS.
Footnotes |
| † Dedicated to the memory of Prof. Kenneth Wade FRS, an inspirational thinker, teacher, mentor and friend. |
| ‡ CCDC 955629 and 955630. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00255a |
| This journal is © The Royal Society of Chemistry 2015 |