DOI:
10.1039/C5DT00216H
(Paper)
Dalton Trans., 2015,
44, 7230-7241
Organo-palladium(II) complexes bearing unsymmetrical N,N,N-pincer ligands: synthesis, structures and oxidatively induced coupling reactions†
Received
16th January 2015
, Accepted 6th March 2015
First published on 9th March 2015
Abstract
The 2-(2′-aniline)-6-imine-pyridines, 2-(C6H4-2′-NH2)-6-(CMe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NAr)C5H3N (Ar = 4-i-PrC6H4 (HL1a), 2,6-i-Pr2C6H3 (HL1b)), have been synthesised via sequential Stille cross-coupling, deprotection and condensation steps from 6-tributylstannyl-2-(2-methyl-1,3-dioxolan-2-yl)pyridine and 2-bromonitrobenzene. The palladium(II) acetate N,N,N-pincer complexes, [{2-(C6H4-2′-NH)-6-(CMeNAr)C5H3N}Pd(OAc)] (Ar = 4-i-PrC6H4 (1a), 2,6-i-Pr2C6H3 (1b)), can be prepared by reacting HL1 with Pd(OAc)2 or, in the case of 1a, more conveniently by the template reaction of ketone 2-(C6H4-2′-NH2)-6-(CMeO)C5H3N, Pd(OAc)2 and 4-isopropylaniline; ready conversion of 1 to their chloride analogues, [{2-(C6H4-2′-NH)-6-(CMeNAr)C5H3N}PdCl] (Ar = 4-i-PrC6H4 (2a), 2,6-i-Pr2C6H3 (2b)), has been demonstrated. The phenyl-containing complexes, [{2-(C6H4-2′-NH)-6-(CMeNAr)C5H3N}PdPh] (Ar = 4-i-PrC6H4 (3a), 2,6-i-Pr2C6H3 (3b)), can be obtained by treating HL1 with (PPh3)2PdPh(Br) in the presence of NaH or with regard to 3a, by the salt elimination reaction of 2a with phenyllithium. Reaction of 2a with silver tetrafluoroborate or triflate in the presence of acetonitrile allows access to cationic [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NCMe)][X] (X = BF4 (4), X = O3SCF3 (5)), respectively; the pyridine analogue of 5, [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NC5H5)][O3SCF3] (5′), is also reported. Oxidation of phenyl-containing 3a with one equivalent of 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor™) in acetonitrile at low temperature leads to a new palladium species that slowly decomposes to give 4 and biphenyl; biphenyl formation is also observed upon reaction of 3a with XeF2. However, no such oxidatively induced coupling occurs when using 3b. Single crystal X-ray diffraction studies have been performed on HL1b, 1a, 1b, 2a, 2b, 3a, 3b and 5′.
NAr)C5H3N (Ar = 4-i-PrC6H4 (HL1a), 2,6-i-Pr2C6H3 (HL1b)), have been synthesised via sequential Stille cross-coupling, deprotection and condensation steps from 6-tributylstannyl-2-(2-methyl-1,3-dioxolan-2-yl)pyridine and 2-bromonitrobenzene. The palladium(II) acetate N,N,N-pincer complexes, [{2-(C6H4-2′-NH)-6-(CMeNAr)C5H3N}Pd(OAc)] (Ar = 4-i-PrC6H4 (1a), 2,6-i-Pr2C6H3 (1b)), can be prepared by reacting HL1 with Pd(OAc)2 or, in the case of 1a, more conveniently by the template reaction of ketone 2-(C6H4-2′-NH2)-6-(CMeO)C5H3N, Pd(OAc)2 and 4-isopropylaniline; ready conversion of 1 to their chloride analogues, [{2-(C6H4-2′-NH)-6-(CMeNAr)C5H3N}PdCl] (Ar = 4-i-PrC6H4 (2a), 2,6-i-Pr2C6H3 (2b)), has been demonstrated. The phenyl-containing complexes, [{2-(C6H4-2′-NH)-6-(CMeNAr)C5H3N}PdPh] (Ar = 4-i-PrC6H4 (3a), 2,6-i-Pr2C6H3 (3b)), can be obtained by treating HL1 with (PPh3)2PdPh(Br) in the presence of NaH or with regard to 3a, by the salt elimination reaction of 2a with phenyllithium. Reaction of 2a with silver tetrafluoroborate or triflate in the presence of acetonitrile allows access to cationic [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NCMe)][X] (X = BF4 (4), X = O3SCF3 (5)), respectively; the pyridine analogue of 5, [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NC5H5)][O3SCF3] (5′), is also reported. Oxidation of phenyl-containing 3a with one equivalent of 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor™) in acetonitrile at low temperature leads to a new palladium species that slowly decomposes to give 4 and biphenyl; biphenyl formation is also observed upon reaction of 3a with XeF2. However, no such oxidatively induced coupling occurs when using 3b. Single crystal X-ray diffraction studies have been performed on HL1b, 1a, 1b, 2a, 2b, 3a, 3b and 5′.
Introduction
Recent years have seen a surge of interest in oxidatively induced coupling reactions involving Pd(III) and Pd(IV) intermediates due, in part, to their potential to promote transformations inaccessible using the conventional low valent Pd(0)/(II) cycle.1–3 For example, the historically challenging arene–fluoride bond forming reaction has become a reality with both types of high valent intermediate isolated and/or proposed in reaction pathways derived from Pd(II) species.3 Central to these developments have been reagents such as Selectfluor™ [1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)] and xenon difluoride that can oxidise the metal centre as a two electron oxidant (from Pd(II) to Pd(IV))4,5 or as a one electron oxidant (from Pd(II) to Pd(III))6,7 and moreover provide a source of F (e.g., as F+ or F˙). In cases where these types of oxidant deliver a fluorine atom direct to the metal centre, selective C–F reductive elimination from the high valent organo–metal intermediate can be challenging as alternative (and potentially desirable) degradation pathways can prove competitive.3d Sanford, for example, has reported that the Pd(IV) mono-aryl complex, [(4,4-t-Bu2bipy)Pd(Ar)(F)2(FHF)] (Ar = 4-FC6H4), only undergoes selective Caryl–F reductive elimination when heated in the presence of excess oxidant, otherwise competitive Ar–Ar coupling occurs through a process described as σ-aryl exchange between metal centres.5 Indeed this type of intermolecular Ar–Ar coupling involving palladium mono-aryl species has some precedent in Pd(II) and Pd(III) chemistry involving complexes bearing a variety of multidentate ligands.2c,8,9
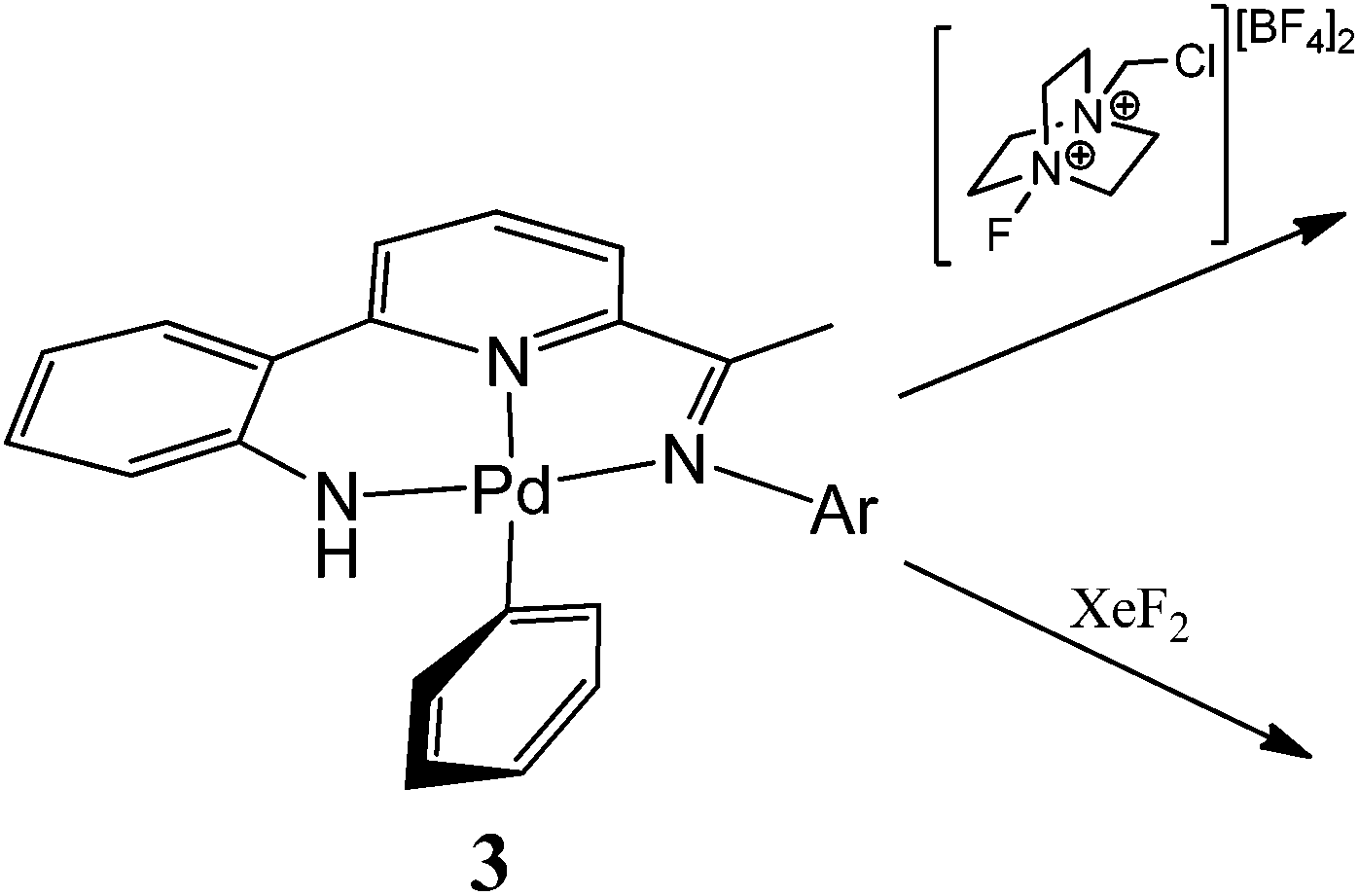
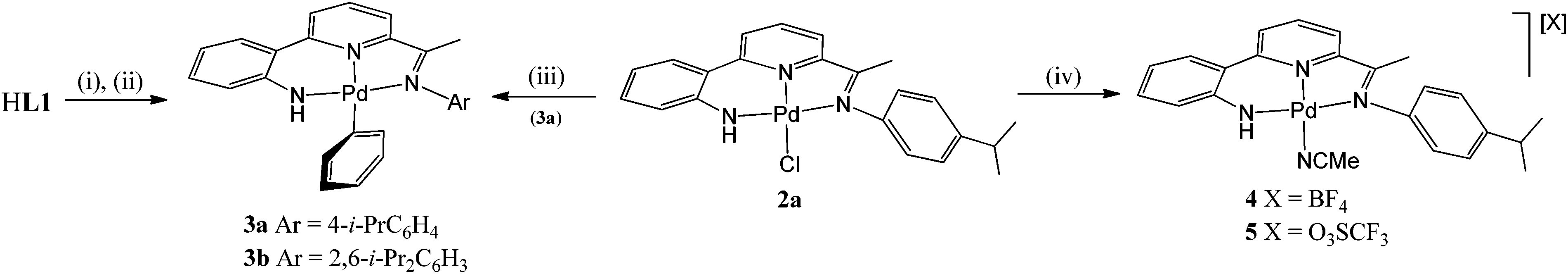
Given the apparent variation in coupling events that can occur from a high valent organo-Pd species,1–7 we have been interested in exploring the influence of a supporting multidentate ligand on the oxidatively induced reaction pathway. Herein, we report the reactivity of a family of N,N,N-pincer bearing Pd(II) mono-phenyl complexes of the type, [{2-(C6H4-2-NH)-6-(CMeNAr)C5H3N}PdPh] (Ar = aryl (3)), towards Selectfluor and XeF2 (Fig. 1);10 as an additional point of interest the effects that steric variation (Ar = 4-i-PrC6H4, 2,6-i-Pr2C6H3) has on the reactivity, will be investigated. Furthermore, we report the full synthetic details for the preparation of the novel pro-ligands (HL1) and their palladium(II) acetate (1), chloride (2) and phenyl (3) derivatives.
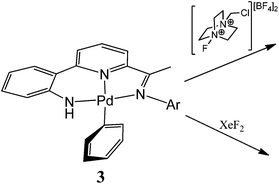 |
| | Fig. 1
N,N,N palladium(II) mono-aryl pincer, 3, and the stoichiometric reactivity to be examined. | |
Results and discussion
Preparation of pro-ligand HL1
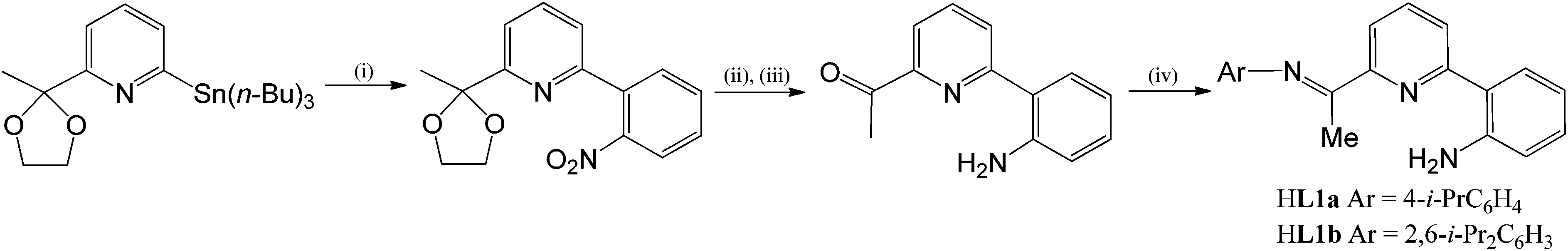
The 2-(2′-aniline)-6-imine-pyridines, 2-(C6H4-2′-NH2)-6-(CMeNAr)C5H3N (Ar = 4-i-PrC6H4 (HL1a), 2,6-i-Pr2C6H3 (HL1b)), have been prepared in reasonable yield via sequential Stille coupling, deprotection and condensation reactions from 6-tributylstannyl-2-(2-methyl-1,3-dioxolan-2-yl)pyridine and 2-bromonitrobenzene (Scheme 1). For both HL1a and HL1b, the condensation step proved sluggish in alcoholic media but proceeded more effectively by running the reaction in the neat aniline at high temperature; nevertheless problems encountered in the work-up of HL1a resulted in its isolation in only a modest yield (see later for a higher yielding template approach to L1a). The precursor ketone and the two N,N,N pro-ligands, HL1a and HL1b, have been characterised using a combination of electrospray mass spectrometry, IR, 1H NMR and 13C NMR spectroscopy (see Experimental section).
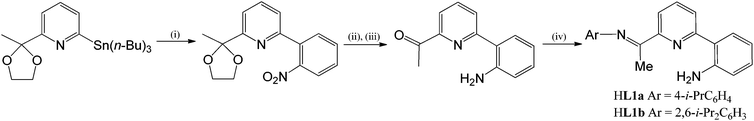 |
| | Scheme 1
Reagents and conditions: (i) 2-BrC6H4NO2, cat. Pd(OAc)2–PPh3, toluene, 100 °C, microwave; (ii) SnCl2, ethanol; (iii) HCl(aq.); (iv) ArNH2, 225 °C. | |
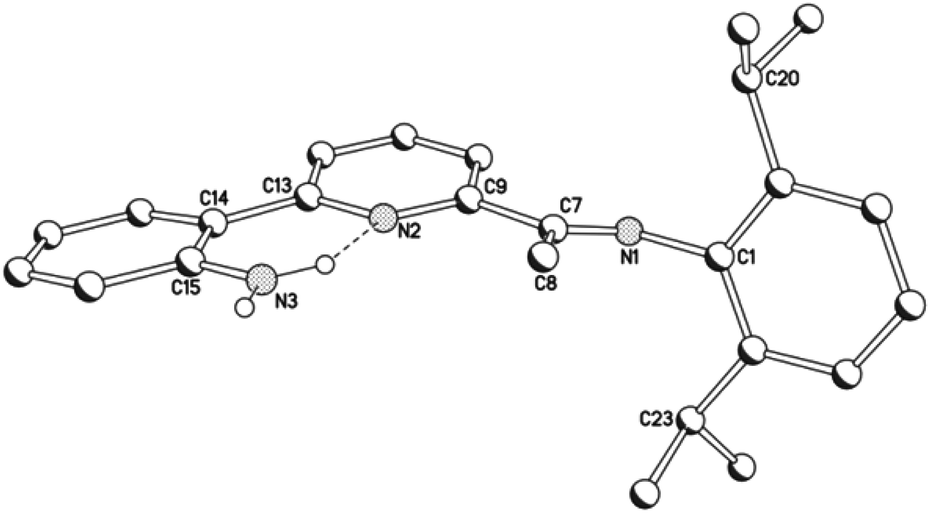
Compounds, HL1a, and HL1b, both display protonated molecular ions peaks in their electrospray mass spectra and downfield shifted signals for the amino protons (range: δ 5.72–5.79) in their 1H NMR spectra. Characteristic imine stretching frequencies of ca. 1638 cm−1 are seen in their IR spectra as are higher wavenumber bands corresponding to the N–H stretches. Further confirmation of the composition of HL1b was achieved in the form of a single crystal X-ray determination.
A perspective view of HL1b is depicted in Fig. 2; selected bond distances and angles are listed in Table 1. The structure consists of a central pyridine ring that is substituted at its 2-position by a phenyl-2′-amine group and at the 6-position by a trans-configured N-arylimine unit [C(7)–N(1) 1.277(3) Å]. The pyridine nitrogen atoms adopt a cis conformation with respect to the neighbouring aniline nitrogen (tors: N(2)–C(13)–C(14)–C(15) 8.1°) as a result of a hydrogen-bonding interaction between one of the amino hydrogen atoms and the pyridine nitrogen [N(3)⋯N(2) 2.675 Å]; a similar arrangement has been reported for a related quinolinyl-substituted aniline.11
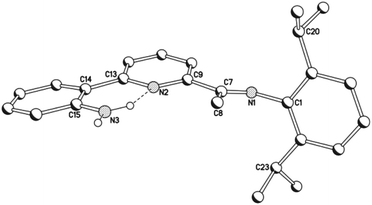 |
| | Fig. 2 Molecular structure of HL1b, including a partial atom numbering scheme. All hydrogen atoms, apart from H3A and H3B, have been omitted for clarity. | |
Table 1 Selected bond distances (Å) and angles (°) for HL1b
|
Bond lengths
|
| C(15)–N(3) |
1.366(4) |
C(13)–C(14) |
1.477(4) |
| C(7)–N(1) |
1.277(3) |
C(7)–C(9) |
1.482(4) |
| C(7)–C(8) |
1.504(4) |
|
|
|
Bond angles
|
| C(8)–C(7)–N(1) |
125.3(2) |
C(9)–C(7)–N(1) |
116.4(3) |
Palladium(II) complexes of L1
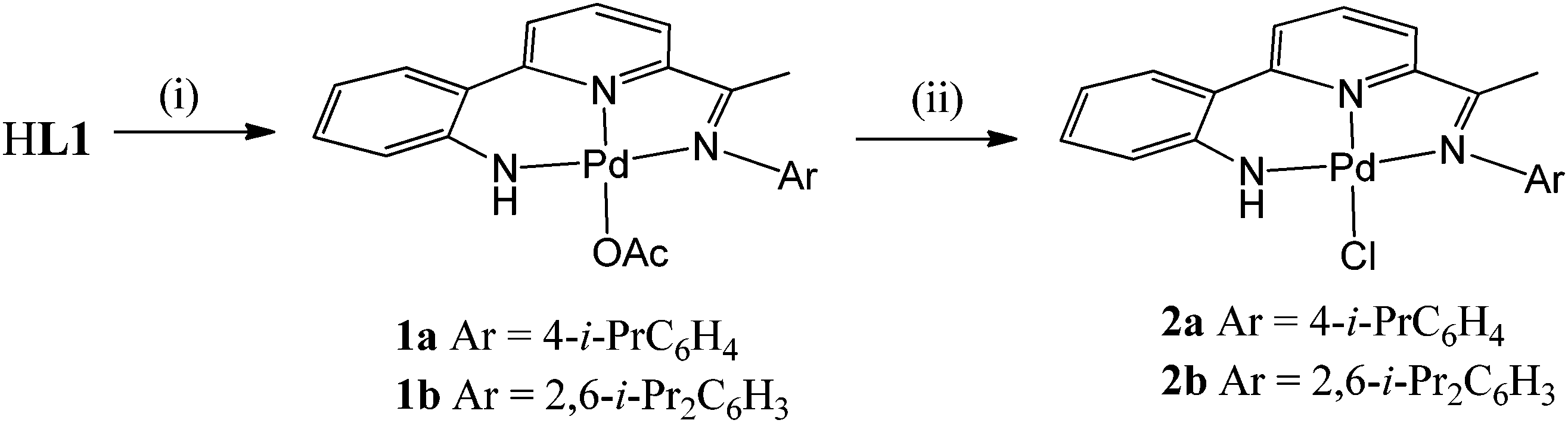
Interaction of HL1b with Pd(OAc)2 at 60 °C in toluene gave on work-up, [{2-(C6H4-2-NH)-6-(CMeN(2,6-i-Pr2C6H3))C5H3N}Pd(OAc)] (1b)), in good yield (Scheme 2). While [{2-(C6H4-2-NH)-6-(CMeN(4-i-PrC6H4))C5H3N}Pd(OAc)] (1a) could also be made by this route, it was more conveniently prepared by the template reaction of ketone 2-(C6H4-2-NH2)-6-(CMeO)C5H3N, Pd(OAc)2 and 4-isopropylaniline. Compounds 1 can be readily converted to their chloride analogues, [{2-(C6H4-2-NH)-6-(CMeNAr)C5H3N}PdCl] (Ar = 4-i-PrC6H4 (2a), 2,6-i-Pr2C6H3 (2b)), by treatment of a dichloromethane solution of 1 with aqueous sodium chloride. All four complexes are air stable and have been characterised using a combination of FAB mass spectrometry, IR and NMR (1H and 13C) spectroscopy and elemental analyses (see Experimental section). In addition, crystals of each complex have been the subject of single crystal X-ray diffraction studies.
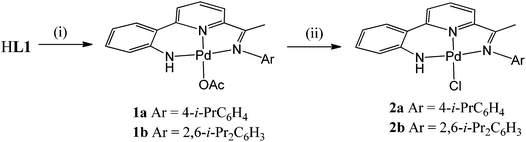 |
| | Scheme 2
Reagents and conditions: (i) Pd(OAc)2, toluene, 60 °C; (ii) NaCl(aq.), CH2Cl2, RT. | |
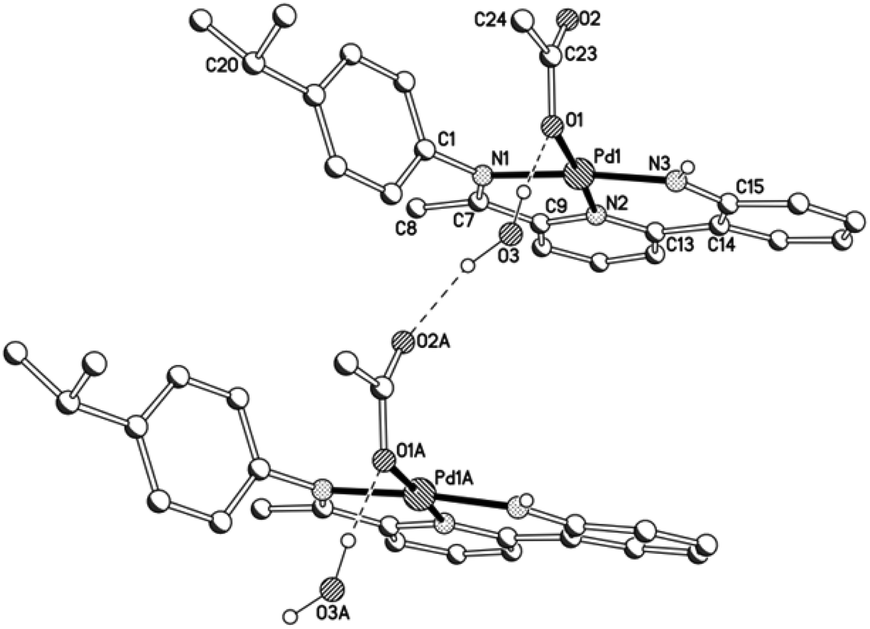
Views of acetate-containing 1a and 1b are given in Fig. 3 and 4; selected bond distances and angles are collected for both structures in Table 2. There are two independent molecules for 1b in the unit cell (A and B) which differ most noticeably in the relative inclination of neighbouring pyridyl and anilido ring planes (vide infra). The structures of 1a and 1b are similar consisting of a four-coordinate palladium centre bound by a tridentate monoanionic 2-(2′-anilido)-6-imine-pyridine ligand and a monodentate O-bound acetate, but contrast in the nature of the hydrogen bonding involving the acetate ligand. In 1a, a water molecule present within the unit cell links the palladium-acetate units to form a hydrogen-bonded network [O(1)acetate⋯O(3)water 2.837, O(3)water⋯O(2A)acetate 2.877 Å], while in 1b the hydrogen bonding is intramolecular in origin involving the pendant acetate oxygen and the anilido proton [N(3)⋯O(2)acetate 2.799A, 2.889B Å]. Within the N,N,N-ligand there are both 5- and 6-membered chelate rings with the bite angle for the 6-membered ring being more compatible with the square planar geometrical requirements of the palladium(II) centre [N(3)–Pd(1)–N(2)6-membered: 93.7(2) (1a), 92.2(3)A, 93.6(2)B (1b) vs. N(2)–Pd(1)–N(1)5-membered 82.2(2) (1a), 82.6(3)A, 82.1(2)B° (1b)]. In both cases some twisting of the anilido unit with respect to the adjacent pyridyl plane is apparent [tors. N(2)–C(13)–C(14)–C(15) 3.6(4) (1a), 4.9(4)A, 9.0(5)B° (1b)]. For a given complex, the Pd–Nimine bond distance is the longest of the three metal–ligand interactions involving the N,N,N-ligand followed by the Pd–Npyridine distance and then by the Pd–Nanilido distance which is best exemplified for 1a [Pd(1)–N(1)imine 2.014(6) > Pd(1)–N(2)pyridine 1.963(5) > Pd(1)–N(3)anilido 1.932(5) Å]. The N-aryl groups are inclined towards orthogonality with regard to the neighbouring CNimine vector [tors. C(7)–N(1)–C(1)–C(6) 87.6(4) (1a), 86.4(4)av° (1b)], with the 2,6-diisopropyl substitution on the N-aryl group in 1b additionally providing some steric protection to the axial sites of the palladium centre. The closest crystallographically characterised comparators to 1 are the phenolate-containing counterparts, [{2-(C6H4-2′-O)-6-(CMeNAr)C5H3N}Pd(OAc)] (Ar = 4-i-PrC6H4, 2,6-i-Pr2C6H3), which display similar bonding characteristics.12
 |
| | Fig. 3 A segment of the network based on 1a linked by water molecules. A partial atom numbering scheme is included while all hydrogen atoms, apart from H3 and those belonging to the water molecule, have been omitted for clarity; dotted lines show the intermolecular hydrogen-bonding interactions. | |
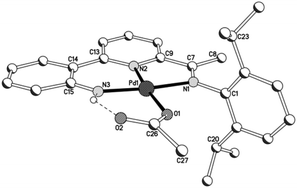 |
| | Fig. 4 Molecular structure of 1b (molecule A) including a partial atom numbering scheme. All hydrogen atoms, apart from H3 have been omitted for clarity. | |
Table 2 Selected bond distances (Å) and angles (°) for 1a and 1b
| |
|
1b
|
| |
1a
|
Molecule A |
Molecule B |
|
Bond lengths
|
| Pd(1)–N(1) |
2.014(6) |
2.017(8) |
2.019(8) |
| Pd(1)–N(2) |
1.963(5) |
1.970(9) |
1.977(7) |
| Pd(1)–N(3) |
1.932(5) |
1.920(9) |
1.922(8) |
| Pd(1)–O(1) |
2.036(5) |
2.011(8) |
2.021(7) |
| C(7)–N(1) |
1.286(8) |
1.285(12) |
1.300(11) |
| C(9)–C(7) |
1.469(9) |
1.468(13) |
1.458(13) |
| C(15)–N(3) |
1.347(9) |
1.316(12) |
1.323(12) |
|
Bond angles
|
| N(1)–Pd(1)–N(2) |
82.2(2) |
82.6(3) |
82.1(2) |
| N(1)–Pd(1)–N(3) |
174.6(3) |
174.3(3) |
174.4(2) |
| N(1)–Pd(1)–O(1) |
93.8(2) |
89.6(3) |
93.8(2) |
| N(2)–Pd(1)–N(3) |
93.7(2) |
92.2(3) |
93.6(2) |
| N(3)–Pd(1)–O(1) |
90.3(2) |
95.6(3) |
90.5(2) |
A view of chloride-containing 2b is given in Fig. 5; selected bond distances and angles are collected for both 2a and 2b in Table 3. The two independent molecules present in the unit cell for 2a (A and B) differ most noticeably in the inclination of the N-aryl plane to the adjacent imine unit. The structures of 2a and 2b are similar to those of their acetate precursors (1) with a tridentate monoanionic 2-(2′-anilido)-6-imine-pyridine filling three coordination sites of the distorted square planar geometry but differ with a chloride now filling the fourth site. Replacing a chloride for an O-bound acetate has little effect on the trans Pd–Npyridine distance [1.976(5)A, 1.984(5)B (2a), 1.987(3) (2b) vs. 1.963(5) (1a), 1.970(9)A 1.977(7)B Å (1b)]. Unlike 1b, the anilido NH proton is not involved in any inter- or intra-molecular contacts of note.
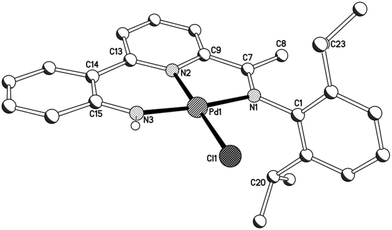 |
| | Fig. 5 Molecular structure of 2b including a partial atom numbering scheme. All hydrogen atoms, apart from H3, have been omitted for clarity. | |
Table 3 Selected bond distances (Å) and angles (°) for 2a and 2b
| |
2a
|
2b
|
| Molecule A |
Molecule B |
|
Bond lengths
|
| Pd(1)–N(1) |
2.022(5) |
2.035(6) |
2.025(3) |
| Pd(1)–N(2) |
1.976(5) |
1.984(5) |
1.987(3) |
| Pd(1)–N(3) |
1.934(5) |
1.931(6) |
1.927(3) |
| Pd(1)–Cl(1) |
2.2971(18) |
2.2931(18) |
2.3123(17) |
| C(7)–N(1) |
1.297(7) |
1.298(7) |
1.289(5) |
| C(15)–N(3) |
1.335(8) |
1.334(8) |
1.336(5) |
|
Bond angles
|
| N(1)–Pd(1)–N(2) |
81.6(2) |
82.2(3) |
82.01(13) |
| N(1)–Pd(1)–N(3) |
174.4(2) |
174.0(2) |
173.63(13) |
| N(2)–Pd(1)–N(3) |
93.1(2) |
92.2(3) |
91.91(13) |
| N(1)–Pd(1)–Cl(1) |
96.66(16) |
96.56(17) |
97.63(10) |
| N(2)–Pd(1)–Cl(1) |
178.21(17) |
175.39(15) |
179.27(9) |
| N(3)–Pd(1)–Cl(1) |
88.54(16) |
89.14(17) |
88.47(10) |
Complexes 1a, 1b, 2a and 2b, all display molecular ion peaks in their FAB mass spectra along with fragmentation peaks corresponding to the loss of an acetate or a chloride, respectively. In their IR spectra the imine stretching frequencies are shifted between 28 and 35 cm−1 to lower wavenumber in comparison with the corresponding free HL1, characteristic of imine-nitrogen coordination.13–15 In 1b and 2b two distinct doublets are seen for the isopropyl methyl groups in their 1H NMR spectra consistent with some restricted rotation about the N-2,6-diisopropylphenyl bond in solution. The acetate methyl groups in 1 can be seen at δ ca. 1.5 in their 1H NMR spectra with the MeC(O)O carbon atoms observable at δ ca. 177.1 in their 13C NMR spectra. The anilido NH proton in 2 is observable at a similar chemical shift (ca. δ 5.8) to that seen in free HL1, but in acetate-containing 1 there is some variation with that observed in 1b being more downfield (δ 5.60 (1a), 7.39 (1b)); this is likely to be due to the influence of the intramolecular NH⋯Oacetate hydrogen bonding seen in 1b (see Fig. 4). As with related monodentate acetate complexes, 1a and 1b both show strong bands assignable to the symmetric and asymmetric ν(COO) vibrations.16
Their phenyl derivatives, [{2-(C6H4-2-NH)-6-(CMeNAr)C5H3N}PdPh] (Ar = 4-i-PrC6H4 (3a), 2,6-i-Pr2C6H3 (3b)), could be readily accessed by treatment of HL1 with NaH followed by (PPh3)2PdPh(Br) (Scheme 3). Alternatively, 3a can be prepared by treating chloride 2a with phenyl lithium; a related salt elimination approach to make 3b has not proved possible. In the case of 2a, chloride abstraction with both silver tetrafluoroborate and triflate in acetonitrile proved facile affording [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NCMe)][X] (X = BF4 (4), X = O3SCF3 (5)) in high yield (Scheme 3). Mono-phenyl 3a and 3b are air and water stable, whereas 4 and 5 proved hygroscopic on prolonged standing. All four complexes have been characterised using a combination of FAB mass spectrometry, IR and NMR (1H and 13C) spectroscopy and elemental analyses (see Experimental section).
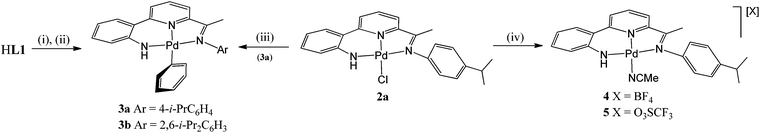 |
| | Scheme 3
Reagents and conditions: (i) xs. NaH, THF, heat; (ii) (PPh3)2PdPh(Br), THF, heat; (iii) LiPh, THF, −78 °C; (iv) AgX (X = BF4, O3SCF3), MeCN, RT. | |
The mass spectra of 3a and 3b exhibit molecular ions while 4 and 5 display peaks corresponding to their cationic units. As with 1 and 2, all four complexes exhibit ν(CN)imine stretches at lower wavenumber (typically by 35 cm−1) when compared with HL1, supporting coordination of L1 to the metal centre.13–15 The imine methyl resonances are seen between δ 2.2 and 2.5 in their 1H NMR spectra, while signals for the imine carbon fall between δ 170.5 and 174.8 in their 13C{1H} NMR spectra. Signals attributable to [BF4]− and [O3SCF3]− counterions could also be seen in the 19F NMR spectra of 4 and 5. In addition, crystals of 3a, 3b and the pyridine analogue of 5, [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NC5H5)][O3SCF3] (5′), have been the subject of single crystal X-ray diffraction studies.
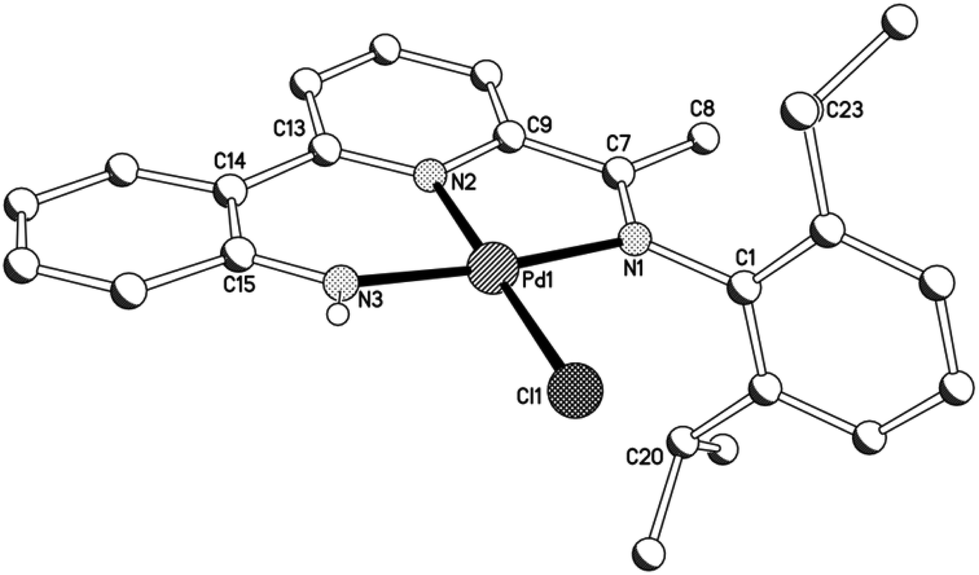
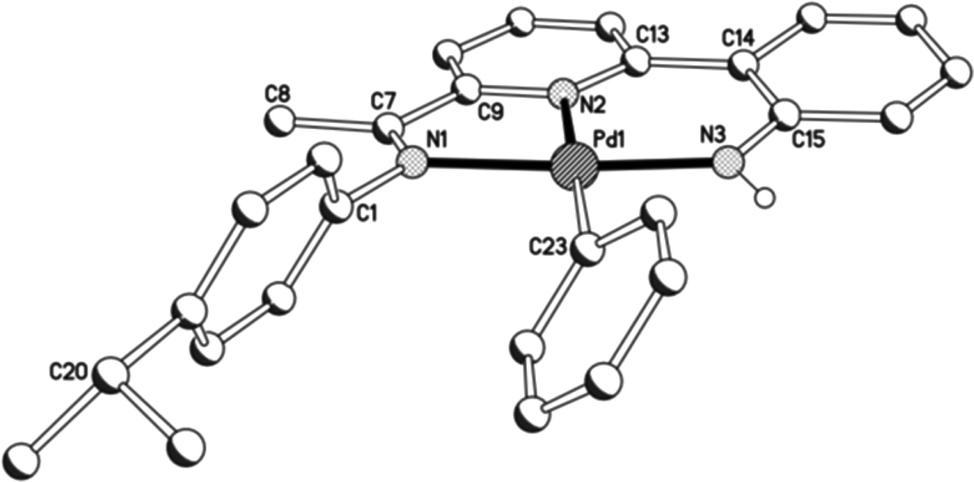
As a representative of the mono-phenyl pair of structures, a view of the molecular structure of 3a is depicted in Fig. 6; selected bond distances and angles are listed in Table 4 for both 3a and 3b. As with 1 and 2, 2-(2′-anilido)-6-imine-pyridine ligand acts a tridentate ligand with the σ-phenyl ligand now occupying the fourth coordination site to complete a distorted square planar geometry. The phenyl ligand in both structures is tilted with respect to the trans-pyridine unit of the N,N,N-ligand and most noticeably for 3a, presumably as a consequence of the variation in steric hindrance imposed by the N-aryl groups [tors. C(13)–N(2)–C(23)–C(24) 46.4(4) (3a), 41.4(4)° (3b)]. When compared to 1 and 2, the presence of a σ-phenyl group in 3 results in an elongation of the trans Pd–Npyridine distance [Pd–N(2) 2.066(6) (3a), 2.069(2) (3b) vs. 1.963(5) (1a), 1.974(8)av. (1b), 1.980(5)av. (2a), 1.987(3) (2b) Å], an observation attributable to the strong trans-influence exhibited by the aryl group. In contrast, the exterior nitrogen-palladium distances remain similar in length to those seen in 1 and 2. To accommodate the increased Pd–N(2)pyridine distance, there is increased twisting of the ligand backbone which is most apparent in 3b [tors. N(2)–C(13)–C(14)–C(15) 25.2(4) and N(1)–C(7)–C(9)–N(2) 13.7(4)°]. As with chloride-containing 2, the anilido NH proton shows no notable intra- or inter-molecular contacts of note.
 |
| | Fig. 6 Molecular structure of 3a including a partial atom numbering scheme. All hydrogen atoms, apart from H3, have been omitted for clarity. | |
Table 4 Selected bond distances (Å) and angles (°) for 3a and 3b
| |
3a
|
3b
|
|
Bond lengths
|
| Pd(1)–N(1) |
2.060(6) |
2.041(2) |
| Pd(1)–N(2) |
2.066(6) |
2.069(2) |
| Pd(1)–N(3) |
1.937(6) |
1.959(3) |
| Pd(1)–C(23) |
2.016(8) |
2.013(3) |
| C(7)–N(1) |
1.302(9) |
1.291(4) |
| C(9)–C(7) |
1.452(10) |
1.472(4) |
| C(15)–N(3) |
1.326(9) |
1.348(4) |
|
Bond angles
|
| N(1)–Pd(1)–N(2) |
81.0(3) |
79.88(10) |
| N(1)–Pd(1)–N(3) |
172.5(3) |
171.38(10) |
| N(1)–Pd(1)–C(23) |
99.2(3) |
99.68(11) |
| N(2)–Pd(1)–C(23) |
178.5(3) |
169.73(11) |
| N(3)–Pd(1)–N(2) |
91.5(3) |
79.88(10) |
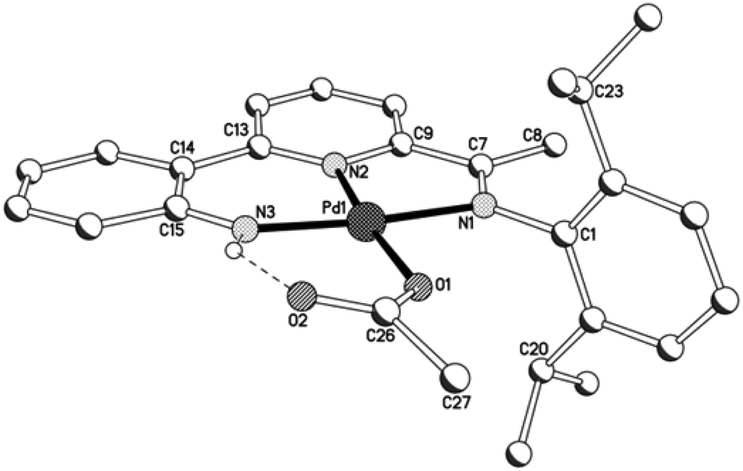
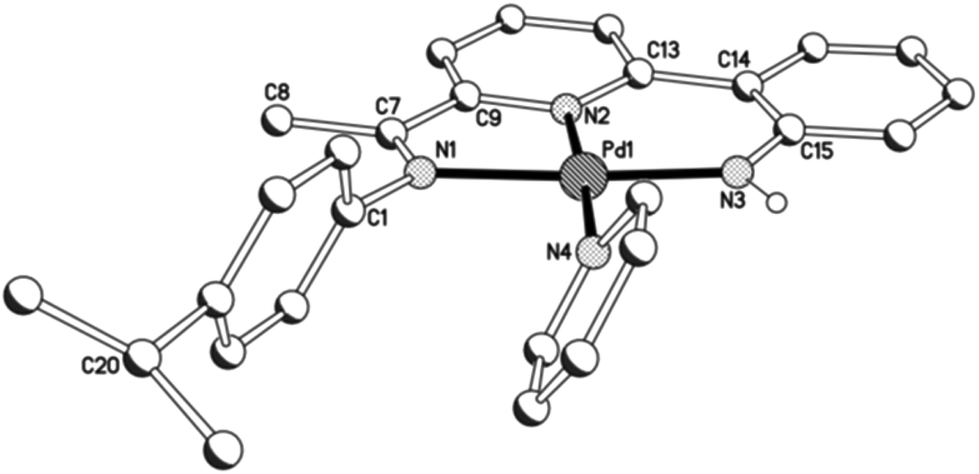
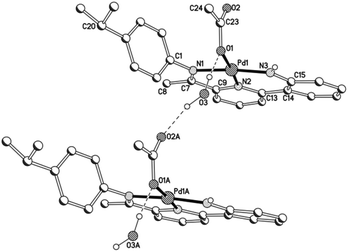
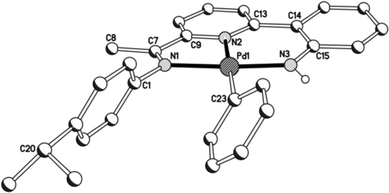
Unfortunately cationic 4 and 5 were not amenable to forming crystals suitable for an X-ray determination. To overcome this practical issue, small amounts of pyridine were added to a solution of 5 in chloroform and hexane slowly diffused forming single crystals of [{2-(C6H4-2′-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NC5H5)][O3SCF3] (5′). The molecular structure of the cationic unit 5′ is depicted in Fig. 7; selected bond distances and angles are listed in Table 5. As with a number of the structures reported in this study, two independent molecules (A and B) were present in the unit cell which, in this case, differ most noticeably in the inclination of the N-aryl groups. The structure of 5′ consists of a palladium(II) cationic unit charge balanced by a non-coordinating triflate counteranion. Within the distorted square planar cationic unit, the 2-(2′-anilido)-6-imine-pyridine ligand acts a tridentate ligand and an N-bound pyridine fills the fourth coordination site. Similar to phenyl-bound 3, the monodentate heteroaromatic in 5′ is not co-planar with the trans-pyridine unit of the tridentate ligand. Instead it adopts a tilted configuration [C(13)–N(2)–N(4)–C(27) 58.1(5)A, 60.0(5)B°] which is ca. 8° greater than that for the aryl group in 3b. Inspection of the trans Pd–N(2)pyridine distance involving the N,N,N-ligand reveals a bond length [Pd(1)–N(2) 1.992(11)A, 1.952(11)B Å] comparable with those seen in 1 and 2, but shorter than that in 3 (vide supra). The NH proton of the anilido unit of the pincer undergoes a modest interaction with a triflate oxygen atom [N(3)⋯O(1) 3.096A, 3.175B Å].
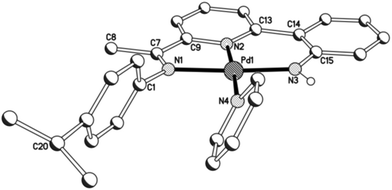 |
| | Fig. 7 Molecular structure of cationic unit in 5′, including a partial atom numbering scheme. All hydrogen atoms, except for H3, have been omitted for clarity. | |
Table 5 Selected bond distances (Å) and angles (°) for 5′
| |
Molecule A |
Molecule B |
|
Bond lengths
|
| Pd(1)–N(1) |
2.049(12) |
2.034(12) |
| Pd(1)–N(2) |
1.992(11) |
1.952(11) |
| Pd(1)–N(3) |
1.937(11) |
1.962(11) |
| Pd(1)–N(4) |
2.010(11) |
2.105(11) |
| C(7)–N(1) |
1.276(16) |
1.279(16) |
| C(15)–N(3) |
1.336(15) |
1.316(16) |
| Range S–O (triflate) |
1.424(11)–1.485(10) |
|
|
Bond angles
|
| N(1)–Pd(1)–N(2) |
82.9(5) |
82.0(5) |
| N(1)–Pd(1)–N(3) |
175.3(5) |
174.9(4) |
| N(1)–Pd(1)–N(4) |
93.7(5) |
94.0(5) |
| N(2)–Pd(1)–N(3) |
92.6(5) |
93.8(5) |
| N(2)–Pd(1)–N(4) |
176.6(5) |
175.7(5) |
| N(3)–Pd(1)–N(4) |
90.8(5) |
90.3(4) |
Reactivity of 3 towards Selectfluor and XeF2
In the first instance the reactivity of mono-phenyl containing 3a towards Selectfluor was explored. Typically 3a was treated with excess Selectfluor at 100 °C in a toluene–MeCN mixture; these higher temperature conditions having been identified as more conducive to formation of the C–F reductive elimination product.4c,3c However, biphenyl was the only aryl-containing organic product identified by GC-MS. Likewise using XeF2 as the oxidant instead of Selectfluor under the same conditions gave only biphenyl.
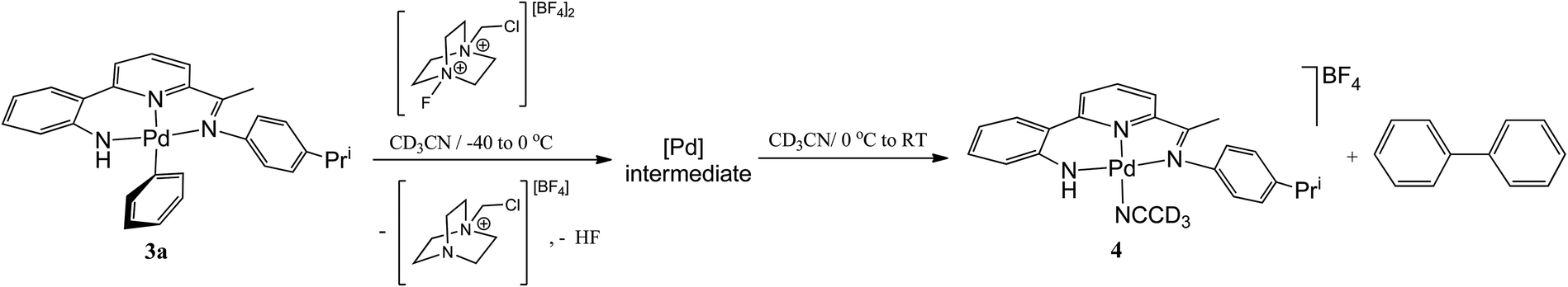
To investigate the reaction further and potentially observe any possible intermediates, a reaction involving an equimolar ratio of 3a and Selectfluor was undertaken in CD3CN at a series of lower temperatures and the reaction monitored by 1H and 19F NMR spectroscopy (Scheme 4). After 15 minutes at room temperature the 19F NMR spectrum revealed full consumption of Selectfluor and a new peak at δ −181 attributable to the formation of hydrogen fluoride.17 The 1H NMR spectrum contained signals consistent with biphenyl, the salt [{2-(C6H4-2-NH)-6-(CMeN(4-i-PrC6H4)C5H3N}Pd(NCCD3)][BF4] (4) (vide supra) and 1-(chloromethyl)-1,4-diazabicyclo[2.2.2]octan-1-ium tetrafluoroborate, the Selectfluor degradation product. In addition, there were signals present attributable to another palladium species that slowly reduced in intensity over time.
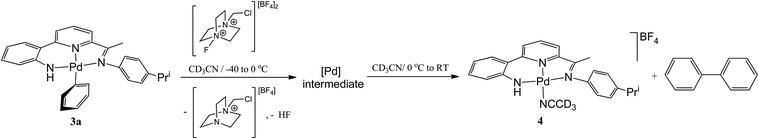 |
| | Scheme 4 Low temperature oxidation of 3a and Ar–Ar coupling on warming. | |
When the reaction was carried out at −40 °C, full consumption of Selectfluor was again evident from the 19F NMR spectrum which also contained a peak attributable to HF, albeit temperature shifted (δ −172). In the 1H NMR spectrum full conversion of 3a to a single palladium species was observed with the aromatic/pyridyl region integrating to sixteen protons; no peaks assignable to biphenyl nor 4 could be identified. As the reaction mixture was warmed to 0 °C, only sharpening of the 1H NMR spectrum was observed with peaks that clearly match those observed for the decomposing palladium species seen at room temperature (Fig. S9 in ESI†). In the 19F NMR spectrum a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 ratio between the HF signal (δ −174) and the BF4 peak (δ −152) accounts for all the fluorine introduced from the Selectfluor (Fig. S10 in ESI†). On warming to room temperature, decomposition of the palladium intermediate ensued generating biphenyl and 4; full conversion being observed after 48 hours (Fig. S11 in ESI†). Unfortunately, further attempts to fully characterise the high valent palladium intermediate were unsuccessful.
:8 ratio between the HF signal (δ −174) and the BF4 peak (δ −152) accounts for all the fluorine introduced from the Selectfluor (Fig. S10 in ESI†). On warming to room temperature, decomposition of the palladium intermediate ensued generating biphenyl and 4; full conversion being observed after 48 hours (Fig. S11 in ESI†). Unfortunately, further attempts to fully characterise the high valent palladium intermediate were unsuccessful.
The 1:1 reaction of 2,6-diisopropylphenyl-containing 3b with Selectfluor was also explored at a range of different temperatures. However, despite consumption of 3b, there was no evidence for the formation of biphenyl, fluorobenzene nor could any characterisable palladium species be identified. It is unclear as to the origin of these differences in reactivity between 3a and 3b towards Selectfluor.
Conclusions
A new family of imino-based monoanionic N,N,N pincer ligands have been developed that can support neutral palladium(II) acetate (1), chloride (2) and phenyl (3) species; the tetrafluoroborate (4) and triflate (5) salts are also reported. The oxidatively induced Ph–Ph coupling reactions involving 3a described in this work highlights the ability of Selectfluor and xenon difluoride to behave as bystanding oxidants.3d Using the more sterically bulky 3b, neither biphenyl nor fluorobenzene were produced under similar oxidative conditions. The identity of the palladium intermediate that generates biphenyl and cationic 4 remains uncertain but investigations into the precise nature of this species are ongoing.
Experimental
General
All operations, unless otherwise stated, were carried out under an inert atmosphere of dry, oxygen-free nitrogen using standard Schlenk and cannular techniques or in a nitrogen purged glove box. Operations involving a Microwave were performed on a CEM Discover Explorer Hybrid instrument. Solvents were distilled under nitrogen from appropriate drying agents18 or were employed directly from a Solvent Purification System (Innovative Technology, Inc.). The electrospray (ESI) mass spectra were recorded using a micromass Quattra LC mass spectrometer with acetonitrile or methanol as the matrix. FAB mass spectra (including high resolution) were recorded on a Kratos Concept spectrometer with NBA as matrix or on a Waters Xevo QToF mass spectrometer equipped with an atmospheric solids analysis probe (ASAP). The infrared spectra were recorded in the solid state with Universal ATR sampling accessories on a Perkin Elmer Spectrum One FTIR instrument. NMR spectra were recorded on a Bruker DRX400 spectrometer at 400.13 (1H), 376.46 (19F) and 100.61 MHz (13C) or a Bruker Avance III 500 spectrometer at 125 MHz (13C), at ambient temperature unless otherwise stated; chemical shifts (ppm) are referred to the residual protic solvent peaks and coupling constants are expressed in hertz (Hz). Melting points (mp) were measured on a Gallenkamp melting point apparatus (model MFB-595) in open capillary tubes and were uncorrected. Elemental analyses were performed at the Science Technical Support Unit, London Metropolitan University. The reagents 4-isopropylaniline, 2,6-diisopropylaniline, tin(II) chloride dihydrate, phenyllithium (1.8 M in n-Bu2O), silver triflate, silver tetrafluoroborate, 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor™) and 2-bromonitrobenzene were purchased from Aldrich Chemical Co. and used without further purification. The compounds 6-tributylstannyl-2-(2-methyl-1,3-dioxolan-2-yl)pyridine19 and [(PPh3)2PdPh(Br)]20 were prepared using literature procedures. All other chemicals were obtained commercially and used without further purification.
Synthesis of 2-(2-methyl-1,3-dioxolan-2-yl)-6-(2-nitrophenyl)pyridine
A 25 mL microwave vial was loaded with 2-bromonitrobenzene (0.536 g, 2.70 mmol), 6-tributylstannyl-2-(2-methyl-1,3-dioxolan-2-yl)pyridine (1.226 g, 2.70 mmol), Pd(OAc)2 (0.025 g, 0.11 mmol) and triphenylphosphine (0.058 g, 0.22 mmol) and the contents dissolved in bench toluene (13 mL). The system was then sealed and stirred for 30 s in a microwave before heating to 100 °C (with 100 W power and 10 bar pressure limits) for 1 h. The resulting dark brown reaction mixture was concentrated under reduced pressure to yield a dark brown oil. This oil was then dry loaded on to a silica column and eluted with a 70:30 mixture of petroleum ether (40–60) and ethyl acetate affording 2-(2-methyl-1,3-dioxolan-2-yl)-6-(2-nitrophenyl)pyridine as a yellow solid (0.517 g, 67%) along with trace amounts of the homocoupled by-product 6,6′-bis(2-methyl-1,3-dioxolan-2-yl)-2,2′-bipyridine. 1H NMR (400 MHz, CDCl3): δ 1.66 (s, 3H, CH3), 3.84 (m, 2H, O–CHH–CHH–O), 4.02 (m, 2H, O–CHH–CHH–O), 2.37 (dd, 3JHH 7.8, 4JHH 1.0, 1H, Ar–H), 7.42–7.46 (m, 1H, Ar–H), 7.50 (dd, 3JHH 7.5, 4JHH 0.9, 1H, Ar–H), 7.54–7.57 (m, 2H, Ar–H), 7.74 (dd, 3JHH 7.8, 3JHH 7.9, Ar–H), 7.78 (dd, 3JHH 8.0, 3JHH 8.1, 1H, Ar–H). 13C {1H} NMR (100 MHz, CDCl3): δ 24.9 (CH3), 65.0 (CH2), 65.1 (CH2), 108.6 (C), 118.7 (CH), 121.7 (CH), 124.4 (CH), 129.1 (CH), 131.1 (CH), 135.3 (C), 137.5 (CH), 149.8 (C), 154.9 (C), 161.1 (C). IR (cm−1): 1587 (CN)pyridine, 1530 (NO2)asymm, 1369 (NO2)symm HRMS (TOFMS, ASAP): calcd for C15H15N2O4 [M + H]+ 287.1032, found 287.1034.
Synthesis of 1-(6-(2-aminophenyl)pyridine-2-yl)ethanone
2-(2-Methyl-1,3-dioxolan-2-yl)-6-(2-nitrophenyl)pyridine (1.040 g, 3.6 mmol) and SnCl2·2H2O (8.220 g, 36.0 mmol) were suspended in bench ethanol (34 mL) and sonicated for 2 h, whereupon a bright yellow slurry was obtained. This slurry was concentrated under reduced pressure and partitioned between 1 M NaOH (100 mL) and CHCl3 (100 mL) until a bright yellow organic phase was observed. The organic phase was separated and the aqueous phase washed with CHCl3 (3 × 25 mL). The combined organic phases were washed with water (2 × 30 mL) and concentrated to a smaller volume under reduced pressure. Aqueous HCl (150 mL, 16% (v/v)) was added to the solution and the resultant biphase stirred for 1 h at ambient temperature. The reaction mixture was neutralised with K2CO3, the organic phase separated and the aqueous phase washed with dichloromethane (2 × 30 mL). The combined organic extracts were washed with water (1 × 30 mL) and brine (1 × 30 mL) and then filtered through Celite layered with magnesium sulphate. The filtrate was concentrated under reduced pressure to afford 1-(6-(2-aminophenyl)pyridin-2-yl)ethanone as a brown/yellow oil which slowly solidifies (0.68 g, 89%). Mp: 95–98 °C. 1H NMR (400 MHz, CDCl3): δ 2.73 (s, 3H, MeCO), 5.77 (br, s, 2H, NH2), 6.82 (m, 2H, Ar–H), 7.22 (ddd, 3JHH 7.3, 3JHH 8.1, 4JHH 1.6, 1H, Ar–H), 7.56 (dd, 3JHH 7.8, 4JHH 1.5, 1H, Ar–H), 7.85–7.95 (m, 3H, Py–H). 13C{1H} NMR (100 MHz, CDCl3): δ 26.1 (Me–CO), 117.4, 118.0, 119.0 (CH), 121.3 (C), 125.7 (CH), 129.6 (CH), 130.5 (CH), 137.9 (CH), 146.5 (C), 151.7 (C), 158.7 (C), 199.4 (CO). IR (cm−1): 3456, 3363 (NH), 1695 (CO)ketone, 1585 (CN)pyridine. ESIMS: m/z 213 [M + H]+. HRMS (FAB): calcd for C13H13N2O [M + H]+ 213.10246, found 213.10252.
Synthesis of 2-(C6H4-2-NH2)-6-(CMe![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) NAr)C5H3N (HL1)
NAr)C5H3N (HL1)
(a) Ar = 4-i-PrC6H34 (HL1a).
To a round bottomed flask equipped with stir bar was added 1-(6-(2-aminophenyl)pyridin-2-yl)ethanone (0.500 g, 2.4 mmol) and 4-isopropylaniline (2.070 g, 15.3 mmol). The reaction vessel was then lowered into a pre-heated heating mantle set at 225 °C and the mixture stirred for 15 min before a catalytic amount of glacial acetic acid was introduced. After 15 min at 225 °C the reaction vessel was allowed to cool to room temperature. The excess aniline was removed by distillation under reduced pressure, the resultant dark residue heated to reflux in ethanol (10 ml) and hot filtered. The filtrate was concentrated to half volume and allowed to cool to room temperature to yield HL1a as a yellow solid (0.040 g, 5%). Mp: 113–115 °C. 1H NMR (400 MHz, CDCl3): δ 1.28 (d, 3JHH 7.0, 6H, CHMe2), 2.37 (s, 3H, MeCN), 2.92 (sept, 3JHH 7.0, 1H, CHMe2), 5.79 (br, s, 2H, NH2), 6.76–6.84 (m, 4H, Ar–H), 7.20 (ddd, 3JHH 8.1, 3JHH 7.4, 4JHH 1.5, 1H, Ar–H), 7.23 (d, 3JHH 8.2, 2H, Ar–H), 7.58 (dd, 3JHH 7.8, 3JHH 1.5, 1H, Ar–H), 7.74 (dd, 3JHH 8.0, 4JHH 0.9, 1H, Py–H), 7.86 (dd, 3JHH 7.93, 3JHH 7.9, 1H, Py–H), 8.76 (dd, 3JHH 7.9, 4JHH 1.0, 1H, Py–H). 13C{1H} NMR (100 MHz, CDCl3): δ 16.6 (MeCN), 24.1 (CHMe2), 33.6 (CHMe2), 117.3, 117.9, 118.8, 119.3 (CH), 122.1 (C), 123.2 (CH), 126.9 (CH), 129.6 (CH), 130.1 (CH), 137.4 (CH), 144.2 (C), 146.5 (C), 148.8 (C), 155.2 (C), 158.2 (C), 166.5 (MeCN). IR (cm−1): 1635 (CN)imine, 1587 (CN)pyridine. ESIMS: m/z 330 [(M + H)]+. HRMS (FAB): calcd C22H24N3 [M + H]+ 330.1970, found 330.1968.
(b) Ar = 2,6-i-Pr2C6H3 (HL1b).
Employing a similar procedure to that described for HL1a with 1-(6-(2-aminophenyl)pyridin-2-yl)ethanone (0.700 g, 3.3 mmol), 2,6-diisopropylaniline (3.800 g, 21.5 mmol) gave following work-up HL1b as a yellow solid (0.54 g, 44%). Crystals suitable for an X-ray determination were grown by slow cooling of an ethanol solution containing the compound. Mp: 139–141 °C. 1H NMR (400 MHz, CDCl3): δ 1.14 (d, 3JHH 6.9, 6H, CHMe2), 1.15 (d, 3JHH 7.0, 6H, CHMe2), 2.21 (s, 3H, MeCN), 2.75 (sept, 3JHH 6.9, 2H, CHMe2), 5.72 (br, s, 2H, NH2), 6.80 (dd, 3JHH 8.2, 4JHH 1.0, 1H, Ar–H), 6.82 (ddd, 3JHH 8.2, 3JHH 8.2, 4JHH 1.3, 1H, Ar–H), 7.08 (dd, 3JHH 8.7, 3JHH 6.3, 1H, Ar–H), 7.17 (dd, 3JHH 6.9, 3JHH 8.5, 2H, Ar–H), 7.21 (ddd, 3JHH 7.3, 3JHH 8.0, 4JHH 1.5, 1H, Ar–H), 7.59 (dd, 3JHH 8.0, 4JHH 1.5, 1H, Ar–H), 7.78 (dd, 3JHH 8.0, 3JHH 0.9, 1H, Py–H), 7.90 (dd, 3JHH 7.9, 3JHH 7.9, 1H, Py–H), 8.26 (dd, 3JHH 7.8, 4JHH 0.9, 1H, Py–H). 13C{1H} NMR (100 MHz, CDCl3): δ 17.5 (MeCN), 22.9 (CHMe2), 23.2 (CHMe2), 28.3 (CHMe2), 117.6, 118.2 (CH), 118.8 (C), 122.2 (CH), 123.0 (CH), 123.4 (CH), 123.7 (CH), 129.6 (CH), 130.1 (CH), 135.8 (C), 137.6 (CH), 146.1 (C), 146.3 (C), 154.5 (C), 158.2 (C), 165.0 (Me–CN). IR (cm−1): 3451, 3282 (br, NH), 1642 (CN)imine, 1584 (CN)pyridine. ESIMS: m/z 372 [(M + H)]+. HRMS (FAB): calcd C25H30N3O [M + H]+ 372.24322, found 372.24310.
Synthesis of [{2-(C6H4-2-NH)-6-(CMeNAr)}Pd(OAc)] (1)
(a) Ar = 4-i-PrC6H4 (1a).
A Schlenk flask equipped with stir bar was evacuated and backfilled with nitrogen and loaded with Pd(OAc)2 (0.740 g, 3.3 mmol), 1-(6-(2-aminophenyl)pyridin-2-yl)ethanone (0.690 g, 0.81 mmol), 4-isopropylaniline (0.660 g, 4.9 mmol) and toluene (70 mL). The reaction vessel was stirred and heated to 80 °C for 3 h. The resultant green/brown solution was evaporated and the resultant solid dissolved in the minimum volume of chloroform before hexane was added to precipitate 1a as a green/brown solid (1.45 g, 89%). Crystals suitable for an X-ray determination were grown by slow diffusion of hexane into a solution of 1a in CHCl3 at room temperature. Mp: >260 °C. 1H NMR (400 MHz, CDCl3): δ 1.22 (d, 3JHH 6.9, 6H, CHMe2), 1.53 (s, 3H, –OC(O)Me), 2.33 (s, 3H, MeCN), 2.98 (sept, 3JHH 6.9, 1H, CHMe2), 5.60 (br, s, 1H, NH), 6.42 (ddd, 3JHH 6.6, 3JHH 8.5, 4JHH 1.2, 1H, Ar–H), 6.86 (dd, 3JHH 8.5, 4JHH 1.2, 1H, Ar–H), 7.02 (ddd 3JHH 6.6, 3JHH 7.9, 4JHH 1.4, 1H, Ar–H), 7.07 (d, 3JHH 8.4, 2H, Ar–H), 7.23 (d, 3JHH 8.3, 2H, Ar–H), 7.59 (dd, 3JHH 7.4, 4JHH 1.0, 1H, Py–H), 7.89 (d, 3JHH 8.0, 1H, Ar–H), 7.99 (dd, 3JHH 7.4, 3JHH 8.8, 1H, Py–H), 8.56 (d, 3JHH 8.8, 1H, Py–H). 13C{1H} NMR (100 MHz, CDCl3): δ 16.3 (MeCN), 21.9 (MeCO2), 22.9 (CHMe2), 32.9 (CHMe2), 111.8 (CH), 112.9 (C), 119.7 (CH), 121.5 (CH), 122.0 (CH), 125.4 (CH), 125.6 (CH), 128.3 (CH), 129.1 (CH), 133.5 (CH), 141.7 (C), 146.9 (C), 148.5 (C), 148.8 (C), 152.7 (C), 169.5 (Me–CN), 177.0 (Me–CO2). IR (cm−1): 1600 (CN)imine, 1591 (COOasymm/CNpyridine), 1367 (COO)symm FABMS: m/z 493 [M]+, 433 [M − OAc]+. Anal Calc. for (C24H25N3O2Pd): C, 58.36; H, 5.10; N, 8.51. Found: C, 58.26; H, 5.23; N, 8.51%.
(b) Ar = 2,6-i-Pr2C6H3 (1b).
A Schlenk flask equipped with stir bar was evacuated and backfilled with nitrogen and loaded with Pd(OAc)2 (0.180 g, 0.81 mmol), HL1b (0.300 g, 0.81 mmol) and toluene (30 mL). After stirring at 60 °C overnight, the green reaction mixture was cooled to room temperature and filtered through Celite and the Celite cake washed thoroughly with dichloromethane. The filtrate was concentrated to ca. 1 mL whereupon hexane (20 mL) was added. The resulting green precipitate was filtered and dried under reduced pressure forming 1b as a dark green powder (0.38 g, 88%). Crystals suitable for an X-ray diffraction study could be grown by slow diffusion of hexane into a chloroform solution of the complex at room temperature. Mp: >260 °C. 1H NMR (400 MHz, CDCl3): δ 1.14 (d, 3JHH 6.8, 6H, CHMe2), 1.39 (d, 3JHH 6.8, 6H CHMe2), 1.51 (s, 3H, –OC(O)Me), 2.41 (s, 3H, MeCN), 3.14 (sept, 3JHH 6.8, 2H, CHMe2), 6.53 (ddd, 3JHH 8.6, 3JHH 6.5, 4JHH 1.4, 1H, Ar–H), 7.06 (dd, 3JHH 8.5, 4JHH 1.2, 1H, Ar–H), 7.13 (ddd, 3JHH 8.5, 3JHH 6.4, 4JHH 1.4, 1H, Ar–H), 7.26 (m, 2H, Ar–H), 7.36 (dd, 3JHH 8.7, 3JHH 6.8, 1H, Ar–H), 7.39 (br, s, 1H, NH), 7.82 (dd 3JHH 7.4, 4JHH 1.0, 1H, Py–H), 8.01 (d, 3JHH 8.6, 1H, Ar–H), 8.10 (dd, 3JHH 8.8, 3JHH 7.3, 1H, Py–H), 6.80 (d, 3JHH 8.7, 1H, Py–H). 13C{1H} NMR (100 MHz, CDCl3): δ 17.0 (CHMe2), 22.3 (CHMe2), 22.7 (MeCO2), 22.8 (MeCN), 27.6 (CHMe2), 112.0 (CH), 112.6 (C), 120.3 (CH), 121.0 (CH), 122.4 (CH), 125.7 (CH), 126.7 (CH), 127.7 (CH), 129.2 (CH), 132.5 (CH), 139.0 (C), 139.4 (C), 149.0 (C), 149.2 (C), 152.7 (C), 170.1 (Me–CN), 177.2 (Me–CO2). IR (cm−1): 1614 (CN)imine, 1583 (COO)asymm/CNpyridine), 1367 (COO)symm ESIMS: m/z 476 [M − OAc]+. TOFMS (ASAP): m/z 536 [M+], 476 [M − OAc]+. Anal Calc. for (C28H32Cl3N3O2Pd): C, 51.32; H, 4.92; N, 6.41. Found: C, 50.92; H, 4.18; N, 7.36%.
Synthesis of [{2-(C6H4-2-NH)-6-(CMeNAr)}PdCl] (2)
(a) Ar = 4-i-PrC6H4 (2a).
A round bottomed flask equipped with stir bar and open to the air was loaded with 1a (0.595 g, 1.20 mmol), dichloromethane (5 mL) and brine (5 mL). The reaction mixture was stirred rapidly for 1 h at room temperature whereupon both phases were diluted and the aqueous layer removed via a separating funnel. The organic phase was washed with water (2 × 20 mL) and concentrated to a smaller volume under reduced pressure. The dark green solution was filtered through a Celite plug and the plug washed thoroughly with dichloromethane. All volatiles were removed under reduced pressure affording 2a as a dark brown solid (0.56 g, 99%). Single crystals suitable an X-ray determination were grown by diffusion of hexane into a solution of 2a in chloroform at room temperature. Mp: >260 °C. 1H NMR (400 MHz, CDCl3): δ 1.23 (d, 3JHH 6.9, 6H, CHMe2), 2.28 (s, 3H, MeCN), 2.89 (sept, 3JHH 6.9, 1H, CHMe2), 5.59 (br, s, 1H, NH), 6.43 (ddd, 3JHH 6.1, 3JHH 8.0, 4JHH 1.20, 1H, Ar–H), 6.81 (dd, 3JHH 8.6, 4JHH 1.0, 1H, Ar–H), 6.99–7.03 (m, 5H, Ar–H), 7.19 (d, 3JHH 8.2, 2H, Ar–H), 7.55 (dd, 3JHH 7.5, 4JHH 0.9, 1H, Py–H), 7.73–7.79 (m, 2H, Py–H/Ar–H), 8.30 (d, 3JHH 8.7, 1H, Py–H). 13C {1H} NMR (125 MHz, CDCl3): δ 17.0 (CH3CN), 22.9 (CHMe2), 32.7 (CHMe2), 112.1 (CH), 112.8 (C), 119.7 (CH), 121.6 (CH), 122.3 (CH), 125.1 (CH), 125.5 (CH), 128.2 (CH), 129.5 (CH), 133.4 (CH), 142.9 (C), 146.6 (C), 148.4 (C), 148.4 (C), 153.1 (C), 171.0 (Me–CN). IR (cm−1): 1603 (CN)imine, 1576 (CN)pyridine. FABMS: m/z 469 (M)+, 434 (M–Cl)+. Anal Calc. for (For C22H22ClN3Pd): C, 56.18; H, 4.71; N, 8.93. Found: C, 56.11; H, 4.69; N, 9.00%.
(b) Ar = 2,6-i-Pr2C6H3 (2b).
Employing a similar procedure to that described for 2a using 1b (0.544 g, 1.02 mmol) gave 2b as a dark green solid (0.520 g, 99%). Single crystals suitable for an X-ray diffraction study could be grown by slow diffusion of hexane into a chloroform solution of 2b at room temperature. Mp: >260 °C. 1H NMR (400 MHz, CDCl3): δ 1.05 (d, 3JHH 6.9, 6H, CHMe2), 1.35 (d, 3JHH 6.9, 6H, CHMe2), 2.28 (s, 3H, MeCN), 2.99 (sept, 3JHH 6.9, 2H, CHMe2), 5.91 (br, s, 1H, NH), 6.49 (ddd, 3JHH 8.5, 3JHH 6.7, 4JHH 1.3, 1H, Ar–H), 6.92 (dd, 3JHH 8.6, 4JHH 1.2, 1H, Ar–H), 7.06 (ddd, 3JHH 8.2, 3JHH 6.5, 4JHH 1.4, Ar–H), 7.19 (m, 2H, Ar–H), 7.28 (dd, 3JHH 8.5, 3JHH 8.5, 1H, Ar–H), 7.77 (dd, 3JHH 7.3, 4JHH 1.1, 1H, Py–H), 7.92 (dd, 3JHH 8.6, 4JHH 1.5, 1H, Ar–H), 8.06 (dd, 3JHH 8.8, 3JHH 8.4, 1H, Py–H), 8.63 (d, 3JHH 8.6, 1H, Py–H). 13C {1H} NMR (125 MHz, CDCl3): δ 18.0 (Me–CN), 23.7 (CHMe2), 23.8 (CHMe2), 28.7 (CHMe2), 113.6 (CH), 113.8 (C), 121.3 (CH), 122.1 (CH), 123.6 (CH), 127.2 (CH), 128.1 (CH), 129.1 (CH), 130.8 (CH), 134.1 (CH), 139.7 (C), 141.6 (C), 150.0 (C), 150.3 (C), 153.7 (C), 172.1 (Me–CN). IR (cm−1): 1608 (CN)imine, 1577 (CN)pyridine. FABMS: m/z 511 [M + H]+, 475 [M − Cl]+. Anal Calc. for (C25H28ClN3Pd): C, 58.60; H, 5.51; N, 8.20. Found: C, 58.49; H, 5.35; N, 8.26%.
Synthesis of [{2-(C6H4-2-NH)-6-(CMeNAr)}Pd(C6H5)] (3)
(a) Ar = 4-i-PrC6H4 (3a).
A Schlenk flask equipped with stir bar was evacuated and backfilled with nitrogen and loaded with 2a (0.208 g, 0.44 mmol) and THF (20 mL). The reaction mixture was stirred and cooled to −78 °C for 15 min. A solution of PhLi (861 μL, 1.55 mmol, 1.8 M in n-Bu2O) was added slowly and the reaction mixture stirred at −78 °C for a further 2 h. One drop of water was added and the solution slowly warmed to room temperature. All volatiles were removed under reduced pressure and the resultant green solid re-dissolved in dichloromethane (20 mL) and washed with water (2 × 20 mL) and brine (10 mL). Following drying over anhydrous magnesium sulphate and filtration, the resulting green solution was concentrated to a smaller volume (ca. 5 mL) and hexane added to precipitate 3a as a green solid (0.161 g, 71%). Single crystals suitable for an X-ray determination were obtained by slow diffusion of hexane into a solution of 3a in chloroform at room temperature. 1H NMR (400 MHz, CDCl3): δ 1.19 (d, 3JHH 6.9, 6H, CHMe2), 2.46 (s, 3H, MeCN), 2.80 (sept, 3JHH 6.9, 1H, CHMe2), 5.48 (br, s, 1H, NH), 6.44 (ddd, 3JHH 6.5, 3JHH 8.0, 4JHH 1.1, 1H, Ar–H), 6.65 (d, 3JHH 8.4, 2H, Ar–H), 6.73–6.75 (m, 3H, Ar–H), 6.90 (dd, 3JHH 8.6, 4JHH 1.3, 1H, Ar–H), 6.95 (d, 3JHH 8.4, 2H, Ar–H), 7.05 (ddd, 3JHH 6.6, 3JHH 8.1, 4JHH 1.4, 1H, Ar–H), 7.07–7.09 (m, 2H, Ar–H), 7.82 (d, 3JHH 7.1, 1H, Py–H), 7.98 (dd, 3JHH 8.5, 4JHH 1.3, 1H, Ar–H), 8.05 (dd, 3JHH 7.5, 3JHH 8.6, 1H, Py–H), 8.58 (d, 3JHH 8.8, 1H, Py–H). 13C{1H} NMR (100 MHz, CDCl3) δ 18.3 (Me–CN), 24.0 (CHMe2), 33.7 (CHMe2), 111.9 (CH), 114.0 (C), 121.3 (CH), 122.2 (CH), 122.6 (CH), 122.9 (CH), 125.9 (CH), 126.0 (CH), 126.1 (CH), 129.9 (CH), 130.0 (CH), 134.2 (CH), 135.9 (CH), 145.1, 146.9, 152.0, 152.1, 153.1, 158.5 (C), 170.5 (MeCN). IR (cm−1): 1602 (CN)imine, 1567 (CN)pyridine. FABMS: m/z 511 [M]+. Anal Calc. for (C28H27N3Pd·1.5OH2): C, 62.40; H, 5.61; N, 7.80. Found: C, 62.04; H, 5.33; N, 8.16%.
(b) Ar = 2,6-i-Pr2C6H3 (3b).
A Schlenk flask equipped with stir bar was evacuated and backfilled with nitrogen and loaded with HL1b (0.100 g, 0.13 mmol), NaH (0.052 g, 2.20 mmol) and THF (10 mL). The resulting slurry was stirred and heated to reflux for 72 h before being allowed to cool to room temperature. The reaction mixture was transferred by cannular filtration to a second Schlenk flask containing [(PPh3)2PdPh(Br)] (0.100 g, 0.13 mmol) and the contents stirred and heated to reflux for a further 72 h. On cooling to room temperature, the resulting green solution was concentrated under reduced pressure and re-dissolved in chloroform (10 mL) before being filtered through a Celite plug. All volatiles were removed under reduced pressure and the resulting residue triturated with hexane (3 × 20 mL) and 3b collected as a green solid (0.067 g, 93%). Crystals suitable for an X-ray determination were grown by slow diffusion of hexane into a solution of 3b in chloroform at room temperature. 1H NMR (400 MHz, CDCl3): δ 0.90 (d, 3JHH 6.7, 6H, CHMe2), 0.95 (d, 3JHH 6.8, 6H, CHMe2), 2.32 (s, 3H, MeCN), 2.94 (sept, 3JHH 6.8, 2H, CHMe2), 6.40 (ddd, 3JHH 8.1, 3JHH 6.6, 4JHH 1.3, 1H, Ar–H), 6.66–6.73 (m, 3H, Ar–H), 6.84 (dd, 3JHH 8.4, 4JHH 1.2, 1H, Ar–H), 6.88–6.93 (m, 2H, Ar–H), 6.99 (ddd, 3JHH 8.3, 3JHH 6.6, 4JHH 1.6, 1H, Ar–H), 6.99 (d, 3JHH 7.8, 2H, Ar–H), 7.12 (dd, 3JHH 7.7, 3JHH 7.7, 1H, Ar–H), 7.80 (dd, 3JHH 7.5, 4JHH 1.0, 1H, PyH), 7.91 (dd, 3JHH 8.6, 4JHH 1.4, 1H, Ar–H), 8.02 (dd, 3JHH 8.8, 3JHH 7.5, 1H, Py–H), 8.55 (d, 3JHH 8.7, 1H, Py–H). 13C{1H} NMR (100 MHz, CDCl3): δ 19.3 (MeCN), 22.9 (CHMe2), 24.2 (CHMe2), 28.2 (CHMe2), 112.1 (CH), 114.5 (C), 121.3 (C), 122.5 (CH), 122.8 (CH), 123.5 (CH), 125.8 (CH), 126.5 (CH), 127.1 (CH), 130.0 (CH), 130.1 (CH), 134.3 (CH), 135.9 (CH), 139.5 (CH), 142.9 (C), 151.5 (C), 152.4 (C), 153.2 (C), 155.5 (C), 172.1 (Me–CN). IR (cm−1): 1605 (CN)imine, 1571 (CN)pyridine. FABMS: m/z 553 [M + H]+.
Synthesis of [{2-(C6H4-2-NH)-6-(CMeN(4-i-PrC6H4)}Pd(NCMe)][X] (4 and 5)
(a) X = BF4 (4).
A Schlenk flask equipped with stir bar was evacuated and backfilled with nitrogen and loaded with 2a (0.200 g, 0.426 mmol), AgBF4 (0.083 g, 0.426 mmol) and MeCN (20 mL). The reaction mixture was stirred at room temperature for 12 h, at which point the suspension was allowed to settle and the solution transferred by cannula filtration into another Schlenk flask. All volatiles were removed under reduced pressure to afford 4 as a dark green solid (0.233 g, 97%). Mp: >260 °C. 1H NMR (400 MHz, CD3CN): δ 1.21 (d, 3JHH 6.9, 3H, CHMe2), 2.27 (s, 3H, CH3CN), 2.93 (sept, 3JHH 6.9, 1H, CHMe2), 6.60 (dd, 3JHH 7.5, 3JHH 7.5, 1H, Ar–H), 6.98 (d, 3JHH 8.4, 1H, Ar–H), 7.04 (d, 3JHH 7.8, 2H, Ar–H), 7.10 (dd, 3JHH 7.6, 3JHH 7.8, 1H, Ar–H), 7.34 (d, 3JHH 8.0, 2H, Ar–H), 7.83 (d, 3JHH 7.5, 1H, Py–H), 7.89 (d, 3JHH 8.0, 1H, Ar–H), 8.08 (dd, 3JHH 8.0, 3JHH 8.0, 1H, Py–H), 8.53 (d, 3JHH 8.6, 1H, Py–H), the coordinated CH3CN ligand was not observed due to rapid exchange with bulk CD3CN. 13C{1H} NMR (100 MHz, CD3CN): δ 17.0 (CH3CN), 22.9 (CHMe2), 33.3 (CHMe2), 115.3 (CH), 117.0 (C), 119.6 (CH), 122.3 (CH), 124.9 (CH), 126.9 (CH), 127.0 (CH), 129.7 (CH), 131.2 (CH), 136.8 (CH), 142.9 (C), 147.1 (C), 148.8 (C), 149.5 (C), 155.0 (C), 174.8 (CN). 19F{1H} NMR (375 MHz, CD3CN): δ −151 (-BF4). ESIMS (+ve) m/z: 475 [M − BF4]+; ESIMS (−ve): m/z 87 [BF4]−. Anal Calc. for (C24H25N4F4PdB): C, 51.23; H, 4.48; N, 9.96. Found: C, 51.13; H, 4.40; N, 9.87%.
(b) X = O3SCF3 (5).
Employing a similar procedure to that described for 4 using 2a (0.205 g, 0.44 mmol), AgOSO2CF3 (0.112 g, 0.44 mmol) and MeCN (20 mL) gave 5 as a dark green solid (0.259 g, 95%). Crystallisation by slow diffusion of petroleum ether (40–60) into a solution of the salt in acetonitrile–dichloromethane (5:95) gave 5 as a microcrystalline powder. Single crystals of pyridine-containing 5′ suitable for X-ray diffraction could be obtained by slow diffusion of hexane into a chloroform solution of 5 that contained a few drops of pyridine. Complex 5: Mp: >260 °C. 1H NMR (400 MHz, CD3CN): δ 1.32 (d, 3JHH 6.9, 6H, CHMe2), 2.51 (s, 3H, MeCN), 3.05 (sept, 3JHH 6.9, 1H, CHMe2), 6.14 (br, s, 1H, NH), 6.74 (app. t, 3JHH 7.7, 1H, Ar–H), 7.17–7.23 (m, 3H, Ar–H), 7.26–7.29 (m, 1H, Ar–H), 7.48 (d, 3JHH 8.3, 2H, Ar–H), 8.16 (m, 2H, Ar–H), 8.33 (dd, 3JHH, 8.0, 3JHH 8.0, 1H, Py–H), 8.54 (d, 3JHH 8.5, 1H, Py–H), the coordinated CH3CN ligand was not observed due to rapid exchange with bulk CD3CN. 13C{1H} NMR (100 MHz, CD3CN): δ 17.0 (MeCN), 22.9 (CHMe2), 33.3 (CHMe2), 115.4 (CH), 119.2 (C), 119.7 (CH), 122.4 (CH), 124.9 (CH), 126.9 (CH), 129.7 (CH), 131.0 (CH), 136.7 (CH), 142.8 (C), 146.7 (C), 148.8 (C), 149.3 (C), 154.8 (C), 174.7 (MeCN), CF3SO3− not observed. 19F{1H} NMR (375 MHz, CD3CN): δ −79 (O3SCF3). IR (cm−1): 1602 (CN)imine, 1570 (CN)pyridine. ESIMS (+ve): m/z 475 [M − O3SCF3]+; ESIMS (−ve): m/z: 149 [O3SCF3]−. Anal Calc. for (C25H25N4O3F3PdS·CH2Cl2): C, 43.99; H, 3.83; N, 7.89. Found: C, 44.13; H, 3.78; N, 7.60%.
Reaction of 3a with Selectfluor in NMR tube at reduced temperatures
(a) −40 to 0 °C.
Complex 3a (0.005 g, 0.0098 mmol) and Selectfluor™ (0.0035 g, 0.0098 mmol) were loaded into a Young's NMR tube open to the air and then cooled to −100 °C before acetonitrile-d3 was added and the system sealed. The NMR tube was inserted into a 400 MHz NMR spectrometer pre-cooled to −40 °C and the 19F and 1H NMR spectra were recorded at −40 °C and then at 10 °C intervals up to 0 °C. At −40 °C, 1H NMR (400 MHz, CD3CN): δ 1.16 (d, 3JHH 6.8, 6H, CHMe2), 2.40 (s, 3H, MeCN), 2.82 (sept, 3JHH 6.8, 1H, CHMe2), 6.70 (d, 3JHH, 8.5, 2H, Ar–H), 6.72–6.77 (m, 2H, Ar–H), 6.84–6.86 (m, 2H, Ar–H), 7.04 (d, 3JHH 8.4, 2H, Ar–H), 7.36–7.43 (m, 2H, Ar–H), 7.47–7.55 (m, 2H, Ar–H), 7.86 (dd, 3JHH 7.7, 4JHH 1.9, 1H, Ar–H), 8.17 (m, 1H, Ar–H), 8.40 (m, 2H, Ar–H). 19F{1H} NMR (375 MHz, CD3CN): δ −152 (BF4), −172 (HF). At −30 °C, 1H NMR δ no change. 19F{1H} NMR: δ −152 (BF4), −172 (HF). At −20 °C, 1H NMR δ no change. 19F{1H} NMR: δ −152 (BF4), −173 (HF). At −10 °C, 1H NMR δ no change. 19F{1H} NMR: δ −152 (BF4), −173 (HF). At 0 °C: 1H NMR δ no change. 19F{1H} NMR: δ −152 (8F, BF4), −174 (1F, HF).
(b) 0 °C to room temperature.
The reaction mixture prepared in (a) was further warmed to room temperature and the 1H NMR spectrum recorded periodically. After 48 h, complete conversion to 4 (data as reported for 4 above) and biphenyl was observed. 19F{1H} NMR δ −152 (BF4), −181 (HF).
Crystallographic studies
Data for HL1b, 1a, 1b, 2a, 2b, 3a, 3b and 5′ were collected on a Bruker APEX 2000 CCD diffractometer. Details of data collection, refinement and crystal data are listed in Table 6. The data were corrected for Lorentz and polarisation effects and empirical absorption corrections applied. Structure solution by direct methods and structure refinement based on full-matrix least-squares on F2 employed SHELXTL version 6.10.21 Hydrogen atoms were included in calculated positions (C–H = 0.96–1.00 Å) riding on the bonded atom with isotropic displacement parameters set to 1.5 Ueq(C) for methyl H atoms and 1.2 Ueq(C) for all other H atoms. All non-H atoms were refined with anisotropic displacement parameters.
Table 6 Crystallographic and data processing parameters for HL1b, 1a, 1b, 2a, 2b, 3a, 3b and 5′a
| Complex |
HL1b |
1a
|
1b
|
2a
|
| Formula |
C25H29N3 |
C24H25Cl3N3O2Pd·CHCl3·OH2 |
C28H32Cl3N3O2Pd |
C22H22ClN3Pd |
|
M
|
371.51 |
631.25 |
655.32 |
470.28 |
| Crystal size (mm3) |
0.24 × 0.17 × 0.12 |
0.27 × 0.05 × 0.04 |
0.27 × 0.17 × 0.11 |
0.23 × 0.20 × 0.04 |
| Temperature (K) |
150(2) |
150(2) |
150(2) |
150(2) |
| Crystal system |
Orthorhombic |
Triclinic |
Triclinic |
Monoclinic |
| Space group |
Pbca
|
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P |
P2(1)/n |
|
a (Å) |
12.906(10) |
6.9770(16) |
14.194(4) |
17.674(4) |
|
b (Å) |
8.308(6) |
12.184(3) |
15.316(4) |
8.9111(19) |
|
c (Å) |
39.43(3) |
15.543(4) |
15.711(4) |
24.267(5) |
|
α (°) |
90 |
87.923(4) |
61.002(5) |
90 |
|
β (°) |
90 |
84.029(5) |
77.050(7) |
93.133(5) |
|
γ (°) |
90 |
84.203(5) |
80.162(7) |
90 |
|
U (Å3) |
4228(5) |
1307.0(5) |
2903.6(14) |
3816.2(14) |
|
Z
|
8 |
2 |
4 |
8 |
|
D
c (Mg m−3) |
1.167 |
1.604 |
1.499 |
1.637 |
|
F(000) |
1600 |
640 |
1336 |
1904 |
|
μ(Mo-Kα)(mm−1) |
0.069 |
1.049 |
0.945 |
1.124 |
| Reflections collected |
28600 |
10332 |
22854 |
28912 |
| Independent reflections |
3715 |
5074 |
11293 |
7469 |
|
R
int
|
0.2201 |
0.1059 |
0.1479 |
0.1336 |
| Restraints/parameters |
0/258 |
20/319 |
0/679 |
1/493 |
| Final R indices (I > 2σ(I)) |
R
1 = 0.0628 |
R
1 = 0.0683 |
R
1 = 0.0873 |
R
1 = 0.0582 |
| wR2 = 0.1155 |
wR2 = 0.1067 |
wR2 = 0.1788 |
wR2 = 0.0914 |
| All data |
R
1 = 0.1397 |
R
1 = 0.1235 |
R
1 = 0.1888 |
R
1 = 0.1237 |
| wR2 = 0.1377 |
wR2 = 0.1232 |
wR2 = 0.2173 |
wR2 = 0.1078 |
| Goodness of fit on F2 (all data) |
0.871 |
0.861 |
0.848 |
0.890 |
| Complex |
2b
|
3a
|
3b
|
5′
|
|
Data in common: graphite-monochromated Mo-Kα radiation, λ = 0.71073 Å; R1 = ∑||Fo| − |Fc||/∑|Fo|, wR2 = [∑w(Fo2 − Fc2)2/∑w(Fo2)2]1/2, w−1 = [σ2(Fo)2 + (aP)2], P = [max(Fo2, 0) + 2(Fc2)]/3, where a is a constant adjusted by the program; goodness of fit = [∑(Fo2 − Fc2)2/(n − p)]1/2 where n is the number of reflections and p the number of parameters.
|
| Formula |
C26H29Cl4N3Pd |
C28H27N3Pd |
C31H33N3Pd |
C28H26F3N4 O3PdS·2CHCl3 |
|
M
|
631.72 |
511.93 |
554.00 |
900.72 |
| Crystal size (mm3) |
0.36 × 0.31 × 0.04 |
0.26 × 0.12 × 0.05 |
0.25 × 0.22 × 0.10 |
0.46 × 0.14 × 0.04 |
| Temperature (K) |
150(2) |
150(2) |
150(2) |
150(2) |
| Crystal system |
Monoclinic |
Monoclinic |
Orthorhombic |
Triclinic |
| Space group |
P2(1)/n |
P2(1)/c |
Pbca
|
P |
|
a (Å) |
11.410(8) |
17.244(11) |
12.118(3) |
14.155(7) |
|
b (Å) |
17.663(13) |
11.142(8) |
10.844(3) |
16.226(8) |
|
c (Å) |
12.991(9) |
12.238(8) |
40.250(10) |
17.503(8) |
|
α (°) |
90 |
90 |
90 |
89.109(9) |
|
β (°) |
94.182(13) |
105.925(14) |
90 |
76.364(11) |
|
γ (°) |
90 |
90 |
90 |
70.267(10) |
|
U (Å3) |
2611(3) |
2261(3) |
5289(2) |
3668(3) |
|
Z
|
4 |
4 |
8 |
4 |
|
D
c (Mg m−3) |
1.607 |
1.504 |
1.391 |
1.631 |
|
F(000) |
1280 |
1048 |
2288 |
1804 |
|
μ(Mo-Kα)(mm−1) |
1.141 |
0.842 |
1.391 |
1.053 |
| Reflections collected |
19939 |
17292 |
41613 |
26756 |
| Independent reflections |
5116 |
4444 |
5763 |
12826 |
|
R
int
|
0.0775 |
0.2311 |
0.0699 |
0.2649 |
| Restraints/parameters |
0/312 |
0/292 |
0/321 |
749/889 |
| Final R indices (I > 2σ(I)) |
R
1 = 0.0446 |
R
1 = 0.0732 |
R
1 = 0.0431 |
R
1 = 0.1055 |
| wR2 = 0.0904 |
wR2 = 0.1210 |
wR2 = 0.0906 |
wR2 = 0.2112 |
| All data |
R
1 = 0.0615 |
R
1 = 0.1615 |
R
1 = 0.0569 |
R
1 = 0.2711 |
| wR2 = 0.0955 |
wR2 = 0.1450 |
wR2 = 0.0955 |
wR2 = 0.2821 |
| Goodness of fit on F2 (all data) |
1.044 |
0.867 |
1.122 |
0.829 |
Acknowledgements
We thank the University of Leicester for financial assistance. Johnson Matthey PLC are thanked for their generous loan of palladium salts.
References
- For reviews, see
(a) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed;
(b) L. M. Mirica and J. R. Khusnutdinova, Coord. Chem. Rev., 2013, 257, 299–314 CrossRef CAS;
(c) D. C. Powers and T. Ritter, Top. Organomet. Chem., 2011, 503, 129–156 CrossRef PubMed.
-
(a) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed;
(b) F. Tang, F. Qu, J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, Dalton Trans., 2012, 41, 14046–14050 RSC;
(c) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2010, 132, 7303–7305 CrossRef CAS PubMed;
(d) J. R. Khusnutdinova, N. P. Rath and L. M. Mirica, J. Am. Chem. Soc., 2012, 134, 2414–2422 CrossRef CAS PubMed;
(e) A. J. Canty, A. Ariafard, M. S. Sanford and B. F. Yates, Organometallics, 2013, 32, 544–555 CrossRef CAS;
(f) M. D. Lotz, M. S. Remy, D. B. Lao, A. Ariafard, B. F. Yates, A. J. Canty, J. M. Mayer and M. L. Sanford, J. Am. Chem. Soc., 2014, 136, 8237–8242 CrossRef CAS PubMed.
-
(a) J. R. Brandt, E. Lee, G. B. Boursalian and T. Ritter, Chem. Sci., 2014, 5, 169–179 RSC;
(b) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed;
(c) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796–3797 CrossRef CAS PubMed;
(d) X. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 7520–7521 CrossRef CAS PubMed;
(e) K. M. Engle, T.-S. Mei, X. Wang and J.-Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478–1491 CrossRef CAS PubMed.
-
(a) T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060–10061 CrossRef CAS PubMed;
(b) T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. Tamg, W. A. Goddard III and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793–3807 CrossRef CAS PubMed;
(c) T. Furuya, H. M. Kaiser and T. Ritter, Angew. Chem., Int. Ed., 2008, 47, 5993–5996 CrossRef CAS PubMed;
(d) E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed.
- N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796–3797 CrossRef CAS PubMed.
- A. R. Mazzotti, M. G. Campbell, P. Tang, J. M. Murphy and T. Ritter, J. Am. Chem. Soc., 2013, 135, 14012–14015 CrossRef CAS PubMed.
-
(a) M. G. Campbell, S.-L. Zheng and T. Ritter, Inorg. Chem., 2013, 52, 13295–13297 CrossRef CAS PubMed;
(b) M. C. Campbell, D. C. Powers, J. Raynaud, M. J. Graham, P. Xie, E. Lee and T. Ritter, Nat. Chem., 2011, 473, 470–477 CrossRef PubMed.
-
(a) V. V. Grushin and W. J. Marshall, J. Am. Chem. Soc., 2006, 128, 4632–4641 CrossRef CAS PubMed;
(b) D. J. Cardenas, B. Martin-Matute and A. M. Echavarren, J. Am. Chem. Soc., 2006, 128, 5033–5040 CrossRef CAS PubMed.
- H. B. Kratz, M. E. van der Boom, Y. Ben-David and D. Milstein, Isr. J. Chem., 2001, 41, 163–171 CrossRef.
- While the N,N,N pincer framework is anticipated to be inert, the possibility of a Caryl–Nanilide coupling reaction involving the anilide moiety of the N,N,N ligand represents an alternative pathway. For Csp3–N coupling via high valent Pd, see for example:
(a) S. R. Whitfield and M. S. Sanford, J. Am. Chem. Soc., 2007, 129, 15142–15143 CrossRef CAS PubMed;
(b) A. Iglesias and K. Muñiz, Helv. Chim. Acta, 2012, 95, 2007–2025 CrossRef CAS;
(c) M. H. Perez-Temprano, J. M. Racowski, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2014, 136, 4097–4100 CrossRef CAS PubMed;
(d) T.-S. Mai, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806–10807 CrossRef PubMed;
(e) P. A. Sibbald and F. E. Michael, Org. Lett., 2009, 11, 1147–1149 CrossRef CAS PubMed;
(f) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945–15951 CrossRef CAS PubMed;
(g) A. Iglesias, R. Alvarez, A. R. de Lera and K. Muiz, Angew. Chem., Int. Ed., 2012, 51, 2225–2228 CrossRef CAS PubMed.
- N. Sakai, K. Annaka and T. Konakahara, J. Org. Chem., 2006, 71, 3653–3655 CrossRef CAS PubMed.
- L. A. Wright, E. G. Hope, G. A. Solan, W. B. Cross and K. Singh, Dalton Trans., 2015 10.1039/c5dt00062a , Advance Article.
-
(a) M. Lopez-Torres, P. Juanatey, J. J. Fernandez, A. Fernandez, A. Suarez, D. Vazquez-Garcia and J. M. Vila, Polyhedron, 2002, 21, 2063–2069 CrossRef CAS;
(b) H. Onoue and I. Moritani, J. Organomet. Chem., 1972, 43, 431–436 CrossRef CAS;
(c) H. Onoue, K. Minami and K. Nakagawa, Bull. Chem. Soc. Jpn., 1970, 43, 3480–3485 CrossRef CAS.
- O. Adeyi, W. B. Cross, G. Forrest, L. Godfrey, E. G. Hope, A. McLeod, A. Singh, K. Singh, G. A. Solan, Y. Wang and L. A. Wright, Dalton Trans., 2013, 42, 7710–7723 RSC.
- W. B. Cross, E. G. Hope, G. Forrest, K. Singh and G. A. Solan, Polyhedron, 2013, 59, 124–132 CrossRef CAS.
-
K. Nakamoto, IR and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 5th edn, 1997, Part B, p. 271 Search PubMed.
- Characteristic HF signal in CD3CN see: M. M. Shaw, R. G. Smith and C. A. Ramsden, ARKIVOC, 2011, 221–228 CAS.
-
W. L. F. Armarego and D. D. Perrin, Purification of Laboratory Chemicals, Butterworth Heinemann, 4th edn., 1996 Search PubMed.
- Y. D. M. Champouret, R. K. Chaggar, I. Dadhiwala, J. Fawcett and G. A. Solan, Tetrahedron, 2006, 62, 79–89 CrossRef CAS.
-
(a) P. Fitton and E. A. Rick, J. Organomet. Chem., 1971, 28, 287–291 CrossRef CAS;
(b) W. R. Moser, A. W. Wang and N. K. Kildahl, J. Am. Chem. Soc., 1988, 110, 2816–2820 CrossRef CAS.
-
G. M. Sheldrick, SHELXTL Version 6.10, Bruker AXS, Inc. Madison, Wisconsin, USA, 2000 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1043191–1043198. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00216h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a