
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamic structure elucidation of chemical reactivity by laser pulses and X-ray probes†
Stuart A.
Bartlett
ab,
Michelle L.
Hamilton
acd and
John
Evans
*aef
aResearch Complex at Harwell, Rutherford Appleton Laboratory, Didcot, OX11 0FA, UK
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK. E-mail: stuart.bartlett@rc-harwell.ac.uk
cSchool of Chemistry, University of Nottingham, Nottingham, NG7 2RD, UK. E-mail: michelle.hamilton@rc-harwell.ac.uk
dRAL Space, Rutherford Appleton Laboratory, Didcot, OX11 0QX, UK
eChemistry, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: je@soton.ac.uk
fDiamond Light Source, Rutherford Appleton Laboratory, Didcot, OX11 0DE, UK. E-mail: john.evans@diamond.ac.uk
First published on 25th February 2015
Abstract
Visualising chemical reactions by X-ray methods is a tantalising prospect. New light sources provide the prospect for studying atomic, electronic and energy transfers accompanying chemical change by X-ray spectroscopy and inelastic scattering. Here we assess how this adventure can illuminate inorganic and catalytic chemistry. In particular X-ray inelastic scattering provides a means of exploiting X-ray free electron lasers, as a parallel to laser Raman spectroscopy.
1. Introduction
Visualising chemical reactions is indeed a tantalising prospect. An excellent illustration of this is the light-induced insulator-metal transition of the organic salt, (EDO-TTF)2PF6, (EDO-TFF = ethylenedioxytetrathiafulvalene) investigated using an ultrabright femtosecond electron source on a crystalline sample.1 An organic metal was formed after a thin crystal of the organic salt was illuminated with 800 nm near infra-red light. The electron diffraction pattern of the insulating phase changed showing that some unit cells of the conducting structure phase were formed within 10–15 ps, and evidence found for the atomic displacements occurring in the first few ps.Deriving similar type of information about the primary steps of the reactions or electron movement within inorganic complexes, compounds and materials would revolutionise our understanding of this field of chemistry. Some years ago, we reviewed the developing opportunities for the use of time resolved X-ray absorption fine structure (XAFS) spectroscopy for structure-reactivity studies in chemistry.2 (XAFS spectra consist of XANES, X-ray absorption near edge structure, and EXAFS, extended X-ray absorption fine structure, regions.) Since then projected developments in X-ray sources3 are in the process of being realised and these are transforming the scope of in situ structure elucidation. In this article we assess what is now possible and what may become feasible over the next few years.
Over the last 100 years, since the report by Bragg,4 X-ray diffraction has been pivotal in developing our understanding of chemical structure. It remains the database of reference structures upon which reaction studies in non-crystalline media are calibrated. In this article we focus on investigating the structures of solutions using X-ray absorption fine structure (XAFS) spectroscopy, and techniques derived from it, as the probe.
2. Structure determination using XAFS
The first observations of an element's X-ray absorption edge by de Broglie5 and the correlation of the energies of X-ray emissions to the Periodic Table by Moseley6 were almost coincident with that of diffraction from crystals.4 Chemical shifts in absorption edge energies were established within a few years, and near-edge structure reported in 1920 by Fricke.7 Over many years, there was uncertainty as to whether XAFS was due to long range order, as for diffraction, or due to local effects. This was resolved by Stern in 1974 as being a local effect,8 thus providing a new structural technique applicable to disordered materials, including solutions, glasses and catalyst surfaces. Once synchrotron radiation provided X-ray sources of the required intensity (generally to get the signal/noise ratio of 105, at least 1010 monochromatic photons are required to impinge on the sampled area), XAFS could then be applied to a range of chemical problems. This requirement is illustrated in Fig. 1. Relative to the start of the EXAFS region, which is visible in the XAFS spectrum, the end of the EXAFS region is weighted by a factor of over 700. With current, third generation sources, such a photon count can be achieved in a focussed (sub mm) spot in the order of 10 ms, and for an X-ray free electron laser, in a single pulse of 20–100 fs. Spectra like this (Fig. 1a), with a 1600 eV range, can be acquired in seconds or minutes.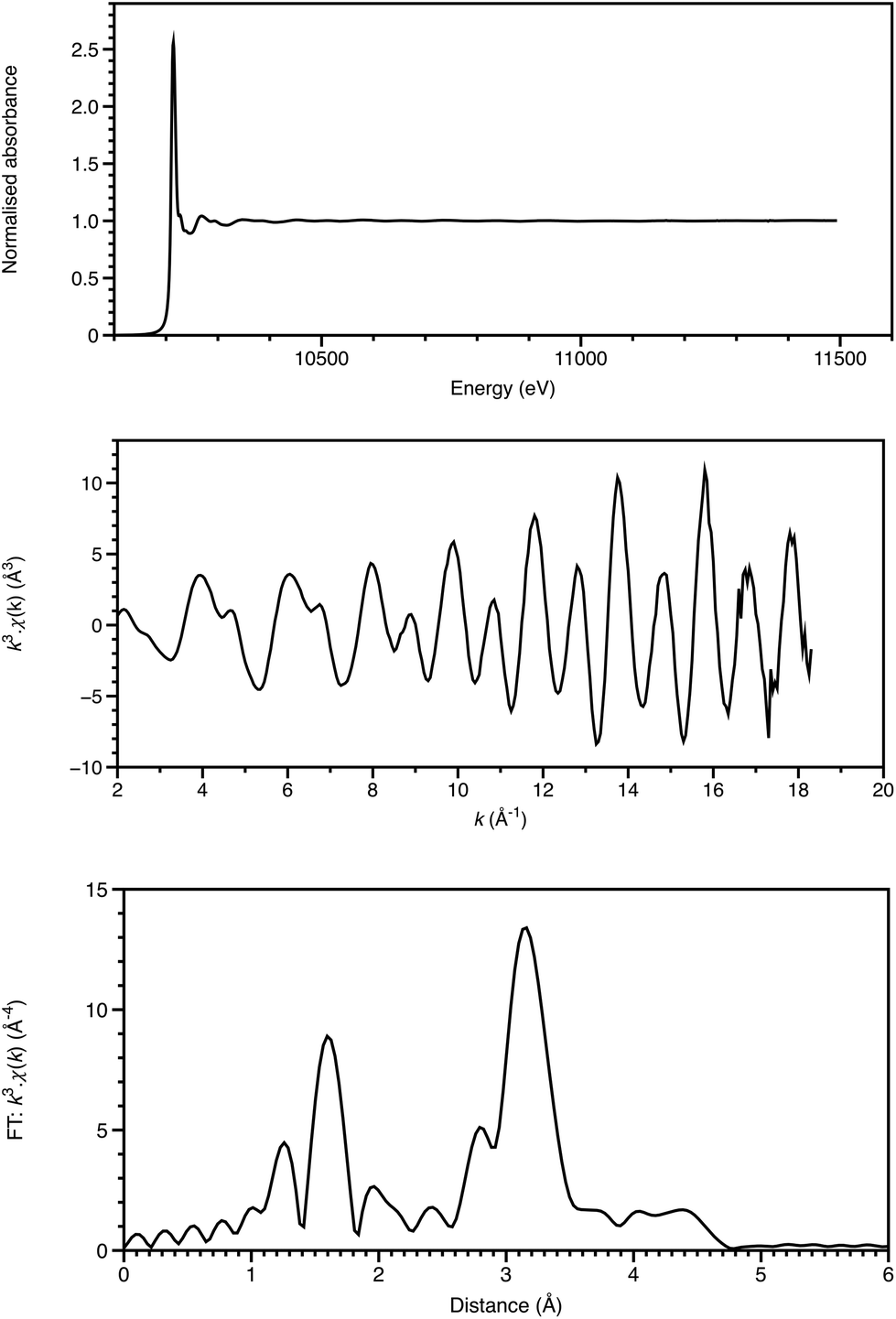 | ||
| Fig. 1 W LIII-edge XAFS of (NBu4)2[W6O19] showing the normalised absorption spectrum (top), the k3-weighted EXAFS (middle) and the Fourier transform (bottom). Data recorded on B18 of the Diamond Light Source. | ||
Analysis of the extended X-ray absorption fine structure (EXAFS) is well established,9 and, depending upon the system, may provide structural detail within 3–5 Å of the absorbing atom (Fig. 1). Single-scattering modelling affords interatomic distances from the absorber, but some bond-angle information is intrinsic to more sophisticated analyses including multiple scattering pathways. This is often essential for a proper analysis, and provides a local cluster structural model.
The features of X-ray absorption near edge structure (XANES) are very sensitive to the coordination geometry, ligand type and spin state of the absorbing atom. Thus they probe the effects of the coordination site on the electronic properties around the absorbing atom. The discrimination of XANES has now been enhanced by utilisation of crystal analysers for X-ray emission spectroscopy (XES) in the following ways (Fig. 2).
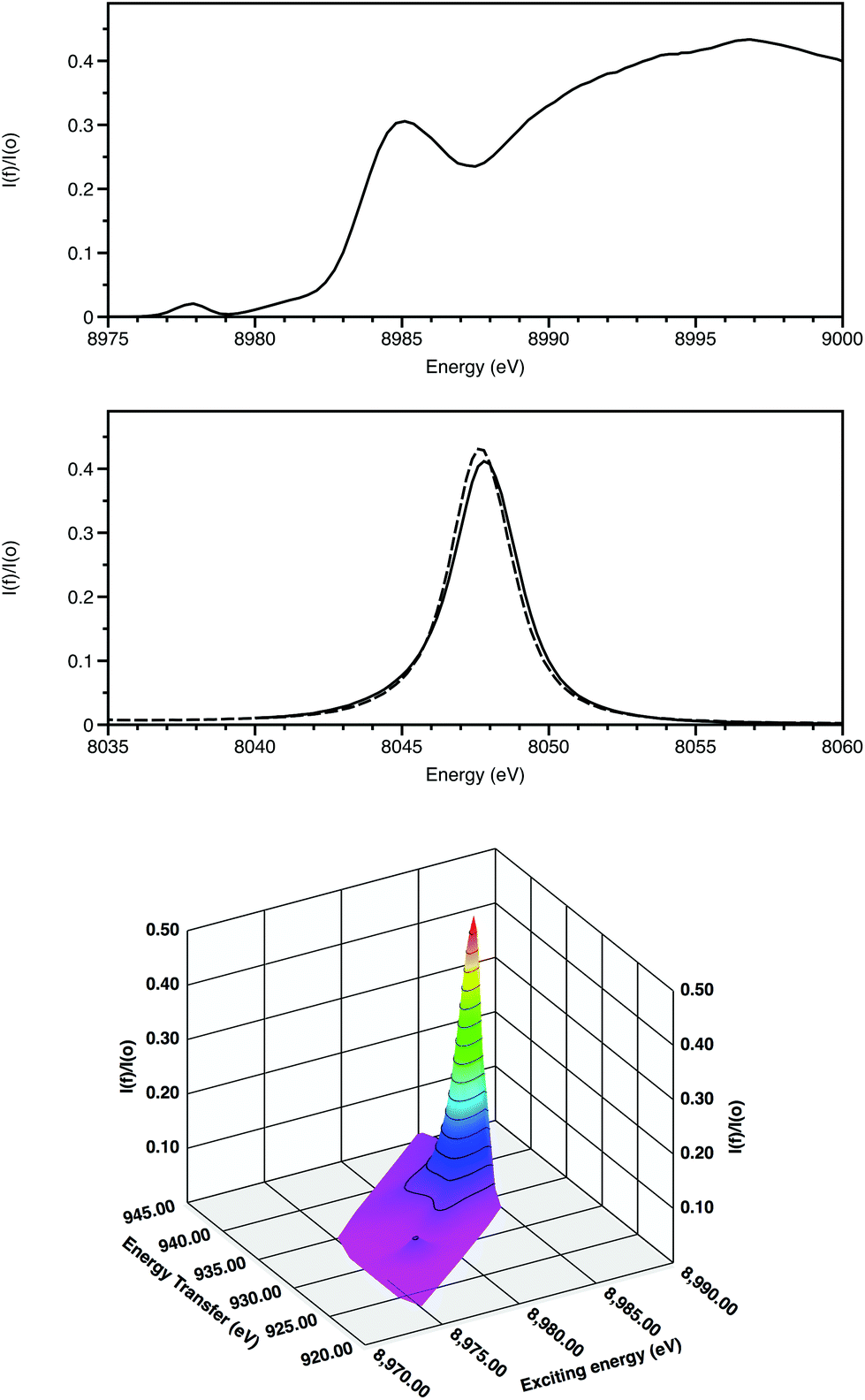 | ||
| Fig. 2 Cu K-edge XANES of CuO showing the HERFD spectrum (top), Kα1 emission spectra of CuO (solid) and Cu (dashed) with the exciting energy above the absorption edge (middle) and Kα1 RIXS (bottom). Energy transfer = Exciting energy – emission energy. Data recorded on I20 of the Diamond Light Source. | ||
(i) High energy-resolution fluorescence detection (HERFD) resolves features within the XANES.10 Firstly this is achieved by monitoring a single channel rather than multiple overlapping ones and, secondly, when following an inelastic scattering process, the core hole lifetime broadening of relevance for Kα is for 2p rather than 1s (0.6 rather than 1.6 eV).
(ii) 2-dimensional maps of absorption and emission features, termed Resonant Inelastic X-ray scattering (RIXS provide greater chemical speciation.11 Here (Fig. 2) the weak 3d Kα1 resonance is clearly separated from the higher energy dipole-allowed transitions to 4p and also to the continuum. The excited state after the RIXS process for CuO has a configuration 2p53d10. Splitting due to spin–orbit coupling is only within the incomplete 2p sub-shell. As a result, this resonance is a doublet, with the second component associated with the Kα2 emission line displaced from the Kα1 features by 20 eV.
Full multiple scattering codes12,13 and density functional methods14,15 can, in many cases, provide a good basis for modelling XANES,16,17 XES17,18 and RIXS.19 Theoretical structure modelling and spectrum simulation are now closely linked,20 and, together with EXAFS structural analysis, afford an iteration cycle.18,21,22 Hence this combination of X-ray spectroscopy, analysis, and theoretical modelling forms a substantial basis for probing chemical reactivity and providing greater understanding of the electronic structure of the absorbing element.
3. Kinetics and dynamics of reactions
The rates of reactions of inorganic systems span a vast timescale. Even restricting the survey to water exchange rates of hexaaqua complexes for divalent 3d metal ions, still provides a range of rate constants between 87 s−1 (VII) to 4.4 × 109 s−1 (CuII).23 Molecular dynamics calculations of water exchange on the 3d divalent ions between Mn and Zn afforded values of ΔG‡ between 17.9 (Mn) and 32.4 (Ni) kJ mol−1. The predicted kexp rates were overestimated, due in part to transmission coefficient, κ, considered as being equal to unity through the transition state.24 In a recent molecular dynamics study of reactions at alkali metal ions, the residence time of water coordinated to Li+ was calculated to be 191 ps, with a ΔG‡ of 17.0 kJ mol−1, giving rise to a rate of transition state formation at 2.38 × 10−10 s, and a transmission coefficient of 0.22.25 In general, metal complexes and clusters will exhibit metal ligand vibrations between ∼300 and 400 cm−1, and metal–metal stretches between 100 and 200 cm−1. The frequencies of these vibrations lie between 3 and 12 × 1012 s−1, giving a time constant range of ∼80–350 fs.These values parameterise the time frames for visualising fast chemical reactions in solution. Species may have lifetimes of the order of ps and longer, and the atomic motion (dynamics) of the transitions between species may be 1000 times faster. The transition time of X-ray absorption is much faster than either of these timescales and thus can afford snapshots. Careful experimental design is required to prepare a significant population of the sample molecules to perform the same transition simultaneously.
4. X-ray spectroscopic structural studies
4.1. Beyond steady-state studies
A general factor is that the acquisition time of a XAFS spectrum for a dilute sample may be of the order of hours, and thus be too slow for the time-resolved studies. This was addressed in the XAFS study of the palladium catalysed Mizoroki–Heck coupling reaction with a pyridine functionalized N-heterocyclic carbene ligand. The pre-catalyst was first formed in situ, and the reactants, butyl acrylate and p-bromoacetophenone, injected at the reaction temperature, allowing stages of the reaction to be investigated by rapid cooling and reheating after XAFS measurement.26 Such a method could identify the major species present despite long acquisition times, but without a forced cooling process, the time resolution was minutes.This approach was developed further when investigating Mo halide complexes for ethylene polymerisation by using stopped-flow instrumentation with a mixing time of ms, and a quench time to liquid nitrogen temperature of ∼1 s.27 The activation and subsequent deactivation mechanism was determined by XAFS analysis showing that the Mo catalyst was firstly methylated by the AlMe3 co-catalyst and subsequently deactivated by Mo particle formation. The same XAFS freeze-quench technique was used for Sc ethylene polymerization catalysis, where Sc was initially methylated by AlMe3, the Sc-Me then abstracted by a Lewis acid with subsequent coordination of an olefin to Sc centre.28 Most recently, probing the active ethylene trimerisation catalyst by the stopped-flow freeze-quench method allowed identification of an unusual 4-coordinate Cr(II) complex [(SNS)CrIICl] {SNS = N(CH2CH2S-Decyl)2−} from the reaction of [CrCl3{SN(H)S}], with AlMe3.29 Without this trapping procedure, the Cr(II) intermediate would not have been observed, since a slower disproportionation process partially reformed Cr(III). The rapid-mixing (ms) and quenching (s) however, still allowed a sequence of reactions to transpire before the trapped species has formed. In this case, it is thought to be N–H deprotonation, and conjugate-base assistance of chloride loss, transmetalation of a methyl group and a bimolecular reductive elimination of ethane.
4.2. Combining techniques
Adding information from complementary technique can be invaluable in differentiating between similar species. Incorporation of UV-visible spectroscopy with stopped-flow XAFS methods, as utilised in the activation of the ethene oligomerisation techniques,27 is relatively straightforward. Indeed the time resolution properties of energy dispersive XAFS closely match those of a diode-array UV-visible spectrometer providing ms time resolution. In a study of the inner-sphere electron transfer between [IrCl6]2− and [Co(CN)5]3−, the UV-visible data indicated that the reaction studied over the a few seconds by Ir LIII energy-dispersive XAFS was the breakup of the of the bridged intermediate, [Cl5Ir-Cl-Co(CN)5]5−, following the electron transfer.30With off-line NMR and EPR studies, the combination of stopped flow XAFS and UV-visible spectroscopies allowed insight into the catalytic cycle for the arylation of imidazole by phenylboronic acid, catalysed by [Cu(OH)(TMEDA)]2Cl2 (TMEDA = tetramethylethylenediamine).31 A proposed cycle could be tested by binary and ternary mixtures of the reagents. It was evident that imidazole (imidH) formed a new, 4-coordinate mononuclear Cu(II) complex of the imid− anion, [CuCl(imid)(TMEDA)], and addition of PhB(OH)2 resulted in the observation of XANES attributable to Cu(I) centres after the arylation of the imidazole. Further PhB(OH)2 was required to reoxidise the copper to complete the cycle; all of the stages could be followed with second time resolution. A key Cu(III) intermediate, [CuX(Ph)(imid)(TMEDA)] could not be observed on this timescale.
4.3. Pump–probe XAFS
All the experiments described so far utilise the storage ring source as pseudo-continuous, rather than their true nature of a train of electron pulses, often of ∼100 ps duration, separated by a few ns, rotating every few μs. Providing that synchronisation can be achieved with low jitter, and the output from a particular bunch detected without distortion from its neighbours, then the time resolution can be improved from 10−3 s to 10−10 s. The appropriate sample preparation method is then a laser pulse, using the storage ring pulse as an X-ray probe.Chen and Coppens provided an exemplar for pump–probe XAFS in their study on the 3MLCT (MLCT = metal-to-ligand charge transfer) state of [Cu(2,9-Me2-phen)2]+, using a differential acquisition mode (X-ray transient absorption spectroscopy, XTA) to optimise the signal to background ratio.32 An example of this is shown in Fig. 3. The Cu K-edge XANES of the excited state was found to be very similar to the electrochemically generated [Cu(2,9-Me2-phen)2]2+, strongly supporting the assignment of this as being a MLCT state. In the ground state CuI has a d10 configuration with a slightly distorted tetrahedral geometry; in the MLCT CuII* complex, d9 centre is susceptible to Jahn–Teller distortion resulting in the excited state having a more flattened tetrahedral geometry (inter-ligand dihedral angle ∼57°). With these relatively small substituents adjacent to the coordinating nitrogen atoms, there was some evidence from the XANES simulations for the coordination of a molecule of the donor solvent, acetonitrile in the excited state. In our study of [Cu(2,9-nBu-3,4,7,8-Me-phen)2]+,33 which was performed using an energy dispersive X-ray spectrometer in an attempt to reduce the real time of the data acquisition, the XANES simulations favoured a 60° angle and no MeCN binding in an exciplex. Having tBu groups on the two carbon atoms adjacent to the nitrogens, in [Cu(2,9-tBu-phen)2]+ was found to essentially lock the coordination geometry as being pseudotetrahedral,34 and this is a likely cause of the long lifetime of the 3MLCT state.
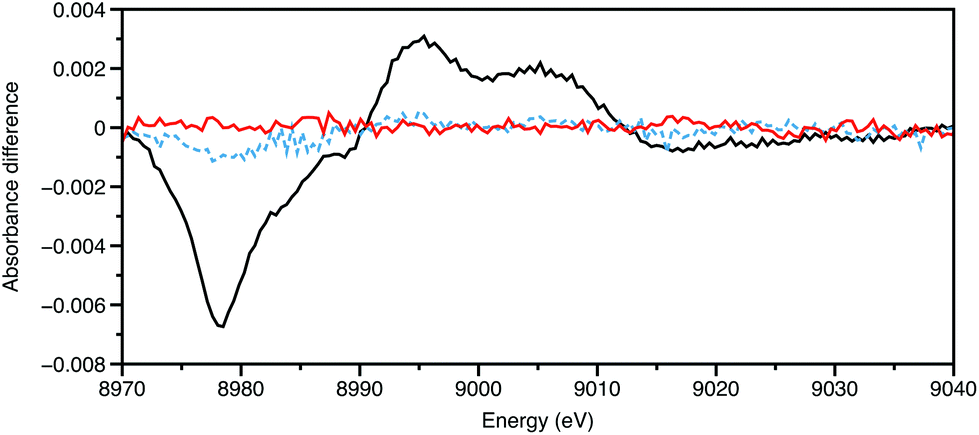 | ||
| Fig. 3 Cu K-edge XTA of [Cu(2,9-Me2-phen)2]+ in MeCN (20 mM) showing the data 5 ns before laser pulse (450 nm) (red), during the pulse (black), and 5 ns after (blue). Data recorded on ID24 of the ESRF. | ||
The majority of synchrotron storage ring sources provide an X-ray pulse of 100 ps, and this marks the normal highest time resolution of such a source. Chen and co workers35 have studied Ni(II)(tetramesitylporphyrin) by photoactivated XTA. This d8 complex has a distorted square-planar geometry with a vacant 3dx2−y2 orbital. Upon photoexcitation, a one electron transition from the ligand π to a π* orbital on the porphyrin with a subsequent transfer from the π* to the empty 3dx2−y2, and there is an associated transfer of an electron from the 3dz2 back into the empty hole in the π orbital, all within 20 ps of the excitation.36 This excited state has a lifetime of ∼200 ps and was observed by Ni K-edge XAFS within 100 ps. The electron transfer resulted in longer Ni–N and Ni–Cα distances (0.08 Å) in the excited state. The transfers generated a singly occupied 3dz2 orbital, shown to promote the binding of an axial ligand that was followed by the 100 ps X-ray pulse structure.
Photo-excited myoglobin studies have also been undertaken using fast XAFS.36 This class of biological compounds have many uses including electron transfer, catalytic oxidation and reduction, and for binding of diatomics such as O2, NO and CO for transport and signalling.37 Chergui has reported a detailed study of myoglobin complexes in physiological conditions,38 and demonstrated that Fe K-edge XANES of an excited state of the protein could be recorded on a 2 mM solution.39 In further XTA studies, Chen40 collected XAFS of the CO derivative every 100 ps, which exhibited a significant change in the Fe K-edge XANES. This showed that photo-dissociation was complete within the first 100 ps and lasted for at least 47 μs. The EXAFS analysis revealed the complex exhibited a square pyramidal geometry, where the Fe–Npor bonds were extended by 0.06 Å.
5. Ultrafast X-ray techniques and dynamics
The results from storage ring pump–probe XAFS have given considerable structural insights on the events occurring after 100 ps of the exciting pulse; in this time regime, they augment optical methods considerably. However, these sources in their standard form cannot serve in the same way for the primary photo-events. Alternatives are available,3 including operation of the storage ring specifically to deliver narrow pulses with low current (low alpha mode), which may afford sub 10 ps time resolution. However, to move from kinetic to dynamic studies, the time resolution will need to be sub 100 fs. This can be achieved either by deflecting a thin slice of an electron bunch with a fs laser or from an X-ray electron laser (X-FEL).41The very low X-ray fluxes from sliced sources renders all experiments to be immensely challenging.41 Nevertheless, some intriguing results have emerged from them. An example is the changes in structure around iodide in aqueous solution following excitation with visible light (400 nm), for which the earliest species observable was a I0(H2O) complex within the solvent cage.42
The opposite circumstance arises from an X-FEL, namely damaging X-ray fluxes (∼1012 photons per pulse) with shot-to-shot intensity variations. The bandwidth of the X-ray source is provided by undulator radiation, which is of the order of 50 eV at 10 keV, thus presently excluding EXAFS structure analysis. At the LCLS facility at Stanford, using a liquid jet, ∼190 fs-resolved Fe K-edge XANES (∼40 eV) using a double-crystal monochromator were recorded on an aqueous solution of [Fe(bipy)3]2+ (50 mM, bipy = 2,2′bipyridine).43 Due to the state-dependence of the XANES features, the time constant for the formation of the 5T2 high spin excited state could be estimated at ∼160 fs.
An alternative approach then to acquire more extensive X-ray spectroscopic information has its parallels in the optical region with Raman spectroscopy. A jet of glycerol-water has been used to provide a medium for studying Mn complexes by energy-dispersive (von Hamos) X-ray emission spectroscopy which is adapted well for shot-to-shot variations in the X-ray properties.44 An oxidation state dependence was evident in the Kβ emission, and this technique was later applied to investigating the oxidation state changes within the Mn4CaO5 cluster of photosystem II.45 Even after a sequence of laser pulses, no evidence for a MnV site was apparent up to 250 μs after the pulses, instead occupying states between MnIV4 and MnIII3MnIV. The diffraction component of this report revealed an absence of O–O bond formation over that 250 μs period too.
Inelastic scattering approach has progressed a stage further at the soft X-ray laser FLASH (Hamburg), which allowed a study of the photochemistry of Fe(CO)5 using the Fe LIII absorption edge.46 The emission spectrometer was used to record RIXS spectra at very high time resolution, such that a feature could be distinguished from those due to the solvated Fe(CO)5 itself and assigned to Fe(CO)4. Using that, the build up of the coordinatively unsaturated intermediate could indeed be monitored.
An alternative use of the dispersive emission spectrometer is to probe the unoccupied states (like XANES) by high-energy resolution off-resonant spectroscopy (HEROS).47 This uses a single probe energy, which is set lower (20–30 eV) than that of the absorption edge and so has anti-Stokes characteristics. It is also complementary to XES, for which the probe energy is set within the XANES region and which probes occupied states. This approach has been tested at LCLS, using Cu and CuO as samples, and has given promising results.48
6. Conclusions
The developments in light sources, X-ray spectroscopic measurements and theory have been very considerable over the last decade. On a synchrotron storage ring source now, there is a clear capability for investigating secondary processes after an initial excitation pulse. These can include fluorescence behaviour, but also thermal reactions from photo-induced transients. Much greater chemical speciation is feasible now, due, in large measure, to the development of emission spectroscopy and inelastic scattering techniques. Examining the primary light-induced processes remains a considerable challenge, but recent results with the first X-ray free electron lasers show that this too is becoming viable. The use of X-ray pulses (∼20 fs) of shorter duration than atomic motion allows X-ray data to be acquired before the coulombic explosion that destroys the irradiated region of the sample, and thus X-ray diffraction is viable as a single shot procedure. That timescale, however, still allows multiphoton events. Some, like multiple ionisation of core orbitals may be problematical for sampling, as observed for H2S.49 However, others point to new technique opportunities involving nonlinear processes.50 A two-photon absorption, using 5.6 keV radiation was used to pump above the K-edge of germanium (11.103 keV), and the event was detected by measuring the Kα1 and Kα2 emissions. One may envisage two-colour versions that might provide inter-element correlations of excited states. In this second century of X-ray science, new classes of experiments are opening up. The challenge for the experimental chemist is to design the incisive experiments, and to love the problem enough to see it through!Acknowledgements
We wish to thank EPSRC for funding to the Dynamic Structural Science (DySS) consortium at the Research Complex at Harwell (EP/ I01974X/1).References
- M. Gao, C. Lu, H. Jean-Ruel, L. C. Liu, A. Marx, K. Onda, S.-y. Koshihara, Y. Nakano, X. Shao, T. Hiramatsu, G. Saito, H. Yamochi, R. R. Cooney, G. Moriena, G. Sciaini and R. J. D. Miller, Nature, 2013, 496, 343–346 CrossRef CAS PubMed.
- M. A. Newton, A. J. Dent and J. Evans, Chem. Soc. Rev., 2002, 31, 83–95 RSC.
- J. Evans, Phys. Chem. Chem. Phys., 2006, 8, 3045–3058 RSC.
- W. H. Bragg, Proc. R. Soc., 1913, 89, 246–248 CrossRef CAS.
- M. de Broglie, C. R Chim., 1913, 157, 924–926 Search PubMed.
- H. G. J. Moseley, Philos. Mag., 1913, 26, 1024–1034 CrossRef CAS; H. G. J. Moseley, Philos. Mag., 1914, 27, 703–713 CrossRef.
- H. Fricke, Phys. Rev., 1920, 20, 201–215 Search PubMed.
- E. A. Stern, Phys. Rev. B: Solid State, 1974, 10, 3027–3037 CrossRef CAS.
- EXCURV(9.301) program: N. Binsted CrossRef CAS, in STFC Daresbury Laboratory computer program, 1998; FEFF programs: M. Newville, J. Synchrotron Radiat., 2001, 8, 322–324 CrossRef CAS; B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef PubMed.
- K. Hämäläinen, D. P. Siddons, J. B. Hastings and L. E. Berman, Phys. Rev. Lett., 1991, 67, 2850–2853 CrossRef.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS PubMed.
- FEFF9 program: J. J. Rehr, J. J. Kas, M. P. Prange, A. P. Sorini, Y. Takimoto and F. Vila, C. R. Phys., 2009, 10, 548–559 CrossRef CAS PubMed; J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
- MXAN program: M. Benfatto and S. Della Longa, J. Synchrotron Radiat., 2001, 8, 1087–1094 CrossRef CAS.
- K. Hermann, L. G. M. Pettersson, M. E. Casida, C. Daul, A. Goursot, A. Koester, E. Proynov, A. St-Amant and D. R. Salahub. Contributing authors: V. Carravetta, H. Duarte, C. Friedrich, N. Godbout, M. Gruber, J. Guan, C. Jamorski, M. Leboeuf, M. Leetmaa, M. Nyberg, S. Patchkovskii, L. Pedocchi, F. Sim, L. Triguero and A. Vela, StoBe–deMon program, http://www.fhi-berlin.mpg.de/KHsoftware/StoBe Search PubMed.
- The ORCA program system: F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- X. Zhang, S. E. Canton, G. Smolentsev, C.-J. Wallentin, Z. Liu, Q. Kong, K. Attenkofer, A. B. Strickrath, M. W. Mara, L. X. Chen, K. Wärnmark and V. Sundström, J. Am. Chem. Soc., 2014, 136, 8804–8809 CrossRef CAS PubMed.
- G. Capano, T. J. Penfold, N. A. Besley, C. J. Milne, M. Renhard, H. Rittmann-Frank, P. Glatzel, R. Abela, U. Rothlisberger, M. Chergui and I. Tavernellis, Chem. Phys. Lett., 2013, 580, 179–184 CrossRef CAS PubMed.
- N. Leidel, P. Chernev, K. G. V. Havelius, L. Schwartz, S. Ott and M. Haumann, J. Am. Chem. Soc., 2012, 134, 14142–14147 CrossRef CAS PubMed.
- M. Lundberg, T. Kroll, S. DeBeer, U. Bergmann, S. A. Wilson, P. Glatzel, D. Nordlund, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2013, 135, 17121–17134 CrossRef CAS PubMed.
- O. M. Roscioni, J. M. Dyke and J. Evans, J. Phys. Chem. C, 2013, 117, 19464–19470 CAS.
- J. C. Swarbrick, T.-C. Weng, K. Schulte, A. N. Khlobystov and P. Glatzel, Phys. Chem. Chem. Phys., 2010, 12, 9693–9699 RSC.
- S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, ACS Catal., 2014, 4, 4201–4204 CrossRef CAS.
- R. J. Deeth and K. Randell, Inorg. Chem., 2008, 47, 7377–7388 CrossRef CAS PubMed , and references therein.
- H. Loffler, A. M. Mohammed, Y. Inada and S. Funahashi, J. Comput. Chem., 2006, 27, 1944–1949 CrossRef CAS PubMed.
- H. V. R. Annapureddy and L. X. Dang, J. Phys. Chem. B, 2014, 118, 8917–8927 CrossRef CAS PubMed.
- S. G. Fiddy, J. Evans, T. Neisius, M. A. Newton, N. Tsoureas, A. A. D. Tulloch and A. A. Danopoulos, Chem. – Eur. J., 2007, 13, 3652–3659 CrossRef CAS PubMed.
- S. A. Bartlett, P. P. Wells, M. Nachtegaal, A. J. Dent, G. Cibin, G. Reid, J. Evans and M. Tromp, J. Catal., 2011, 284, 247–258 CrossRef CAS PubMed.
- S. A. Bartlett, G. Cibin, A. J. Dent, J. Evans, M. J. Hanton, G. Reid, R. P. Tooze and M. Tromp, Dalton Trans., 2013, 42, 2213–2223 RSC.
- S. A. Bartlett, J. Moulin, M. Tromp, G. Reid, A. J. Dent, G. Cibin, D. S. McGuinness and J. Evans, ACS Catal., 2014, 4, 4201–4204 CrossRef CAS.
- S. Diaz-Moreno, D. T. Bowron and J. Evans, Dalton Trans., 2005, 3814–3817 RSC.
- M. Tromp, G. P. F. van Strijdonck, S. S. van Berkel, A. van der Hoogenband, M. C. Feiters, B. de Bruin, S. G. Fiddy, A. M. J. van der Eerden, J. A. van Bokhoven, P. W. N. M. van Leeuwen and D. C. Koningsberger, Organometallics, 2010, 29, 3085–3097 CrossRef CAS.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022–7034 CrossRef CAS PubMed.
- M. Tromp, A. J. Dent, J. Headspith, T. L. Easun, X.-Z. Sun, M. W. George, O. Mathon, G. Smolentsev, M. L. Hamilton and J. Evans, J. Phys. Chem. B, 2013, 117, 7381–7387 CrossRef CAS PubMed.
- N. A. Gothard, M. W. Mara, J. Huang, J. M. Szarko, B. Rolcynski, J. V. Lockard and L. X. Chen, J. Phys. Chem. A, 2012, 116, 1984–1992 CrossRef CAS PubMed.
- L. X. Chen, X. Y. Zhang, E. C. Wasinger, K. Attenkofer, G. Jennings, A. Z. Muresan and J. S. Lindsey, J. Am. Chem. Soc., 2007, 129, 9616–9618 CrossRef CAS PubMed; X. Y. Zhang, E. C. Wasinger, A. Z. Muresan, K. Attenkofer, G. Jennings, J. S. Lindsey and L. X. Chen, J. Phys. Chem. A, 2007, 111, 11736–11742 CrossRef PubMed.
- M. L. Shelby, M. W. Mara and L. X. Chen, Coord. Chem. Rev., 2014, 277–278, 291–299 CrossRef CAS PubMed.
- U. B. Hendgen-Cotta, U. Flögel, M. Kelm and T. Rassaf, J. Exp. Biol., 2010, 213, 2734–2740 CrossRef CAS PubMed; U. B. Hendgen-Cotta, M. Kelm and T. Rassaf, Free Radicals Biol. Med., 2014, 73, 252–259 CrossRef PubMed.
- F. A. Lima, T. J. Penfold, R. M. van der Veen, M. Reinhard, R. Abela, I. Tavernelli, U. Rothlisberger, M. Benfatto, C. J. Milne and M. Chergui, Phys. Chem. Chem. Phys., 2014, 16, 1617–1631 RSC.
- F. A. Lima, C. J. Milne, D. C. V. Amarasinghe, M. H. Rittmann-Frank, R. M. van der Veen, M. Renhard, V.-T. Pham, S. Karlsson, S. L. Johnson, D. Grolimund, C. Borca, T. Huthwelker, M. Janousch, F. van Mourik, R. Abela and M. Chergui, Rev. Sci. Instrum., 2011, 82, 063111 CrossRef PubMed.
- M. W. Mara, M. Shelby, A. Stickpath, M. Harpham, J. Huang, X. Y. Zhang, B. M. Hoffman and L. X. Chen, J. Phys. Chem. B, 2013, 117, 14089–14098 CrossRef CAS PubMed.
- C. J. Milne, T. J. Penfold and M. Chergui, Coord. Chem. Rev., 2014, 277–278, 44–68 CrossRef CAS PubMed.
- V.-T. Pham, T. J. Penfold, R. M. van der Veen, F. Lima, A. El Nahha, S. L. Johnson, P. Beard, R. Abela, C. Bressler, I. Tavernelli, C. J. Milne and M. Chergui, J. Am. Chem. Soc., 2011, 133, 12740–12748 CrossRef CAS PubMed.
- H. T. Lemke, C. Bressler, L. X. Chen, D. M. Fritz, K. J. Gaffney, A. Galler, W. Gawelda, K. Haldrup, R. W. Hartsock, H. Ihee, J. Kim, K. H. Kim, J. H. Lee, M. M. Nielson, A. B. Strickrath, W. Zhang, D. Zhu and M. Cammarata, J. Phys. Chem. A, 2013, 117, 735–740 CrossRef CAS PubMed.
- R. Alonso-Mori, J. Kern, R. J. Gildea, D. Sokaras, T.-C. Weng, B. Lassalle-Kaiser, R. Tran, J. Hattne, H. Laksmono, J. Hellmich, C. Glöckner, N. Echols, R. G. Sierra, D. W. Schafer, J. Sellberg, C. Kenney, R. Herbst, J. Pines, P. Hart, S. Herrmann, R. W. Grosse-Kunstleve, M. J. Latimer, A. R. Fry, M. M. Messerschmidt, A. Miahnahri, M. M. Seibert, P. H. Zwart, W. E. White, P. D. Adams, M. J. Bogan, S. Boutet, G. J. Williams, A. Zouni, J. Messinger, P. Glatzel, N. Sauter, V. K. Yachandra, J. Yano and U. Bergmann, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 19103–19107 CrossRef CAS PubMed.
- J. Kern, R. Tran, R. Alonso-Mori, S. Koroidov, N. Echols, J. Hattne, M. Ibrahim, S. Gul, H. Laksmono, R. G. Sierra, R. J. Gildea, G. Han, J. Hellmich, B. Lassalle-Kaiser, R. Chatterjee, A. S. Brewster, C. A. Stan, C. Glöckner, A. Lampe, D. DiFiore, D. Milathianaki, A. R. Fry, M. M. Seibert, J. E. Koglon, E. Gallo, J. Uhig, D. Sokaras, T.-C. Weng, P. H. Zwart, D. E. Skinner, M. J. Bogan, M. Messerschmidt, P. Glatzel, G. J. Williams, S. Boutet, P. D. Adams, A. Zouni, J. Messinger, N. K. Sauter, U. Bergmann, J. Yano and V. K. Yachandra, Nat. Commun., 2014, 5, 4371 CAS.
- P. Werner, M. Beye, F. de Groot, S. Düsterer, K. Gaffney, S. Grübel, R. Hartsock, F. Hennies, I. Josefsson, B. Kennedy, K. Kunnus, T. Leitner, T. Mazza, M. Meyer, D. Nordlund, M. Odelius, W. Quevedo, P. Radcliffe, I. Rajkovic, B. Schlotter, M. Scholz, E. Schreck, E. Suljoti, S. Techert, J. Turner, C. Weniger, W. Zhang and A. Föhlisch, EPJ Web Conf., 2013, 41, 05025 CrossRef.
- W. Blachucki, J. Szlachetko, J. Hoszowska, J.-C. Dousse, Y. Kayser, M. Nachtegaal and J. Sá, Phys. Rev. Lett., 2014, 112, 173003 CrossRef CAS.
- J. Szlachetko, C. J. Milne, J. Hoszowska, J.-C. Dousse, W. Bachucki, J. Sà, Y. Kayser, M. Messerschmidt, R. Abela, S. Boutet, C. David, G. Williams, M. Pajek, B. D. Patterson, G. Smolentsev, J. A. van Bokhoven and M. Nachtegaal, Struct. Dyn., 2014, 1, 021101 CrossRef PubMed.
- B. F. Murphy, L. Fang, M.-H. Chen, J. D. Bozek, E. Kukk, E. P. Kanter, M. Messerschmidt, T. Osipov and N. Berrah, Phys. Rev. A, 2012, 86, 053423 CrossRef.
- K. Tamasaku, E. Shigemasa, Y. Inubushi, T. Katayama, K. Sawada, H. Yumoto, H. Ohashi, H. Mimura, M. Yabishi, K. Yamauchi and T. Ishilawa, Nat. Photonics., 2014, 8, 313–316 CrossRef CAS.
Footnote |
| † Dedicated to Professor Ken Wade, in memory of his chemical imagination, his leadership, and his humour. |
| This journal is © The Royal Society of Chemistry 2015 |