
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iminoborylene complexes: evaluation of synthetic routes towards BN-allenylidenes and unexpected reactivity towards carbodiimides†‡
J.
Niemeyer
*a,
M. J.
Kelly
b,
I. M.
Riddlestone
b,
D.
Vidovic§
b and
S.
Aldridge
*b
aInstitute of Organic Chemistry, Department of Chemistry, University of Duisburg-Essen, Universitätsstrasse 7, 45141 Essen, Germany. E-mail: jochen.niemeyer@uni-due.de
bInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: SimonAldridge@chem.ox.ac.uk
First published on 11th February 2015
Abstract
The synthetic and reaction chemistries of cationic iminoborylene complexes [LnM![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BNCR2]+, which feature a unique heterocumulene structure, have been systematically investigated. Precursors of the type CpFe(CO)2B(Cl)NCAr2 (Ar = p-Tol/Mes, 5c/d) have been generated by B-centred substitution chemistry using CpFe(CO)2BCl2 and suitable lithiated ketimines – a reaction which is found to be highly sensitive to the steric bulk at both the metal fragment and the ketimino group. Carbonyl/phosphine exchange (using PCy3 or PPh3), followed by halide abstraction allows for the generation of the cationic iminoborylenes [CpFe(PR3)(CO)(BNCAr2)]+[BArX4]− (R = Cy, Ar = p-Tol/Mes, 12c/d; R = Ph, Ar = Mes, 13d; ArX = 3,5-X2C6H3 where X = Cl, CF3) which have been characterized spectroscopically and by X-ray crystallography. The reactivity of these iminoborylene systems towards a range of nucleophiles and unsaturated substrates has been investigated. The latter includes the first examples of MB metathesis reactivity with a carbodiimide, and results in FeB cleavage and formation of the isonitrile complexes [CpFe(PCy3)(CO)(CNR)]+[BArCl4]− (R = iPr/Cy, 16/17).
BNCR2]+, which feature a unique heterocumulene structure, have been systematically investigated. Precursors of the type CpFe(CO)2B(Cl)NCAr2 (Ar = p-Tol/Mes, 5c/d) have been generated by B-centred substitution chemistry using CpFe(CO)2BCl2 and suitable lithiated ketimines – a reaction which is found to be highly sensitive to the steric bulk at both the metal fragment and the ketimino group. Carbonyl/phosphine exchange (using PCy3 or PPh3), followed by halide abstraction allows for the generation of the cationic iminoborylenes [CpFe(PR3)(CO)(BNCAr2)]+[BArX4]− (R = Cy, Ar = p-Tol/Mes, 12c/d; R = Ph, Ar = Mes, 13d; ArX = 3,5-X2C6H3 where X = Cl, CF3) which have been characterized spectroscopically and by X-ray crystallography. The reactivity of these iminoborylene systems towards a range of nucleophiles and unsaturated substrates has been investigated. The latter includes the first examples of MB metathesis reactivity with a carbodiimide, and results in FeB cleavage and formation of the isonitrile complexes [CpFe(PCy3)(CO)(CNR)]+[BArCl4]− (R = iPr/Cy, 16/17).
Introduction
The investigation of boron-transition metal complexes has attracted widespread attention in recent years. A number of novel classes of compounds featuring conventional 2-centre 2-electron metal–boron bonds have been studied, not only with respect to their structural and bonding properties, but also with a view to targeting new modes of reaction chemistry.1 Within this area, boryl complexes, LnM(BX2), featuring a disubstituted boron-fragment coordinated at M were the first to be discovered,2 and have subsequently been implicated in a number of unprecedented transformations, such as the borylation of unactivated hydrocarbon substrates.3More recently, reliable synthetic routes to subvalent transition metal borylene complexes, (LnM)x(BX), have also been developed.4 These species feature a mono-substituted boron fragment, and are of particular interest due to their close relationship with archetypal organometallic complexes.5 Along these lines, fluoroborylene (LnMBF) and aminoborylene (LnMBNR2) species have been have been synthesized, representing isolobal analogues of classical carbonyl (LnMCO)6 and vinylidene (LnMCCR2) complexes.7,8
Reactivity-wise the chemistry of many borylene complexes is dominated by the electrophilicity of the boron centre, which underpins their use in C–H activation9 or cycloaddition reactions.7a,10 One possibility, with precedent in organometallic systems, to further broaden the scope of reactivity of transition-metal boron complexes is by the introduction of further elements of unsaturation into the boron ligand. Thus, for example, Braunschweig and co-workers have achieved this by use of boryl ligands containing B–X double or triple bonds (X = NR, O or CR2, Scheme 1).11 Taking this idea further, we have recently communicated12 the first examples of cationic imino-borylene complexes [LnMBNCR2]+ featuring an extended array of unsaturated bonds (Scheme 1).13 Such complexes can be viewed as hetero-analogues of well-known allenylidene complexes,14 which show a highly versatile reaction chemistry resulting from their dual α, γ-electrophilicity and β-nucleophilicity. With this in mind, we set out to uncover new patterns of reactivity for iminoborylene complexes which are otherwise inaccessible to known alkyl- or aminoborylene systems.4,7
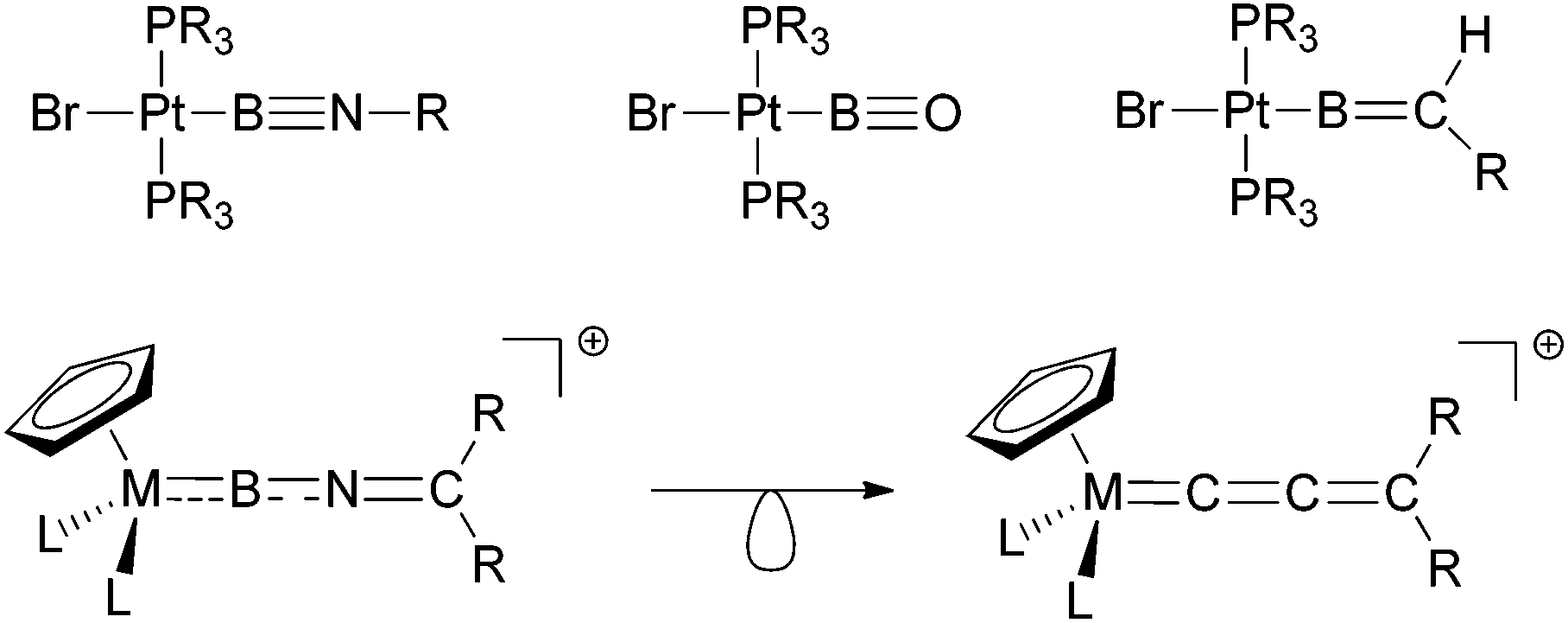 | ||
| Scheme 1 Highly unsaturated metal–boron complexes featuring iminoboryl, oxoboryl, alkylideneboryl (top) and iminoborylene ligands (bottom, also showing the isolobal relationship with allenylidenes). | ||
Herein, we now report in full on synthetic approaches towards iminoborylene systems, and their reaction chemistry both with respect to anionic nucleophiles and unsaturated substrates. A key finding is the discovery of novel metathesis-type reactivity towards carbodiimides, RNCNR.
Results and discussion
The synthesis of terminal borylene complexes has been achieved using a variety of different approaches, including double salt elimination,4f,g metal-to-metal borylene transfer,15 and dehydrogenation of σ-borane complexes.16 Moreover, halide abstraction from haloboryl complexes has been shown to give access to cationic borylenes in a reliable fashion.17 Based on this approach, we envisaged the use of suitable imino-functionalized haloboryl complexes as precursors, which upon halide abstraction with sodium tetra-arylborates would give the desired cationic iminoborylenes (Scheme 2).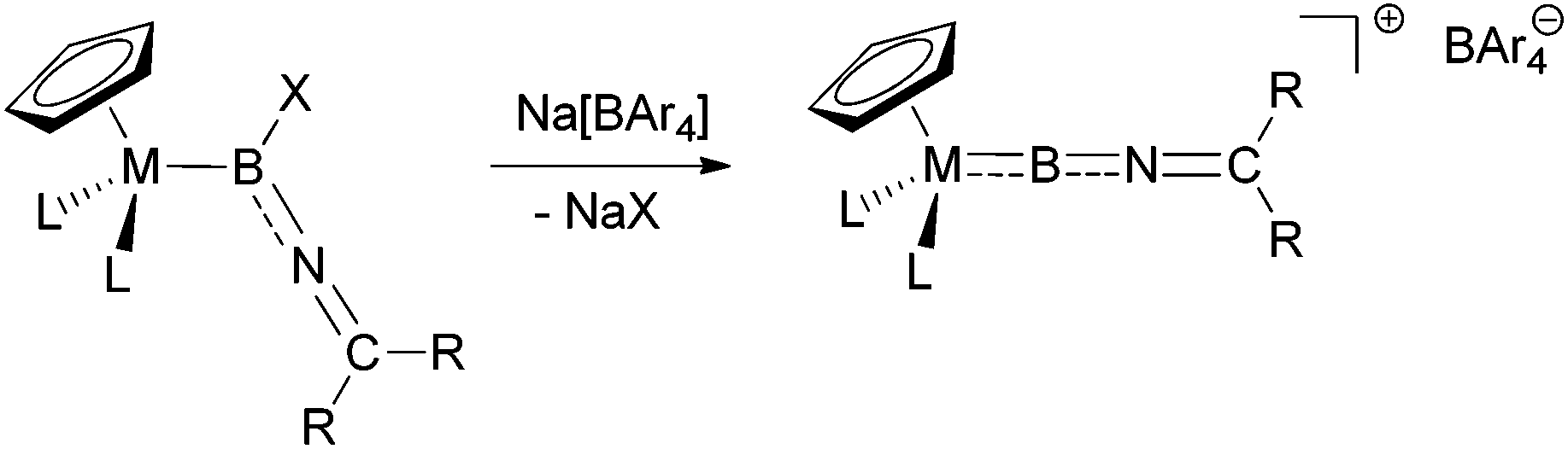 | ||
| Scheme 2 Target synthesis of iminoborylene complexes by halide abstraction from halo(imino)boryl complexes. | ||
Synthesis of iminoboryl complexes
In order to put our synthetic efforts towards imino-substituted systems on a comparable basis to known complexes, we initially decided to target the [CpFe(CO)2] unit as the metal fragment, given its successful use for the generation of related cationic aminoborylenes.7a,b For the construction of precursors featuring the necessary array of consecutive Fe–B–N–C bonds, we evaluated two synthetic approaches, differing in the order of formation of the relevant bonds to the boron centre (Scheme 3).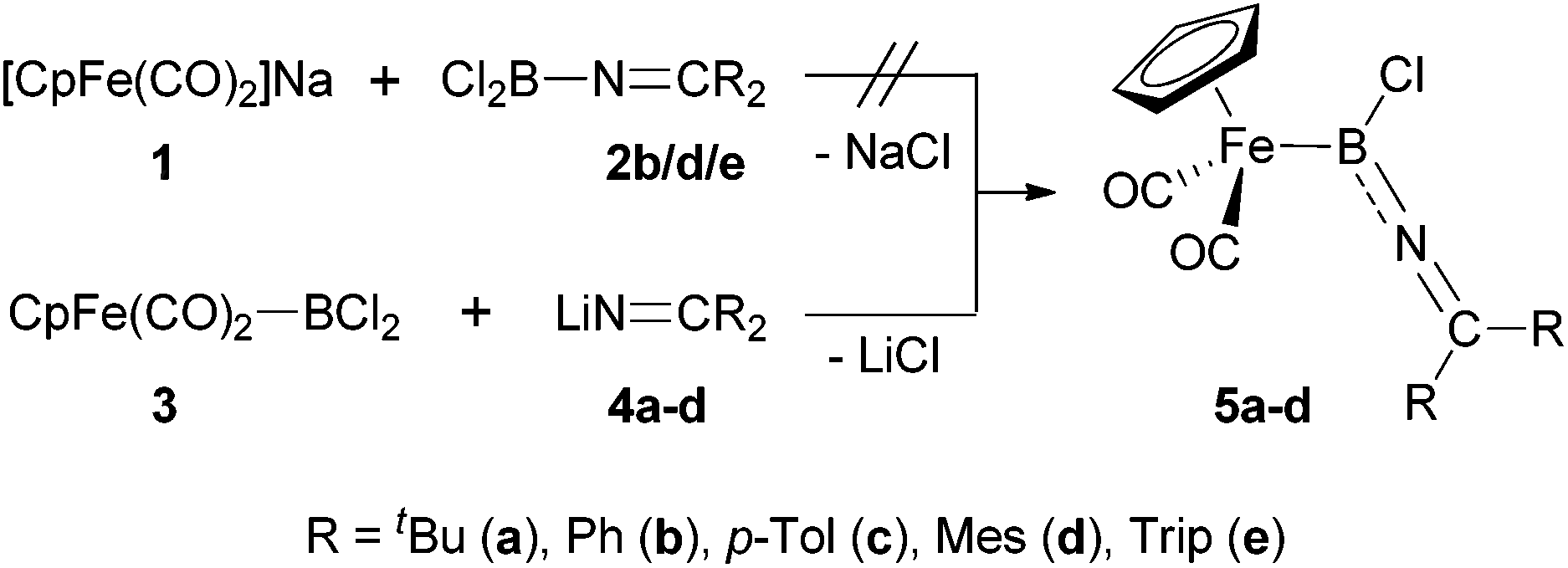 | ||
| Scheme 3 Synthesis of iminoboryl complexes 5via dichloro-iminoboranes (top) or dichloroboryl iron precursors (bottom). | ||
Mirroring existing synthetic routes to [CpFe(CO)2] boryl complexes,17c we initially attempted the generation of complexes of type 5 by reaction of the anionic [CpFe(CO)2]− reagent 1 (as the sodium salt) with the corresponding dichloro-(imino)boranes 2, thus establishing the B–N connectivity prior to the formation of the Fe–B bond (Scheme 3, upper). While Cl2B(NCPh2), 2a, was readily synthesized according to Wade's original procedure,18 it showed no reactivity towards ferrate 1. Assuming that the dimeric nature of 2a (indicated by its 11B NMR shift of δB = −7 ppm) is responsible for its low reactivity, we attempted to generate monomeric dichloro-(imino)boranes by the use of bulkier ketimino substituents (e.g. R = Mes or Trip). These syntheses were initially frustrated by a ligand redistribution reaction which apparently occurs on exposure to continuous vacuum [yielding ClB(NCR2)2], and which prevents isolation of the pure dichloro(ketimino)-boranes.19 This problem could be circumvented by in situ generation (see ESI‡), which generates the corresponding monomeric compounds Cl2B(NCR2) (R = Mes/Trip (2d/e), δB = 26/27 ppm). However, these systems do not show clean reactivity towards 1, with the starting borane being the predominant species in the reaction mixtures even under forcing conditions.
For this reason, we shifted our synthetic strategy towards a reversed order of bond formation reactions at boron, employing the known reaction of 1 with BCl3 to generate the iron dichloroboryl complex 3in situ (δB = 91 ppm).20 Complex 3 was then treated with a series of ketiminolithium reagents LiNCR2 [R = tBu/Ph/p-Tol/Mes/Trip (4a–e)],21 to install the B–N linkage (Scheme 3, lower). Accordingly, the reactions with less bulky lithium salts (e.g.4a–d) lead to clean formation of the desired iminoboryl complexes 5a–d (as judged by 1H and 11B NMR spectroscopy), which could be purified by precipitation from hexane in case of the p-tolyl- and mesityl-substituted complexes (5c/d, 38–52%); the high solubility of complexes 5a/b, on the other hand, prevented their isolation as pure compounds. By contrast, the reaction of the lithium salt LiNCTrip2 (4e) with 3 gives a different type of boron-containing product, with the high field 11B chemical shift (δB = 27 ppm) arguing against Fe–B bond formation. The product is tentatively assigned as borane 2e, resulting from the nucleophilic displacement of the [CpFe(CO)2]− anion (rather than chloride) from precursor 3. Such a transformation has recent precedent,22 and is presumably induced by the large steric bulk of the bis(triisopropylphenyl)ketimino group.
Complexes 5c/d have been characterized spectroscopically, showing the expected NMR resonances for the [CpFe(CO2)] fragment (Cp: δH = 4.26/4.30 ppm, δC = 84.4/84.6 ppm, CO: δC = 215.3/215.3 ppm) and [B–NC] fragments (δC = 150.7/153.1 ppm, δB = 50/47 ppm). Additionally, in the case of 5c structural authentication could be achieved by X-ray crystallography (Fig. 2). In the solid state, 5c exhibits a near-linear arrangement of the B–C–N unit [∠ B(11)–N(13)–C(14) = 175.6(3)°], consistent with a significant degree of N → B π-donation, a finding also reflected in the short B–N [1.349(4) Å] and relatively long Fe–B bond lengths [2.016(4) Å].
Having established a viable synthetic route for the generation of complexes of type 5 by boron-centred substitution chemistry, and with the steric constraints of the ketimino nucleophile now apparent, we set out to investigate the scope of this approach by variation of the metal fragment. Thus, we generated the previously described tungsten dichloroboryl complex 6 (δB = 91 ppm) alongside its bromo analogue 7 (δB = 84 ppm) by reaction of the tungstate Na[CpW(CO3)] with the respective trihaloboranes.23
Although 6 has previously been reported by Schmid and Nöth, it has not been structurally characterized, and given the dearth of structural data available for dihaloboryl systems we sought to investigate it crystallographically. Accordingly, the solid-state structure of 6 (Fig. 1) features a W–B bond [2.22(2) Å] which is considerably longer than in the corresponding CpFe(CO)2BCl2 complex 3 [1.942(3) Å] (even taking into account the larger van der Waals radius of tungsten vs. iron: 2.10 vs. 2.05 Å),20a,24 while the B–Cl bonds are in the expected range [e.g. 1.78(1) and 1.79(1) Å for 6, cf. 1.781(6) and 1.783(4) Å for 3].20a Due to the presence of three carbonyl co-ligands, complex 6 is sterically rather congested when compared to 3, as can be seen from the close B–CO contacts [B(13)–C(11) 2.37(2), B(13)–C(2) 2.53(2) Å, cf. B–C(1) 2.574(5), B–C(2) 2.638(6) Å for 3], a factor which presumably also leads to the (near parallel) orientation of the BCl2 unit with respect to the Cp(centroid)-W-B plane [∠ Cp(centroid)–W(1)–B(13)–Cl(14) = 9.5(9)°, cf. ∠ Cp(centroid)–Fe–B–Cl(1) = 100.7(2)° for 3].
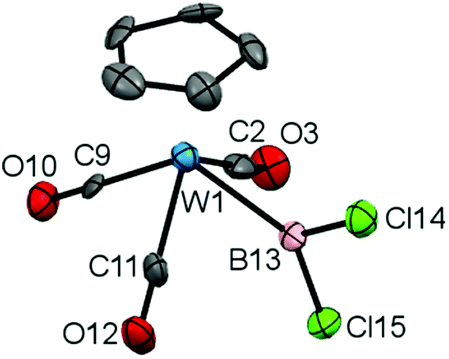 | ||
| Fig. 1 Molecular structure of 6 in the solid state, hydrogen atoms omitted for clarity and thermal ellipsoids set at the 40% probability level. Key bond lengths [Å] and angles [°]: W(1)–B(13) 2.22(2), B(13)–Cl(14) 1.78(1), B(13)–Cl(15) 1.79(1), Cl(14)–B(13)–Cl(15) 110.5(7). | ||
While 6 could be structurally characterized, its reactivity – in terms of boron-centred substitution processes – proves to be much less facile than the corresponding chemistry for 3. Thus, in contrast to the clean reactivity observed in the iron case, no M-B containing products could be observed upon reaction of the representative ketiminolithium salts 4a/d with either of the dihaloboryl-tungsten complexes 6 or 7. As judged by 11B NMR spectroscopy, breakage of the W–B bond and extrusion of the [CpW(CO)3]− unit generates instead the corresponding dihalo(ketimino)boranes 2a/d (δB = 21/26 ppm) (Scheme 4).
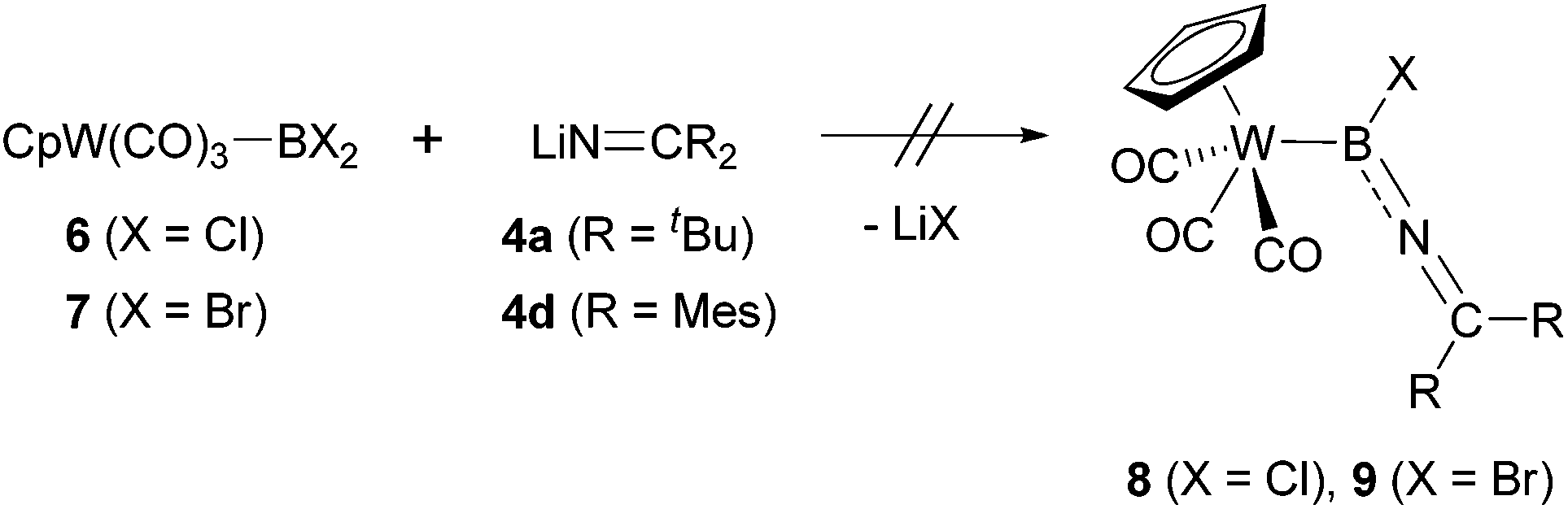 | ||
| Scheme 4 Attempted synthesis of tungsten iminoboryl complexes. | ||
These results further suggest that the boron-centred substitution reaction using a metal dihaloboryl complex is very sensitive to the steric bulk of the substituents both on the metal fragment and on the incoming nucleophile, with the partnership of the less sterically demanding iron boryl complex 3 and the less bulky iminolithium salts 4a–d uniquely bringing about substitution at boron without breakage of the metal–boron bond.
Synthesis of iminoborylene complexes
With the iminoboryl-complexes 5c/d in hand, we next attempted the synthesis of the corresponding borylene complexes by halide abstraction. Reaction of 5d with Na[BArf4] [Arf = 3,5-(CF3)2C6H3] leads to the formation of the corresponding cationic borylene [CpFe(CO2)(BNCMes2)]+, as indicated by a downfield shift in the 11B NMR signal (δB = 75 ppm, cf. 47 ppm for 5d). While this borylene complex could be shown to be stable at −30 °C in solution over a period of several days, it decomposes rapidly at room temperature. This led us to investigate the use of more electron-rich metal fragments in order to generate borylene species stabilized by more efficient M → B π-backbonding. Thus, we attempted the photolytic displacement of the π-acidic carbonyl-ligands in 5c/d by strong σ-donor phosphine ligands. While attempts to substitute both carbonyl ligands by reaction with chelating bis-phosphines (dppe/dmpe for example) failed to yield the desired products,25 reaction of 5c/d with monodentate donors cleanly gave the corresponding mixed phosphine/carbonyl complexes (Scheme 5).26 Assuming that bulky trialkylphosphines would lead to an additional kinetic stabilization of the corresponding borylene complexes, we first used PCy3 in this substitution chemistry, leading to the formation of the desired complexes 10c/d in moderate yields (48–60%). In order to further investigate the influence of the steric/electronic properties of the phosphine ligands, we also employed PPh3 in the reaction with 5d, giving the triphenylphosphine-substituted boryl complex 11d (43%).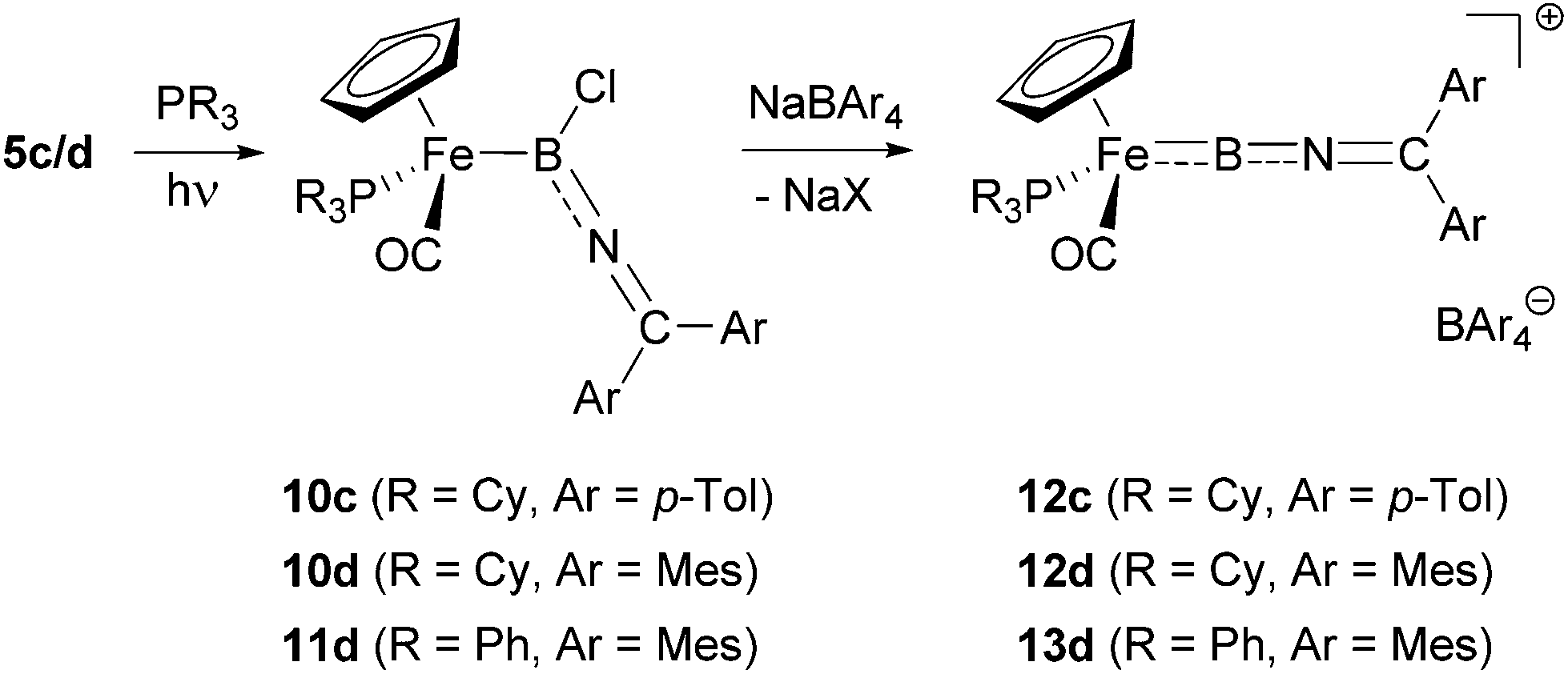 | ||
| Scheme 5 Synthesis of phosphine-substituted iminoboryl complexes 10 and 11 and halide abstraction to give borylenes 12 and 13. | ||
Spectroscopic characterization of 10c/d and 11d clearly signals the successful introduction of the phosphine co-ligands via31P NMR spectroscopy (δP = 77.1/75.0/78.8 ppm for 10c/10d/11d), while little change is observed in the respective 11B spectra (δB = 47/50/51 ppm). In addition, diastereotopic splitting is observed for the aryl substituents of the axially prochiral ketimino fragments, brought about by the formation of a chiral metal centre (e.g. p-CH3 groups in 10c/10d/11c: δH = 2.09, 2.06/2.11, 2.09/2.12, 2.11 ppm). In addition, the formation of a more electron-rich metal centre leads to the expected red-shift of the CO stretches in the respective IR-spectra [ν(CO) = 1902/1905/1909 cm−1 for 10c/10d/11d, cf. ν(CO) = 2002, 1922/2005, 1937 cm−1 for 5c/d].
Crystallographically, complexes 10c/d (Fig. 2) feature a piano-stool geometry around the central metal atom in the solid state, with the M–C(O) distances reflecting a higher degree of π-backbonding compared to 5c [1.716(2)/1.716(3) Å for 10c/d, cf. 1.758(3), 1.753(3) Å for 5c]. Interestingly (and in contrast to dicarbonyl-ligated 5c), complex 10c features a non-linear arrangement of the B–N–C unit [∠ B–N–C = 144.8(2)°], implying a reduced degree of N → B donation. Consistently, 10c features a relatively long B–N [1.396(4) Å], with the accompanying shortening of the Fe–B bond [1.980(4) Å] presumably reflecting augmented Fe → B donation. This balance of competing π-donation to boron is clearly a fine one, however, as the closely related dimesityl system 10d features a linear B–N–C arrangement [∠ B–N–C = 174.7(2)°] and bond lengths consistent with dominant N → B donation [B–N 1.368(3) Å, Fe–B 2.015(2) Å].
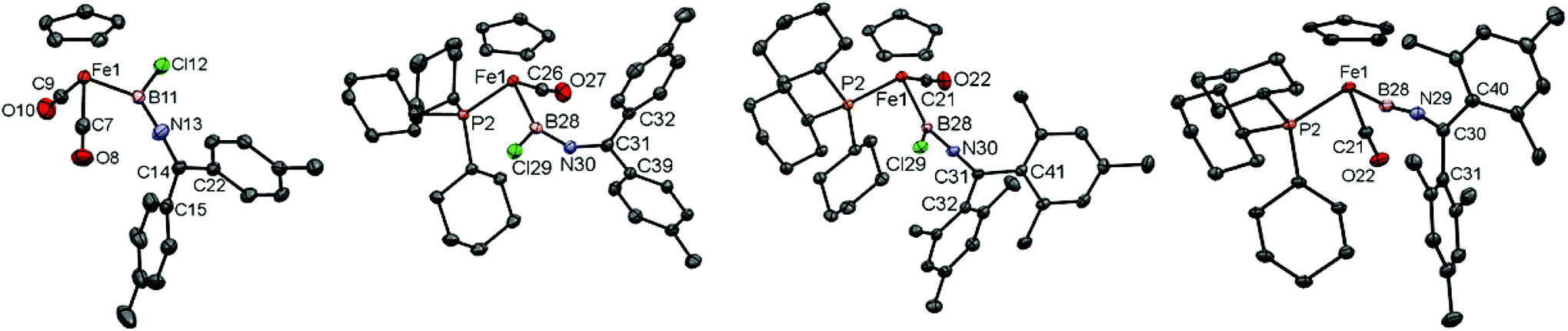 | ||
| Fig. 2 Molecular structures of 5c, 10c, 10d and 12d in the solid state. Hydrogen atoms and counter-ion omitted for clarity and thermal ellipsoids set at the 40% probability level (20% for 12d). Key bond lengths [Å] and angles [°]: (for 5c) Fe(1)–B(11) 2.016(4), B(11)–N(13) 1.349(4), N(13)–C(14) 1.277(4), Fe(1)–B(11)–N(13) 126.0(2), B(11)–N(13)–C(14) 175.6(3); (for 10c/d) Fe(1)–B(28) 1.980(4)/2.015(2), B(28)–N(30) 1.397(4)/1.368(3), N(30)–C(31) 1.269(3)/1.263(3), B(28)–N(30)–C(31) 144.8(2)/174.7(2); (for 12d) Fe(1)–B(28) 1.835(6), B(28)–N(29) 1.302(8), N(29)–C(30) 1.287(7), Fe(1)–B(28)–N(29) 170.9(5), B(28)–N(36)–C(37) 175.3(2). | ||
Utilising the monophosphine boryl complexes 10c/d and 11d as precursors, halide abstraction with Na[BArCl4] (ArCl = 3,5-Cl2C6H3) leads to the clean formation of the desired borylene complexes 12c/d and 13 in yields of 55–77% (Scheme 5). In comparison with their dicarbonyl-supported analogues, these complexes are more stable at room temperature, at least when handled under inert atmosphere conditions. Borylene formation can be followed by the downfield shifts in the respective 11B signals (δB = 82/85/85 ppm for 12c/12d/13d), while the shifts of the 31P resonances are less informative (δP = 84.9/75.0/69.3 ppm). In the 1H and 13C NMR spectra, the two sets of distinct signals for the ketimino aryl substituents merge to give a single set of resonances, indicating fast rotation of the BNCAr2 unit (e.g. δH = 2.48/2.31/2.34 ppm for the p-CH3 signal in 12c/12d/13c), which is not frozen out even at low temperatures (down to −75 °C). The IR spectra of these new compounds are also informative. These feature not only a BNC stretch consistent with the analogous mode observed for allenylidenes [ν(BNC) = 1763/1753/1779 cm−1], but also blue-shifted carbonyl stretching frequencies in comparison with their chloroboryl precursors [ν(CO) = 1962/1969/1984 cm−1 for 12c/12d/13c] consistent with weaker Fe → CO π-backbonding in the cationic systems.
Attempts to obtain crystals of complexes 12c and 13d revealed instead the tendency of each complex to slowly decompose over several days to [CpFe(PR3)(CO)2]+[BArCl4]− (R = Cy, Ph, respectively); the combined steric bulk of the mesityl and tricyclohexyl substituents, however, render complex 12d stable enough to be characterized by both X-ray crystallography and by positive-ion ESI-MS, the latter being consistent with the presence of the [CpFe(PCy3)(CO)2(BNCMes2)]+ cation (ESI‡). Moreover, the solid state structure (Fig. 2) reveals two crystallographically independent species with almost identical structural features. The cationic borylene component features a cumulene-type linear arrangement of the Fe–B–N–C unit [∠ Fe(1)–B(28)–N(29) = 170.9(5)°, ∠ B(28)–N(36)–C(37) = 175.3(2)°]. In the solid state at least, the ketimino-group is orientated near-parallel to the Cp(centroid)–Fe–B plane [∠ Cp(centroid)–Fe(1)–C(30)–C(40) = 7.1°], and the Fe–B bond [1.835(6) Å] is noticeably shorter than in the precursor 10d [2.015(2) Å], being comparable to that observed in monophosphine-substituted aminoborylene-complexes (e.g. 1.821(4) Å in [CpFe(CO)(PMe3)(BNCy2)]+).26 However, the observed B–N distance is rather short and the N–C distance is long [B–N 1.314(6) Å, N–C 1.292(6) Å, cf. 1.368(3) Å and 1.263(3) Å for 10d], consistent with a significant contribution from a resonance form containing a Fe–B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N–CMes2+ unit. Such a contribution is also consistent with the low-field shift of the ketimino-carbon 13C resonance, a feature also characteristic of allenylidene complexes (δC = 187.0 ppm, cf. δC = 150.2 ppm for 10d).
N–CMes2+ unit. Such a contribution is also consistent with the low-field shift of the ketimino-carbon 13C resonance, a feature also characteristic of allenylidene complexes (δC = 187.0 ppm, cf. δC = 150.2 ppm for 10d).
Reactivity of the iminoborylene complexes
With the crystallographic and spectroscopic analysis of iminoborylene complex 12d hinting at a partial contribution from a carbo-cationic resonance form, we set out to determine experimentally whether selectivity for nucleophilic addition at either the α- or γ-position would be observed. With this in mind, we further sought to compare the addition chemistry of both the mesityl- and p-tolyl substituted systems (12c/d, Scheme 6) in order to investigate the influence of the steric loading at the ketimino group.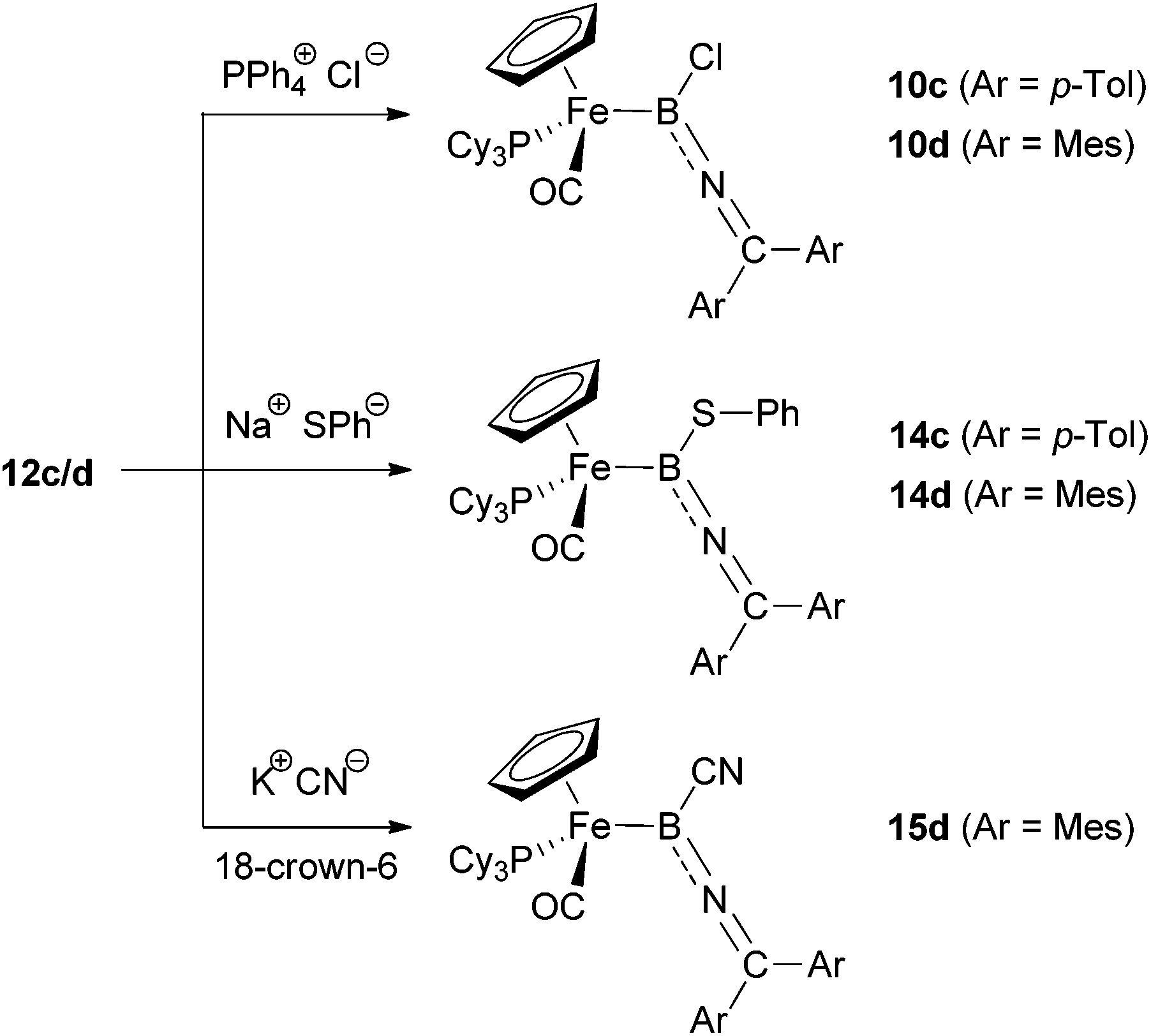 | ||
| Scheme 6 Reactivity of the borylene-complexes 12c/d towards anionic nucleophiles. | ||
In the first instance, we investigated whether reactions with a chloride source (e.g. [PPh4]Cl) could be used to generate products of the type CpFe(CO)(PCy3){BNC(Cl)Mes2}, thus allowing a formal α, γ-isomerization of the precursors 10c/dvia iminoborylene intermediates (i.e. a formal reversal of the conversion of [LnMCC–CR2OH]+ to [LnMC(OH)CCR2]+via the corresponding allenylidene27). However, exclusive α-attack led to the re-formation of the precursors 10c/d. In similar fashion, the reaction of 12c/d with sodium thiophenolate leads to the products of boron-centred nucleophilic attack, exclusively giving the B(SPh) complexes 14, independent of the steric bulk at the ketimino group. The syntheses of the thiolate-functionalized boryl complexes 14c/d could also be achieved directly by reaction of 10c/d with NaSPh in a boron-centred substitution reaction, thus providing independent verification of compound identity.
The situation is slightly different, however, when using cyanide (KCN, 18-crown-6) as a nucleophile. In this case, boryl precursors 10c/d are completely resistant towards substitution at boron, so we investigated the reactivity of the corresponding borylenes 12c/d towards CN−. On mixing KCN and 18-crown-6 with 12c/d generated in situ by the reaction of 10c/d with Na[BArCl4], re-formation of the chloroboryls 10c/d is observed. This suggests that in the presence of NaCl (from the initial salt metathesis) and KCN, in conjunction with 18-crown-6 as a solubilizing agent, the addition of chloride is preferred over the addition of cyanide. Presumably such an observation reflects thermodynamic control due the more favourable B–Cl bond enthalpy (ca. 128 vs. 107 kcal mol−1).28 The reaction of the pure complex 12d with KCN does, however, lead to addition of cyanide to the borylene. Once again, α-selectivity is observed, yielding the corresponding cyano-substituted boryl-complex 15d. Unfortunately, reaction of KCN with the less sterically encumbered borylene 12c gives only decomposition products, so that the influence of the aryl substituents on the regio-selectivity could not be fully investigated in this case.
Complexes 14c/d and 15d were fully characterized by spectroscopic, mass spectrometric and, in case of 14d, by crystallographic methods.12 The 11B and 31P resonances (δB = 56/52/41 ppm, δP = 76.3/74.1/73.3 ppm for 14c/14d/15d) are similar to those of the corresponding chloroboryl complexes (δB = 47/50 ppm, δP = 77.1/75.0 ppm for 10c/10d), which together with the CN ketimino-resonances (δC = 147.7/149.9/150.5 ppm) verify the postulated structures resulting from α-attack at boron. The observed high α-selectivity is presumably brought about by the high electrophilicity of the boron centre in each case, bearing in mind the fact that γ-selectivity has been observed in the addition of a variety of nucleophiles (including thiolate and cyanide) to cationic allenylidene complexes.14,29
Hoping to uncover more diverse patterns of reactivity, we targeted a study of the reactivity of the iminoborylenes towards unsaturated substrates. It has been shown that neutral borylene complexes undergo borylene transfer reactions with alkynes,10 insertion reactions with isonitriles and carbodiimides,30 and metathesis-type reactions with ketones,30 while cationic borylenes oftentimes show contrasting reactivity, displaying hydride transfer reactivity towards ketones,31 insertion reactions with carbodiimides,32 and metathesis-type reactivity with isocyanates and phosphine sulfides.7b
In order to investigate the reactivity of our iminoborylene complexes towards unsaturated substrates, we used the mesityl-substituted complex 12d which shows the highest resistance towards undesired hydrolysis and decomposition reactions. Mixing of 12d with non-polar substrates such as 2,3-dimethyl-butadiene and trimethylsilyl-acetylene in dichloromethane leads to no conversion, even at 40 °C, and over prolonged periods of time. While this result is consistent with the fact that other cationic borylenes show little affinity for alkenes or alkynes, we were surprised to find that mixing of 12d with isopropyl-isocyanate also did not lead to any conversion (as judged from in situ1H and 11B NMR measurements). This contrasts with the chemistry of cationic aminoborylenes, which react with isocyanates, RNCO, cleanly and under mild conditions to give the corresponding isonitrile complexes [CpFe(CO)2(NCR)]+via a metathesis-type reaction.26
By contrast, the reaction of 12d with an excess of either diisopropyl- or dicyclohexylcarbodiimide (RNCNR, R = iPr/Cy) gives clean conversion within hours at room temperature, to a single 31P containing species (δP = 76.4/76.5 ppm, respectively) and a compound giving rise to a 11B signal at δB = 29/30 ppm. Rather than the carbodiimide insertion products found for related aminoborylene complexes30,32 and organic boranes,33in situ spectroscopic analysis of the reaction mixture in this case supports an alternative pathway. Thus, as opposed to a characteristic low-field carbene 13C resonance seen for either a mono- or a bis-carbodiimide insertion product (e.g. [CpFe(CO)2C(NCy)2BNCy2]+, δC = 251.5 ppm or [CpFe(CO)2 = C(NCy)2B(NCy)2CNCy2]+, δC = 224.0 ppm),32b we observe the corresponding quaternary carbon resonance at δC = 153.6/153.8 ppm (for R = iPr/Cy). This observation suggests the formation of the isonitrile complexes [CpFe(CO)(PCy3)(NCR)]+[BArCl4]− (16/17, R = iPr/Cy, Scheme 7), which could also be detected by positive-ion ESI MS (SI).
 | ||
| Scheme 7 Reaction of iminoborylene complex 12d with carbodiimides to give isonitrile complexes 16/17. | ||
In case of 16, we were also able to isolate the metal-containing species by crystallization and unambiguously confirm its structure by X-ray crystallography (Fig. 3). In the solid state, complex 16 shows a piano-stool geometry, with the isonitrile unit featuring a linear geometry [∠ C(26)–N(27)–C(28) = 175.0(2)°], brought about by the presence of the C–N triple bond [C(26)–N(27) 1.163(3) Å]. In solution, complexes 16 and 17 show very similar spectroscopic features, e.g. resonances in the 1H and 13C NMR spectra for the Cp (δH = 4.92/4.92 ppm, δC = 84.2/84.3 ppm for 16/17) and CN–CHR2 units (CH: δH = 4.08/3.85 ppm, δC = 51.3/57.3 ppm for 16/17).
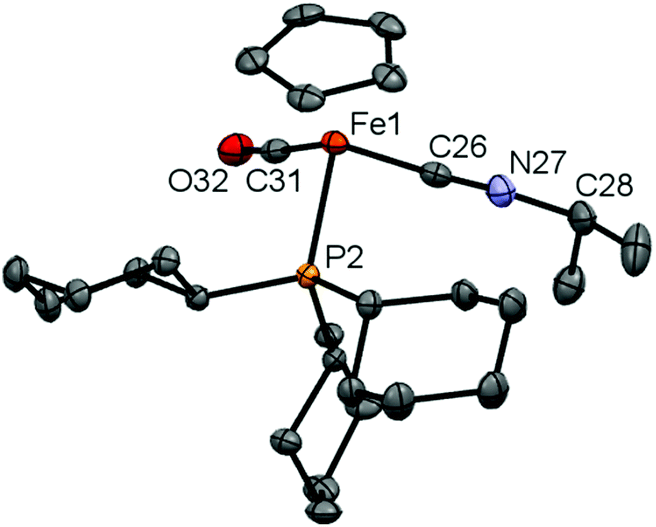 | ||
| Fig. 3 Molecular structure of 16 in the solid state, hydrogen atoms and counter-ion omitted for clarity and thermal ellipsoids set at the 40% probability level. Key bond lengths [Å] and angles [°]: Fe(1)–C(26) 1.850(2), C(26)–N(27) 1.163(3), N(27)–C(28) 1.461(3), Fe(1)–C(26)–N(27) 176.7(2), C(26)–N(27)–C(28) 175.0(2). | ||
This chemistry represents, to our knowledge, the first example of metathesis-type reactivity of a borylene complex towards a carbodiimide, and we therefore performed further investigations in order to better understand the reaction mechanism and to probe the fate of the boron-containing [BNCMes2] heterocumulene fragment.
Upon mixing of the 12d with the respective carbodiimide RNCNR at −60 °C in CD2Cl2, we observe the immediate formation of an intermediate (18/19 for R = iPr/Cy), which is stable at temperatures below 0 °C. Accordingly, we were able to characterize these species by multinuclear NMR spectroscopy. In the 1H and 13C NMR spectra we observe a splitting of the resonances for the ketimino aryl substituents (e.g. mesityl p-CH3 in 18/19: δH = 2.30, 2.25/2.31, 2.25 ppm), as is also seen for the boryl precursor 10d. In addition, we also observe two sets of resonances for the carbodiimide iPr/Cy substituents (e.g. for the N–CHR2 protons in 18/19: δH = 3.73, 3.01/3.24, 2.54 ppm), consistent with desymmetrization of the RNCNR unit. These spectroscopic features are consistent with the formation of either a Lewis acid–base adduct between the electrophilic boron and one of the carbodiimide nitrogens,32a,b,33b or with the formation of a [2 + 2]-cycloaddition product,30a both of which have been observed as intermediates in the reactions of carbodiimides with borylene complexes.
Somewhat unexpectedly, the 11B and 31P NMR resonances for 18/19 are shifted downfield in comparison to those observed for the free borylene (δB = 91/91 ppm, δP = 80.8/80.4 ppm for 18/19, cf. δB = 85 ppm, δP = 75.0 ppm for 12d). While these shifts imply retention of the Fe–B linkage at this stage in the reaction, they appear counter-intuitive for the formation of either a B-bound Lewis acid–base adduct or a [2 + 2]-cycloaddition product, both of which would be expected to lead to an upfield shift in the 11B NMR resonance. Thus, adducts of [CpFe(CO)2(BNR2)]+ (δB = 94 ppm) with carbodiimides or imines (adducts: δB = 71/54 ppm)7b,32b and the [2 + 2] cyclo-addition product of CpMn(CO)2(BtBu) (δB = 144 ppm) with carbodiimide (product: δB = 62 ppm) show upfield shifts in the 11B signal, consistent with an increased coordination number at boron.30c To an even greater extent, the 11B resonances measured for 18 and 19 contrast with those observed for the FeB insertion products formed in the reaction of the same carbodiimides with cationic iron aminoborylene complexes (e.g. δB = 25 ppm for [CpFe(CO)2{C(NCy)2BNCy2}]+).32b
In the 13C spectra the downfield shifts observed for the carbodiimide quaternary carbons (δC = 168.8/168.3 ppm for 18/19, cf. δC = 140.2/139.9 ppm for free RNCNR with R = iPr/Cy)34 are consistent with the formation of a direct metal-carbon interaction [cf. δC = 151.0, 162.0 for CpMn(CO)2{κ2-B(tBu)N(Cy)CNCy} and CpMn(CO)2{κ2-B(tBu)OCPh2}, respectively], although not with complete insertion into the FeB bond (cf. δC = 251.0 ppm for [CpFe(CO)2{C(NCy)2BNCy2}]+, which features partial FeC carbenoid character). Moreover, the observation of resonances at δc ≈ 180 ppm for the γ-carbons of the FeBNC units (along with the downfield 11B shifts for 18/19), suggests retention of a substantial degree of delocalization along the hetero-cumulene framework. With this in mind, we suggest the formation of an unsymmetrical [2 + 2]-cycloaddition product featuring a strong interaction between the metal and the central carbodiimide carbon and a relatively weak N → B interaction (Scheme 8).
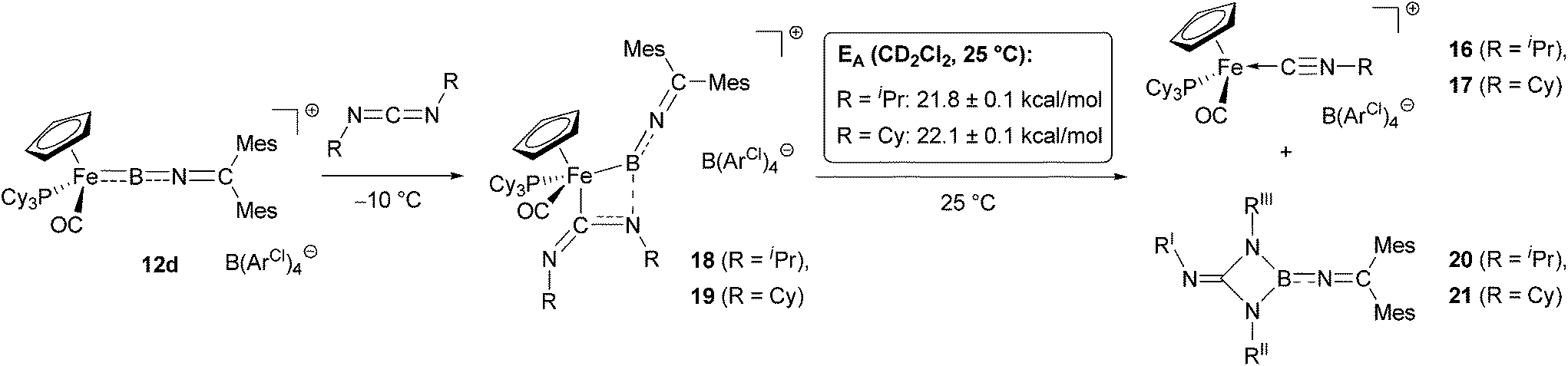 | ||
| Scheme 8 Pathway of the reaction of borylene complex 12d with carbodiimides. Formation of the intermediates 18/19 and subsequent reaction to give the final products 16/17 and 20/21 (tentative structures). | ||
Repeated attempts to obtain structural information on 18/19 by crystallization at low temperatures failed to give crystals suitable for X-ray analysis, and in contrast to the [2 + 2] cycloaddition products of carbodiimides with CpMn(CO)2(BtBu),30c solutions of 18/19 are labile, yielding isonitrile complexes 16/17 within a few hours at room temperature. The activation barriers for this step could be determined in each case by following of the intensity of the cyclopentadienyl 1H signals as a function of time. Values of 21.8 ± 0.1 kcal mol−1 and 22.1 ± 0.1 kcal mol−1 (at T = 25 °C) are thus obtained for 18 and 19, respectively (Scheme 8).
Finally, we sought to establish the fate of the boron-containing fragment in the final reaction mixture. When the reaction is performed with a stoichiometric amount of either carbodiimide, the 1H NMR spectra show the isonitrile complexes 16/17, together with a number of products containing mesityl- or isopropyl/cyclohexyl groups, respectively. Only in the presence of an excess of carbodiimide, could well-defined boron-containing products be isolated. The 11B resonances (δB = 29/30 ppm for R = iPr/Cy) indicate a three-coordinate boron centre without any metal–boron interaction, while the 1H NMR spectra show the presence of three inequivalent iPr- or Cy-groups [e.g. CHR2-units: δH = 3.90/3.39/3.61 for iPr-(I/II/III), 3.04/2.96/3.45 for Cy-(I/II/III), (for numbering see Scheme 8)]. The 13C-NMR and GHMBC-data indicate that all three alkylamino-substituents are bound to a central quaternary carbon (δC = 152.4/ 155.9 ppm), with two of the alkyl-groups (I and III, respectively) being in close proximity as seen from NOE difference spectra. Taken together, these observations suggest that in the presence of excess carbodiimide, RIINCNRIII, coordinative trapping of the initial metathesis product [RINBNCMes2] leads to the formation of a trialkyl-guanidinate, which is bound to the BNCMes heterocumulene fragment. The resulting triaminoboranes of the type RNC(NR)2BNCMes2 [with R = iPr (20) and R = Cy (21), Scheme 8] thus resemble the metalla-amidinates [CpFe(CO)2{C(NCy)2BNCy2}]+ formed by mono-carbodiimide insertion in the case of aminoborylene systems.32
Conclusions
Our investigation of the possible synthetic routes to iminoborylene complexes (12/13) has given insight into the scope of metal-fragments, ketimino substituents and ancillary ligands which allow for successful formation of the desired cationic heterocumulenes. For the synthesis of the iminoboryl-precursors, it is found that an optimal level of overall steric bulk, in combination with the correct order of bond formation (Fe–B prior to B–N bond formation), is required for the generation of the boryl complexes CpFe(CO)2{B(Cl)NCAr2} (Ar = p-Tol/Mes, 5c/d). The use of reagents with increased steric bulk on either the metal [CpW(CO)3vs. CpFe(CO)2] or the ketimino side (Trip vs. Mes) leads primarily to products resulting from M–B bond breakage, illustrating the sensitivity of the boron-centred substitution reaction to steric factors.While direct halide abstraction from complexes 5c/d leads to thermally unstable borylene species, the substitution of one carbonyl ligand for a tertiary phosphine drastically increases complex stability, leading to the isolation of the cationic hetero-allenylidenes [CpFe(PR3)(CO)(BNCAr2)]+ as borate salts (12c/d, 13d). The reactivity of these complexes towards nucleophilic substrates is dominated by the high electrophilicity of the boron centre, leading exclusively to α-attack, while the reactivity towards unsaturated substrates leads to unprecedented transformations. While no reactivity is observed towards isocyanates, we observe clean metathesis-type reactivity with carbodiimides. This contrasts with the insertion-type reactivity of closely related amino- and alkylborylene complexes towards the same substrates. Spectroscopic analysis of the reaction mixtures leads to identification of the boron-containing reaction products as the coordinatively trapped heteroallenes (20/21), with the metal-containing products being unambiguously identified as the isonitrile complexes (16/17). This reactivity is unprecedented and represents the first example of a productive metathesis-type reaction of a borylene compound with a carbodiimide.
Experimental
General considerations
All reactions involving air- or moisture-sensitive compounds were carried out under an inert atmosphere by using Schlenk-type glassware or in a glovebox. UV photolysis experiments were carried out using a Spectral Energy mercury arc lamp (1 kW) with samples contained within quartz Schlenk vessels. Solvents were dried using an MBraun SPS800 prior to use. NMR-solvents were dried over molecular sieves and degassed before use when necessary. Solid starting materials were dried on high vacuum before use when necessary. Unless otherwise noted, all starting materials were commercially available and were used without further purification. The following compounds were synthesized according to literature procedures (for references see ESI‡): Na[B(3,5-Cl2C6H3)4, Na[B(3,5-(CF3)2C6H3)4], Na[CpW(CO)3], Na[CpFe(CO)2] (1), (Ph2CNBCl2)2 (2b), CpFe(CO)2BCl2 (3), ketimino lithium salts 4a/b/c/d, boryl complexes 5c/d, 10c/d, 14c/d and 15d, borylene complexes 12c/d. For the synthesis of 4e see ESI.‡The following instruments were used for physical characterization of novel compounds: IR: Nicolet Magna-IR 560; NMR: Bruker AVC500 (1H: 500 MHz; 13C: 125 MHz); Bruker DRX500 (11B: 160 MHz), Varian Unity500 (1H: 500 MHz; 13C: 125 MHz, 11B: 160 MHz), Varian Mercury VX-300 (31P: 122 MHz, 19F: 282 MHz, 11B: 96 MHz). Mass spectra of compounds 12d, 16 and 17 were recorded on a Bruker Microtof mass-spectrometer. All other mass spectra were measured by the EPSRC National Mass Spectrometry Service Centre, Swansea University. For all crystallographic studies, diffraction data were collected at 150 K using an Enraf Nonius Kappa CCD diffractometer or an Oxford Diffraction/Agilent Technologies SuperNova instrument. For complete analytical data (including 2D-NMR data) and for details concerning the determination of the activation energies, see the ESI.‡
Syntheses
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 681 reflections collected, 2427 independent [R(int) = 0.0069], which were used in all calculations. R1 = 0.0578, wR2 = 0.1389 for observed unique reflections [F2 > 2σ(F2)] and R1 = 0.0834, wR2 = 0.1558 for all unique reflections. Max. and min. residual electron densities 3.87 and −4.01 e Å−3. CSD reference: 1037787.
500 reflections collected, 11969 independent [R(int) = 0.000], which were used in all calculations. R1 = 0.0384, wR2 = 0.0905 for observed unique reflections [F2 > 2σ(F2)] and R1 = 0.0590, wR2 = 0.0982 for all unique reflections. Max. and min. residual electron densities 0.97 and −0.65 e Å−3. CSD reference: 1037786.
C), 152.4 (NC(NR2)), 139.4 (p-Mes), 139.1 (i-Mes), 136.4 (o-Mes), 130.1 (m-Mes), 46.3 (CH(III)), 46.2 (CH(I)), 42.9 (CH(II)), 25.5 (CH3(I)), 23.2 (CH3(III)), 22.3 (CH3(II)), 21.5 (o-CH3), 21.0 (p-CH3); 11B NMR (96 MHz, [D2]dichloromethane, 298 K): δ = 29 (ν1/2 = 390 Hz).
681 reflections collected, 2427 independent [R(int) = 0.0069], which were used in all calculations. R1 = 0.0578, wR2 = 0.1389 for observed unique reflections [F2 > 2σ(F2)] and R1 = 0.0834, wR2 = 0.1558 for all unique reflections. Max. and min. residual electron densities 3.87 and −4.01 e Å−3. CSD reference: 1037787.
500 reflections collected, 11969 independent [R(int) = 0.000], which were used in all calculations. R1 = 0.0384, wR2 = 0.0905 for observed unique reflections [F2 > 2σ(F2)] and R1 = 0.0590, wR2 = 0.0982 for all unique reflections. Max. and min. residual electron densities 0.97 and −0.65 e Å−3. CSD reference: 1037786.
C), 152.4 (NC(NR2)), 139.4 (p-Mes), 139.1 (i-Mes), 136.4 (o-Mes), 130.1 (m-Mes), 46.3 (CH(III)), 46.2 (CH(I)), 42.9 (CH(II)), 25.5 (CH3(I)), 23.2 (CH3(III)), 22.3 (CH3(II)), 21.5 (o-CH3), 21.0 (p-CH3); 11B NMR (96 MHz, [D2]dichloromethane, 298 K): δ = 29 (ν1/2 = 390 Hz).
1H NMR (500 MHz, [D2]dichloromethane, 293 K): δ = 6.85 (s, 4 H, m-Mes), 3.45 (m, Cy(III)-1), 3.04 (m, Cy(I)-1), 2.96 (m, Cy(II)-1), 2.24 (s, 6 H, p-CH3), 2.13 (s, 12 H, o-CH3), 1.75, 1.53, 1.42, 1.07, 0.80 (each m, 10 H, Cy(I)-2, Cy(I)-3, Cy(I)-4), 1.65, 1.49, 1.41, 1.08, 0.77 (each m, 10 H, Cy(II)-2, Cy(II)-3, Cy(II)-4), 1.70, 1.58, 1.27, 1.18 (Cy(III)-2, Cy(III)-3 Cy(III)-4); 13C NMR (126 MHz, [D2]dichloromethane, 293 K): δ = 172.1 (BNC), 155.9 (NC(NR2)), 139.7 (i-Mes), 139.1 (p-Mes), 136.6 (o-Mes), 130.2 (m-Mes), 54.7 (Cy(III)-1), 54.6 (Cy(I)-1), 50.8 (Cy(II)-1), 21.6 (o-CH3), 21.0 (p-CH3); 11B NMR (96 MHz, [D2]dichloromethane, 293 K): δ = 30 (ν1/2 = 350 Hz).
Acknowledgements
We thank the EPSRC for funding and for access to the National Mass Spectrometry Facility, Swansea University. J. Niemeyer thanks the DFG for a postdoctoral fellowship.Notes and references
-
(a) H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed
; (b) H. Braunschweig, C. Kollann and D. Rais, Angew. Chem., Int. Ed., 2006, 45, 5254–5274 CrossRef CAS PubMed
-
(a) R. T. Baker, D. W. Ovenall, J. C. Calabrese, S. A. Westcott, N. J. Taylor, I. D. Williams and T. B. Marder, J. Am. Chem. Soc., 1990, 112, 9399–9400 CrossRef CAS
-
(a) J. F. Hartwig, Chem. Soc. Rev., 2011, 40, 1992–2002 RSC
-
(a) H. Braunschweig, Q. Ye and K. Radacki, Chem. Commun., 2012, 48, 2701–2703 RSC
-
(a) H. Braunschweig, R. D. Dewhurst and V. H. Gessner, Chem. Soc. Rev., 2013, 42, 3197–3208 RSC
-
(a) D. Vidovic and S. Aldridge, Chem. Sci., 2011, 2, 601–608 RSC
-
(a) S. Aldridge, C. Jones, T. Gans-Eichler, A. Stasch, D. L. Kays, N. D. Coombs and D. J. Willock, Angew. Chem., Int. Ed., 2006, 45, 6118–6122 CrossRef CAS PubMed
-
(a) G. Alcaraz, A. B. Chaplin, C. J. Stevens, E. Clot, L. Vendier, A. S. Weller and S. Sabo-Etienne, Organometallics, 2010, 29, 5591–5595 CrossRef CAS
- H. Braunschweig, R. D. Dewhurst, T. Herbst and K. Radacki, Angew. Chem., Int. Ed., 2008, 47, 5978–5980 CrossRef CAS PubMed
-
(a) H. Braunschweig, R. D. Dewhurst, K. Radacki, C. W. Tate and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 6263–6266 CrossRef CAS PubMed
-
(a) J. Brand, H. Braunschweig, F. Hupp, A. K. Phukan, K. Radacki and S. S. Sen, Angew. Chem., Int. Ed., 2014, 53, 2240–2244 CrossRef CAS PubMed
- J. Niemeyer, D. A. Addy, I. Riddlestone, M. Kelly, A. L. Thompson, D. Vidovic and S. Aldridge, Angew. Chem., Int. Ed., 2011, 50, 8908–8911 CrossRef CAS PubMed
- For isomeric B–N–C–M heterocumulenes see:
(a) A. J. V. Marwitz, S. P. McClintock, L. N. Zakharov and S.-Y. Liu, Chem. Commun., 2010, 46, 779–781 RSC
-
(a) V. Cadierno and J. Gimeno, Chem. Rev., 2009, 109, 3512–3560 CrossRef CAS PubMed
-
(a) S. Bertsch, R. Bertermann, H. Braunschweig, A. Damme, R. D. Dewhurst, A. K. Phukan, C. Saalfrank, A. Vargas, B. Wennemann and Q. Ye, Angew. Chem., Int. Ed., 2014, 53, 4240–4243 CrossRef CAS PubMed
-
(a) D. A. Addy, J. I. Bates, M. J. Kelly, I. M. Riddlestone and S. Aldridge, Organometallics, 2013, 32, 1583–1586 CrossRef CAS
-
(a) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, K. Kraft, T. Kramer and K. Radacki, Chem. – Eur. J., 2013, 19, 13402–13407 CrossRef CAS PubMed
-
(a) J. R. Jennings, I. Pattison and K. Wade, J. Chem. Soc. A, 1969, 565–568 RSC
- D. L. Kays, A. Rossin, J. K. Day, L.-L. Ooi and S. Aldridge, Dalton Trans., 2006, 399–410 RSC
-
(a) H. Braunschweig, K. Radacki, F. Seeler and G. R. Whittell, Organometallics, 2006, 25, 4605–4610 CrossRef CAS
- I. Pattison, K. Wade and B. K. Wyatt, J. Chem. Soc. A, 1968, 837–842 RSC
- D. A. Addy, J. I. Bates, D. Vidovic and S. Aldridge, J. Organomet. Chem., 2013, 745–746, 487–493 CrossRef CAS PubMed
- G. Schmid and H. Nöth, J. Organomet. Chem., 1967, 7, 129–134 CrossRef CAS
- S. S. Batsonov, Inorg. Mater., 2001, 37, 1031–1046 Search PubMed
-
(a) D. A. Addy, N. Phillips, G. A. Pierce, D. Vidovic, T. Kramer, D. Mallick, E. D. Jemmis, G. Reid and S. Aldridge, Organometallics, 2012, 31, 1092–1102 CrossRef CAS
- G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, A. L. Thompson, E. D. Jemmis, S. De and S. Aldridge, Organometallics, 2009, 28, 2947–2960 CrossRef CAS
- M. A. Esteruelas, A. V. Gómez, F. J. Lahoz, A. M. López, E. Oñate and L. A. Oro, Organometallics, 1996, 15, 3423–3435 CrossRef CAS
-
CRC Handbook of Chemistry and Physics 1999-2000: A Ready-Reference Book of Chemical and Physical Data, ed. D. R. Lide, CRC Press, Boca Raton, Florida, USA, 2000 Search PubMed
-
(a) M. I. Bruce, P. J. Low and E. R. T. Tiekink, J. Organomet. Chem., 1999, 572, 3–10 CrossRef CAS
-
(a) J. Bauer, H. Braunschweig, A. Damme, J. O. Carlos, J.-H. T. Kramer, K. Radacki, R. Shang, E. Siedler and Q. Ye, J. Am. Chem. Soc., 2013, 135, 8726–8734 CrossRef CAS PubMed
- D. L. Kays, J. K. Day, S. Aldridge, R. W. Harrington and W. Clegg, Angew. Chem., Int. Ed., 2006, 45, 3513–3516 CrossRef CAS PubMed
-
(a) S. De, G. A. Pierce, D. Vidovic, D. L. Kays, N. D. Coombs, E. D. Jemmis and S. Aldridge, Organometallics, 2009, 28, 2961–2975 CrossRef CAS
-
(a) M. Findlater, N. J. Hill and A. H. Cowley, Polyhedron, 2006, 25, 983–988 CrossRef CAS PubMed
- I. Yavari and J. D. Roberts, J. Org. Chem., 1978, 43, 4689–4693 CrossRef CAS
Footnotes |
| † Dedicated to the memory of Professor Ken Wade FRS. |
| ‡ Electronic Supplementary Information (ESI) available: Synthetic details and analytical data for all compounds. CCDC 1037786 and 1037787 (crystallographic data for 6 and 16). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5dt00131e |
| § Current address: Division of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological, University, 21 Nanyang Link, 637371 Singapore. |
| This journal is © The Royal Society of Chemistry 2015 |