
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecoverable and recyclable water-soluble sulphonated salicylaldimine Rh(I) complexes for 1-octene hydroformylation in aqueous biphasic media†
Leah C.
Matsinha
a,
Selwyn F.
Mapolie
b and
Gregory S.
Smith
*a
aDepartment of Chemistry, University of Cape Town, P. Bag X3, Rondebosch 7701, Cape Town, South Africa. E-mail: Gregory.Smith@uct.ac.za
bDepartment of Chemistry and Polymer Science, Stellenbosch University, Matieland 7602, South Africa
First published on 21st November 2014
Abstract
A series of water-soluble Rh(I) mononuclear complexes of general formula: [Rh(sulphsal-X-R)(COD)] [sulphsal = sulphonated salicylaldimine, COD = cyclooctadiene; where R = H, Cl, CH3 and X = H, tBu] have been synthesized. All the compounds were characterised using various spectroscopic and analytical techniques such as nuclear magnetic resonance spectroscopy, infrared spectroscopy, single crystal X-ray diffraction (for complex 10) and mass spectrometry. All the compounds display excellent water-solubility at room temperature and were tested as catalyst precursors in the aqueous biphasic hydroformylation of 1-octene. The catalysts could be easily recovered by phase separation and were used up to 5 times without any significant loss in activity and 1-octene conversion. Very high yields of the expected aldehydes were obtained without addition of any phase transfer agents, co-solvents or hydrophobic ligands. Excellent aldehyde chemoselectivity is observed for all the catalysts but this varied each time the catalysts were recycled, with the formation of a small amount of internal olefins. ICP-OES and mercury poisoning experiments show that a combination of homogeneous catalysis and catalysis mediated by nanoparticles is taking place in these systems.
Introduction
The hydroformylation reaction is an important reaction for the synthesis of aldehydes in the chemical industry. The process involves the transition metal-catalysed reaction of olefins with hydrogen and carbon monoxide to afford aldehydes which can further be processed to produce detergents and plasticizers.1 For this reaction to be economically viable and sustainable, it is important that the active metal catalyst be recoverable and recyclable. One strategy that enables both recovery and recyclability is aqueous biphasic catalysis.2a–cPioneering work into aqueous biphasic catalysis can be traced back to the Ruhrchemie/Rhône-Poulenc (RCH/RP) process.2a–f The process employs a highly water-soluble TPPTS-modified Rh-hydrido carbonyl complex as catalyst for the hydroformylation of propene. Aqueous biphasic catalysis has been widely explored for the easy recovery of catalysts by phase separation and this technique is currently in operation in five plants around the world.3 This technique is also a strategy to achieve environmentally friendly, active, selective and highly economically viable catalysts in line with Green Chemistry Practices.4–12 This technique has been used widely including applications in various olefin transformation reactions.13–24Fig. 1 shows an illustration of the aqueous biphasic hydroformylation of 1-octene.
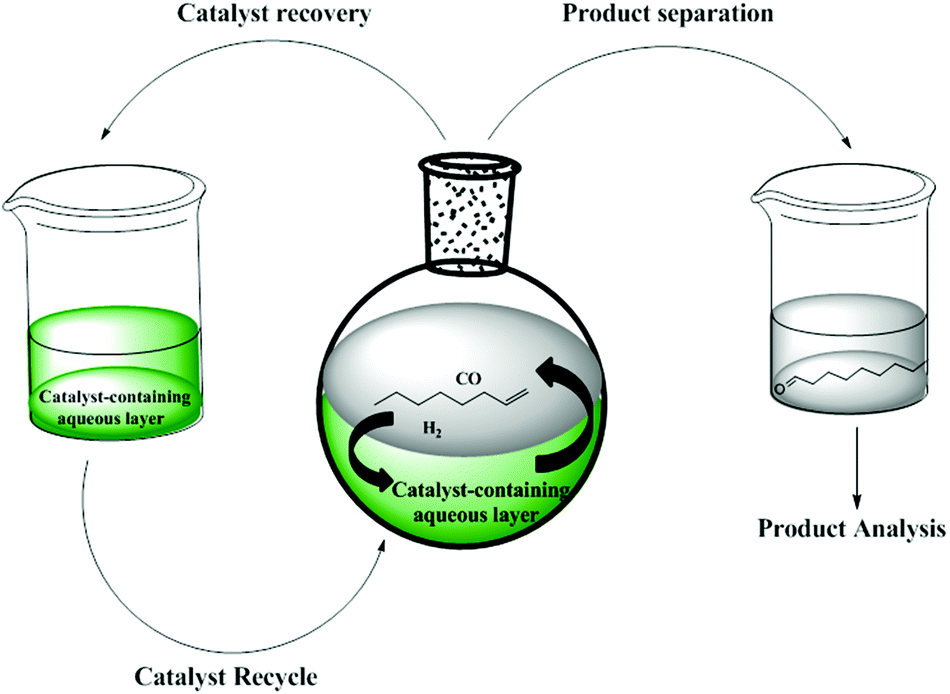 | ||
| Fig. 1 Aqueous biphasic hydroformylation. | ||
The concept of aqueous biphasic hydroformylation involves a catalyst-containing aqueous layer and a substrate-containing organic layer which form two immiscible layers. The active catalyst remains in the aqueous layer so that the reactants and reaction products which are entirely organic can easily be phase separated from the catalyst. Besides easy catalyst recovery, this technique is advantageous as it makes use of water, a green solvent, which is non-toxic, non-flammable, odourless and readily available in huge quantities at low cost.2a,4,5,12a Various ligands can be used in order to fine-tune the selectivity and activity of the catalysts and ligand basicity has been shown to have a pronounced influence on the hydroformylation rates. Previously, we have reported the synthesis and aqueous biphasic hydroformylation of 1-octene using sulphonated Rh(I) mononuclear complexes together with their dendritic analogues.4 In our previous work, the mononuclear derivatives display better activity and chemoselectivity for the desired aldehyde products in comparison with the polynuclear analogues. The metallodendrimers could not be isolated and they did not give better catalytic results in comparison to the mononuclear derivatives. These results prompted us to expand our evaluation of the mononuclear analogues. In this paper, we report the synthesis and characterisation of a series of new water-soluble sulphonated mononuclear Rh(I) complexes and their evaluation in the aqueous biphasic hydroformylation of 1-octene.
Results and discussion
Synthesis and characterisation of water-soluble sulphonated ligands
The sulphonated imine ligands were prepared by stirring the known sulphonated aldehydes 1 and 24 with equimolar equivalents of various amines at room temperature overnight (Scheme 1). All ligands 3–5 were isolated as bright yellow solids that are stable and readily soluble in water at room temperature.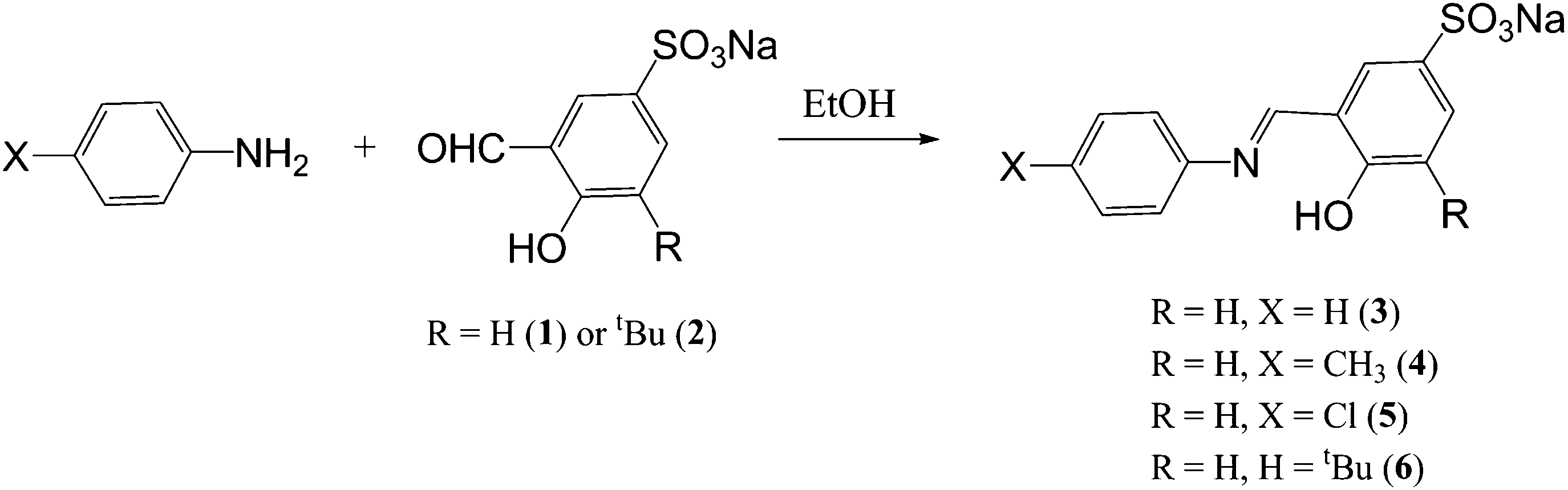 | ||
| Scheme 1 Synthesis of water-soluble sulphonated salicylaldimine ligands (3–5). | ||
The 1H NMR spectra of the ligands (3–5) show a characteristic imine singlet between 8.55 ppm and 9.06 ppm. The presence of this signal confirms a successful Schiff base condensation reaction to form a new imine bond which is similar to what has been reported previously for similar compounds.4 For ligand 4 a singlet at 7.96 ppm for the proton ortho to the imine is seen and a doublet for the proton para to the imine at 7.65 ppm (3J = 8.3 Hz) is observed. This doublet is observed since this proton is coupled to the proton meta to the imine. Similar trends in the 1H NMR spectra of 3 and 5 are observed. The imine functionality is also seen in the infrared spectra of the compounds and appears as an intense absorption band between 1615 cm−1 and 1621 cm−1 for these compounds. The ESI-MS spectra show peaks for [M]− in the negative mode at m/z = 310 (4) and 290 (5) where M is the anion.
Synthesis and characterisation of water-soluble sulphonated Rh(I) complexes
The sulphonated salicylaldimine ligands (3–5) were dissolved or suspended in a minimum amount of water and/or ethanol. Deprotonation of the phenolic proton was achieved using an equimolar equivalent of KOH. The metal precursor [Rh(COD)Cl]2 was then allowed to react with the ligands as depicted in Scheme 2. The complexes were obtained as bright yellow solids. The synthesis of ligand 6 and complex 10 (R = tBu and X = H) was performed in a one-pot reaction because the ligand is a sticky hygroscopic oil. The compounds are stable at room temperature and are insoluble in hexane, ethanol and diethyl ether but display excellent water solubility at room temperature (0.4 mg mL−1–5 mg mL−1).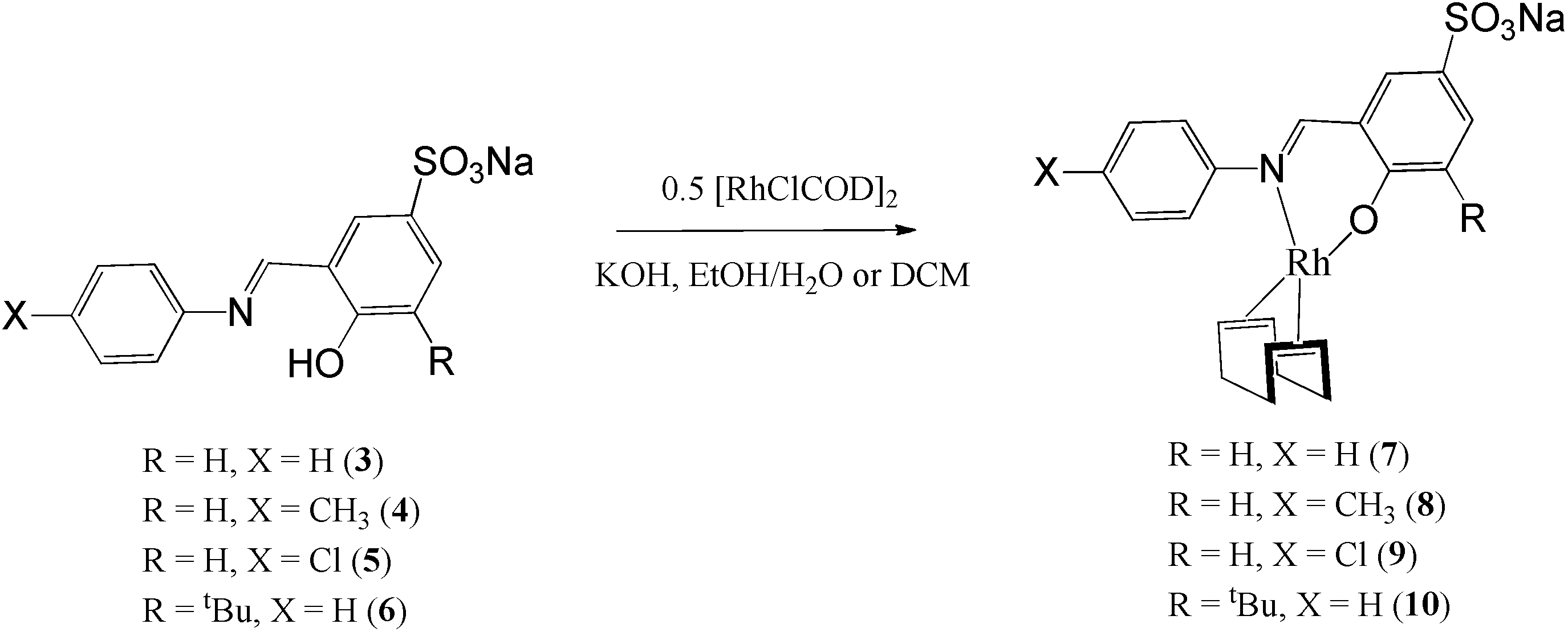 | ||
| Scheme 2 Synthesis of water-soluble 5-sulphonato Rh(I) complexes (7–10). | ||
In the 1H NMR spectra of the compounds (7–10), a distinct imine signal is observed. Of interest, is the upfield shift of the signal to chemical shifts between 8.13 ppm and 7.36 ppm, in contrast to downfield chemical shifts between 8.55 ppm and 9.06 ppm in the metal-free ligands. The upfield shift of the signals assigned to the proton of the imine functionality upon coordination of the metal is due to increased shielding of this proton due to back-donation of the Rh metal via the imine nitrogen. Two multiplets are observed between 2.35 ppm and 1.84 ppm for the cyclooctadiene methylene protons whilst the olefinic protons appear between 4.28 ppm and 4.07 ppm. In the 13C NMR spectra of these compounds, the number of signals observed agrees with the number of carbon atoms in the compounds. The infrared spectra of the compounds show a characteristic imine absorption band at lower wavenumbers (1606 cm−1–1602 cm−1) compared to those observed in the metal-free ligands (1615 cm−1–1621 cm−1). These shifts to lower wavenumbers are due to the synergic effect. The process occurs because the imine nitrogen lone pair of electrons is withdrawn to the empty orbitals of the metal. This together with the release of electrons from the metal d-orbitals (back-donation) into the empty π-anti bonding orbitals of the ligand results in weakening of the imine bonds and consequently lowers the imine stretching frequency. This together with the disappearance of the OH vibration in the infrared spectra is evidence of coordination of the ligand to the Rh metal centre in a bidentate fashion. Electrospray ionisation mass spectra of these water-soluble complexes were recorded in the negative ion mode and show peaks at m/z = 521 (8), 500 (9) and 488 (10) respectively for [M]− where M is the sulphonated anionic complex. Complex 10 was also characterised using single-crystal X-ray diffraction. The crystals were obtained by slow diffusion of diethyl ether into a concentrated solution of the compound dissolved in acetonitrile. The ORTEP drawing with the atom labelling scheme for this complex is shown in Fig. 2.
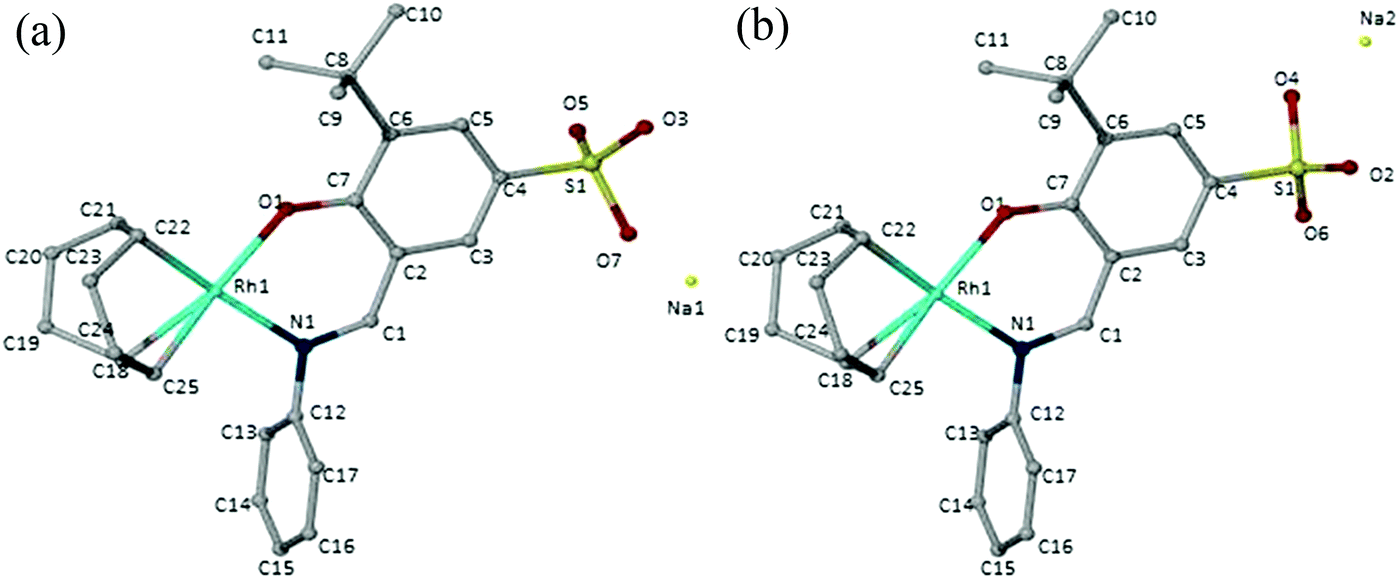 | ||
| Fig. 2 Molecular structure of 10 determined by single crystal X-ray diffraction. | ||
Solvent molecules are also observed as these co-crystallized with the complex during formation of the crystals. These have been omitted in Fig. 2 for clarity. The oxygen atoms on sulphonate group (O2–O7) and the sodium atoms (Na1 and Na2) are disordered over two positions (a) and (b) and these were refined with 50% site occupancy factors. The molecular structure of complex 10 shows a square planar geometry at the Rh metal centre, with the metal coordinated to the cyclooctadiene moiety and the N,O chelating ligand. The bond angles around the Rh metal centre are between 81° and 96° and this is similar to what has been reported for similar compounds in the literature.25 A slightly distorted tetrahedral geometry is observed around the sulphur atom with bond angles between 105° and 120°. From the data obtained, O7–S1 and O4–S1 are the longest bonds around the sulphur atom and hence the single bonds of the sulphonate moiety. Selected crystallographic data, bond angles and bond distances are summarised in Tables 1 and 2.
| Bond lengths (Å) | |||
| Rh1–N1 | 2.062(4) | O3–S1 | 1.516(8) |
| Rh1–O1 | 2.037(3) | O5–S1 | 1.315(8) |
| Rh1–C18 | 2.137(5) | O7–S1 | 1.568(8) |
| Rh1–C21 | 2.129(6) | O2–S1 | 1.329(12) |
| Rh1–C22 | 2.147(6) | O6–S1 | 1.400(8) |
| Rh1–C25 | 2.110(5) | O4–S1 | 1.598(8) |
| Na1–O7 | 1.484(9) | Na2–O4 | 1.711(10) |
| Bond angles (°) | |||
| N1–Rh1–O1 | 89.85(15) | O5–S1–O3 | 115.6(5) |
| N1–Rh1–C25 | 95.89(18) | O3–S1–O7 | 105.4(6) |
| O1–Rh1–C21 | 87.34(19) | O7–S1–O5 | 113.8(5) |
| O1–Rh1–C22 | 86.85(18) | O4–S1–O2 | 109.9(6) |
| C25–Rh1–C22 | 82.5(2) | O2–S1–O6 | 119.7(7) |
| C18–Rh1–C22 | 90.8(2) | O6–S1–O4 | 108.6(6) |
| Complex 10 | |
|---|---|
| Chemical formula | C28H35.50N1.50NaO4.50RhS |
| Formula weight | 623.04 |
| Crystal system | Monoclinic |
| Space group | C2/c |
| Crystal color and shape | Red block |
| Crystal size | 0.18 × 0.12 × 0.08 |
| a/Å | 31.483(3) |
| b/Å | 20.1500(17) |
| c/Å | 10.3174(8) |
| α/° | 90.00 |
| β/° | 90.00 |
| γ/° | 90.00 |
| V/Å3 | 6286.4(9) |
| Z | 8 |
| T/K | 173(2) |
| D c/g cm−3 | 1.317 |
| μ/mm−1 | 0.656 |
| Unique reflections | 6915 |
| Reflections used [I > 2s(I)] | 3913 |
| R int | 0.072 |
| Final R indices [I > 2s(I)] | 0.0555, wR2 0.1657 |
| R indices (all data) | 0.1186 |
| Goodness-of-fit | 0.988 |
| Max, Min Δρ/e A−3 | 0.66, −0.51 |
Aqueous biphasic hydroformylation of 1-octene
The complexes (7–10) were tested as catalyst precursors in the aqueous biphasic hydroformylation of 1-octene. To obtain the best working conditions for the catalysts, optimisation was performed using the simplest catalyst precursor 7 (where X = R = H). Scheme 3 shows the reaction of 1-octene with syngas (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CO–H2) in the presence of Rh(I) catalyst to form aldehydes as the major products and internal olefins as the minor products. The experiments were carried out at 30 bar and 50 bar while the temperature was varied from 75 °C to 95 °C. All the reactions were performed for 8 h. The organic layer was analysed using gas chromatography with n-decane as the internal standard.
:1 CO–H2) in the presence of Rh(I) catalyst to form aldehydes as the major products and internal olefins as the minor products. The experiments were carried out at 30 bar and 50 bar while the temperature was varied from 75 °C to 95 °C. All the reactions were performed for 8 h. The organic layer was analysed using gas chromatography with n-decane as the internal standard.
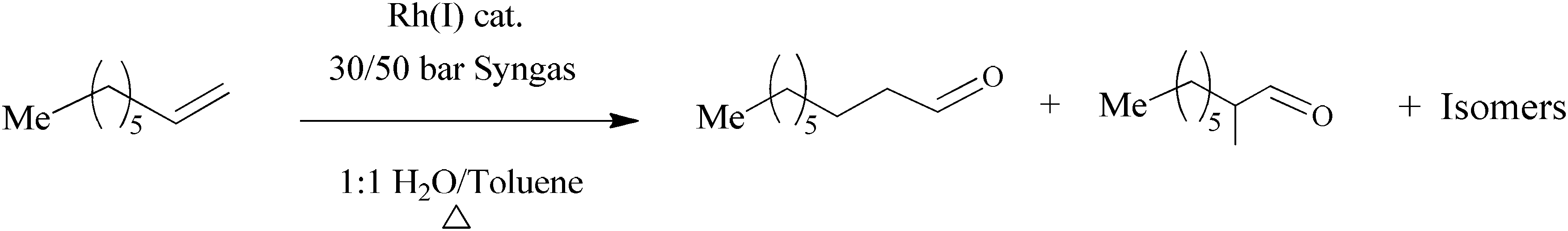 | ||
| Scheme 3 Hydroformylation of 1-octene. | ||
Chemoselectivity and regioselectivity of the catalysts
The catalysts display excellent aldehyde chemoselectivity, with only a slight formation of internal olefins for catalyst 7 (Table 3). Over 99% of the products formed are aldehydes and this is very similar to what has been reported for similar catalysts by Hager and coworkers.4 The presence of a chloro-group and a methyl substituent in catalysts 8 and 9 does not seem to affect the chemoselectivity of these catalysts.| Catalyst | Pressure (bar) | Temperature (°C) | Conversion (%) | Aldehydes (%) | Iso-octenes (%) | n:iso | TOF/h |
|---|---|---|---|---|---|---|---|
|
a The reactions were performed in a 90 mL stainless steel pipe reactor. The reactor was charged with 1:1 toluene–H2O (10 mL), 1-octene (6.37 mmol), internal standard n-decane (1.26 mmol) and catalyst precursor (2.87 × 10−3 mmol). The reactor was flushed with nitrogen three times, followed by flushing twice with syngas (1:1 CO–H2).
|
|||||||
| 7 | 50 | 95 | 98 | 99 | 0.6 | 0.75 | 276 |
| 8 | 50 | 95 | >99 | >99 | — | 0.61 | 276 |
| 9 | 50 | 95 | >99 | >99 | — | 0.16 | 277 |
| 10 | 50 | 75 | >99 | >99 | — | 2.37 | 276 |
Catalyst precursors 7, 8 and 9, all favour the formation of the branched aldehydes (>55%). A closer look at the results shows that almost 60% of the aldehydes formed with catalyst 10 are linear aldehydes whilst almost 60% of the aldehydes formed with the other catalysts are branched aldehydes. This is expected for catalysts with bulky substituents.26 These N,O based chelating systems show inferior regioselectivity for the linear products when compared to previously reported N,N and N,P based catalysts that have been previously reported for the hydroformylation of 1-octene.27,28
Recyclability of the catalysts
The recovery of the catalysts was done by decanting the organic layer. The recovered catalyst-containing aqueous layer was reused in a new catalytic run. All the catalysts displayed excellent recyclability and could be used up to 5 times without significant drop in catalyst activity and 1-octene conversions. The chemoselectivity of the catalysts did not vary significantly as shown in Fig. 3.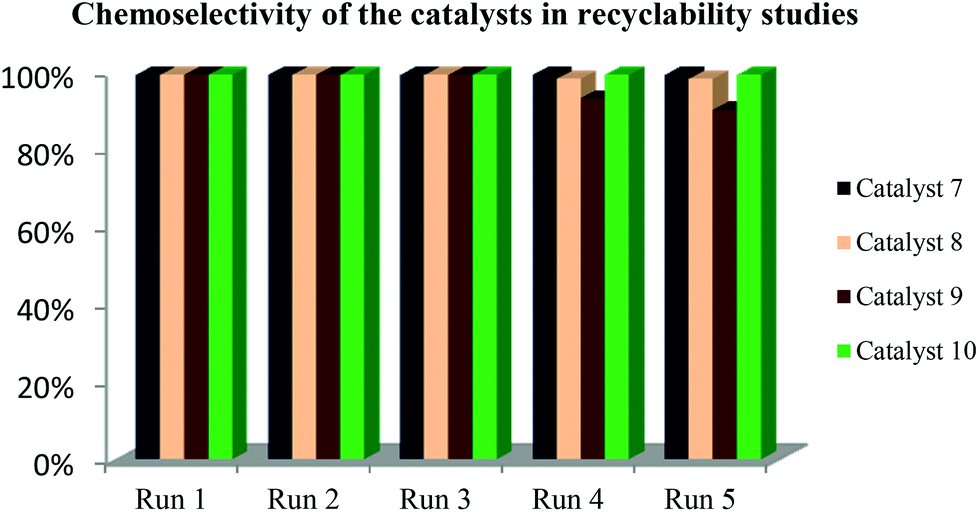 | ||
| Fig. 3 Chemoselectivity of the catalysts in recyclability studies. The reactions were performed in a 90 mL stainless steel pipe reactor. Solvent 1:1 toluene–water (10 mL), 1-octene (6.37 mmol), internal standard n-decane (1.26 mmol), catalyst (2.87 × 10−3 mmol), Syngas (1:1 CO: H2), 8 h. | ||
There is a slight decrease in aldehyde production with increase in the number of recycles with catalyst 9 (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H, XCl) in the fourth and fifth recycle. From the results, catalyst 8 (RH, XCH3) performs better than 9 since it maintains good selectivity for aldehydes throughout the five cycles. It has been reported that the more electron-withdrawing the substituents in the ligand, the more basic the catalyst becomes and hence the less it favours high hydroformylation rates.13,29,30 This is not observed in the results except the slight drop in the aldehyde chemoselectivity with 9 in the fourth recycle, which could be due to altering of structure of the catalyst.
H, XCl) in the fourth and fifth recycle. From the results, catalyst 8 (RH, XCH3) performs better than 9 since it maintains good selectivity for aldehydes throughout the five cycles. It has been reported that the more electron-withdrawing the substituents in the ligand, the more basic the catalyst becomes and hence the less it favours high hydroformylation rates.13,29,30 This is not observed in the results except the slight drop in the aldehyde chemoselectivity with 9 in the fourth recycle, which could be due to altering of structure of the catalyst.
The regioselectivity of the each catalyst varies slightly each time the catalyst is recycled as shown in Fig. 4. This could be attributed to changes in the structure of the active catalyst as it is recycled. Catalyst 10 produces more of the linear product (nonanal). This is expected since bulky substituents on the catalysts favour the formation of the linear products.
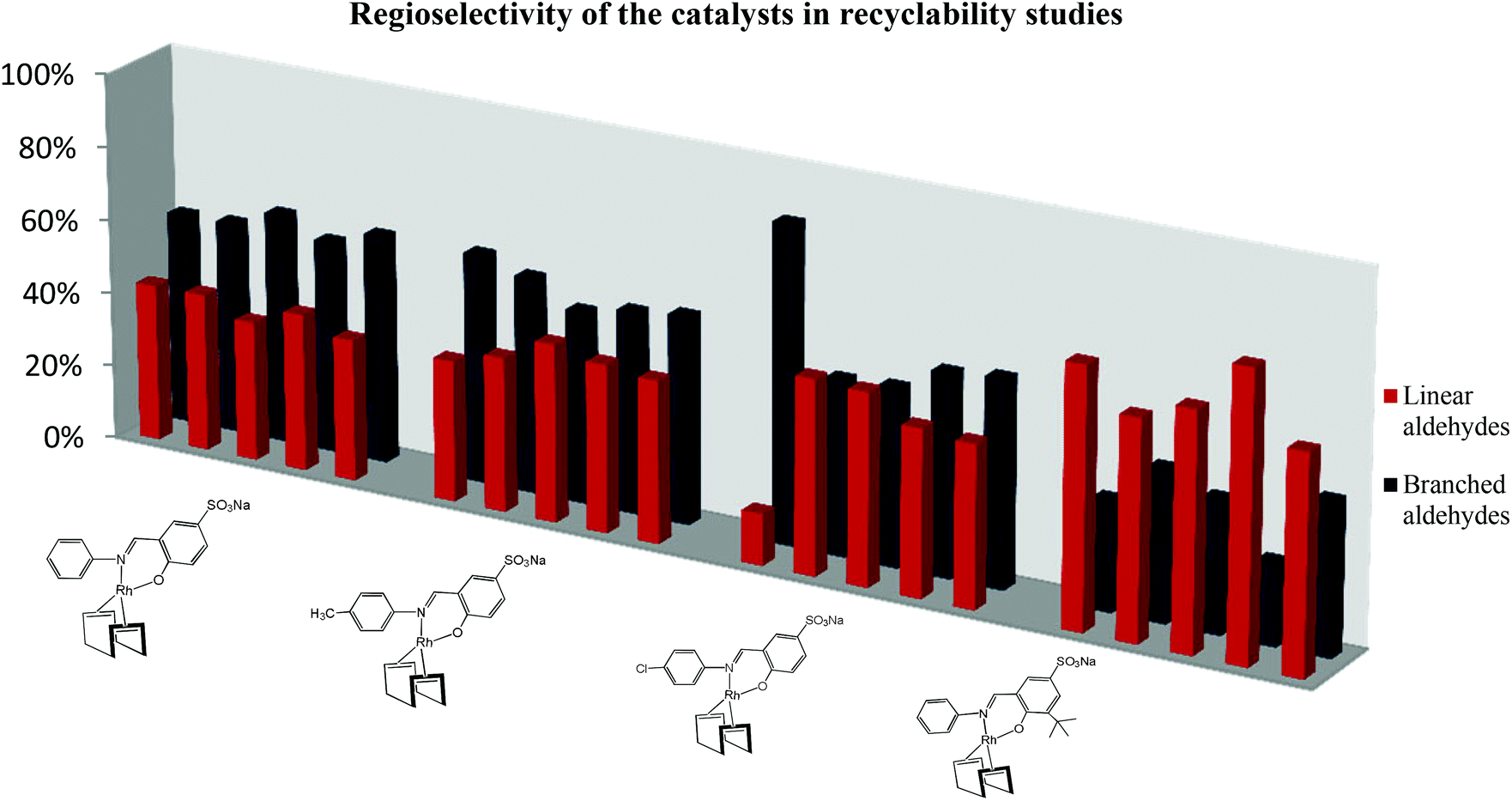 | ||
| Fig. 4 Regioselectivity of the catalysts in five recycles. The reactions were performed in a 90 mL stainless steel pipe reactor. Solvent 1:1 toluene–water (10 mL), 1-octene (6.37 mmol), internal standard n-decane (1.26 mmol), catalyst (2.87 × 10−3 mmol), Syngas (1:1 CO: H2), 8 h. | ||
Despite this, a considerable amount of linear aldehydes is also formed when catalysts 7, 8 and 9 are employed and this trend is maintained in all the recycles. Catalyst 9 shows an unusual trend, with almost 80% of the aldehydes in the first run are the linear aldehydes. There is a significant drop of nonanal production in the second recycle and after this almost 1:1 ratio of linear to branched aldehydes are formed. In all experiments, it was observed that the colour of the aqueous layer changed from bright yellow in the first cycle to almost colourless in the fifth cycle. The presence of black particles is observed and the amount of these black species increases with the number of recycles. To determine whether the original catalyst was still present in the aqueous solution, inductively coupled plasma optical spectrometry was performed on both the aqueous and organic layers at the beginning of the first cycle and at the end of the fifth cycle for each catalyst. The organic layers was analysed to see if leaching of the catalyst into the toluene layer was occurring.
Inductively coupled plasma optical spectrometry experiments
The analyses show that a significant amount of the metal complexes formed Rh nanoparticles towards the end of the third cycle which were suspended in the aqueous layer as a black species. Over 99% of the metal complex was no longer available in solution in the case of catalysts 9 and 10. For catalysts 8 and 10 less than 0.5% Rh metal was detected in the aqueous phase and no traces of metal were present in the organic layer. However, 1-octene conversion to products is still observed even though very little Rh is present in solution. The black Rh particles present as a suspension in the aqueous layer are therefore responsible for the activity observed. These together with the little metal complex in solution result in conversion of 1-octene to aldehydes and iso-octenes. The increase in the amount of iso-octenes formed with each recycle, could be due to these Rh particles which seem appear to favour isomerisation of 1-octene. As the formation of the particles becomes more pronounced in the third recycle of the catalysts, at this point isomerisation products become significant. To confirm the role of the Rh particles in the systems, a mercury drop test was performed.Mercury poisoning
Suppressing unwanted heterogeneous catalysts is a very important way to determine to what extent a catalyst is entirely homogeneous or whether it is a combination of both homogeneous and heterogeneous catalysis taking place. The mercury poisoning experiment was performed to establish whether the presence of Rh nanoparticles is responsible for substrate conversion to products. This was done using catalyst 9 which shows a significant amount of nanoparticle formation in the second recycle. Table 4 below shows a summary of the results obtained.| Cycle | Pressure (bar) | Temperature (°C) | Conversion (%) | Aldehydes (%) | Iso-octenes (%) | n:iso | TOF/h |
|---|---|---|---|---|---|---|---|
|
a The reactions were performed in a 90 mL stainless steel pipe reactor. The reactor was charged with 1:1 toluene–water (10 mL), 1-octene (6.37 mmol), internal standard n-decane (1.26 mmol) and suitable catalyst precursor (2.87 × 10−3 mmol). The reactor was flushed with nitrogen three times, followed by flushing twice with syngas (1:1 CO–H2). Each catalyst was recycled 5 times and all reactions were performed for 8 hours.
|
|||||||
| No mercury | |||||||
| 1 | 50 | 95 | >99 | >99 | — | 0.61 | 276 |
| 2 | 50 | 95 | 96 | >99 | — | 0.71 | 276 |
| 3 | 50 | 95 | 99 | >99 | — | 0.93 | 273 |
| 4 | 50 | 95 | 98 | 98 | 2 | 0.83 | 268 |
| 5 | 50 | 95 | 92 | 98 | 2 | 0.78 | 257 |
| With mercury | |||||||
| 1 | 50 | 95 | 91 | 48 | 52 | 2.55 | 121 |
| 2 | 50 | 95 | 64 | 47 | 53 | 1.45 | 62 |
| 3 | 50 | 95 | 60 | 35 | 65 | — | 52 |
| 4 | 50 | 95 | — | — | — | — | — |
| 5 | 50 | 95 | — | — | — | — | — |
In the presence of mercury, the catalyst can only be recycled 3 times. There is a decrease in conversion, aldehyde chemoselectivity and activity (TOF) in the presence of mercury. Initially, the homogeneous catalyst is responsible for the high conversion. However, in the second recycle conversion and activity drop significantly. At this stage both the homogeneous catalyst and heterogeneous catalysts are responsible for the conversions observed. Of interest is the change in catalyst chemoselectivity with increase in the number of recycles. The species formed favour the formation of internal olefins (in some cases) and this is evidence of a different active catalyst.
Conclusions
A series of new water-soluble sulphonated monomeric ligands was synthesised. These were reacted with the dimeric rhodium-chloro-COD precursor to afford a series of mononuclear complexes with varying substituents which were fully characterised using various spectroscopic and other analytical techniques. The complexes display excellent water-solubility at room temperature. The complexes were tested as catalyst precursors in the aqueous biphasic hydroformylation of 1-octene. All the catalysts tested could be used up to 5 times without significant drop in activity and 1-octene conversions. The catalysts display very good activity, chemoselectivity and recyclability at 50 bar syngas pressure and 95 °C temperature. However, the chemoselectivity varied with each recycle and the most significant observation was the formation of more iso-octenes each time the catalysts were reused. The catalyst that gives the best results is 7 (X = H = R) under the test conditions. The electron-withdrawing effects in 8 (X = CH3, R = H) maintains excellent aldehyde chemoselectivity in all 5 recycles. Catalyst 9 (X = Cl, R = H) favours the production of iso-octenes and the amount increases with increase in the number of recycles. The presence of a bulky tert-butyl substituent in 10 increases the steric crowding around the Rh metal centre and therefore favours the production of the linear product, nonanal. On analysis of the toluene and the aqueous layers after recycling, very little Rh is found in solution with almost all of the Rh present in the form of Rh nanoparticles suspended in the water layer. The mercury drop test confirms that these species are also responsible for the results obtained and therefore we can conclude that a combination of homogeneous catalysis and catalysis mediated by nanoparticles is taking place in these systems.Experimental
General details
All reagents and solvents were purchased from a commercial source (Sigma-Aldrich) and were used as received. Rhodium trichloride salt was received as a kind donation from Anglo-Platinum Corporation/Johnson Matthey Limited. The rhodium dimeric precursor,31 sodium sulphonate aldehydes and ligand 34 were prepared according to previously reported literature methods. Nuclear magnetic resonance (NMR) spectra were recorded on either a Varian XR300 MHz (1H at 300.08 MHz, 13C at 75.46 MHz) or a Bruker Biospin GmbH (1H at 400.22 MHz, 13C at 100.65 MHz) spectrometer at ambient temperature. Elemental analysis for C, H, N and S were carried out using a Thermo Flash 1112 Series CHNS-O Analyser. Some of the data is outside the accepted limit and this can be ascribed to presence of water molecules due to the slight hygroscopic nature of the compounds. Infrared absorptions were measured using a Perkin-Elmer Spectrum 100 FT-IR spectrometer as KBr pellets. Mass spectrometry was carried out on a Waters API Quattro Micro Triple Quadrupole electrospray ionisation mass spectrometer. Data were recorded in the negative mode. Hydroformylation samples were analysed on a Perkin Elmer Clarus 580 GC. Inductively coupled plasma optical emission spectroscopy experiments were carried out on an ICP-OES Varian 730-ES spectrophotometer.
Synthesis and characterisation of water-soluble sulphonate ligands
N). δH (400 MHz, DMSO-d6, 30 °C) (ppm) = 8.94 (s, 1 H, Himine), 7.96 (s, 1 H, Ar), 7.65 (d, 3J = 8.3 Hz, 1 H, Ar), 7.49–7.39 (m, 4 H, Ar), 7.00–6.88 (m, 1 H, Ar). δC (75 MHz, DMSO-d6, 30 °C) (ppm) = 164.1, 160.7, 147.5, 140.4, 131.7, 131.5, 130.1, 129.8, 123.7, 118.5, 116.3. Elemental Analysis (calculated for C13H9NO4ClSNa): C, 46.92; H, 2.61; N, 4.21; S, 9.63. Found C, 46.65; H, 2.65; N, 3.07; S, 9.42. ESI-MS (negative): m/z 310 [M]−, where M is the anion. S20 °C = 0.35 mg mL−1 in water.
N). δH (400 MHz, DMSO-d6, 30 °C) (ppm) = 13.34 (s, 1 H, OH), 8.99 (s, 1 H, Himine), 7.92 (d, 3J = 2.3 Hz, 1 H, Ar), 6.30–7.59 (m, 1 H, Ar), 7.37–7.22 (m, 4 H, Ar), 6.98 (d, 3J = 8.7 Hz, 1 H, Ar), 2.32 (s, 3 H, CH3). δC (75 MHz, DMSO-d6, 30 °C) (ppm) = 162.8, 160.9, 145.8, 140.3, 137.0, 131.0, 130.4, 121.8, 118.5, 116.2, 114.5, 21.1. Elemental Analysis (calculated for C14H12NNaO4S·2.5H2O): C, 46.92; H, 4.78; N, 3.91; S, 8.95. Found C, 47.41; H, 4.76; N, 3.44; S, 8.42. ESI-MS (negative): m/z 290 [M]−, where M is the anion. S20 °C = 8 mg mL−1 in water.
Synthesis and characterisation of water-soluble sulphonate Rh(I) complexes
N). δH (400 MHz, DMSO-d6, 30 °C) (ppm) = 7.36 (s, 1 H, Himine), 6.83 (s, 1 H, Ar), 8.83 (d, 3J = 8.8 Hz, 1 H, Ar), 6.60 (t, 3J = 7.6 Hz, 2 H, Ar), 6.49–6.38 (m, 2H, Ar), 6.30 (d, 3J = 7.6 Hz, 2 H, Ar), 5.83 (d, 3J = 8.80 Hz, 1H, Ar), 4.39 (m, 4 H, CHCOD), 1.70 (m, 4 H, CH2COD), 1.50 (m, 4 H, CH2COD). δC (75 MHz, DMSO-d6, 30 °C) (ppm) = 161.8, 135.0, 134.9, 134.0, 130.1, 123.8, 122.5, 120.4, 118.5, 118.1, 116.3, 84.7, 33.7, 28.0. Elemental Analysis (calculated for C21H21NO4SNaRh): C, 49.48; H, 4.12; N, 2.74; S, 6.28. Found C, 49.15; H, 4.37; N, 2.28; S, 4.47. ESI-MS (negative): m/z 486 [M]−, where M is the anion. S20 °C = 5 mg mL−1 in water.
:1 mixture of water and ethanol (20 mL). This was followed by addition of KOH (0.25 mL) and this was left to stir at room temperature for 30 min. Rhodium precursor [Rh(COD)Cl]2 (0.049 g, 0.099 mmol) was added and the mixture was left to stir at room temperature for 1 h. The clear solution formed was filtered by gravity and solvent was removed from the filtrate under reduced pressure. The product obtained was dried under vacuum to afford a yellow brown powder as the product. Yield (0.039 g, 76%). Mp: Decomposed without melting, onset at 262 °C. FT-IR (νmax/cm−1, KBr): 1604 (CN). δH (400 MHz, DMSO-d6, 30 °C) (ppm) = 8.31 (s, 1 H, Himine), 7.62 (d, 3J = 2.4 Hz, 1 H, Ar), 7.55 (m, 1 H, Ar), 7.19 (d, 3J = 7.9 Hz, 2 H, Ar), 6.98 (m, 2 H, Ar), 6.64 (d, 3J = 8.8 Hz, 1 H, Ar), 4.32 (m, 4 H, CHCOD), 2.32 (m, 4 H, CH2COD) 1.87 (m, 4 H, CH2COD), 1.76 (s, 3 H, CH3). δC (75 MHz, DMSO-d6, 30 °C) (ppm) = 166.3, 149.7, 137.7, 135.5, 133.5, 129.4, 123.4, 122.6, 121.7, 120.6, 117.1, 74.3, 30.6, 29.5, 20.9. Elemental Analysis (calculated for C21H20ClNNaO4NaRhS): C, 46.38; H, 3.71; N, 2.58; S, 5.90. Found C, 46.07; H, 3.87; N, 3.77; S, 5.12. ESI-MS (negative): m/z 500 [M]−, where M is the anion. S20 °C = 5 mg mL−1 in water.
N). δH (400 MHz, DMSO-d6, 30 °C) (ppm) = 8.21 (s, 1 H, Himine), 7.76–7.80 (m, 2 H, Ar), 7.38–7.34 (m, 2 H, Ar), 7.02–7.07 (m, 2 H, Ar), 6.81–6.78 (m, 1 H, Ar), 4.75 (br s, 4 H, CHCOD), 2.36–230 (m, 4 H, CH2COD) 1.83 (m, 4 H, CH2COD). δC (75 MHz, DMSO-d6, 30 °C) (ppm) = 164.2, 131.5, 130.1, 129.8, 128.9, 125.7, 123.8, 120.4, 118.5, 116.3, 115.7, 87.7, 30.7, 27.5. Elemental Analysis (calculated for C21H20NO4ClSNaRh): C, 46.37; H, 3.68; N, 2.58; S, 5.89. Found C, 46.07; H, 3.87; N, 3.77; S, 5.12. ESI-MS (negative): m/z 521 [M]−, where M is the anion. S20 °C = 4.7 mg mL−1 in water.
N). δH (400 MHz, DMSO-d6, 30 °C) (ppm) = 8.13 (s, 1 H, Himine), 7.54 (m, 2 H, Ar), 7.37 (t, 3J = 1.8 Hz, 1 H, Ar), 7.22 (m, 1 H, Ar), 7.09 (d, 3J = 7.7 Hz 2H, Ar), 6.98 (m, 1 H, Ar), 4.27 (m, 4 H, CHCOD), 2.36 (m, 4 H, CH2COD), 1.81 (m, 4 H, CH2COD). δC (75 MHz, DMSO-d6, 30 °C) (ppm) = 165.1, 152.2, 138.7, 134.5, 132.1, 129.9, 129.1, 126.4, 123.7, 117.2, 114.7, 73.9, 39.4, 27.5, 30.0 C, 26.4. Elemental Analysis (calculated for C25H29NNaO4NaRhS): C, 53.10; H, 5.17; N, 2.48; S, 5.67. Found C, 53.07; H, 5.87; N, 3.77; S, 5.12. ESI-MS (negative): m/z 543 [M]−, where M is the anion. S20 °C = 4 mg mL−1 in water.
X-ray crystallography
Single-crystal X-ray diffraction data were collected with a Bruker Kappa APEX II DUO diffractometer with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data collection was performed at 173(2) K. The temperature was controlled by an Oxford Cryostream cooling system (Oxford Cryostat). Cell refinement and data reduction were performed by using the program SAINT.32 The data were scaled, and absorption correction was performed by using SADABS.33 The structure was solved by direct methods by using SHELXS-9733 and refined by full-matrix least-squares methods based on F2 by using SHELXL-9733 and the graphics interface program X-Seed.34,35 The programs X-Seed and POV-Ray were both used to prepare molecular graphic images. CCDC 1008938 for 10 contains the supplementary crystallographic data for this paper.General method for the hydroformylation reactions
The reactions were performed in a 90 mL stainless steel pipe reactor. The reactor was charged with 1:1 toluene–H2O (10 mL), 1-octene (6.37 mmol), internal standard n-decane (1.26 mmol) and catalyst precursors (2.87 × 10−3 mmol). The reactor was flushed with nitrogen three times, followed by flushing twice with syngas (1:1 CO–H2). This was then pressurised and heated to the desired pressure and temperature. All reactions were done for 8 hours and samples were collected at the beginning and at the end of each reaction. Samples were analysed on a GC and products were confirmed in relation to authentic iso-octenes and aldehydes. Catalyst recycling was performed by decanting the organic layer followed by addition of a fresh substrate and the hydroformylation procedure was repeated.
Acknowledgements
We greatly acknowledge the financial support from the University of Cape Town, The Department of Science and Technology South Africa, Canon Collins Trust and NRF-DST Centre of Excellence in Catalysis – c*change. We are also grateful for a generous donation of hydrated rhodium trichloride from Anglo-Platinum Corporation/Johnson Matthey Limited.References
- (a) G. T. Whiteker and C. J. Cobley, Top. Organomet. Chem., 2012, 42, 35 CrossRef CAS.
- (a) E. G. Kuntz, Chem. Tech., 1987, 17, 570 CAS; (b) N. Pinault and D. W. Bruce, Coord. Chem. Rev., 2003, 241, 1 CrossRef CAS; (c) E. Wiebus and B. Cornils, Chem. Ing. Tech., 1994, 66, 916 CrossRef CAS; (d) R. A. Sheldon, Green Chem., 2005, 7, 267 RSC; (e) B. Cornils and E. G. Kuntz, J. Organomet. Chem., 1995, 502, 177 CrossRef CAS; (f) E. G. Kuntz, FR Patent, 1976, 2 314 910, 2 349 562, 2 338 253, 2 366 237, Rhône-Poulenc Search PubMed.
- (a) E. Wiebus and B. Cornils, in Catalyst Separation, Recovery and Recycling, Springer, Dordrecht, 2006, 105 Search PubMed; (b) L. Obrecht, P. C. J. Kamer and W. Laan, Catal. Sci. Technol., 2013, 3, 541 RSC.
- E. B. Hager, B. C. E. Makhubela and G. S. Smith, Dalton Trans., 2012, 41, 13927 RSC.
- K. H. Shaughnessy, Chem. Rev., 2009, 109, 643 CrossRef CAS PubMed.
- N. T. S. Phan, C. S. Gill, J. V. Nguyen, Z. J. Zhang and C. W. Jones, Angew. Chem., Int. Ed., 2006, 45, 2209 CrossRef CAS PubMed.
- M. E. Hanhan, C. Cetinkaya and M. P. Shaver, Appl. Organomet. Chem., 2013, 27, 570 CAS.
- A. Buhling, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 1995, 98, 69 CrossRef CAS.
- B. Cornils, Top. Curr. Chem., 1999, 206, 133 CrossRef.
- A. A. Dabbawala, H. C. Bajaj, H. Bricout and E. Monflier, Catal. Sci. Technol., 2012, 2, 2273 CAS.
- Q. Peng, Y. Yang, C. Wang, X. Liao and Y. Yuan, Catal. Lett., 2003, 88, 219 CrossRef CAS.
- (a) P. T. Anastas and J. C. Warner, Green Chemistry Theory and Practice, Oxford University Press, New York, 1998, 30 Search PubMed; (b) L. Xiaozhong, L. Hongmei and K. Fanzhi, J. Organomet. Chem., 2002, 664, 1 CrossRef.
- L. C. Matsinha, P. Malatji, A. T. Hutton, G. A. Venter, S. F. Mapolie and G. S. Smith, Eur. J. Inorg. Chem., 2013, 4218 Search PubMed.
- A. Solsona, J. Suades and R. Mathieu, J. Organomet. Chem., 2003, 669, 172 CrossRef CAS.
- S. M. Mercer, T. Robert, D. V. Dixon and P. G. Jessop, Catal. Sci. Technol., 2012, 2, 1315 CAS.
- L. Bai, L. Zhang, J. Pan, J. Zhu, Z. Cheng and X. Zhu, Macromolecules, 2013, 46, 2060 CrossRef CAS.
- A. Behr, Y. Brunsch and A. Lux, Tetrahedron Lett., 2012, 53, 2680 CrossRef CAS PubMed.
- Y. Brunsch and A. Behr, Angew. Chem., Int. Ed., 2013, 52, 1586 CrossRef CAS PubMed.
- R. Chen, X. Liu and Z. Jin, J. Organomet. Chem., 1998, 571, 201 CrossRef CAS.
- Z. Jin, X. Zheng and B. Fell, J. Mol. Catal. A: Chem., 1997, 116, 55 CrossRef CAS.
- S. Tilloy, C. Binkowski-Machut, S. Menuel, H. Bricout and E. Monflier, Molecules, 2012, 17, 13062 CrossRef CAS PubMed.
- F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, C. R. Chim., 2011, 14, 149 CrossRef CAS PubMed.
- D. N. Tran, F. X. Legrand, S. Menuel, H. Bricout, S. Tilloy and E. Monflier, Chem. Commun., 2012, 48, 753 RSC.
- F. Hapiot, H. Bricout, S. Tilloy and E. Monflier, Eur. J. Inorg. Chem., 2012, 2012, 1571 CrossRef CAS.
- (a) M. Enamullah, A. Sharmin, M. Hasegawa, T. Hoshi, A. Chamayou and C. Janiak, Eur. J. Inorg. Chem., 2006, 2146 CrossRef CAS; (b) M. Enamullah, A. K. M. Royhan Uddin, A. Chamayou and C. Janiak, Z. Naturforsch., B: Chem. Sci., 2007, 62, 807 CAS.
- A. V. Rooy, J. N. H. de Bruijin, K. F. Roobeek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, J. Organomet. Chem., 1996, 507, 69 CrossRef.
- B. C. E. Makhubela, A. M. Jardine and G. S. Smith, Green Chem., 2012, 14, 338 RSC.
- B. C. E. Makhubela, A. M. Jardine, G. Westman and G. S. Smith, Dalton Trans., 2012, 41, 10715 RSC.
- E. R. Tucci, Ind. Eng. Chem. Prod. Res. Dev., 1970, 9, 516 CAS.
- V. K. Srivastava, R. S. Shukla, H. C. Bajaj and R. V. Jasra, Appl. Catal., A, 2005, 282, 31 CrossRef CAS PubMed.
- J. Chatt and L. M. Venanzi, Olefin Coord. Comp. Part IV, 1957, 4735 CAS.
- SAINT, v. 7.60a, Bruker AXC Inc., Madison, WI, 2006 Search PubMed.
- G. M. Sheldrick, SHELXS-97, SHELXL-97 and SADABS, v. 2.05, University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Barbour, J. Supramol. Chem., 2001, 1, 189 CrossRef CAS.
- J. L. Atwood and L. Barbour, Cryst. Growth Des., 2003, 3, 3 CAS.
Footnote |
| † CCDC 1008938. For crystallographic data in CIF or other electronic format see DOI: 10.1039/c4dt02740j |
| This journal is © The Royal Society of Chemistry 2015 |