
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCatalytic upgrading of α-angelica lactone to levulinic acid esters under mild conditions over heterogeneous catalysts†
Mohammad G.
Al-Shaal
,
Wirawan
Ciptonugroho
,
Fabian J.
Holzhäuser
,
Joel B.
Mensah
,
Peter J. C.
Hausoul
and
Regina
Palkovits
*
Institut für Technische und Makromolekulare Chemie, RWTH Aachen University, Worringerweg 2, D-52074, Aachen, Germany. E-mail: palkovits@itmc.rwth.aachen.de; Fax: +49 (0)241 80 22177; Tel: +49 (0)241 80 26497
First published on 19th August 2015
Abstract
Butyl levulinate was prepared starting from α-angelica lactone and butanol over Amberlyst® 36. Different reaction conditions were optimized, which resulted in full conversion and 94% selectivity toward the ester at 75 °C. A reaction network analysis reveals pseudo-butyl levulinate and levulinic acid as intermediates in the preparation of butyl levulinate. The mild protocol was successfully applied for different alcohols and compared with the esterification of levulinic acid. Overall, this study identifies α-angelica lactone as a better candidate than levulinic acid for the heterogeneously catalysed preparation of levulinic acid esters. A catalyst screening shows that also zeolites and zirconia-based catalysts are able to catalyse the reaction. However, the transformation of the intermediate pseudo-butyl levulinate into butyl levulinate requires acid sites of sufficient strength to proceed.
Introduction
As a result of the massive exploitation of petroleum and its foreseeable depletion, the search for renewable resources is becoming increasingly urgent.1 Biomass presents a promising example of a sustainable source of energy since it can be utilized to produce liquid fuels or fuel additives.2 Particularly, levulinic acid esters (LAE) are a group of compounds with high potential for use, not only, in the fuel sector,3–6 but also for polymers,7–9 green solvents,10 and fragrances.11 LAE can be produced through a catalytic conversion of various lignocellulosic-derived intermediates (Scheme 1).4 The direct conversion of polysaccharides in a one-pot reaction to LAE, although highly attractive,12 is severely limited by the harsh conditions (e.g. 175 °C for 20 h) required to achieve this reaction.13,14 The one-pot conversion of cellulose to methyl levulinate (ML) is the only example thus far.15 Other studies have focused on mono- and disaccharides as starting material.15,16 These sugars can be obtained through hydrolysis of polysaccharides17 and less by-products and milder conditions are expected. LAE can be prepared from glucose through acid-catalysed dehydration via 5-hydroxymethylfurfural (HMF) and levulinic acid (LA) and esterification in alcoholic medium.18 For example, using TiO2–SO3H as a catalyst, 80% yield of ML was obtained after 1 h at 175 °C in methanol.15 Ethyl levulinate (EL) has been prepared from fructose with yields up to 84% at 120 °C for 24 h over sulfonated carbon nanotubes.16 LAE have also been prepared from furfuryl alcohol (FA). EL and butyl levulinate (BL) were prepared in high yields from FA in alcoholic medium over different heterogeneous catalysts at temperatures between 110 and 140 °C.19 However, considering that biorefinery processes likely use water as solvent or generate water during the processes,20 LA and FA become the final products of the hydrolysis of HMF.21,22 | ||
| Scheme 1 Possible routes for the preparation of LAE using lignocellulosic compounds. | ||
Many studies indicate that the esterification reaction of pure LA in alcoholic medium delivers LAE in high yields.23–25 Nevertheless, the formation of water during esterification prohibits complete conversion. For an efficient esterification of LA obtained from biomass streams, the removal of water is required both before and after the reaction, respectively. This stresses the need for new protocols in which LAE can be prepared in high yield without costly separation steps. In this regard, the conversion of α-angelica lactone (α-AL) to LAE via the addition of alcohols is a promising alternative.6,26–28 α-AL can be readily generated from biomass-derived LA via an acid catalysed dehydration reaction (Scheme 1).27 The synthesis of LAE from α-AL and alcohols has been reported as early as 1948.29,30 Using HCl, Langlois et al. observed pseudo-LAE in high yield before obtaining LAE as a final product. Patents awarded to Du Pont report the use of solid acid catalysts for this reaction.6,26–28 Different LAE were efficiently prepared in high selectivity at 100 °C over Amberlyst® 15 (A15).28
Nevertheless, only little is known about the influence of the alcohol, the acidic catalyst or the reaction mechanism. Recently, we have introduced α-AL as an alternative to LA for the preparation of γ-valerolactone (γ-VL).31–33 Herein we study the preparation of LAE starting from α-AL and LA using different alcohols. The influence of temperature and reaction time were investigated for different catalysts and alcohols. The data reveal a strong dependency of the catalytic activity and selectivity on number and nature of available acid sites.
Results and discussion
In a first set of experiments, the influence of the temperature was investigated over time. The preparation of BL was conducted using α-AL and butanol (BuOH) over Amberlyst® 36 (A36). Fig. 1 shows that at 50 °C, initially pseudo-butyl levulinate (pseudo-BL) (identified by GC-MS, Fig. S2†) is the main product of the reaction. Over time a trans-esterification of pseudo-BL to BL takes place (Scheme 2, pathway 1). 330 min were required for full conversion and a final yield of 89.3% BL (Table 1, entry 1), while 100 min were required to obtain similar results using the homogenous catalyst p-toluenesulfonic acid (see Fig. S1†).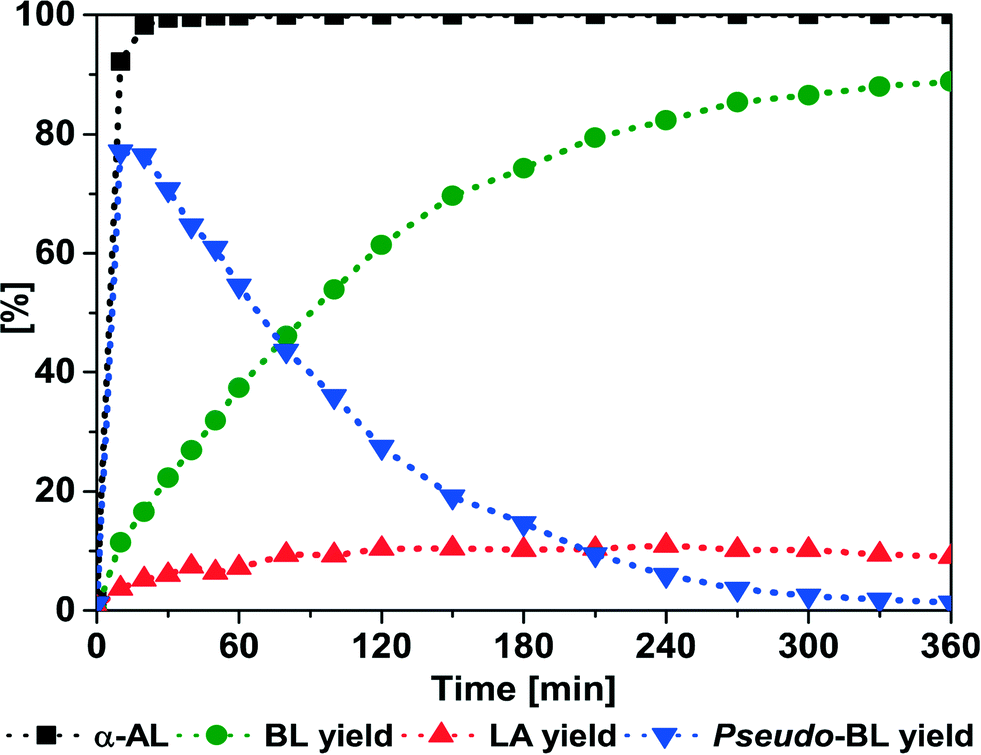 | ||
| Fig. 1 A36-catalyzed addition of BuOH to α-AL. Conditions: 50 °C; α-AL (5.00 g, 51.00 mmol); BuOH (3.78 g, 51.00 mmol); A36 (250 mg). | ||
 | ||
| Scheme 2 Proposed network for the reaction of α-AL and alcohols over solid acid catalysts. | ||
| Entry | T [C°] | t [min] | α-AL conv.b [%] | Yieldsb [%] | ||
|---|---|---|---|---|---|---|
| BL | Pseudo-BL | LA | ||||
| a Conditions: α-AL (5.00 g, 51.00 mmol); BuOH (3.78 g, 51.00 mmol) A36 (250 mg). b Determined by GC. | ||||||
| 1 | 50 | 30 | 99.4 | 18.1 | 67.2 | 4.8 |
| 120 | 100 | 62.0 | 27.8 | 10.5 | ||
| 330 | 100 | 89.3 | 1.8 | 9.5 | ||
| 2 | 75 | 10 | 99.7 | 42.1 | 50.1 | 6.9 |
| 60 | 100 | 79.8 | 10.0 | 9.3 | ||
| 240 | 100 | 94.0 | 1.0 | 5.2 | ||
| 3 | 100 | 10 | 99.7 | 68.5 | 30.4 | 2.2 |
| 30 | 100 | 91.4 | 5.3 | 2.8 | ||
| 60 | 100 | 98.4 | 0.1 | 2.1 | ||
A similar behaviour was observed at 75 °C (93.1% yield after 120 min) and 100 °C (98.4% yield after 60 min) (Table 1, entries 2 and 3). In all cases, minor amounts of LA are formed during the reaction. Interestingly the maximum amount of LA decreases from 10.5% at 50 °C to 2.8% at 100 °C. Reference experiments in the presence of 5 and 10 mol% water relative to α-AL confirm the significant effect of water causing LA formation and decreased LAE production (ESI:† Fig. S8). To exclude impurities of water in the reactants or the reaction vessel, all materials were dried prior to the reaction and reactions conducted under inert gas and water-free conditions. Despite of this, LA persisted in the reaction. In addition, GC-analysis also revealed the presence of trace amounts of β-angelica lactone (β-AL), γ-methylene-γ-butyrolactone (γ-BL), and 2,4-pentadienoic acid in the reaction mixtures.32,34,35 To study the stability of α-AL in the presence of acidic catalyst, a blank reaction was performed with A36 (Fig. 2). Interestingly, 45.3% conversion of α-AL was observed after 180 min at 75 °C. While the yield of α-AL isomers and LA only reaches 7.5%, a browning of the reaction mixture and formation of precipitates occurred suggesting the conversion to polymeric species. The formation of LA only reaches a maximum yield of 3.1% (Fig. 2). It is reasonable to argue that the remaining water formation and hydrolysis process are related to the isomerization of α-AL. In presence of an acidic catalyst β-AL can suffer ring opening to yield 2,4-pentadienoic acid, which in the presence of BuOH undergoes esterification with concomitant water formation (Scheme 2, pathway 3). The presence of LA can then be explained by hydrolysis of α-AL, BL, or pseudo-BL, respectively (Scheme 2, pathway 2). A test using neat BL and A36 showed that BL remains stable under these conditions. Alternatively, the hydrolysis of pseudo-BL can yield the acetal form of LA which after elimination of the alcohol ultimately yields LA.36 Similarly, addition of water to α-AL may yield pseudo-LA which can readily convert back to LA. Nevertheless, the results show, that in the presence of BuOH, increasing the temperature results in a decrease of LA formation. Thus, the more rapid conversion of α-AL to pseudo-BL prevents isomerization and ring-opening thereby minimizing water formation.
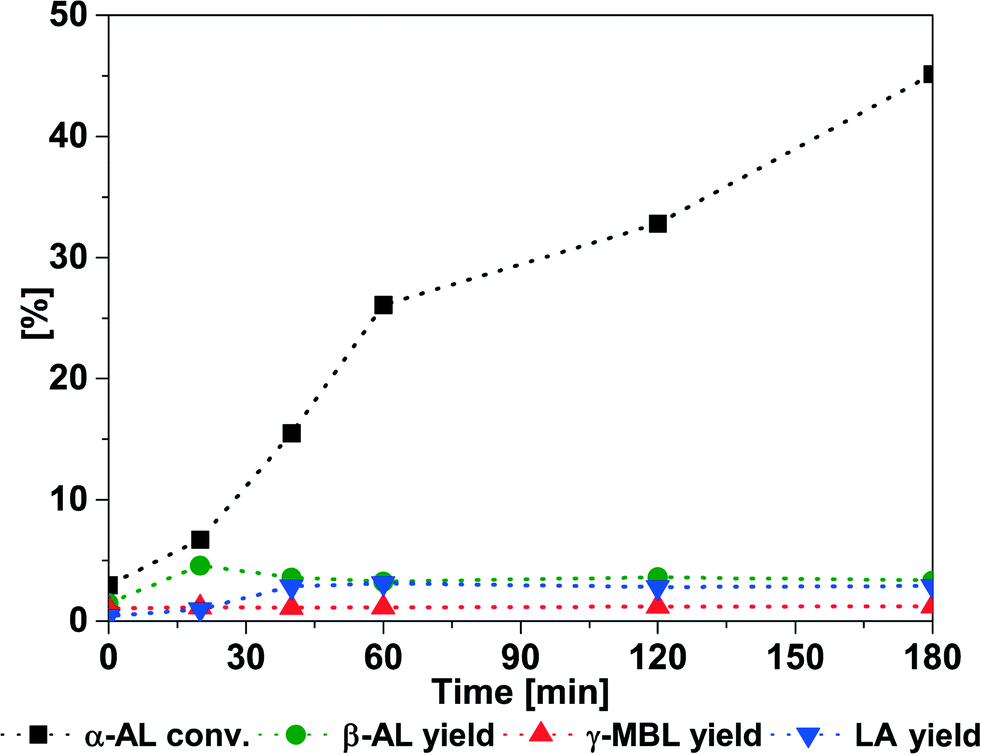 | ||
| Fig. 2 Isomerization and polymerization of α-AL. Conditions: 75 °C; α-AL (5.00 g, 51.00 mmol); A36 (250 mg). | ||
The reactions of ethanol (EtOH), octanol (OcOH), and 2-propanol (iPrOH) with α-AL were tested to gain more insight into the role of the alkyl group on the addition reaction. The data (Table 2) confirm a clear structure activity trend for linear alkyl chains following the order: EtOH > BuOH > OcOH. In case of EtOH, up to 91.3% EL is obtained after 180 min whereas for BuOH and OcOH, 240 and 300 min are required for 94.0% BL and 93.8% octyl levulinate (OL), respectively (Table 2, entries 1 and 2). In case of iPrOH the reaction proceeds significantly slower than for BuOH, as 40 min are required for reaching 99.4% conversion of α-AL. These differences show that the steric properties of the alkyl chain influence the reaction.37
| Entry | ROH | t [min] | α-AL conv.b [%] | Yieldsb [%] | ||
|---|---|---|---|---|---|---|
| LAE | Pseudo-LAE | LA | ||||
| a Conditions: 75 °C; α-AL (5.00 g, 51.00 mmol); alcohol (51.00 mmol); A36 (250 mg). b Determined by GC. | ||||||
| 1 | EtOH | 10 | 99.1 | 43.0 | 45.0 | 11.1 |
| 180 | 99.2 | 91.3 | 0.1 | 7.8 | ||
| 2 | OcOH | 60 | 99.8 | 33.1 | 52.9 | 13.9 |
| 300 | 100 | 93.8 | 0.5 | 5.0 | ||
| 3 | iPrOH | 40 | 98.9 | 40.5 | 40.5 | 11.6 |
| 120 | 99.3 | 75.8 | 3.4 | 19.7 | ||
Nevertheless, good to near full conversion to LAE is possible in all cases. For comparison, we studied the esterification of LA under the same conditions (Fig. 3 and Table 3). In case of EL, the yield only increases to 66.1%. This suggests that the reaction moves to equilibrium. BuOH and OcOH show a similar behaviour (Table 3, entries 2 and 3). Esterification of BuOH reaches equilibrium after 240 min with 58.2% yield of BL after 300 min. In case of OcOH, the equilibrium is reached after 210 min with 41.3% yield of OL. Finally, iPrOH facilitates the poorest conversion and only 8.8% of the ester is obtained (Table 3, entry 4). These data clearly demonstrate the superiority of the addition reaction in comparison to the esterification of LA.
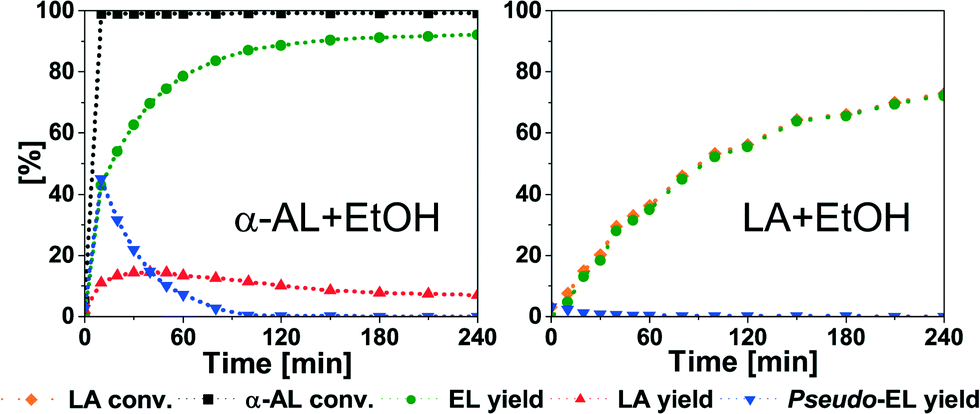 | ||
| Fig. 3 Addition of EtOH to α-AL (left) and esterification of LA (right). Conditions: 75 °C; α-AL or LA (51.00 mmol); EtOH (2.34 g, 51.00 mmol); A36 (250 mg). | ||
Focusing on BL formation, we studied the activity and selectivity of different classes of commercially available solid acid catalysts under the same reaction conditions (Fig. 4). Their specific surface area and pore volume are summarized in Table S1.† Amberlyst® catalysts, A36 and A15 exhibit almost identical behaviour toward the formation of BL over time. Both catalysts enable full conversion after 10 min and above 90% yield of BL after 120 min. This observation is comparable to the results of Manzer who reported full conversion of α-AL and 99% selectivity toward the ester after 1 h at 100 °C.6 ZrO2-based catalysts exhibit high thermal and mechanical stability and have been successfully applied as solid acid catalysts for various reactions.38–42 However, one has to note that the acid site density of inorganic materials usually is one order of magnitude lower compared to ion exchange resins such as Amberlyst (ESI:† Table S2). The activity of WO3/ZrO2 (WZ) and SO42−/ZrO2 (SZ) was tested for the addition reaction of α-AL. For comparison a bare monoclinic ZrO2 was included in the study. After 360 min, ZrO2 gave 16.2% conversion of α-AL whereas WZ gave 85.1% conversion. Interestingly, in both cases pseudo-BL is obtained as the main product, together with small amounts of LA and BL. In contrast, in the presence of SZ, α-AL is already fully converted within 80 min accompanied by a higher yield of 11.5% BL and a comparable yield of 3.6% LA. Hence, the different activity of SZ is likely caused by its increased number of acid sites and acid strength compared to WZ.43 The results of NH3-TPD are displayed in Fig. S3 and Table S2† summarizes the quantitative analysis of these data. Indeed, SZ exhibits both a higher concentration of acid sites as well as more acid sites of high strength facilitating α-AL conversion. Interestingly, WZ does not enable BL production and even in the presence of SZ pseudo-BL presents the dominant product.
 | ||
| Fig. 4 Synthesis of BL over different solid acid catalysts. Conditions: 75 °C; α-AL (5.00 g, 51.00 mmol); BuOH (3.78 g, 51.00 mmol); catalyst (250 mg). | ||
Additionally, two zeolites were investigated as catalysts for the synthesis of LAE.44,45 The commercial zeolites, HBEA-25 and 150 (Si/Al ratio = 25 and 150) were employed and gave full conversion of α-AL after 100 min. Over HBEA-150, 93% yield of BL were achieved after 360 min, whereas for HBEA-25, 63% yield were obtained. As can be seen, only trace amounts of pseudo-BL (0.5%) remained after 360 min when HBEA-150 was applied. On the other hand, for HBEA-25 still 31% pseudo-BL are present. NH3-TPD confirms a higher concentration of acid sites in case of HBEA-25 in agreement with the higher aluminium content of this material. It would be expected that this leads to higher activity; however, our study emphasizes that the activity of both catalysts is quite comparable. In fact HBEA-150 facilitates higher yields of LAE than HBEA-25. A potential explanation relates to higher acidity of the individual acid sites for lower aluminium contents in the zeolite framework. Future studies will aim at a comprehensive analysis of the influence of concentration and nature of acid sites on the addition reaction with special emphasis on the impact of acidity and the difference between Lewis and Broensted acid sites.
Overall, the data above suggest that strong acidic sites are required for trans-esterification of pseudo-LAE. Moreover, these results also suggest that the trans-esterification of the pseudo-ester is the rate limiting step of the reaction. The proposed mechanism for the conversion of α-AL to LAE via pseudo-LAE is shown in Scheme 3. Protonation of α-AL leads to a stabilized cation which can undergo nucleophilic addition of an alcohol to give pseudo-LAE as product. Protonation of the ester functionality of pseudo-LAE in turn allows another nucleophilic addition and ring opening occurs resulting in the hemi-acetal of LAE. Elimination of the alcohol yields LAE.
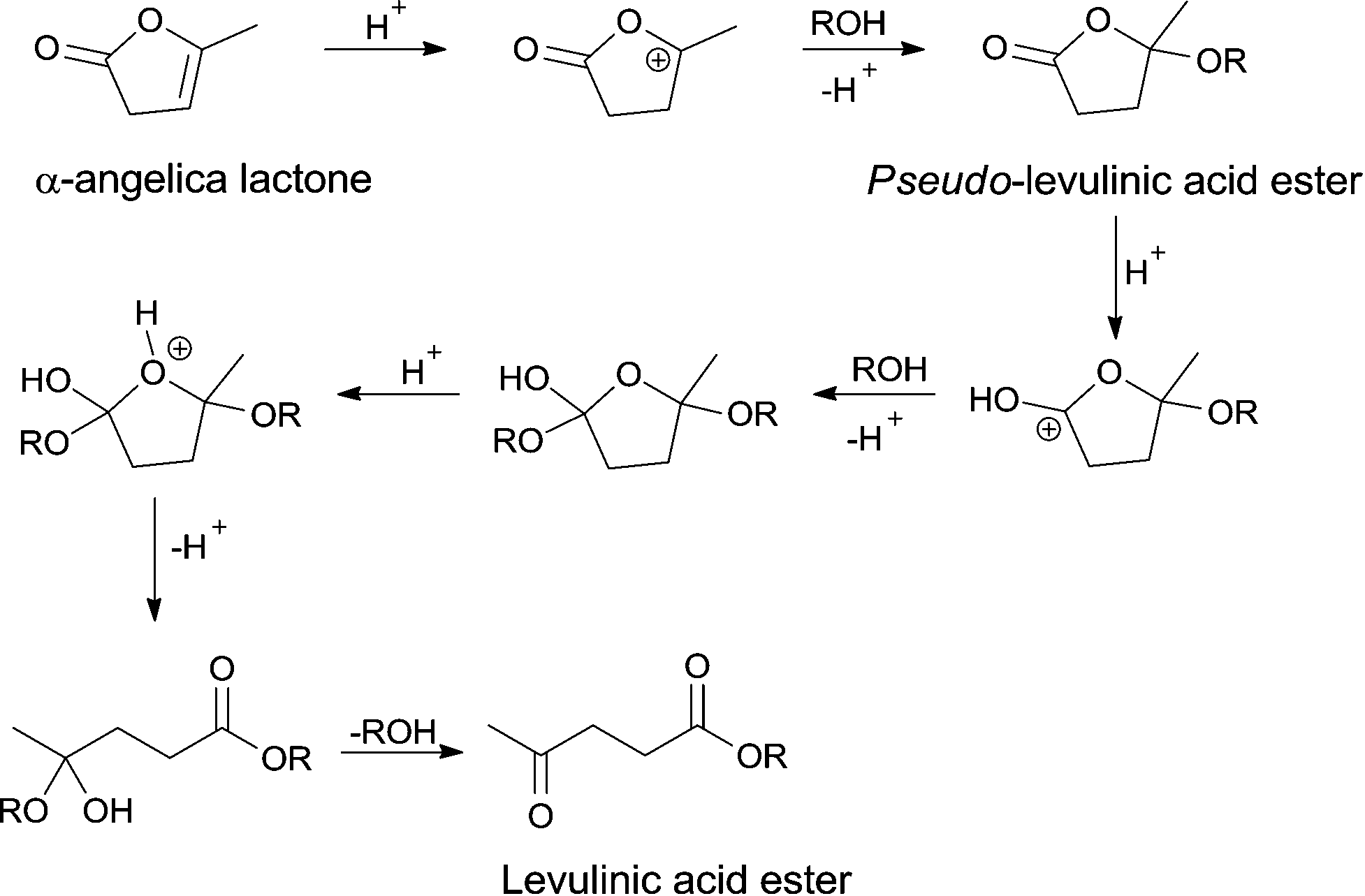 | ||
| Scheme 3 Proposed mechanism for the conversion of α-AL into LAE via pseudo-LAE. | ||
Finally, kinetic studies at different reaction temperatures were carried out (ESI:† Fig. S4–6, Table S3) to obtain the activation energy for a consecutive formation of pseudo-LAE and LAE. Data fitting was performed using pseudo-BL and BL as model compounds.
From the estimation, the activation energy for the formation of pseudo-BL and BL are 3 and 50 kJ mol−1 respectively (ESI:† Fig. S7 and Table S3). The data confirm that the protonation of α-AL and addition of the alcohol are an almost barrier-less process. Consequently, weak acidic sites should be sufficient to catalyse this transformation. In contrast, the barrier for trans-esterification of pseudo-LAE is considerably higher. The step requires activation of an ester toward nucleophilic addition and the data show that the acidity of ZrO2 and WZ are not suitable for this reaction. On the other hand, SZ, Amberlyst® and the studied zeolites possess strong acidic sites enabling a smooth conversion of pseudo-LAE to LAE. In addition, both HBEA zeolites and zirconia-based catalysts give rise to much lower amounts of LA compared to the Amberlyst® materials. It appears therefore, that these two classes of catalysts possess a lower ability to promote the isomerization of α-AL to β-AL.
In summary, transformation routes via α-AL bear the potential to facilitate e.g. γ-VL as well as LAE formation overcoming water release during these reactions. Consequently, α-AL has to be considered as important platform chemical in the frame of efficient future biorefinery concepts.
Conclusions
In conclusion, we were able to demonstrate the successful conversion of α-AL to LAE over solid acids. Full conversion and high yields of different LAE were achieved under relatively mild conditions. Comparing the addition of alcohols to α-AL and the esterification of LA shows the superiority of the addition reactions under mild conditions. A kinetic investigation enables a better understanding of the reaction network and the stability of intermediates and products. Finally, a catalytic screening shows a dependency of the reactivity and the selectivity toward LAE on the acidity and acid strength of the catalyst. While α-AL conversion and the formation of pseudo-LAE proceeds rapidly over all investigated catalysts, trans-esterification of pseudo-LAE to LAE appears to require acid sites of sufficient acid strength.Experimental section
General procedure
A dried 50 mL three-neck round flask, fitted with a condenser was charged with α-angelica lactone or levulinic acid (51.00 mmol), alcohol (51.00 mmol), and the catalyst (250 mg) under inert gas. The reactor was heated using an oil bath and agitation was provided by magnetic stirrer at 350 rpm. Samples of the reaction mixture (≃50 mg) were collected by syringe and centrifuged. The resulting clear samples were diluted with 1,4-dioxane and analysed by GC after adding diethylene glycol dimethyl ether as an internal standard.Sample analysis
The identity of reaction products (e.g. pseudo-alkyl levulinates, β-angelica lactone, γ-methylene-γ-butyrolactone, 2,4-pentadienoic acid, and levulinic acid) was confirmed by GC-MS analysis and compared with authentic samples. Quantitative analysis of samples of the addition reactions was performed by GC-FID using a CP-Wax-52 column (60 m × 250 μm × 0.25 μm). Qualitative analysis was performed by GC-MS on a Trace GC chromatograph 1310 equipped with a Restek Rxi-1 MS column (60 m × 250 μm × 0.5 μm) and a Thermo Scientific ISQ mass spectrometer (EI+, 70 eV, 250 °C).Catalysts characterization
N2 physisorption measurement was performed using Micromeritic ASAP 2000. Before being measured, the samples were evacuated at 120 °C overnight and measurements were conducted at −196 °C (Table S1†). NH3-TPD was measured by ChemBET Pulsar TPR/TPD Automated Chemisorption Analyser. Approximately 40–70 mg sample was firstly subjected to 20 mL min−1 of He flow for 30 min at 400 °C to remove any contaminant on its surface. The preheated sample was further cooled down to room temperature. Subsequently, the adsorption of NH3 was carried out for 20 minute at 100 °C by passing a 50%NH3–50%Ar gas mixture. The physisorbed NH3 molecules were evacuated by switching the flow to He and held for 20 min. Then the chemisorbed NH3 was gradually desorbed by passing He gas with a ramping rate 20 °C min−1.Acknowledgements
We wish to thank the Higher Institute of Applied Science and Technology of Syria for the studentship of M. G. Al-Shaal and the support of Indonesian Ministry of National Education Scholarship for W. Ciptonugroho. We acknowledge financial support by the Robert Bosch Foundation. Part of this work was performed within the Cluster of Excellence “Tailor-Made Fuels from Biomass” funded by the Excellence Initiative by the German federal and state governments to promote science and research at German universities.Notes and references
- BP Energy Outlook 2035, 2014 Search PubMed.
-
J. R. van Ommen and W. de Jong, Biomass as a Sustainable Energy Source for the Future: Fundamentals of Conversion Processes, Wiley-AIChE, 1st edn, 2014 Search PubMed
.
- E. Christensen, A. Williams, S. Paul, S. Burton and R. L. McCormick, Energy Fuels, 2011, 25, 5422–5428 CrossRef CAS
- A. Démolis, N. Essayem and F. Rataboul, ACS Sustainable Chem. Eng., 2014, 2, 1338–1352 CrossRef
-
P. J. Fagan, E. Korovessi, L. E. Manzer, R. Mehta and S. M. Thomas, WO2003/085071A1, 2003
-
L. E. Manzer, US2006/0063948A1, 2005
-
M. A. Harmer, V. Kapur and S. R. Williams, WO2012/162645A2, 2012
-
B. D. Mullen, V. Badarinarayana, E. S. Hall, M. J. Tjossas and C. M. Leibig, WO2013/055781A1, 2013
-
P. Bloom, WO2007/094922A2, 2007
- L. Lomba, B. Giner, I. Bandrés, C. Lafuente and M. R. Pino, Green Chem., 2011, 13, 2062 RSC
-
D. J. Yontz, US2011/0274643A1, 2011
-
E. S. Olson and C. Heide, US2010/0312028A1, 2010
- F. Rataboul and N. Essayem, Ind. Eng. Chem. Res., 2011, 50, 799–805 CrossRef CAS
- S. Saravanamurugan and A. Riisager, ChemCatChem, 2013, 5, 1754–1757 CrossRef CAS PubMed
- C.-H. Kuo, A. S. Poyraz, L. Jin, Y. Meng, L. Pahalagedara, S.-Y. Chen, D. A. Kriz, C. Guild, A. Gudz and S. L. Suib, Green Chem., 2014, 16, 785–791 RSC
- R. Liu, J. Chen, X. Huang, L. Chen, L. Ma and X. Li, Green Chem., 2013, 15, 2895–2903 RSC
- A. M. Ruppert, K. Weinberg and R. Palkovits, Angew. Chem., Int. Ed., 2012, 51, 2564–2601 CrossRef CAS PubMed
- J. M. R. Gallo, D. M. Alonso, M. A. Mellmer and J. A. Dumesic, Green Chem., 2012, 15, 85–90 RSC
- Z. Zhang, K. Dong and Z. Zhao, ChemSusChem, 2011, 4, 112–118 CrossRef CAS
-
D. J. Hayes, S. Fitzpatrick, M. H. B. Hayes and J. R. H. Ross, in Biorefineries-Industrial Processes and Products: Status Quo and Future Directions, 2008, vol. 1, pp. 139–164 Search PubMed
- A. M. Hengne, S. B. Kamble and C. V. Rode, Green Chem., 2013, 15, 2540 RSC
- M. J. Climent, A. Corma and S. Iborra, Green Chem., 2014, 16, 516–547 RSC
- J. A. Melero, G. Morales, J. Iglesias, M. Paniagua, B. Hernández and S. Penedo, Appl. Catal., A, 2013, 466, 116–122 CrossRef CAS PubMed
- S. Dharne and V. V. Bokade, J. Nat. Gas Chem., 2011, 20, 18–24 CrossRef CAS
- A. Caretto and A. Perosa, ACS Sustainable Chem. Eng., 2013, 1, 989–994 CrossRef CAS
-
L. E. Manzer, US2005/0210738A1, 2005
-
L. E. Manzer, US2005/0171374A1, 2005
-
L. E. Manzer, WO2005/097723A2, 2005
- D. P. Langlois and H. Wolff, J. Am. Chem. Soc., 1948, 70, 2624–2626 CrossRef CAS
-
D. P. Langlois and H. Wolff, US2493676, 1950
- M. G. Al-Shaal, W. R. H. Wright and R. Palkovits, Green Chem., 2012, 14, 1260 RSC
- M. G. Al-Shaal, P. J. C. Hausoul and R. Palkovits, Chem. Commun., 2014, 50, 10206–10209 RSC
- W. R. H. Wright and R. Palkovits, ChemSusChem, 2012, 5, 1657–1667 CrossRef CAS PubMed
- E. Guntrum, W. Kuhn, W. Spoenlein and V. Jäger, Synthesis, 1986, 921–925 CrossRef CAS
- E. Taskinen, Tetrahedron, 1994, 50, 1885–1888 CrossRef CAS
- C.-K. Shu and B. M. Lawrence, J. Agric. Food Chem., 1995, 43, 782–784 CrossRef CAS
- K. Dutta and V. V. Dasu, J. Mol. Catal. B: Enzym., 2011, 72, 150–156 CrossRef CAS PubMed
- W. Chu, T. Echizen, Y. Kamiya and T. Okuhara, Appl. Catal., A, 2004, 259, 199–205 CrossRef CAS PubMed
- A. Ulgen and W. Hoelderich, Catal. Lett., 2009, 131, 122–128 CrossRef CAS
- N. R. Shiju, M. Anilkumar, W. F. Hoelderich and D. R. Brown, J. Phys. Chem. C, 2009, 113, 7735–7742 CAS
- S. T. Wong, T. Li, S. F. Cheng, J. F. Lee and C. Y. Mou, J. Catal., 2003, 215, 45–56 CrossRef CAS
- K. Saravanan, B. Tyagi and H. C. Bajaj, Catal. Sci. Technol., 2012, 2, 2512–2520 CAS
- D. E. López, J. G. Goodwin, D. A. Bruce and S. Furuta, Appl. Catal., A, 2008, 339, 76–83 CrossRef PubMed
- K. Maheria, J. Kozinski and A. Dalai, Catal. Lett., 2013, 143, 1220–1225 CrossRef CAS
- D. R. Fernandes, A. S. Rocha, E. F. Mai, C. J. A. Mota and V. Teixeira da Silva, Appl. Catal., A, 2012, 425–426, 199–204 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Textural properties of the catalysts and kinetic studies of the addition reaction. See DOI: 10.1039/c5cy00446b |
| This journal is © The Royal Society of Chemistry 2015 |