
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic chemistry with nitrous oxide
Kay
Severin
Institut des Sciences et Ingénierie Chimiques, Ecole Polytechnique Fédérale de Lausanne (EPFL), 1015 Lausanne, Switzerland. E-mail: kay.severin@epfl.ch; Fax: +41-21-693-9305
First published on 24th June 2015
Abstract
This review article summarizes efforts to use nitrous oxide (N2O, ‘laughing gas’) as a reagent in synthetic chemistry. The focus will be on reactions which are carried out in homogeneous solution under (relatively) mild conditions. First, the utilization of N2O as an oxidant is discussed. Due to the low intrinsic reactivity of N2O, selective oxidation reactions of highly reactive compounds are possible. Furthermore, it is shown that transition metal complexes can be used to catalyze oxidation reactions, in some cases with high turnover numbers. In the final part of this overview, the utilization of N2O as a building block for more complex molecules is discussed. It is shown that N2O can be used as an N-atom donor for the synthesis of interesting organic molecules such as triazenes and azo dyes.
1 Introduction
Since its discovery in 1772 by Joseph Priestley, nitrous oxide has had a remarkable career.1 First, it became a popular recreational drug among the British upper class. In the second part of the 19th century, N2O was employed as an anesthetic by dentists. This application is less common today, but in some countries, N2O is given as a pain relief during childbirth.2 Very preliminary results indicate that N2O could be used as a drug for patients with treatment-resistant depression.3 More technical applications include its utilization as a whipping agent for cream or as a fuel additive for rockets and motors.1 But there is also a ‘dark side’ of N2O, and that is its environmental impact. In fact, N2O has been identified as the most potent ozone-depleting substance emitted in the 21st century.4 In addition, N2O is a very effective greenhouse gas.5 The concentration of N2O in the atmosphere is increasing, and human activities contribute significantly to N2O emissions.5 The extensive use of fertilizers fosters the formation of N2O during enzymatic nitrification and denitrification.6 Furthermore, there are industrial processes in which N2O is produced as side product, and part of this N2O is still released in the atmosphere.7For a synthetic chemist, N2O is of interests because it is a very strong oxidant from a thermodynamic point of view.8 Oxidation reactions typically result in the release of dinitrogen, which is an environmentally benign side product. However, reactions with N2O are hampered by the highly inert character of this gas. The utilization of high pressure and temperature in combination with heterogeneous catalysts allows performing oxidation reactions with N2O. These kinds of reactions have been summarized before,9 and they are not discussed in this overview. Instead, the focus will be on reactions which are carried out in homogeneous solution under (relatively) mild conditions. As described in Section 2, N2O can be used as a very selective oxidant for highly reactive compounds such as disilenes or low-valent metallocenes. Transition metal-catalyzed oxidation reactions are also possible (Section 3), and recent results have shown that high turnover numbers can be achieved.
Another interesting avenue is the utilization of N2O as a nitrogen atom donor for the synthesis of more complex organic molecules. Since many years, it is known that reactions of N2O with organometallic reagents can give nitrogen-containing products. However, efficient synthetic procedures which employ N2O as N-atom donor have only been discovered recently. Section 4 provides an overview of these transformations.
To better define the scope of this review article, it should be noted that the extensive bioinorganic chemistry of N2O will not be discussed. For a description of the ligand properties of N2O, the reader is referred to an excellent review article by Tolman.10
2 N2O as O-atom donor
Chemical reactions with N2O typically proceed via oxygen atom transfer and release of N2. Due to the very inert character of N2O, only highly reactive compounds are able to react with N2O under mild conditions. For plain organic compounds such as olefins, on the other hand, rather harsh conditions are required. For example, it is possible to perform a solution-based oxidation of cyclohexene and cyclopentene to the corresponding cyclic ketones, but temperatures above 200 °C and elevated pressures (>25 bar) are needed to achieve good conversions.11,12The low intrinsic reactivity of N2O can be advantageous because it allows performing very selective oxidation reactions, which would be difficult to achieve with other oxidants such as O2. Selective O-atom transfer reactions with N2O have been used in particular in the context of synthetic inorganic chemistry. Low valent silicon compounds are suited substrates. For example, N2O has been used to oxidize disilenes (Scheme 1a),13 silanimines,14 silaethenes,15 silylenes (Scheme 1b),16 and carbene-stabilized Si(0) compounds.17 It is worth noting that a metallosilylene18 and an osmium silylene complex19 were also found to react with N2O. Recently, it was shown that low-valent germanium compounds can be oxidized with N2O as well.20 A β-diketiminato germanium(II) hydride, for example, was converted into a hydroxide compound (Scheme 1c),20b and donor-stabilized germylenes were oxidized to give germanones.16a,20a,c Other main group compounds which react with N2O under mild conditions are basic phosphines,21,22 methylenetriphenylphosphorane (PPh3![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2),23 sodium sulphite,24 and boranes.25 However, these reactions are less interesting from a synthetic point of view.
CH2),23 sodium sulphite,24 and boranes.25 However, these reactions are less interesting from a synthetic point of view.
 | ||
| Scheme 1 Oxidation of low-valent silicon and germanium compounds with N2O. | ||
Reaction of transition metal complexes with N2O are hampered by the fact that N2O is a very poor ligand.10 Accordingly, there are very few well-characterized LnM(N2O) complexes described in the literature.26–28 A first example was reported by Armor and Taube in 1969.26 They showed that N2O can displace the water ligand in [Ru(NH3)5(OH2)]2+ to give the adduct [Ru(NH3)5(N2O)]2+ in a reversible fashion. Despite this early success, it was not until recently that a high-resolution crystallographic analysis of an N2O vanadium complex was reported (Scheme 2).28,29 As in the case of [Ru(NH3)5(N2O)]2+, the coordination of N2O is weak and ligand release is triggered by applying a vacuum. The intermediate formation of an osmium–N2O complex was proposed for the reaction of (PNP)OsH3 (PNP = N(SiMe2CH2PtBu2)2) with N2O, which ultimately leads to the formation of a dinitrogen complex and water (hydrogenation of N2O).30
 | ||
| Scheme 2 Reversible binding of N2O to a vanadium complex. | ||
Even though the coordination of intact N2O to a metal complex is a rarely observed phenomenon, there are numerous reports about transition metal complexes which react with N2O in a stoichiometric fashion.10 In the majority of these cases, N2O acts as oxygen atom donor. Selected examples are summarized below.
Early studies by Bottomley et al. focused on cyclopentadienyl titanium complexes. The reaction of the Ti(III) complex (Cp2TiCl)2 (Cp = η5-C5H5) with N2O was shown to give (Cp2TiCl)2O, whereas the Ti(II) complex Cp2Ti gave the dinuclear complex (Cp2Ti)2O (Scheme 3a).31,32 Subsequently, other cyclopentadienyl complexes of the early transition metals were oxidized with N2O. The reaction of Cp2Cr gave the tetramer (CpCrO)4, which features a heterocubane structure (Scheme 3b).33 A similar reaction was observed for the pentamethylcyclopentadienyl complex Cp*2Cr.34 Oxygen atom transfer was also demonstrated for cyclopentadienyl complexes of vanadium,35 tantalum,36 zirconium,37 and hafnium.38
 | ||
| Scheme 3 Oxygen atom transfer reactions with transition metal complexes. | ||
Oxygen atom transfer reactions are not restricted to complexes with cyclopentadienyl co-ligands. The Mindiola group has shown that vanadium39 and titanium40 alkyl complexes can be oxidized with N2O to give complexes with terminal oxo ligands. A representative example is depicted in Scheme 3c. LnM(O) complexes were also obtained by reaction of N2O with a titanium tellurido complex,41 with a niobium hydride complex,42 or with the V(III) complex V[(Me3SiNCH2CH2)3N].43
A detailed kinetic study of the O-atom transfer reaction from N2O to the V(III) complex V[N(tBu)(3,5-C6H3Me2)]3 revealed that the reaction at room temperature is second order in concentration of the vanadium complex and first order in concentration of N2O.44 At low temperature, however, an overall second order was observed. These data suggest that the oxygen atom transfer proceeds via a bimetallic LnV(N2O)VLn complex with a bridging N2O ligand.
The transfer of multiple oxygen atoms was observed for the reaction of Ar′CrCrAr′ (Ar′ = C6H3-2,6-(C6H3iPr2)) with an excess of N2O (Scheme 3d).45 This reaction is good evidence for the utility of N2O as a mild and selective oxidant because the product, Ar′Cr(μ-O)2Cr(O)Ar′, is extremely air and moisture sensitive. Accordingly, no defined product could be isolated when O2 was used instead of N2O.
Reactions of N2O with complexes of the late transition metal nickel were examined by the group of Hillhouse. They observed that complexes of the general formula L2NiR2 (L = neutral P- or N-donor; R = alkyl, aryl) give alkoxide or aryloxide complexes of the formula L2Ni(OR)R.46 For example, the metallacyclopentane (bipy)Ni(C4H8) can be converted to the oxametallacycle (bipy)Ni(C4H8O) upon reaction with N2O (Scheme 3e). Chemically induced demetallation of the latter results in the formation of 1-butanol, tetrahydrofuran or δ-valerolactone, respectively.46b,d It should be noted that the oxametallocycle cannot be prepared with O2, because cyclobutane is formed instead. More recently, it was shown that a Ni–carbene complex is able to react with N2O to give an oxametallacyclopropane.47 The oxidation of a Ni(0) carbonyl complex with N2O was reported to give a complex with a chelating carbonate ligand.48
As mentioned above, some ruthenium complexes are able to bind intact N2O in a reversible fashion.26,27 However, oxygen atom transfer has also been observed. Caulton et al. have shown that the Ru(IV) nitride complex (PNP)RuN is converted into the corresponding nitrosyl complex (PNP)RuNO upon exposure to N2O.49 Insertion of oxygen into a Ru-hydride bond was observed by Kaplan and Bergman.50 They found that RuH2(DMPE) (DMPE = Me2PCH2CH2PMe2) reacts with N2O in a step-wise fashion to give first the hydroxo complex RuH(OH)(DMPE) and then the dihydroxo complex Ru(OH)2(DMPE). We have examined the reaction of dinuclear organometallic Ru complexes with N2O.51 When a solution of (p-cymene)Ru(μ-Cl)3Ru(IMes)(C2H4)Cl (IMes = 1,3-dimesitylimidazol-2-ylidene) was subjected to an atmosphere of N2O, we observed the formation of a mixed-valence Ru(II)–Ru(III) complex with a chelating alkoxy ligand (Scheme 3f). The dinitrogen complex (p-cymene)Ru(μ-Cl)3Ru(IMes)(N2)Cl was identified as a reactions intermediate, providing indirect evidence that N2 is released during the reaction. The Chang group has reported that a complex of the lighter homologue iron can also activate N2O.52 The reaction of a four-coordinate Fe(II) complex with N2O was shown to give an iron hydroxo complex, presumably via an intermediate Fe(IV)–oxo complex.
Bleeke and Behm have examined the reaction of an iridium metallacycle with N2O.53 As initial product, an iridacyclohexadienone complex was observed. The latter isomerizes slowly at room temperature (Scheme 3g).
The oxidation of low-valent lanthanide54 and actinide55 complexes with N2O is a convenient method for the preparation of complexes with bridging or terminal oxo ligands. The μ-oxo complex (Cp*Sm)2(μ-O) (Cp* = η5-C5Me5), for example, can be obtained by reaction of (Cp*Sm)2(THF)2 with N2O (Scheme 3h).54 The Meyer group has shown that U(III) tris(aryloxide) complexes react with N2O to give terminal U(V) oxo complexes (Scheme 3i).55a
3 Metal-catalyzed reactions with N2O
The fact that transition metal complexes are able to activate N2O suggests that metal-catalyzed oxidation reactions with N2O can be performed. A generic catalytic cycle is shown in Scheme 4. Reactions of this kind have been realized with heterogeneous catalysts9 or in the gas phase,56 but these systems are not discussed here. This section summarizes catalytic oxidation reactions with N2O which are performed in homogeneous solution. | ||
| Scheme 4 Metal-catalyzed oxidation reactions with N2O. | ||
Initial attempts to use N2O as an oxidant in metal-catalyzed reactions have focused on a rather ‘easy’ reaction: the oxidation of phosphines to phosphine oxides. It was shown that the hydride complex CoH(N2)(PPh3)3 is able to catalyze the oxidation of PPh3 to give OPPh3 (Scheme 5a).57 The reaction was performed under ambient conditions and at least six turnovers were achieved. These findings are in line with observations by Pratt et al., who showed that Co(I) complexes are able to reduce N2O to N2.58 The cobalt-catalyzed oxidation of PPh3 was recently re-investigated by Beloglazkina et al. using different Co complexes and higher turnover numbers were obtained (≤73).59 Another competent catalyst system for the conversion of phosphines to phosphine oxides by N2O is a mixture of NiCl2(DPPP) (DPPP = 1,3-bis(diphenylphosphino)propane) and n-BuLi (Scheme 5b).60 The active catalyst is assumed to be a low-valent Ni complex which is formed upon reduction of NiCl2(DPPP) with n-BuLi.
 | ||
| Scheme 5 Metal-catalyzed oxidation of phosphines by N2O. | ||
The oxidation of different of organic substrates in the presence of Ru–porphyrin complexes was investigated by Yamada et al. First, they were able to show that the Ru(VI) complex Ru(TMP)(O)2 (TMP = tetramesitylporphyrinato) is a catalyst for the epoxidation of olefins, including structurally complex substrates such a steroids (Scheme 6a).61,62 The reactions were performed under rather forcing conditions (140 °C, 10 bar) and aromatic solvents, in particular fluoro- and chlorobenzene, gave the best results. Soon after, the same group reported that Ru(TMP)(O)2 can be used as a catalyst for the oxidation of secondary and primary benzylic alcohols (Scheme 6b),63,64 as well as for the oxidation of 9,10-dihydroanthracene derivatives (Scheme 6c).65 Again, rather harsh reactions conditions were applied. In this context, a study by Groves and Roman is worth mentioning. They have shown that the Ru(II) complex Ru(TMP)(THF)2 can be oxidized with N2O to give Ru(TMP)(O)(THF) or Ru(TMP)(O)2, depending on the reaction conditions.66
 | ||
| Scheme 6 Oxidation of different organic substrates by N2O in the presence of Ru–porphyrin catalysts (TMP = tetramesitylporphyrinato). | ||
The utilization of polyoxometalates as catalysts for N2O-based oxidation reactions was investigated by the group of Neumann. The vanadium-containing polyoxometalate [PV2Mo10O40]5− was shown to catalyze the oxidation of alcohols (Scheme 7a) and alkylarenes (Scheme 7b).67 The reactions were performed at ambient pressure and a temperature of 150 °C. The combination of H5PV2Mo10O40 with a Pd complex featuring a phenanthroline ligand decorated with a crown ether allowed to perform Wacker-type oxidation reactions of olefins with N2O (Scheme 7c).68 Again, an elevated temperature of 150 °C was employed for these reactions. More recently, the Neumann group has shown that H4PSbMo11O40, H4PVMo11O40 or H3PMo12O40 can be used for the oxidation of dihydrophenanthrene to phenanthrene (1 bar N2O, 110 °C).69 However, better results were obtained when O2 was used instead of N2O.
 | ||
| Scheme 7 Oxidation of different organic substrates by N2O in the presence of polyoxometalate catalysts (phen* = crown ether-functionalized phenanthroline ligand). | ||
The Sita group has investigated oxygen atom transfer reactions mediated by organometallic Mo complexes.70 They were able to demonstrate the catalytic oxidation of an isocyanide to an isocyanate (Scheme 8). During the reaction, the catalysts cycles between Mo(II) and Mo(IV). At present, the reaction is less interesting from a synthetic point of view because low turnover numbers and frequencies (1 per week) were achieved. Still, the reaction is quite remarkable because catalysis occurs under ambient conditions (1 bar N2O, 25 °C).
 | ||
| Scheme 8 Oxidation of an isocyanide by N2O in the presence of an organometallic Mo catalyst. | ||
We have recently reported that N2O can be used as an oxidant for the metal-catalyzed homo-coupling of Grignard reagents (Scheme 9).71 Simple metal salts such as Fe(acac)3, CoCl2, or Li2CuCl4 were employed as catalyst precursors. For most reactions, catalyst concentrations of 0.1–1.0 mol% were sufficient to obtain good yields. Coupling reactions of some arylmagnesium compounds could be performed with less than 0.01 mol% under very mild conditions. The corresponding turnover numbers of up to 9400 are unprecedented for solution-based oxidation reactions with N2O. Compared to alternative procedures which utilize O2 as oxidant, our method offers some important advantages. First, it is possible to use lower amounts of catalyst since N2O is less prone to undergo metal-independent side reactions. Second, sterically demanding aryl Grignard reagents as well as highly reactive alkyl Grignard reagents can be used as substrates. Another noteworthy feature is the fact that aryl–alkyl and alkenyl–alkyl cross-coupling reactions can be achieved with good selectivity. All these characteristics should make the method attractive for applications in organic synthesis.
 | ||
| Scheme 9 Metal-catalyzed homo-coupling of Grignard reagents with N2O. | ||
Interestingly, it is also possible to perform metal-catalyzed reductions in the presence of N2O. A system of this kind was recently described in a communication by Higuchi.72 They were able to show that alkenes can be reductively dimerized in the presence of metalloporphyrin catalysts using simultaneously the reductant NaNH4 and the oxidant N2O (Scheme 10). The best results were obtained with the iron tetraphenylporphyrinato complex Fe(TPP)Cl. For the dimerization of 2,3-dimethyl-2,3-diphenylbutane, a turnover number of 1380 was obtained. The following mechanism is proposed: reduction of Fe(TPP)Cl by NaBH4 in the presence of the alkene gives the dimerization product along with a highly reduced [Fe(TPP)]− complex. The latter is oxidized by N2O to regenerate an Fe(III) porphyrin complex, and close the catalytic cycle. This proposition is supported by the fact that a reduced form of myoglobin containing Fe(I) can be oxidized by N2O.73
 | ||
| Scheme 10 Reductive coupling of olefins with NaBH4, N2O and the Fe porphyrin catalyst Fe(TPP)Cl. | ||
4 N2O as N-atom donor
This section describes reactions with N2O in which nitrogen atoms are incorporated into the final product. A first reaction of this kind was reported in 1892 by Wislicenus.74 He showed that sodium azide is obtained upon exposure of NaNH2 to N2O at elevated temperatures (Scheme 11a). KNH2 and Zn(NH2)2 were found to react in a similar fashion. The ‘Wislicenus reaction’ is nowadays used by industry to produce sodium azide on a larger scale.75 The mechanism of the reaction has been investigated by Clusius et al.76 Using 15N-labelled nitrous oxide (N15NO and 15NNO can be prepared by decomposition of either NH415NO3 or 15NH4NO3), they were able to show that two reaction pathways are operational. An attack of the amide at the terminal and the central nitrogen atom were proposed.76a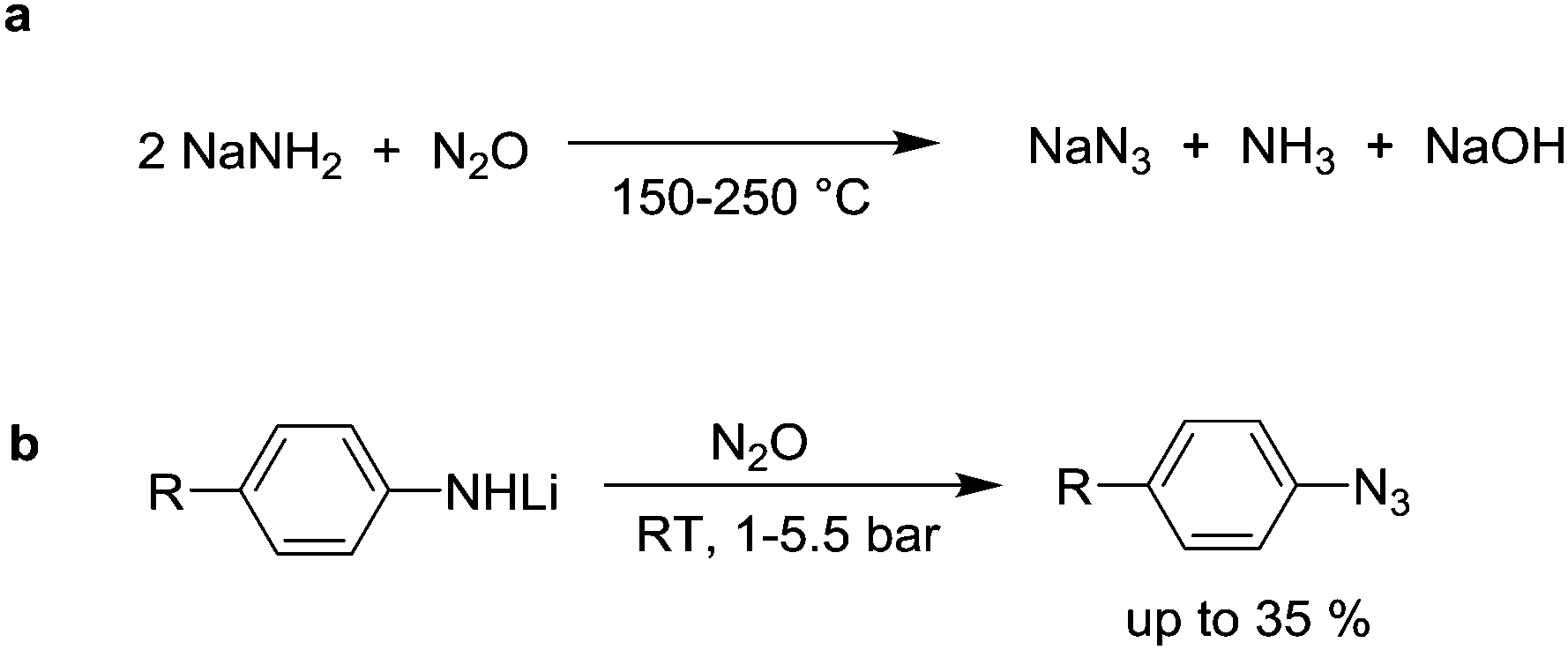 | ||
| Scheme 11 Synthesis of azides by reaction of amides with N2O. | ||
Amides of aromatic amines can also be converted into azides. Meier showed that lithium anilide reacts with N2O to give azobenzene, biphenyl, and a small amount of a yellow oil, which he assumed to be phenyl azide.77 The reaction was later reinvestigated by Koga and Anselme.78 By optimizing the reaction conditions, they were able to increase the amount of phenyl azide to 35% (Scheme 11b). Amides derived from p-toluidine, p-anisidine, and cyclohexylamine were found to react in a similar fashion, but the yields were likewise low. Apart from simple amides, hydrazine anions are also able to react with N2O, giving rise to a mixture of products.79
Organometallic compounds of the alkali and the alkaline-earth metals often react with N2O under mild conditions.80 Already in 1928, it was shown that the sodium salt of triphenylmethane adds N2O to give a diazotate.81 The latter is converted into triphenylcarbinol upon reaction with ethanol (Scheme 12a). A first comprehensive study about the reaction of organolithium compounds with N2O was published by Beringer et al.82 They showed that primary, secondary, and tertiary alkyllithium compounds and most aryllithium compounds are able to react with N2O. For example, the reaction of nBuLi with N2O gave a hydrazone, which could be isolated with a yield of 24% (Scheme 12b). For phenyl lithium, they observed a complex mixture of products including biphenyl, azobenzene, triphenylhydrazine and phenol. The mechanism this reaction was investigated by Meier.77,83 He proposed that the initially formed phenyldiazotate reacts with a second equivalent of PhLi to give azobenzene and Li2O. Side products arise from decomposition of the diazotate and from the fact that azobenzene can react further with PhLi.
 | ||
| Scheme 12 Reaction of organometallic compounds of the alkali and the alkaline-earth metals with N2O. | ||
The reaction of N2O with the simplest organolithium compound, CH3Li, was investigated by the group of Müller.84 They were able to show that diazomethane is formed after basic workup (Scheme 12c). Under optimized conditions, a yield of 70% can be obtained.
The reaction of lithiated ferrocene with N2O allows the preparation of azoferrocene in 25% yield (Scheme 12d).85 A similar reaction was used to synthesize azo-bridged ferrocene oligomers, albeit in very low yield.86 For the preparation of simple aromatic azo compounds, aryl calcium reagents appear to be best suited. This was first shown by Meier and Rappold, who isolated azobenzene along with larger amounts of biphenyl from the reaction of PhCaI with N2O in diethyl ether.87 More recently, the reaction was reinvestigated by Hays and Hanusa.88 Under optimized reaction conditions, they were able to increase the yield of azobenzene to 61% (Scheme 12e), but they mentioned problems with reproducibility and the substrate scope was very narrow.
In contrast to organocalcium reagents, Grignard reagents were believed to be inert towards N2O. An early attempt to combine an organomagnesium compound with N2O was reported by Zerner in 1913.89 He observed that solutions of MeMgI in Et2O did not react with N2O, even upon heating. Since then, statements about the unreactivity of Grignard reagents towards N2O have appeared in several articles.8,87,88 We have recently demonstrated that this generalization is not correct. Some primary and secondary aliphatic Grignard reagents such as EtMgCl, BnMgCl or iPrMgCl (Scheme 12f) are converted to hydrazones when THF solutions are subjected to an atmosphere of N2O. When the reactions are combined with an acidic work-up, it is possible to obtain alkylhydrazinium salts on a preparative scale.
As mentioned in Section 2, olefins are able to react with N2O under forcing conditions to give ketones.11,12 Calculations suggest that these reactions proceed via 1,3-diploar cycloadditions of N2O to the double bond of the olefins.90 The latter decomposes to give the ketone and dinitrogen. Banert and Plefka have shown that cyclic alkynes are much more reactive towards N2O than simple olefins.91 Reactions were found to proceed at temperatures between −25 °C and RT using pressures between 15 and 50 bar. Interestingly, they were able to obtain products which contain all three atoms of nitrous oxide (Scheme 13).
 | ||
| Scheme 13 Reactions of cyclic alkynes with N2O. | ||
The incorporation of all three atoms of N2O into the final product was also observed for reactions with some transition metal complexes. The diphenylacetylene complexes of permethyltitanocene and zirconocene react with N2O to give azoxymetallacyclopentene complexes (Scheme 14a).92 The Zr complex is thermally labile and undergoes extrusion of dinitrogen to give an oxametallacyclobutane complex. The carbon–metal bond of the latter can be cleaved with a variety of substrates processing acidic protons. It is noteworthy that the oxametallacyclobutane complex can be obtained in quantitative yield by exposure of the solid Zr diphenylacetylene complex to N2O. Recently, it was shown that the labile azoxymetallacyclopentene complex can be trapped by reactions with MeO3SCF3 (alkylation of the β-N-atom).93 The reaction product can be isolated and is stable as a solid if stored at −35 °C. In case of Ti, the initial N2O adduct is more stable and can be used for further reactions. Apart from an alkylation reaction with MeO3SCF3 (O- and β-N-alkylation), it was shown that the complex can be reduced to give a stable radical anion.93
 | ||
| Scheme 14 Insertion of N2O into metal–carbon bonds. | ||
Some organometallic samarium complexes are also able to insert N2O into metal–carbon bonds. When a solution of Cp*2Sm(CH2Ph)(THF) was subjected to an atmosphere of N2O, a dinuclear complex was formed (Scheme 14b).94 A related reaction was observed for allyl complexes of the formula Cp*2M(η3-C3H5) (M = Y, Sm, La).95
A rarely observed phenomenon is the addition of N2O to transition metal complexes with concomitant cleavage of the N–N bond.96–100 Cummins has shown that three-coordinate Mo(III) complexes are able to react with N2O in this fashion to give a nitrosyl complex along with a nitride complex (Scheme 15a).96,97 It is interesting to note that the reverse reaction, the formation of N2O from a metal nitride complex and nitric oxide, has been observed for an osmium101 and a ruthenium49 nitride complex. More recently, the Sita group has shown that a Mo carbonyl complex (generated in situ by photolysis of a dicarbonyl complex) can add N2O to give a nitrosyl, isocyanate complex (Scheme 15b).98,99
 | ||
| Scheme 15 Metal-induced rupture of the N–N bond of N2O. | ||
The formation of stable covalent adducts of N2O can be achieved without transition metals. In 2009, the group of Stephan has shown that the frustrated Lewis pair (FLP) tBu3P/B(C6F5)3 reacts with N2O at ambient conditions to give the adduct tBu3P(N2O)B(C6F5)3 (Scheme 16a).102,103 A crystallographic analysis of the product revealed that the tBu3P and the OB(C6F5)3 group are oriented trans with respect the NN double bond. Upon thermal or photochemical activation, the adduct liberates dinitrogen to give (tBu3PO)B(C6F5)3. The B(C6F5)3 group in the adduct tBu3P(N2O)B(C6F5)3 is labile and can be replaced by the Lewis acid Zn(C6F5)2.104 Subsequent studies showed that the adduct tBu3P(N2O)B(C6H4F)3 is particularly well suited for exchange reactions, since it features the relatively weak Lewis acid B(C6H4F)3. Clean exchange reactions were observed for boron-based Lewis acids such as PhB(C6F5)2 and MesB(C6F5)2, as well as for cationic metallocene complexes and the tritylium cation (Scheme 16b).22 The Al analogue tBu3P(N2O)Al(C6F5)3 can be prepared by slow addition of N2O to a cooled (−78 °C) solution containing tBu3P (2 eq.) and Al(C6F5)3·tol.105 Subsequent reaction with additional Al(C6F5)3·tol results in cleavage of the N–O bond to generate the highly reactive radical ion pair (tBu3P˙)[(C6F5)3Al(O˙)Al(C6F5)3] that can activate C–H bonds. First steps towards the preparation of N2O sensors based on FLPs were recently reported by the groups of Aldridge and Tamm.106 They showed that FLPs containing organometallic sandwich complexes (e.g. ferrocene, Scheme 16c) change their color upon binding to N2O.
 | ||
| Scheme 16 Covalent capture of N2O by frustrated Lewis pairs. | ||
Covalent capture of intact N2O can also be achieved by N-heterocyclic carbenes (NHCs).107–110 This was first demonstrated by our group in 2012.107 The reactions occur at room temperature and ambient pressure to give the adducts NCH–N2O in (mostly) good yields (Scheme 17). Conveniently, the carbenes can be prepared in situ by deprotonation of the corresponding imidazolium salts.108 Owing to the strong C–N bond, most adducts show a good stability at room temperature. On heating, however, they decompose to give dinitrogen and the corresponding urea. The decomposition reaction was found to depend strongly on the nature of the carbene and the solvent. Aqueous solutions of the adduct derived from 1,2-dimethylimidazol-2-ylidene (R = R′ = Me), for example, could be heated to 100 °C for a prolonged period of time without significant decomposition.108 A noteworthy characteristic of NHC–N2O adducts is the long N–N bond (1.27–1.33 Å), which is in contrast to what has been observed for N2O adducts of frustrated Lewis pairs (N–N ∼ 1.25 Å). This structural feature suggests that the activation of N2O by carbenes might facilitate N–N bond rupture. In fact, when NHC–N2O adducts were allowed to react with MeI, HCl (Scheme 18a), or acetyl chloride, rupture of the N–N bond was observed.107,108 Reaction with the tritylium tetrafluoroborate, on the other hand, led to the formation of the adduct (IMes–N2O–CPh3)(BF4) (Scheme 18b).
 | ||
| Scheme 17 Covalent capture of N2O by N-heterocyclic carbenes. | ||
 | ||
| Scheme 18 Reactions of IMes–N2O with HCl (a), Ph3CBF4 (b), V(Mes)3(THF) (c), and Ni(COD)2 (d). | ||
Reactions of IMes–N2O with different transition metal complexes have been investigated. When combined with simple 3d transition metal salts, IMes–N2O can act as an N-donor, as an O-donor, or as a chelating N,O-donor.108 More interesting results were obtained with the low-valent vanadium complex V(Mes)3(THF) and the Ni(0) complex Ni(COD)2. In case of the highly oxophilic V(Mes)3(THF), the addition of IMes–N2O resulted in N–O bond cleavage with oxygen atom transfer to the metal center and formation of a deprotonated hydrazone ligand (Scheme 18c).111 With Ni(COD)2, on the other hand, insertion of the metal in the N–N bond was observed.112 The product of the reaction is an unusual three-coordinate Ni nitrosyl complex with bridging imidazolin-2-iminato ligands (Scheme 18d).
Recently, we found that AlCl3 is able to induce cleavage of the N–O bond of NHC–N2O adducts. When the reactions are performed in the presence of an aromatic compound, azoimidazolium salts are formed in good yields (Scheme 19).113 These kinds of salts are of interest because they are strongly colored dyes. They are produced industrially (e.g. Basic Red 51) and used for a variety of applications such as dying of synthetic and natural fibers. An advantage of the N2O-based procedure is its flexibility. The heterocyclic coupling partner can have aliphatic as well as aromatic substituents on the nitrogen atoms. Furthermore, it is possible to use a wide range of aromatic coupling partners including deactivated arenes such as C6H5F, heterocycles, and polycyclic arenes such as pyrene and azulene (Scheme 19). As such, the method complements existing procedures for the synthesis of these dyes, each of which has its own limitations.
 | ||
| Scheme 19 Synthesis of azoimidazolium dyes with N2O. | ||
Triazenes are compounds of the general formula R–NN–NR′R′′. They have been used extensively in synthetic organic chemistry.114 Furthermore, triazenes have been examined as potential anti-tumor drugs, and the triazenes dacarbazine and temozolomide are currently used in the clinic for the treatment of cancer.115 We have recently shown that triazenes can be prepared by coupling of lithium amides and organomagnesium compounds with N2O.116 The reaction is best performed in a sequential fashion, with initial addition of N2O to a solution of the amide, followed by reaction with the Grignard reagent (Scheme 20). A key advantage of the new procedure is the ability to access triazenes with alkenyl and alkynyl substituents (some selected examples are show in Scheme 20). These compounds are difficult to synthesize by conventional methods because the required starting materials are unstable. Interestingly, some of the new alkynyltriazenes were found to display high cytotoxicity in in vitro tests on ovarian and breast cancer cell lines. Recent results from our laboratory show that alkynyltriazenes are versatile starting materials for subsequent reactions, and details about these reactions will be reported in due course.
 | ||
| Scheme 20 Synthesis of triazenes with N2O. | ||
5 Conclusions
The examples discussed above show that nitrous oxide is an interesting reagent for synthetic organic and inorganic chemistry. The inert character of N2O allows selective oxygen atom transfer reactions to highly reactive species, while minimizing the risk of over-oxidation. Stoichiometric reactions of this kind have been used in particular for the oxidation of low-valent silicon compounds and for transition metal complexes.Catalytic reactions with N2O have been examined extensively because N2O is a cheap and environmentally begin oxidant. Initially, solution-based reactions with transition metal catalysts have shown only very limited success. High temperatures and/or pressures were needed, and low turnover numbers were achieved. However, recent results demonstrate that efficient catalytic processes at ambient conditions are possible. Based on these initial results, it appears that catalytic reactions which involve low-valent transition metal complexes are particularly well suited.
For reactions with N2O, the transfer of oxygen is the most commonly observed mode of reactivity. Nevertheless, it is possible to use N2O as a donor of nitrogen atoms. Reactions of this kind are known for many decades, but applications in organic synthesis were sparse. The formation of side products, low yields, and the existence of more attractive alternative procedures have hampered the utilization of N2O in N-atom transfer reactions. But this might change in the future. As demonstrated by synthesis of triazenes and azoimidazolium dyes, it is possible to perform high yield N-atom transfer reactions with N2O. Notably, the N2O-based methods can offer distinct advantages over more established synthetic procedures.
Acknowledgements
This work was supported by funding from the Swiss National Science Foundation and the Ecole Polytechnique Fédérale de Lausanne (EPFL). I am grateful to my coworkers and collaborators Alexander Tskhovrebov, Gregor Kiefer, Loïc Jeanbourquin, Euro Solari, Florian Perrin, Léonard Eymann, Lara Naested, Basile Vuichoud, Matthew Woodrich, Tina Riedel, Paul Dyson, and Rosario Scopelliti, who have contributed to some of the N2O chemistry described in this overview.Notes and references
- D. Zuck, P. Ellis and A. Dronsfield, Educ. Chem., 2012, 26 CAS.
- J. Speth, A. Biedler and F. G. Mathers, Gynaekologe, 2013, 46, 129 CrossRef CAS.
- P. Nagele, A. Duma, M. Kopec, M. A. Gebara, A. Parsoei, M. Walker, A. Janski, V. N. Panagopoulos, P. Cristancho, J. P. Miller, C. F. Zorumski and C. Conway, Biol. Psychiatry, 2015, 78, 10 CrossRef CAS PubMed.
- (a) M. Dameris, Angew. Chem., Int. Ed., 2010, 49, 489 CrossRef CAS PubMed; (b) A. R. Ravishankara, J. S. Daniel and R. W. Portmann, Science, 2009, 326, 123 CrossRef CAS PubMed.
- D. S. Reay, E. A. Davidson, K. A. Smith, P. Smith, J. M. Melillo, F. Dentener and P. J. Crutzen, Nat. Clim. Change, 2012, 2, 410 CrossRef CAS.
- S. R. Pauleta, S. Dell’Acqua and I. Moura, Coord. Chem. Rev., 2013, 257, 332 CrossRef CAS.
- J. Pérez-Ramírez, F. Kapteijn, K. Schöffel and J. A. Moulijn, Appl. Catal., B, 2003, 44, 117 CrossRef.
- A. V. Leont’ev, O. A. Fomicheva, M. V. Proskurnina and N. S. Zefirov, Russ. Chem. Rev., 2001, 70, 91 CrossRef.
- (a) G. I. Panov, K. A. Dubkov and A. S. Kharitonov, in Modern Heterogeneous Oxidation Catalysis, ed. M. Noritaka, Wiley-VCH, Weinheim, 2009, p. 217 Search PubMed; (b) V. N. Parmon, G. I. Panov and A. S. Noskov, Catal. Today, 2005, 100, 115 CrossRef CAS.
- W. B. Tolman, Angew. Chem., Int. Ed., 2010, 49, 1018 CrossRef CAS PubMed.
- (a) E. V. Starokon, K. A. Dubkov, D. E. Babushkin, V. N. Parmon and G. I. Panov, Adv. Synth. Catal., 2004, 346, 268 CrossRef CAS; (b) G. I. Panov, K. A. Dubkov, E. V. Starokon and V. N. Parmon, React. Kinet. Catal. Lett., 2002, 76, 401 CrossRef CAS; (c) K. A. Dubkov, G. I. Panov, E. V. Starokon and V. N. Parmon, React. Kinet. Catal. Lett., 2002, 77, 197 CrossRef CAS; (d) F. S. Bridson-Jones, G. D. Buckley, L. H. Cross and A. P. Driver, J. Chem. Soc., 1951, 2999 RSC.
- For related reactions see: (a) D. P. Ivanov, K. A. Dubkov, D. E. Babushkin, S. V. Semikolenov and G. I. Panov, Adv. Synth. Catal., 2009, 351, 1905 CrossRef CAS; (b) S. V. Semikolenov, K. A. Dubkov, D. P. Ivanov, D. E. Babushkin, M. A. Matsko and G. I. Panov, Eur. Polym. J., 2009, 45, 3355 CrossRef CAS; (c) K. A. Dubkov, S. V. Semikolenov, D. P. Ivanov, D. E. Babushkin, M. A. Matsko and G. I. Panov, J. Appl. Polym. Sci., 2009, 114, 1241 CrossRef CAS; (d) E. P. Romanenko, E. V. Starokon, G. I. Panov and A. V. Tkachev, Russ. Chem. Bull., Int. Ed., 2007, 56, 1239 CrossRef CAS; (e) S. V. Semikolenov, K. A. Dubkov, E. V. Starokon, D. E. Babushkin and G. I. Panov, Russ. Chem. Bull., Int. Ed., 2005, 54, 948 CrossRef CAS.
- (a) B. Maity and D. Koley, J. Mol. Graphics Modell., 2014, 51, 50 CrossRef CAS PubMed; (b) S. Khan, R. Michel, D. Koley, H. W. Roesky and D. Stalke, Inorg. Chem., 2011, 50, 10878 CrossRef CAS PubMed; (c) N. Wiberg, W. Niedermayer, K. Polborn and P. Mayer, Chem. – Eur. J., 2002, 8, 2730 CrossRef CAS PubMed; (d) H. B. Yokelson, A. J. Millevolte, G. R. Gillette and R. West, J. Am. Chem. Soc., 1987, 109, 6865 CrossRef CAS.
- N. Wiberg and K. Schurz, Chem. Ber., 1988, 121, 581 CrossRef CAS.
- N. Wiberg, G. Preiner and K. Schurz, Chem. Ber., 1988, 121, 1407 CrossRef CAS.
- (a) Y. Xiong, S. Yao and M. Driess, Angew. Chem., Int. Ed., 2013, 52, 4302 CrossRef CAS PubMed; (b) R. Azhakar, K. Pröpper, B. Dittrich and H. W. Roesky, Organometallics, 2012, 31, 7568 Search PubMed; (c) R. Azhakar, R. S. Ghadwal, H. W. Roesky, H. Wolf and D. Stalke, Chem. Commun., 2012, 48, 4561 RSC; (d) A. Jana, R. Azhakar, S. P. Sarish, P. P. Samuel, H. W. Roesky, C. Schulzke and D. Koley, Eur. J. Inorg. Chem., 2011, 5006 CrossRef CAS; (e) S. S. Sen, G. Tavčar, H. W. Roesky, D. Kratzert, J. Hey and D. Stalke, Organometallics, 2010, 29, 2343 CrossRef CAS; (f) S. Yao, Y. Xiong and M. Driess, Chem. – Eur. J., 2010, 16, 1281 CrossRef CAS PubMed; (g) Y. Xiong, S. Yao and M. Driess, J. Am. Chem. Soc., 2009, 131, 7562 CrossRef CAS PubMed; (h) S. Yao, Y. Xiong, M. Brym and M. Driess, J. Am. Chem. Soc., 2007, 129, 7268 CrossRef CAS PubMed; (i) C. A. Arrington, R. West and J. Michl, J. Am. Chem. Soc., 1983, 105, 6176 CrossRef CAS.
- (a) Y. Wang, M. Chen, Y. Xie, P. Wei, H. F. Schaefer III, P. von R. Schleyer and G. H. Robinson, Nat. Chem., 2015, 7, 509 CrossRef CAS PubMed; (b) K. C. Mondal, P. P. Damuel, M. Tretiakov, A. P. Singh, H. W. Roesky, A. C. Stückl, B. Niepötter, E. Carl, H. Wolf, R. Herbst-Irmer and D. Stalke, Inorg. Chem., 2013, 52, 4736 CrossRef CAS PubMed.
- A. F. Filippou, B. Baars, O. Chernov, Y. N. Lebedev and G. Schnakenburg, Angew. Chem., Int. Ed., 2013, 52, 1 CrossRef PubMed.
- P. B. Glaser, P. W. Wanandi and T. D. Tilley, Organometallics, 2004, 23, 693 CrossRef CAS.
- (a) S. Yao, Y. Xiong, W. Wang and M. Driess, Chem. – Eur. J., 2011, 17, 4890 CrossRef CAS PubMed; (b) A. Jana, H. W. Roesky and C. Schulzke, Dalton Trans., 2010, 39, 132 RSC; (c) S. Yao, Y. Xiong and M. Driess, Chem. Commun., 2009, 6466 RSC.
- (a) S. Poh, R. Hernandez, M. Inagaki and P. G. Jessup, Org. Lett., 1999, 1, 583 CrossRef CAS; (b) H. Staudinger and E. Hauser, Helv. Chim. Acta, 1921, 4, 861 CrossRef CAS.
- R. C. Neu, E. Otten, A. Lough and D. W. Stephan, Chem. Sci., 2011, 2, 170 RSC.
- W. Kundel and P. Kästner, Liebigs Ann. Chem., 1965, 686, 88 CrossRef.
- M. L. Nichols and I. A. Derbigny, J. Phys. Chem., 1926, 30, 491 CrossRef CAS.
- P. Paetzhold and G. Schimmel, Z. Naturforsch., 1980, 35b, 568 Search PubMed.
- J. N. Armor and H. Taube, J. Am. Chem. Soc., 1969, 91, 6874 CrossRef CAS.
- (a) F. Paulat, T. Kuschel, C. Näther, V. K. K. Praneeth, O. Sander and N. Lehnert, Inorg. Chem., 2004, 43, 6979 CrossRef CAS PubMed; (b) C. B. Pamplin, E. S. F. Ma, N. Safari, S. J. Rettig and B. R. James, J. Am. Chem. Soc., 2001, 123, 8596 CrossRef CAS PubMed; (c) F. Bottomley and W. V. F. Brooks, Inorg. Chem., 1977, 16, 501 CrossRef CAS; (d) F. Bottomley and J. R. Crawford, J. Am. Chem. Soc., 1972, 94, 9092 CrossRef CAS; (e) J. N. Armor and H. Taube, Chem. Commun., 1971, 287 RSC; (f) J. N. Armor and H. Taube, J. Am. Chem. Soc., 1971, 93, 6476 CrossRef CAS; (g) A. A. Diamantis and G. J. Sparrow, Chem. Commun., 1970, 819 RSC; (h) J. N. Armor and H. Taube, J. Am. Chem. Soc., 1970, 92, 2560 CrossRef CAS.
- N. A. Piro, M. F. Lichterman, W. H. Harman and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 2108 CrossRef CAS PubMed.
- For other structural investigations of M(N2O) complexes see: (a) D. J. Xiao, E. D. Bloch, J. A. Mason, W. L. Queen, M. R. Hudson, N. Planes, J. Borycz, A. L. Dzubak, P. Verma, K. Lee, F. Bonino, V. Crocellà, J. Yano, S. Bordiga, D. G. Truhlar, L. Gagliardi, C. M. Brown and J. R. Long, Nat. Chem., 2014, 6, 590 CrossRef CAS PubMed; (b) A. Pomowski, W. G. Zumft, P. M. H. Kroneck and O. Einsle, Nature, 2011, 477, 234 CrossRef CAS PubMed.
- J.-H. Lee, M. Pink, J. Tomaszewski, H. Fan and K. G. Caulton, J. Am. Chem. Soc., 2007, 129, 8706 CrossRef CAS PubMed.
- (a) F. Bottomley, I. J. B. Lin and M. Mukaida, J. Am. Chem. Soc., 1980, 102, 5238 CrossRef CAS; (b) F. Bottomley and H. Brinzinger, J. Chem. Soc., Chem. Commun., 1978, 234 RSC.
- For related reactions with Ti complexes see: (a) M. D. Walter, C. D. Sofield and R. A. Andersen, Organometallics, 2008, 27, 2959 CrossRef CAS; (b) F. Bottomley, G. O. Egharevba, I. J. B. Lin and P. S. White, Organometallics, 1985, 4, 550 CrossRef CAS; (c) F. Bottomley, I. J. B. Lin and P. S. White, J. Am. Chem. Soc., 1981, 103, 703 CrossRef CAS.
- F. Bottomley, D. E. Paez and P. S. White, J. Am. Chem. Soc., 1981, 103, 5581 CrossRef CAS.
- F. Bottomley, J. Chen, S. M. MacIntosh and R. C. Thompson, Organometallics, 1991, 10, 906 CrossRef CAS.
- (a) M. R. Smith III, P. T. Matsunaga and R. A. Andersen, J. Am. Chem. Soc., 1993, 115, 7049 CrossRef; (b) F. Bottomley, C. P. Magill and B. Zhao, Organometallics, 1991, 10, 1946 CrossRef CAS; (c) F. Bottomley, C. P. Magill and B. Zhao, Organometallics, 1990, 9, 1700 CrossRef CAS; (d) F. Bottomley and J. Darkwa, J. Chem. Soc., Dalton Trans., 1983, 399 RSC; (e) F. Bottomley, D. E. Paez and P. S. White, J. Am. Chem. Soc., 1982, 104, 5651 CrossRef CAS; (f) F. Bottomley and P. S. White, J. Chem. Soc., Chem. Commun., 1981, 28 RSC.
- D. M. Antonelli, W. P. Schaefer, G. Parkin and J. E. Bercaw, J. Organomet. Chem., 1993, 462, 213 CrossRef CAS.
- (a) K. MvNeill and R. G. Bergman, J. Am. Chem. Soc., 1999, 121, 8260 CrossRef; (b) W. A. Howard, T. M. Trnka, M. Waters and G. Parkin, J. Organomet. Chem., 1997, 528, 95 CrossRef CAS; (c) A. M. Berenger, T. A. Hanna and R. G. Bergman, J. Am. Chem. Soc., 1995, 117, 10041 CrossRef; (d) W. A. Howard and G. Parkin, J. Am. Chem. Soc., 1994, 116, 606 CrossRef CAS; (e) W. A. Howard, M. Waters and G. Parkin, J. Am. Chem. Soc., 1993, 115, 4917 CrossRef CAS; (f) A. G. Vaughan, G. L. Hillhouse, R. T. Lum, S. L. Buchwald and A. L. Reingold, J. Am. Chem. Soc., 1988, 110, 7215 CrossRef.
- A. G. Vaughan, P. B. Rupert and G. L. Hillhouse, J. Am. Chem. Soc., 1987, 109, 5538 CrossRef.
- (a) J. G. Andino, U. J. Kilgore, M. Pink, A. Ozarowski, J. Krzystek, J. Telser, M.-H. Baik and D. J. Mindiola, Chem. Sci., 2010, 1, 351 RSC; (b) U. J. Kilgore, C. A. Sengelaub, H. Fan, J. Tomaszewski, J. A. Karty, M.-H. Baik and D. J. Mindiola, Organometallics, 2009, 28, 843 CrossRef CAS.
- (a) M. G. Crestani, A. Olasz, B. Pinter, B. C. Bailey, S. Fortier, X. Gao, C.-H. Chen, M.-H. Baik and D. J. Mindiola, Chem. Sci., 2013, 4, 2543 RSC; (b) M. G. Crestani, A. K. Hickey, X. Gao, B. Pinter, V. N. Cavaliere, J.-I. Ito, C.-H. Chen and D. J. Mindiola, J. Am. Chem. Soc., 2013, 135, 14754 CrossRef CAS PubMed; (c) V. N. Cavaliere, M. G. Crestani, B. Pinter, M. Pink, C.-H. Chen, M.-H. Baik and D. J. Mindiola, J. Am. Chem. Soc., 2011, 133, 10700 CrossRef CAS PubMed.
- J. L. Kisko, T. Hascall and G. Parkin, J. Am. Chem. Soc., 1997, 119, 7609 CrossRef CAS.
- J. S. Figueroa and C. C. Cummins, J. Am. Chem. Soc., 2003, 125, 4020 CrossRef CAS PubMed.
- C. C. Cummins, R. R. Schrock and W. M. Davis, Inorg. Chem., 1994, 33, 1448 CrossRef CAS.
- T. D. Palluccio, E. V. Rybak-Akimova, S. Majumdar, X. Cai, M. Chui, M. Temprado, J. S. Silvia, A. F. Cozzolino, D. Tofan, A. Velian, C. C. Cummins, B. Captain and C. D. Hoff, J. Am. Chem. Soc., 2013, 135, 11357 CrossRef CAS PubMed.
- C. Ni, B. D. Ellis, G. J. Long and P. P. Power, Chem. Commun., 2009, 2332 RSC.
- (a) K. Koo and G. L. Hillhouse, Organometallics, 1998, 17, 2942 CrossRef; (b) P. T. Matsunaga, J. C. Mavropoulus and G. L. Hillhouse, Polyhedron, 1995, 14, 175 CrossRef CAS; (c) K. Koo, G. L. Hillhouse and A. L. Reingold, Organometallics, 1995, 14, 456 CrossRef CAS; (d) P. T. Matsunaga, G. L. Hillhouse and A. L. Reingold, J. Am. Chem. Soc., 1993, 115, 2075 CrossRef CAS.
- N. D. Harrold, R. Waterman, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2009, 131, 12872 CrossRef CAS PubMed.
- B. Horn, C. Limberg, C. Herwig, M. Fiest and S. Mebs, Chem. Commun., 2012, 48, 8243 RSC.
- A. Walstrom, M. Pink, H. Fan, J. Tomaszewski and K. G. Caulton, Inorg. Chem., 2007, 46, 7704 CrossRef CAS PubMed.
- A. W. Kaplan and R. G. Bergman, Organometallics, 1998, 17, 5072 CrossRef CAS.
- A. G. Tskhovrebov, E. Solari, R. Scopelliti and K. Severin, Organometallics, 2012, 31, 7235 CrossRef CAS.
- W. H. Harman and C. Chang, J. Am. Chem. Soc., 2007, 129, 15128 CrossRef CAS PubMed.
- J. R. Bleeke and R. Behm, J. Am. Chem. Soc., 1997, 119, 8503 CrossRef CAS.
- (a) D. J. Berg, C. J. Burns, R. A. Andersen and A. Zalkin, Organometallics, 1989, 8, 1865 CrossRef CAS; (b) W. J. Evans, J. W. Gate, I. Bloom, W. E. Hunter and J. L. Atwood, J. Am. Chem. Soc., 1985, 107, 405 CrossRef CAS.
- (a) S. M. Franke, B. L. Tran, F. W. Heinemann, W. Hieringer, D. J. Mindiola and K. Meyer, Inorg. Chem., 2013, 52, 10552 CrossRef CAS PubMed; (b) O. P. Lam, S. C. Bart, H. Kameo, F. W. Heinemann and K. Meyer, Chem. Commun., 2010, 46, 3137 RSC; (c) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448 CrossRef CAS; (d) L. R. Avens, D. M. Barnhart, C. J. Burns, S. D. McKee and W. H. Smith, Inorg. Chem., 1994, 33, 4245 CrossRef CAS; (e) J.-C. Berthet, J.-F. Le Maréchal, M. Nierlich, M. Lance, J. Vignier and M. Ephritikhine, J. Organomet. Chem., 1991, 408, 335 CrossRef CAS.
- For examples see: (a) S. Hirabayashi and M. Ichihashi, Phys. Chem. Chem. Phys., 2014, 16, 26500 RSC; (b) J.-B. Ma, Z.-C. Wang, M. Schlangen, S.-G. He and H. Schwarz, Angew. Chem., Int. Ed., 2013, 52, 1226 CrossRef CAS PubMed; (c) Z.-C. Wang, S. Yin and E. R. Bernstein, Phys. Chem. Chem. Phys., 2013, 15, 10429 RSC; (d) Z.-C. Wang, N. Dietl, R. Kretschmer, T. Weiske, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2011, 50, 12351 CrossRef CAS PubMed; (e) M. Schlangen and H. Schwarz, Catal. Lett., 2012, 142, 1265 CrossRef CAS; (f) V. Blagojevic, G. Orlova and D. K. Bohme, J. Am. Chem. Soc., 2005, 127, 3545 CrossRef CAS PubMed; (g) I. Balteanu, O. P. Balaj, M. K. Beyer and V. E. Bondybey, Phys. Chem. Chem. Phys., 2004, 6, 2910 RSC; (h) O. P. Balaj, I. Balteanu, T. T. J. Roßteuscher, M. K. Beyer and V. E. Bondybey, Angew. Chem., Int. Ed., 2004, 43, 6519 CrossRef CAS PubMed; (i) M. Brönstrup, D. Schröder, I. Kretschmar, H. Schwarz and J. N. Harvery, J. Am. Chem. Soc., 2001, 123, 142 CrossRef; (j) V. Baranov, G. Javahery, A. C. Hopkinson and D. K. Bohme, J. Am. Chem. Soc., 1995, 117, 12801 CrossRef CAS; (k) M. M. Kappes and R. H. Staley, J. Am. Chem. Soc., 1981, 103, 1286 CrossRef CAS.
- (a) A. Yamamoto, S. Kitazume, L. S. Pu and S. Ikeda, J. Am. Chem. Soc., 1971, 93, 371 CrossRef CAS; (b) L. S. Pu, A. Yamamoto and S. Ikeda, Chem. Commun., 1969, 189 RSC.
- (a) R. G. S. Banks, R. J. Henderson and J. M. Pratt, J. Chem. Soc. A, 1968, 2886 RSC; (b) R. G. S. Banks, R. J. Henderson and J. M. Pratt, Chem. Commun., 1967, 387 RSC.
- (a) A. N. Chernysheva, E. K. Beloglazkina, A. A. Moiseeva, R. L. Antipin, N. V. Zyk and N. S. Zefirov, Mendeleev Commun., 2012, 22, 70 CrossRef CAS; (b) E. K. Beloglazkina, A. G. Majouga, A. A. Moiseeva, N. V. Zyk and N. S. Zefirov, Mendeleev Commun., 2009, 19, 69 CrossRef CAS.
- T. Yamada, K. Suzuki, K. Hashimoto and T. Ikeno, Chem. Lett., 1999, 1043 CrossRef CAS.
- T. Yamada, K. Hashimoto, Y. Kitaichi, K. Suzuki and T. Ikeno, Chem. Lett., 2001, 268 CrossRef CAS.
- H. Tanaka, K. Hashimoto, K. Suzuki, Y. Kitaichi, M. Sato, T. Ikeno and T. Yamada, Bull. Chem. Soc. Jpn., 2004, 77, 1905 CrossRef CAS.
- K. Hashimoto, Y. Kitaichi, H. Tanaka, T. Ikeno and T. Yamada, Chem. Lett., 2001, 922 CrossRef CAS.
- For the solution-based oxidation of alcohols with supported Ru catalysts see: T. L. Stuchinskaya and I. V. Kozhevnikov, Catal. Commun., 2003, 4, 609 CrossRef CAS.
- K. Hashimoto, H. Tanaka, T. Ikeno and T. Yamada, Chem. Lett., 2002, 582 CrossRef CAS.
- J. T. Groves and J. S. Roman, J. Am. Chem. Soc., 1995, 117, 5594 CrossRef CAS.
- R. Ben-Daniel and R. Neumann, Angew. Chem., Int. Ed., 2003, 42, 92 CrossRef CAS.
- J. Ettedgui and R. Neumann, J. Am. Chem. Soc., 2009, 131, 4 CrossRef CAS PubMed.
- H. Goldberg, D. Kumar, G. N. Sastry, G. Leitus and R. Neumann, J. Mol. Catal. A: Chem., 2012, 356, 152 CrossRef CAS.
- B. L. Yonke, J. P. Reeds, P. Y. Zavalij and L. R. Sita, Angew. Chem., Int. Ed., 2011, 50, 12342 CrossRef CAS PubMed.
- G. Kiefer, L. Jeanbourquin and K. Severin, Angew. Chem., Int. Ed., 2013, 52, 6302 CrossRef CAS PubMed.
- S. Saito, H. Ohtake, N. Umezawa, Y. Kobayashi, N. Kato, M. Hirobe and T. Higuchi, Chem. Commun., 2013, 49, 8979 RSC.
- M. Mayachou, L. Elkbir and P. J. Farmer, Inorg. Chem., 2000, 39, 289 CrossRef.
- W. Wislicenus, Ber. Dtsch. Chem. Ges., 1892, 25, 2084 CrossRef.
- J. Haase, in Organic Azides, Syntheses and Applications, ed. S. Bräse and K. Banert, John Wiley & Sons, Weinheim, 2010, pp. 29–51 Search PubMed.
- (a) K. Clusius and H. Schuhmacher, Helv. Chim. Acta, 1958, 41, 972 CrossRef CAS; (b) K. Clusius and H. Knopf, Chem. Ber., 1956, 89, 681 CrossRef; (c) K. Clusius and E. Effenberger, Helv. Chim. Acta, 1955, 38, 1834 CrossRef.
- R. Meier, Chem. Ber., 1953, 86, 1483 CrossRef CAS.
- G. Koga and J.-P. Anselme, Chem. Commun., 1968, 446 RSC.
- G. Koga and J.-P. Anselme, J. Org. Chem., 1970, 35, 960 CrossRef CAS.
- The discussion focusses on solution-based reactions. For the reaction of anionic C- and Si-nucleophiles with N2O in the gas phase see: (a) C. H. DePuy and R. Damrauer, Organometallics, 1984, 3, 362 CrossRef CAS; (b) J. H. J. Dawson and N. M. M. Nibbering, J. Am. Chem. Soc., 1978, 100, 1928 CrossRef CAS; (c) V. M. Bierbaum, C. H. dePuy and R. H. Shapiro, J. Am. Chem. Soc., 1977, 99, 5800 CrossRef CAS.
- W. Schlenk and E. Bergmann, Liebigs Ann. Chem., 1928, 464, 1 CrossRef CAS.
- F. M. Beringer, J. A. Farr, Jr. and S. Sands, J. Am. Chem. Soc., 1953, 75, 3984 CrossRef CAS.
- R. Meier and W. Frank, Chem. Ber., 1956, 89, 2747 CrossRef.
- (a) E. Müller and W. Rundel, Chem. Ber., 1957, 90, 1302 CrossRef; (b) E. Müller, D. Ludsteck and W. Rundel, Angew. Chem., 1955, 67, 617 CrossRef.
- A. N. Nesmeyanov, E. G. Perevalova and T. V. Nikitina, Dokl. Akad. Nauk SSSR, 1961, 138, 1118 CAS.
- M. Kurusawa, T. Nankawa, T. Matsuda, K. Kubo, M. Kurihara and H. Nishihara, Inorg. Chem., 1999, 38, 5113 CrossRef.
- R. Meier and K. Rappold, Angew. Chem., 1953, 65, 560 CrossRef CAS.
- M. Hays and T. P. Hanusa, Tetrahedron Lett., 1995, 36, 2435 CrossRef CAS.
- E. Zerner, Monatsh. Chem., 1913, 34, 1609 CrossRef.
- (a) I. Hermans, B. Moens, J. Peeters, P. Jacobs and B. Sels, Phys. Chem. Chem. Phys., 2007, 9, 4269 RSC; (b) V. I. Avdeev, S. P. Ruzankin and G. M. Zhidomirov, Chem. Commun., 2003, 42 RSC.
- K. Banert and O. Plefka, Angew. Chem., Int. Ed., 2011, 50, 6171 CrossRef CAS PubMed.
- (a) G. A. Vaughan, G. L. Hillhouse and A. L. Rheingold, J. Am. Chem. Soc., 1990, 112, 7994 CrossRef CAS; (b) G. A. Vaughan, C. D. Sofield and G. L. Hillhouse, J. Am. Chem. Soc., 1989, 111, 5491 CrossRef CAS.
- D. J. Mindiola, L. A. Watson, K. Meyer and G. L. Hillhouse, Organometallics, 2014, 33, 2760 CrossRef CAS PubMed.
- T. Labahn, A. Mandel and J. Magull, Z. Anorg. Allg. Chem., 1999, 625, 1273 CrossRef CAS.
- S. Demir, E. Montalvo, J. W. Ziller, G. Meyer and W. J. Evans, Organometallics, 2010, 29, 6608 CrossRef CAS.
- (a) J.-P. F. Cherry, A. R. Johnson, L. M. Baraldo, Y.-C. Tsai, C. C. Cummins, S. V. Kryatov, E. V. Rybak-Akimova, K. B. Capps, C. D. Hoff, C. M. Haar and S. P. Nolan, J. Am. Chem. Soc., 2001, 123, 7271 CrossRef CAS PubMed; (b) A. R. Johnson, W. M. Davis, C. C. Cummins, S. Serron, S. P. Nolan, D. G. Musaev and K. Morokuma, J. Am. Chem. Soc., 1998, 120, 2071 CrossRef CAS; (c) C. E. Laplaza, A. L. Odom, W. M. Davis and C. C. Cummins, J. Am. Chem. Soc., 1995, 117, 4999 CrossRef CAS.
- For a computational study see: C. Cavigliasso, A. Criddle, H.-S. Kim, R. Stranger and B. F. Yates, Dalton Trans., 2014, 43, 4631 RSC.
- J. P. Reeds, B. L. Yonke, P. Y. Zavalij and L. R. Sita, J. Am. Chem. Soc., 2011, 133, 18602 CrossRef CAS PubMed.
- For a computational study see: H. Xie, L. Yang, X. Ye and Z. Cao, Organometallics, 2014, 33, 1553 CrossRef CAS.
- For a metal-induced N–N bond cleavage in the gas phase see: C. Heinemann and H. Schwarz, Chem. – Eur. J., 1995, 1, 7 CrossRef CAS.
- M. R. McCarthy, T. J. Crevier, B. Bennett, A. Dehestani and J. M. Mayer, J. Am. Chem. Soc., 2000, 122, 12391 CrossRef CAS.
- E. Otten, R. C. Neu and D. W. Stephan, J. Am. Chem. Soc., 2009, 131, 9918 CrossRef CAS PubMed.
- For a computational study see: T. M. Gilbert, Dalton Trans., 2012, 41, 9046 RSC.
- R. C. Neu, E. Otten and D. W. Stephan, Angew. Chem., Int. Ed., 2009, 48, 9709 CrossRef CAS PubMed.
- G. Ménard, J. A. Hatnean, H. J. Cowley, A. J. Lough, J. M. Rawson and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 6446 CrossRef PubMed.
- (a) E. Theuergarten, A. C. T. Kuate, M. Freytag and M. Tamm, Isr. J. Chem., 2015, 55, 202 CrossRef CAS; (b) M. J. Kelly, J. Gilbert, R. Tirfoin and S. Aldrigde, Angew. Chem., Int. Ed., 2013, 52, 14094 CrossRef CAS PubMed.
- A. G. Tskhovrebov, E. Solari, M. Wodrich, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2012, 51, 232 CrossRef CAS PubMed.
- A. G. Tskhovrebov, B. Vuichoud, E. Solari, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2013, 135, 9486 CrossRef CAS PubMed.
- M. Göhner, P. Haiss, N. Kuhn, M. Stöbele and K.-P. Zeller, Z. Naturforsch., 2013, 68b, 539 Search PubMed.
- E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651 RSC.
- A. G. Tskhovrebov, E. Solari, M. D. Wodrich, R. Scopelliti and K. Severin, J. Am. Chem. Soc., 2012, 134, 1471 CrossRef CAS PubMed.
- A. G. Tskhovrebov, E. Solari, R. Scopelliti and K. Severin, Inorg. Chem., 2013, 52, 11688 CrossRef CAS PubMed.
- A. G. Tskhovrebov, L. C. E. Neasted, E. Solari, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 1289 CrossRef CAS PubMed.
- (a) D. K. Kölmel, N. Jung and S. Bräse, Aust. J. Chem., 2014, 67, 328 CrossRef; (b) D. B. Kimball and M. M. Haley, Angew. Chem., Int. Ed., 2002, 41, 3338 CrossRef CAS.
- D. R. Newell, B. J. Foster, J. Carmichael, A. L. Harris, K. Jenns, L. A. Gumbrell and A. H. Calvert, Triazenes-Chemical, Biological and Clinical Aspects, Springer, Berlin, Heidelberg, 1990, p. 119 Search PubMed.
- G. Kiefer, T. Riedel, P. Dyson, R. Scopelliti and K. Severin, Angew. Chem., Int. Ed., 2015, 54, 302 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |