
Ultrasonically treated liquid interfaces for progress in cleaning and separation processes
Darya
Radziuk
* and
Helmuth
Möhwald
Max-Planck Institute of Colloids and Interfaces, Science Park Golm, D-14476 Potsdam, Germany. E-mail: darya.radziuk@mpikg.mpg.de
First published on 22nd September 2015
Abstract
Ultrasound and acoustic cavitation enable ergonomic and eco-friendly treatment of complex liquids with outstanding performance in cleaning, separation and recycling of resources. A key element of ultrasonic-based technology is the high speed of mixing by streams, flows and jets (or shock waves), which is accompanied by sonochemical reactions. Mass transfer across the phase boundary with a great variety of catalytic processes is substantially enhanced through acoustic emulsification. Encapsulation, separation and recovery of liquids are fast with high production yield if applied by ultrasound. Here we discuss the state of knowledge of these processes by ultrasound and acoustic cavitation from a perspective of a physico-chemical model in order to predict and control the outcome. We focus on the physical interpretation and quantification of ultrasonic parameters and properties of liquids to understand the chemistry of liquid/liquid interfaces in acoustic fields. The roles of thermodynamic enthalpy and entropy (incl. Laplace and osmotic pressure) in the context of sonochemical reactions (separation, catalysis, degradation, cross-linking, ion exchange and phase transfer) are outlined. The synergy of ultrasound and electric fields or continuous flow chemistry for cleaning and separation via emulsification is highlighted by specific strategies involving polymers and ultrasonic membranes.
 Darya Radziuk |
 Helmuth Möhwald |
1 Introduction
Awareness of the contaminated environment has been considerably increased, as large amounts of pollutants (e.g. industrial chemicals, agrotoxics, hormones, dyes, pharmaceutical substances, etc.) accumulate in the aquatic environment. Many of these substances exhibit carcinogenic and mutagenic properties, decreasing the health of living beings. For example, contamination can be effected by dyes through color change, odor alteration, eutrophication, underoxygenation and bioaccumulation,1 which can be hardly degraded by physico-chemical or biological methods.2 Often, industrial wastewater contains high concentrations of toxic ions of heavy metals (e.g. Pb, Cu, Hg and Cr), which can be hardly removed by biodegradation due to the inhibition or prevention of the oxidation of organic waste by bacteria.2Water is a renewable resource, which can be converted from waste into environmentally acceptable and drinking water (wastewater treatment). Water cleaning requires science and engineering by applying mechanical, physical, biological and chemical methods. Most of them involve the removal of solids, bacteria, algae, plants, inorganic and organic compounds. Among the biological and physicochemical technologies filtration, coagulation and flocculation, ion exchange, electrodialysis, aeration, clarification and removal of insoluble contaminants have been widely applied.2 However, some of them (e.g. aeration) cannot remove nonvolatile organic compounds (e.g. phenols and creosols) and require strong chemical oxidants such as chlorine (Cl2) or potassium permanganate (KMnO4). In ion exchange systems poor effluent quality, decreased lengths of service run, or increased consumption of the regenerant are typical drawbacks. Oil can block the passage of ions to and from exchange sites and requires surfactants for its removal. Polymerization of silica can occur in a weak base resin at low pH being a problem in an exhausted strong base anion resin. Many particulate or weakly ionized contaminants cannot be removed by membrane technology. Overall, the above mentioned cleaning processes only transfer the contaminants from one medium to the other, producing secondary wastes/sludge and leaving the problem unsolved.
Recycling is often laborious and expensive in comparison to the synthesis of the same product from raw materials or other sources, which can be limited in amount, if they are not renewable. There is a need to prevent the waste of useful materials, reduce the consumption of fresh raw materials, and decrease the amount of chemical compounds in catalysis and material production. At present the economic and environmental benefits of recycling suffer due to their costs and require multiple steps of treatment processes of heating, hydrolysis, purification, polymerization and complicated removal of residues.
Solvent extraction is one of the most important separation methods, which involve porous membranes with amphiphilic/oleophilic properties.3 The mass transfer and separation processes are controlled by the chemical composition, thickness and porosity of a membrane.4 A larger interfacial area of a membrane surface significantly accelerates the mass transfer and reduces the residence time yielding a higher volume of the extracted liquid phase.3,5 Liquid/liquid phase separation can be improved in a microflow as the aqueous phase flows from the downstream of the organic phase and dissolves in the material. In conventional systems the counter-current flow is accomplished by gravitational segregation and involves droplets.6 In microfluidic systems, viscosity and surface wetting are more effective at controlling flow than gravity and inertia. Therefore, in such systems, laminar rather than microdroplet countercurrent flow is more feasible. Although laminar flow has a disadvantage in mixing compared to a droplet system,7 the laminar countercurrent flow allows the versatile design of chemical processes.8
Conventional methods, such as gravity separation, skimming, and flotation, are limited to low efficiency and high operation cost,9 and are unable to separate oil/water emulsions.10 Although hydrophobic/oleophilic membranes have received a wide practical application, they display poor separation selectivity and efficiency due to pore clogging and surface fouling by oil or grease.11 Because water and many oils are intrinsically immiscible, many efforts are focused on the development of new technology with effective mixing and design of superhydrophobic/superoleophilic and underwater superoleophobic surfaces.12
Ultrasound and acoustic cavitation substantially improve cleaning and separation processes through the efficient mixing and emulsification,13 acceleration of reaction rates,14 extraction,15 selective separation and exchange,16 enhanced mass transfer,17 degradation18 and recovery.19 For example, acoustic emulsification of oil droplets with organic substances in aqueous solution within seconds yields stable microspheres.20 Ultrasonic conversion of compounds and phase transfer reactions occur at faster rates with a higher yield of products than by conventional procedures.21 Acoustic separation in the mixture of suspended species is fast, nondestructive, environmentally clean and requires only a standing field of ultrasound.22
Acoustic cavitation is more efficient in terms of total power consumed per mole of contaminant (e.g. p-nitrophenol) degraded per liter of water being far superior to conventional UV-photolytic degradation. The sonochemical degradation of chemical contaminants in water can be performed through oxidation by hydroxyl radicals, pyrolytic decomposition and supercritical water oxidation during acoustic cavitation.23 In water disinfection inactivation of bacteria and deagglomeration of bacterial clusters or flocs can be induced during acoustic cavitation through high pressure gradients, strong shear forces acting within bacterial walls, chemical attack by radicals (OH˙ and H˙) and H2O2 as an effective bactericide.24
The strength and efficiency of the ultrasound-based technology depends on our ability to comprehend the interaction of ultrasound and acoustic cavitation (formation, growth and collapse of bubbles) at interfaces. At present the ultrasonic studies of liquid/liquid systems are extended by excellent empirical results, but are limited to the explanation of influencing parameters (ultrasonic intensity, irradiation time and properties of liquids). Most findings reveal higher mass transfer and attribute it to an intense mixing due to ultrasonically formed streams and flows, but lack quantitative prediction. Still shock waves and liquid jets are difficult to be distinguished and controlled. Little is known about the conditions at the interfacial zone at the liquid/liquid boundary in the acoustic field versus surface tension, density, viscosity and dissolved gas, critical for the initial formation of emulsion droplets.
In this review we address the above points from two perspectives: (i) ultrasound propagation mechanisms at liquid interfaces with different complexities (complex liquid solution) and (ii) acoustic cavitation processes at liquid interfaces in the presence of an electric field and continuous liquid flows. These are extended with a great variety of approaches for cleaning, separation and ion exchange processes to be performed by ultrasound as next. For the first time we introduce a new method to quantify the acoustic pressure dissipation within a droplet volume (jets or shock wave impact) by using only ultrasound and electric field. Overall, here we describe the current state of knowledge and outline approaches and strategies to predict conditions and outcome at liquid interfaces in acoustic fields.
2 Physical principles of ultrasound at liquid interfaces
2.1. Ultrasound propagation at liquid interfaces
In a liquid ultrasound propagates as a longitudinal wave of acoustic pressure with periodic compression and rarefaction cycles and develops a certain velocity (Fig. 1A). The latter is inversely proportional to the square root of the material density (ρ) multiplied by the compressibility (k). In liquids with higher density (e.g. water has a higher density than oil) ultrasound propagates with lower velocity (v), therefore it develops a smaller wavelength. In comparison, solids rather than liquids have a higher potential to experience a direct action of ultrasound with stronger affinity for reflection and formation of standing waves. | ||
| Fig. 1 Schematic illustration of ultrasound interaction in a mixture of two unlike liquids, which form an interfacial zone between a non-polar phase (liquid A) and a polar phase (liquid B). (A) Ultrasound is produced by a horn-type system with an irradiating tip positioned in the liquid A nearby an interfacial zone with liquid B, which have different densities (ρ), velocities of sound (v), viscosities (η) and relaxation times (τ). (B) Physical model of ultrasound propagation along the surface normal x with a wavelength λ and an amplitude ξ in the interfacial zone. The perturbation of the latter is stronger at higher ultrasonic amplitude ξ. The schematic model is adapted with permission from the reference M. K. Lit and H. S. Fogler, J. Fluid Mech., 1978, 88, 499. | ||
In ultrasound propagation the acoustic pressure is time dependent with the square amplitude proportional to the intensity.25 By analogy with electrical vibrations, the intensity of ultrasound increases in liquids with low density and small speed of propagation at a constant pressure amplitude.25 The velocity of sound in liquids varies from about 900 to 2000 m s−1 depending on the temperature and pressure. In most pure liquids the value of velocity decreases as the temperature increases being far from the critical values. As an exception, in water the sound velocity increases by ∼2 m s−l for each 1 K rise until it reaches a maximum at 73 K. In almost all liquids the value of v linearly increases with higher pressure as a consequence of a squeezed closer contact between the molecules.26 The propagation of ultrasound in a liquid is characterized by the acoustic impedance, Z, which is a product of the density of a liquid (ρ) multiplied by the sound velocity (v) in this liquid.
Let us consider the propagation of ultrasound across the interface between two unlike liquids, e.g. liquid A (non-polar or organic phase) and liquid B (polar or aqueous phase) (Fig. 1B). The interfacial zone between these two liquid phases acts as an acoustic boundary with an acoustic impedance characteristic for each of the liquids. The latter is a resistance measure of a liquid and is directly proportional to the density of the medium (ρ) and the ultrasound velocity (v) in it (Fig. 1A). At the liquid boundary ultrasound (λ) disturbs the interface with amplitude (ξ) and forms an emulsion, if ξ exceeds a certain threshold value. The diameter of the dispersed phase (emulsion droplets) is proportional to the ultrasonic amplitude being one third of the surface wavelength.27 This dependence is explained by a model, in which the crests of the interfacial wave are considered to be cylindrical jets during breakup.
In general, by analogy with light, the incident ultrasound undergoes reflection and scattering, refraction and absorption at the liquid/liquid interface.28 Two types of reflection can occur, specular (the interfacial boundary is smooth and larger than the wavelength of ultrasound) and non-specular (or scattering, the dimension of an interfacial boundary is much smaller than the wavelength of ultrasound). In specular reflection the difference in the acoustic impedance, Z-value, at an interface determines the reflection coefficient, the proportion of the reflected intensity at the interface. High Z-values at an interface create large echoes, which is widely used in ultrasonic imaging. The scattering of ultrasound depends on the dimensions of the target (particle, impurity, etc.), the diameter of the ultrasonic beam and its frequency.
The frequency of ultrasound, the viscosity (η) and the relaxation time (τ) of the liquid determine the absorption of ultrasound by a medium (Fig. 1A). Other parameters include thermal conduction and molecular effects. The latter are increased at higher frequency of ultrasound as molecules move at a faster rate, producing stronger molecular vibrations followed by more frictional heat. Viscosity is a measure of these frictional forces between molecules, i.e. higher absorption of ultrasound occurs in liquids with high values of viscosity. In contrast, the time necessary for a reversion of molecular displacements by ultrasound to the original mean positions (relaxation time, τ) is inversely related to the absorption of ultrasound. This can be explained by reoriented molecular motion at the next compression pressure cycle resulting in additional dissipation of the ultrasonic energy. In most cases, the intensity of ultrasound decreases continuously while propagating through a liquid. This propagation loss consists of spreading (geometrical configuration of waves) and attenuation (characteristic of a liquid) contributions. The latter is a combination of scattering and absorption of ultrasound in a liquid.
Ultrasound absorption and dispersion can be calculated from chemical relaxation during the stepwise dissociation of an electrolyte, involving formation of intermediate aquo complexes of metal ions.29 To remind, metal aquo complexes are coordination compounds containing metal ions with only water as a ligand. These complexes are the predominant species in aqueous solutions of many metal salts, such as metal nitrates, sulfates and perchlorates. By measuring the ultrasound absorption cross-sections and the relaxation frequencies one can find out the mechanism of the metal complex formation (e.g. different ions of alkaline rare earth metals). For example, one of the mechanisms consists of a gradual substitution of the solvent molecules in the coordination shells of metal ions. This process involves ion–dipole or dipole–dipole interactions, field stabilisation of the ligands and stereospecific effects.
2.2. Injected liquid droplet model in acoustic cavitation
In a liquid above a certain threshold value of the intensity in the negative half-cycle of acoustic pressure ultrasound creates cavitation: formation, growth and collapse of gaseous or vapor filled bubbles (Fig. 2). The latter are composed of a hot core (above 5000 K and 1000 atm) and a liquid-like surrounding shell (about 1900 K and hundreds atm) with heating/cooling rates higher than 1010 K s−1.30 In the bulk the bubble collapse can follow the spherical symmetry with the formation of shock waves.31 The symmetry of this collapse can be perturbed nearby a larger boundary followed by a jet impinging outwards through the bubble interior.32 The mechanism of bubble collapse can be understood in the frame of the adiabatic heating model with no time left for heat dissipation in the bulk.33 Turbulent flows and streams are the physical consequences of the bubble collapse in each type of symmetry.34 A rapid gas-phase compression of a cavitation bubble creates pyrolysis of water molecules and free radical species (e.g. H˙ and OH˙) escaping from the gas–liquid interface for redox reactions with liquid molecules.35 In complex liquid solution a cavitation bubble can be explained by an injected liquid droplet model.36 In this model tiny drops of nonvolatile substances can enter the hot gaseous bubble core through the capillary waves of a perturbed liquid-like shell. The latter can be modified by charged additives (e.g. surfactants, polymers, particles, solvent molecules) adsorbed at the cavitation interface and may contribute to electrostatic/electrosteric interactions.37 | ||
| Fig. 2 Schematic illustration of a model of heterogeneous acoustic cavitation – injected liquid droplet model – in a complex liquid solution consisting of liquid A (organic phase) and liquid B (aqueous phase) in the presence of foreign matter. The latter, which is depicted as a red sphere, represents nonvolatile metal ions or cations, hydrophobic proteins or lipids, carbohydrates or dyes, volatile or biological substances (e.g. bacteria) amongst many others. The acoustic cavitation bubble is sketched with a perturbed spherical curvature due to the acting surface capillary waves, which bring liquid droplets inside the hot gaseous interior. Other processes at the liquid-like cavitation shell include formation of radical species for redox reactions in the bulk, high heating/cooling rates and electrostatic/electrosteric interaction with additives. Shock waves and jets upon bubble collapse are followed by acoustic streams and turbulent flows. | ||
Very often the polar liquid is water, which involves ionic and hydrogen bonding in solution (liquid B). Oil or any other organic solvent (e.g. toluene) can be considered as a nonpolar liquid with hydrophobic molecular interactions (liquid A). A foreign matter, which is depicted as a red sphere in the scheme, can be represented by nonvolatile metal ions or cations, hydrophobic proteins or lipids, hydrophilic carbohydrates and dyes, volatile or biological substances (e.g. bacteria) amongst many others. Under ambient conditions two liquids A and B, that are brought into contact, create an interfacial zone of two separated phases characterized by physico-chemical differences in polarity, density, viscosity, solubility and surface tension. In acoustic cavitation ultrasound does not directly interact with liquid molecules as its wavelength is far above molecular (atomic) dimensions (e.g. about several cm in liquids). Ultrasonic interaction of two different liquids A and B in an acoustic pressure field mainly occurs through cavitation bubbles and subsequent physical phenomena (shock waves, jets, turbulence flows and streams).
2.3. Ultrasound and acoustic cavitation in cleaning and separation
Ultrasound and acoustic cavitation can substantially advance cleaning and separation processes assisting conventional methods for water treatment (e.g. aeration, clarification, filtration, ion exchange, precipitation and recovery). In aeration flowing streams and turbulence can increase sufficient contact time and the ratio of air to water. This yields an effective removal of the unwanted gas (CO2) and its reduction (decarbonation), oxidation of iron and manganese to insoluble precipitates, reduction (stripping) of ammonia and hydrogen sulfide and control of bacteria (disinfection).38 Carbon dioxide can be formed from bicarbonate and carbonate ions and removed at higher temperature depending on aeration time, volume of air in contact with water, and surface area of water exposed to air. Acoustic cavitation can significantly advance the processes in aeration reducing time and cost of chemical consumption. For example, CO2 can be degassed by ultrasound at lower temperatures (below 60 °C), significantly reducing the degradation processes in the desorption unit.39In general, iron, chromium and manganese have a negative potential (or a small positive value) and cannot be sonochemically reduced to a metallic state, but oxides can be prepared. For example, sonochemical oxidation of air-saturated Fe2+ ions to Fe3+ ions is an indirect process of reaction with OH radicals due to the degassing of solution by ultrasound.40 Sonochemical oxidation of iron yields products with different morphologies at the nanometer scale and exhibits super paramagnetic properties in the presence of ultrasonically formed oxidants (e.g. hydrogen peroxide, nitrite and nitrate ions).41 Low frequency acoustic cavitation (e.g. 20 kHz or 24 kHz) effectively oxidizes iron(II), iron(III) and soluble iron–organic complexes with higher content at an increased intensity.42 Effective destabilization and removal of iron complexes can be performed at lower ultrasonic intensity. Permanganate can be transformed into manganese dioxide due to the reaction with sonochemically formed H2O2, H2 and H in the presence of 1-propanol acting as a scavenger of OH radicals.43
Sonochemical treatment of bicarbonate and carbonate ions considerably enhances the degradation of hydrophilic dyes (e.g. rhodamine B) with higher reaction rates in water.44 This process is selective in the sonochemical reaction between carbonate and OH radicals, which significantly reduces the chemical oxygen demand and carbon dioxide formation. In another example, cavitation bubbles saturated with argon effectively remove hydrogen from the magnesium alloy surface and smoothen its surface during degassing, not accessible by aeration.45 In the absence of an inert gas the dissolved oxygen concentration reaches an equilibrium steady state due to the simultaneous action of degassing and absorption of cavitation bubbles.46 Neither the sonicated volume nor ultrasonic intensity perturbs this dissolved oxygen equilibrium. In disinfection volatile chlorinated compounds cannot be degraded by aeration and require the addition of chemicals.47 Acoustic cavitation can effectively destroy chlorinated compounds in contaminated ground water following pseudo-first order kinetics after only 30 min of treatment.
3 Ultrasound at the organic/water interface
3.1. Physico-chemical model of ultrasonic emulsification
Propagation of ultrasound through two immiscible liquids under conditions above a certain cavitation threshold value causes acoustic emulsification followed by the formation of droplets and an emulsion of a continuous phase (Fig. 3). The initial droplet formation is controlled by the density (ρ) and viscosity (η) of these unlike liquids48 (Fig. 1). Another parameter in emulsification is irradiation time, which leads to a reduction of the droplet diameter with prolonged duration. For example, short sonication (few seconds) leads to larger droplets (70 μm) and longer irradiation yields submicron emulsions depending on the liquid jet, wavelength and amplitude of ultrasound (Fig. 3A). Acoustic emulsification can be explained as a two stage process, which involves the initial droplet formation by a type of Rayleigh–Taylor instability mechanism and a break up process (Fig. 3B).49 The characteristic droplet diameter produced by the instability is related to the induced capillary wavelength at the interface. The droplet diameter is related to the interfacial wavelength by a rough model, in which the crests of the interfacial wave are considered to be cylindrical jets during breakup. Such a jet in the initial phase of acoustic emulsification may be related to the jet wavelength lmax at the maximum instability and to the interfacial ultrasound wavelength λ. If the drops are formed from a jet that breaks at a distance of one wavelength lmax, the droplet volume is equal to the cylinder volume of the broken jet with a diameter d and a length lmax.50 The interfacial instability leads to the eruption of the oil phase into the water phase. The threshold amplitude of the vibration ξ (Fig. 1B) can reach 10–80 μm, which is close to the displacement amplitude of the vibration of the transducer, and agrees well with the theoretically predicted values. At the second stage droplets are assumed to break off from the oil–water interface in the form of ‘capillary-jets’, once the instability criterion (the threshold amplitude) is exceeded. | ||
| Fig. 3 Schematic illustration of liquid/liquid emulsification by ultrasound and acoustic cavitation. (A) A model explaining the initial formation of a liquid droplet by ultrasound with a wavelength λ. Ultrasound perturbs an interfacial zone of liquid A (non-polar phase) and liquid B (polar phase) by a jet with a length lmax, which forms a droplet with a diameter d. The latter is created if the interface becomes unstable and satisfies the instability criterion: higher difference in densities (ρB − ρA) or a smaller value of (ηA + ηB). (B) A model explaining the formation of droplets with different diameters in two stages of ultrasonic treatment. The 1st stage includes perturbation of the interfacial zone by ultrasound (acoustic wave) resulting in an initial formation of larger droplets. The 2nd stage is acoustic cavitation, in which preformed droplets continuously decrease their diameter with a prolonged treatment. The scheme in B is adapted with permission from reference J. P. Canselier, H. Delmas, A. M. Wilhelm and B. Ablsrnail, J. Dispersion Sci. Technol., 2002, 23, 333 and M. K. Li and H. S. Fogler, J. Fluid Mech., 1978, 88, 499. | ||
Acoustic emulsification occurs only in the presence of dissolved gas such as air, N2, O2, H2 or Ar (any other inert gas) and at a certain value of an external pressure.51 Dissolved gases favor the formation of nuclei and bubbles, but at high concentration gases cause a buffering effect on the bubble collapse. If the vapor or gas pressure in bubbles is in equilibrium with the external hydrostatic pressure, collapse does not occur and emulsification does not take place. At a certain external pressure the concentration of an acoustic emulsion increases with pressure up to a high value and then decreases due to the oversaturation by a gas. The value of this pressure depends on the ultrasonic energy, the temperature of the mixture and the type of liquids (density and viscosity). As a consequence, acoustic energy, which is transmitted per sq. cm through a liquid, increases at a higher pressure gradient, but can be lowered in liquids with high density.52,53
As we have shown above, the diameter of the emulsion droplets is determined by the length of the acoustic liquid jet and the density and viscosity of liquids (A and B). As another parameter, high interfacial tension (γi) increases the Laplace pressure leading to smaller droplets at lower concentration of surfactants. For a uniform diameter distribution of droplets the osmotic pressure inside droplets counteracts the Laplace pressure by increasing the hydrophobicity in the dispersed phase. The latter can be performed through introduced additives, for example proteins. Let us examine an acoustic emulsion in the presence of additives (proteins, saccharides or polymers) in the mixture of oil (liquid A) and aqueous phases (liquid B). Specific examples of ultrasonic emulsions are explained in the context of cleaning and separation by introduced encapsulation, mixing of oil/alcohol (transesterification) and ion exchange.
3.2. Ultrasonic emulsions for cleaning by encapsulation
Acoustic emulsification of the aqueous phase in the presence of human serum albumin (HSA) leads to microspheres with diameter depending on the acoustic active zones and ultrasonic parameters (intensity or power and duration) (Fig. 4A).58 The nonhomogeneous dissipation of ultrasonic energy near the irradiating horn probe yields microspheres with different diameters being larger in low acoustic zones (A and E) and smaller in highly intense cavitation field (C) (Fig. 4A). The initial diameter is about 30 μm during the first minutes of sonication and only several μm after longer treatment, which is explained in the frame of the acoustic emulsification model.59
 | ||
| Fig. 4 Acoustic emulsification of oil/water liquid phases in the presence of an aqueous solution of proteins. (A) Schematic illustration of the acoustic pressure field zones for the formation of emulsions with a proteinaceous human serum albumin (HAS) shell of various diameters. Optical phase contrast images of ultrasonically prepared proteinaceous microspheres from A, B and C acoustic cavitation zones. (B) True color photo images of acoustic emulsions with increased concentration of proteins (a – no proteins, b – 0.1 wt%, c – 0.5 wt% and d – 1 wt%). (C) Confocal fluorescence microscopy images of CY3-t-RNA (red) encapsulated within FITC-BSA (green). The upper row shows the reconstructed 3D image of the selected proteinaceous microsphere with encapsulated CY3-t-RNA. The lower row depicts the selected microspheres, shortly after the emulsification (a) and after 24–25 hours (b) and another type consisting of the mixed shell structure (c). Reprinted with permission from references Y. Han, D. Radziuk, D. Shchukin and H. Möhwald, J. Mater. Chem., 2008, 18, 5162 and U. Shimanovich, I. D. Tkacz, D. Eliaz, A. Cavaco-Paulo, S. Michaeli and A. Gedanken, Adv. Funct. Mater., 2011, 21, 3659. | ||
The concentration of proteins in the shell determines the stability of microspheres (Fig. 4B). Freshly prepared emulsions are milky suspensions independent of the concentration of protein. After one week of aging the emulsion develops a two phase separation with a clear oil/water interface (no proteins, in a) and increased turbidity with higher concentration of proteins (from b to d) (Fig. 4B). The precipitant is composed of stable homogeneous small microspheres (about 1 μm) with a thick proteinaceous shell, while the supernatant contains light aggregates, indicating droplets with lower density and a thinner shell.
Ultrasonic cleaning of water through encapsulation has been productively extended by different drug compounds60 (e.g. antibiotic tetracycline and anticancer taxol)54 and biomolecules (e.g. DNA,61 RNA,62 hemoglobin63) at various chemical composition of shells (e.g. protein/protein,64 protein/silica,65 layer by layer polymer/protein/silica66 and calcium phosphate/protein67). This process is fast as it takes only several minutes for emulsification and seconds for encapsulation by ultrasound. For example, acoustic emulsification of aqueous solutions in the presence of bovine serum albumin protein (BSA) and t-RNA yields encapsulation of the latter as an oil-core inside the BSA-shell (3D image in Fig. 4C).68 Freshly prepared RNA-encapsulating emulsions have a distinct red core (t-RNA) and a green shell (BSA) (image a in Fig. 4C). The t-RNA core is delocalized closer to the walls of the sphere within 24–25 hours (image b in Fig. 4C). Another type of droplets acquires a mixed sphere consisting of a yellow shell and a dark core due to simultaneous contribution of red t-RNA and green BSA to the construction of the droplet walls. The diameter of emulsion droplets increases with the increased number of nucleotides of the RNA from about 630 nm to 2.7 μm.
Ultrasonic treatment of an aqueous solution of gum acacia (complex mixture of glycoproteins and polysaccharides) and α-tocopherol/vitamin E from vegetable oil yields stable microspheres.72 The latter can be modified with poly-L-lysine and embedded into polymer Ca-alginate film, excluding high temperature and toxic organic solvents. The precipitate of chitosan and xanthan gum as a polyelectrolyte complex in the shell of emulsion droplets is stable due to the ultrasonic cross-linking. The latter is independent of the type of the oil phase (e.g. miglyol, soybean oil, hexane, cyclohexane or toluene).70
In agreement with the model (Fig. 3) the diameter of droplets decreases with higher volume fraction of the aqueous phase (lower oil/water ratio) followed by a higher surface area and an increase of the entropy in the emulsion. To remind, the entropy (designated as ΔS) is a measure of the extent of diameter decrease of the organic phase with a positive value for an increased number of droplets and higher stability. The enthalpy (designated as ΔH), as another factor, enhances the binding energy of the organic phase material and requires the counter balance with the entropy for a minimal positive value of the free Gibbs energy. If the volume changes of droplets are small (approaching zero), the enthalpy corresponds to the internal energy, which is the sum of the work (designated as ΔW) and energy losses due to heat (designated as ΔQ). The value of ΔW being required to expand the interfacial area decreases with the lower value of the surface tension, contributing to the stability against coalescence. In this way, a nearly homogeneous emulsion with stable larger droplets can be produced by ultrasound.
To note, surface tension can be significantly lowered at higher concentration of surfactants, and lower Laplace pressure leads to a larger diameter. Another factor, osmotic pressure inside droplets, can counteract the Laplace pressure by increasing the hydrophobicity through the additive introduced into the dispersed phase. The dispersion phase (with higher volume fraction) favors transport processes between the droplets, for example, in styrene or methyl methacrylate in the presence of the anionic surfactant SDS and hexadecane as a cosurfactant.78 On the other hand, sonochemical reactions with radical species yield facile oxidation and formation of iron oxide and magnetite (Fe2O3 and Fe3O4), zirconia (ZrO2), calcium carbonate (CaCO3)79 and polymer/silica nanocomposites.80
Acoustic emulsification in the presence of polyelectrolytes in the aqueous phase forms microdroplets with a liquid core (organic phase) and a polymeric shell.81 For example, ultrasonic treatment of aqueous polyglutamate solution and toluene containing a hydrophobic compound (e.g. dye 5,10,15,20-tetraphenylporphin, TPP) forms a negatively charged emulsion.56 Next, these emulsion droplets are modified by the layer by layer electrostatic assembly of polyethyleneimine (PEI)/polyacrylic acid (PAA). Upon reaction suspended micropsheres acquire properties with permeability being controlled by varying pH82 or ionic strength.83 This method is successfully extended to a vegetable oil with a lipophilic drug (rifampicin)84 and other polyelectrolytes (e.g. PAH/PSS).57
As we have shown above the acoustic emulsification involves the successful synergy of mechanical action (shock waves, jets, turbulent flows and streams with shear forces) and sonochemically formed radicals. The latter are formed due to the pyrolytic decomposition of liquid molecules inside hot spots (cavitation bubbles at a collapse state). These features advance not only ultrasonic emulsification, but also phase transfer catalysis across the interface and liquid/liquid separation and extraction.
3.3. Ultrasound phase transfer catalysis
In general, acoustic emulsification is an effective tool for phase transfer reactions and processes at the liquid interface between the organic and aqueous phases (e.g. transesterification for the production of biodiesel, hydrolysis for degradation of compounds85 including polymers,86 acid or base-catalyzed methanolysis,87 oxidative desulfurization,88 demulsification89 and nano-emulsification,90 amongst others). Consequently the reaction rate in phase transfer catalysis increases with higher interfacial liquid/liquid area. As we have learned, ultrasonic treatment of the latter at low frequency forms an emulsion between immiscible fluids by disruption of the phase boundary according to the model described in Fig. 3. The higher values of the surface to the volume interfacial area of a preformed emulsion can be increased by a subsequent increase of the ultrasound frequency. Few tens of nm of emulsion droplets can be obtained by using the tandem 20 kHz–1.6 MHz–2.4 MHz sequential processing, which increases the phase transfer reaction rate between chloroform and potassium carbonate aqueous solution.91 To note, the ultrasonic treatment only at high frequency (MHz) does not emulsify the organic/aqueous liquid phases, as the small amplitude is insufficient for phase separation.Acoustic emulsification improves the miscibility between phases,92 enhances the interaction of alcohol and oil phases, eliminating external heating due to local temperature increase of cavitation hot spots. For example, the transesterification of soybean oil and methanol is accelerated by ultrasound, but requires Brønsted acidic ionic liquid as a catalyst.93 The quantity of a product is controlled by the alcohol to oil ratio: higher ratio (e.g. 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) results in higher yield. Ultrasonic conversion of triglycerides in nonedible feedstock (e.g. S. Marianum seed oil) occurs with higher yield and a faster reaction rate with methanol and ethanol in the presence of potassium hydroxide.94 As the transesterification reaction is reversible, the equilibrium toward the formation of esters can be shifted by an excess of alcohol, which acts as an extracting solvent and an esterification reagent.
:1) results in higher yield. Ultrasonic conversion of triglycerides in nonedible feedstock (e.g. S. Marianum seed oil) occurs with higher yield and a faster reaction rate with methanol and ethanol in the presence of potassium hydroxide.94 As the transesterification reaction is reversible, the equilibrium toward the formation of esters can be shifted by an excess of alcohol, which acts as an extracting solvent and an esterification reagent.
In a reverse process, i.e. demulsification, water can be separated from crude oil solution by ultrasound at low frequency without chemical demulsifiers.89 The demulsification efficiency can reach up to 65% with higher rates at lower temperature (45 °C instead of industrial 65 °C) reducing the energy consumption in heating. However, little is known about the demulsification mechanism of this system and existing quantitative approaches are limited to direct visualization of oil flocculation only at high frequencies (e.g. 2 MHz).95
The presence of phase transfer catalysts, i.e. onium salts (ammonium and phosphonium salts), macrocyclic polyethers (crown ethers) and aza-macrobicyclic ethers (cryptands) produces an emulsion with a smaller droplet diameter and a higher surface area.97 This is due to the accumulation of these catalysts at the interface acting as surface active compounds with ionic properties. The mass transfer across the interfacial area is enhanced by ultrasound with faster rates at higher methanol-to-oil and OH−-to-oil molar ratios.
On the other hand the mass transport can be increased by higher pressure gradient across the interface due to the coexistence of both the gas and liquid phases, and this effect is enhanced by cavitation.17 The latter also forms strong oxidants of primary and secondary radicals, which accelerate the oxidative desulfurization.98 In this process an oxidant forms a complex with a phase transfer agent for transportation across the interface. Ultrasound reduces the undesired consumption of an oxidant by sonochemically formed reducing species. For example, quaternary ammonium salts as phase transfer agents form a complex with the oxidant in the aqueous phase99 and transport it across the interface to the organic phase.100
Acoustic energy can be supplied via transducers, piezoelectric microdevices or ultrasonic bath or horn (probe type) for versatile ways of sound propagation and attenuation.6 For example, droplet surface acoustic waves, employing shock waves with amplitudes of a few nanometers, are generated by a piezoelectric inter-digital transducer.102 As a consequence the movement of liquid droplets is forced by the acoustic pressure eliminating cover or slit.103
The phase transfer efficiency is increased if the diameter of liquid droplets is significantly decreased by forcing liquid at a high velocity through a small aperture (ultrasonic atomization). A thin liquid film flows at the tip of the surface, which vibrates at the frequency of ultrasound and causes film disruption into fine droplets. One mechanism of liquid disintegration is explained by the capillary wave hypothesis,104 in agreement with the model in Fig. 3, and the other one by cavitation.105 Consequently the droplet diameter in ultrasonic atomization is a function of ultrasound frequency and amplitude, surface tension, density, viscosity, including non-Newtonian behavior and liquid flow parameters (rate, surface area and its geometry). For atomization to occur a certain threshold acceleration value α should be reached in order to give rise to the ejection of droplets from the crests of the capillary wave.106,107
Microfluidic transport flow can be advanced by acoustic streaming in the presence of air bubbles if a piezoelectric disk is glued to a microchamber.108 The bubble oscillations enhance a circulatory flow with efficient convection, and this process is controlled by the frequency of ultrasound and the bubble radius. The coalescence, clustering and fragmentation of acoustic bubbles can be hindered in the presence of surface active additives (e.g. n-propanol or sodium dodecyl sulfate).109
The facile integration of piezoelectric actuators into microchips is used for separation110 and in synthetic chemistry.111 The continuous separation of mixed particle suspensions in an ultrasonic field can be performed by standing acoustic waves.22,36 The latter generates stationary pressure gradients with acoustic forces for separation and fractionation at different ionic strengths.111,112
The physical principle involves interfacial parameters (i.e. gravity-to-tension ratio, difference in density, interfacial tension between two phases) and is described by the pressure difference between two phases.113 Microflows are considered to be laminar because of the low Reynolds number, indicating the dominance of diffusion over convection,114 the latter enhances mixing and separation.
The driving force in liquids is a pressure gradient, ΔPF, which is a function of the contact length and the flow velocity due to the difference in the viscosities (Fig. 5A). The interface boundary within a point is determined by the balance between the ΔPL Laplace pressure (interfacial tension between two phases) and the ΔPF pressure difference.115 The latter occurs between two phases and is a function of the contact length and the flow velocity. The Laplace pressure is described by the Young–Laplace equation and increases at lower curvature radii of the liquid–liquid interface, but at higher surface tension.115
 | ||
| Fig. 5 (A) Schematic illustration of the shape of the liquid–liquid interface, whose contact line is pinned at the boundary between the hydrophilic and the hydrophobic surfaces. (B) Illustration of a particle mixture in an enclosed channel in the medium exchange chip under silent conditions (upper image) and under ultrasound (lower image). The suspended particles have negative (yellow) and positive (blue) acoustic contrast factors (ϕ-factor). An ultrasonic standing wave is depicted in green with the pressure node in the center of the channel between the walls. The particle separation occurs either at the pressure node or antinode according to the ϕ-factor. (C) Schematic illustration of the medium exchange principle. The particles in the contaminated medium (orange) enter through the side inlets and exit through the side outlets under silent conditions (left). In the presence of ultrasound (standing wave in green color) the particles are acoustically forced to the clean medium (yellow) leaving the contaminated medium to flow to the side outlets. On the right, a schematic close-up of the outlet channel zone of the medium exchange chip. A buffer zone of clean medium in the side channels reduces the diffusion and convection by a carry-over across the laminated fluid interface. Reprinted with permission from references A. Aota, K. Mawatari and T. Kitamori, Lab Chip, 2009, 9, 2470 and F. Petersson, A. Nilsson, H. Johnsson and T. Laurell, Anal. Chem., 2005, 77, 1216. | ||
In the ultrasonic field particles are separated by acoustic forces followed by removal in a microfluidic flow (Fig. 5B).16 The primary acoustic forces dominate the motion of suspended particles. These forces increase with a larger volume of particles and a higher acoustic contrast factor.116 The latter is a function of density and compressibility of particle and medium. If the acoustic contrast factor is positive acoustic separation occurs towards a pressure node and vice versa (Fig. 4B), and this effect is stronger for larger particles (Fig. 5C). For example, under silent conditions (no ultrasound) the particles, which are in the contaminated medium, enter the channel through the side inlets and exit through the side outlets. In the presence of ultrasound the particles are switched over to the clean medium and exit through the center outlet with the clean medium, leaving the contaminated medium to flow to the side outlets. By this approach the particles are separated with high exchange efficiency (up to 95%), at low activation voltage and small particle concentration at high quantity of contaminant species.
3.4. Ultrasonic liquid/liquid extraction and ion exchange
In liquid/liquid extraction ultrasound shortens the diffusion length and increases the specific interfacial area, in which separation occurs through the interaction with the immiscible acceptor.113 This is widely applied in solvent extraction of a cobalt complex,8 Co2+,117 pesticides,118 enzyme reaction,119 hydrocarbon,120 formaldehyde,121 ammonia122 and also aroma compounds from must, wine,123 aged brandies and aqueous alcoholic wood extracts124 or specific monoterpenoids from wine,125 volatiles126 and methylmercury from biological materials.127 Ultrasonic extraction of chemical compounds other than oil is extended by herbal, protein, polyphenolics, anthocyanins and polysaccharides.128 In extraction ultrasound increases the mass transfer due to the higher vapor pressure of the organic phase, and this process is more significant if accompanied by sonochemical reactions. Other methods are introduced by an ultrasound-assisted microwave extraction of soybean-germ oil,129 pressurized liquid,130 micro-extraction131 and acoustophoretic separation in a standing acoustic field.22,132Metal ions are ultrasonically extracted if the aqueous phase contains acid or ligands133 or none of them.134 Ultrasound selectively removes cations Pb2+, Cd2+, Cu2+ and Fe2+ from aqueous phase through the complexation of metal ions by amino groups in the matrix of poly-(methyl methacrylate).135 Ultrasonic adsorption is thermodynamically faster at low temperature (higher viscosity) and preserves the stable kinetics after five cycles of adsorption/desorption. Other methods can involve polyelectrolyte complexes for controlled ion exchange and precipitation.
3.5. Polyelectrolyte complexes for ion exchange and separation
In general, the main distinguishing features of polyelectrolyte complexes are their multifunctional and versatile modification on the nanometer scale. They can be formed through the interaction of oppositely charged polyelectrolytes by layer by layer deposition136 or in a one-step complex surface precipitation.137 Important for cleaning and separation, polyelectrolyte complexes can be used for inorganic crystallization within polyelectrolyte shells138 assessing the ion exchange reaction for precipitation of water-insoluble complexes139 (Fig. 6). The nanoshell containing PAH/citrate-PAH/PSS layers allows substitution of citrate ions by OH−, PO43− or F− ions for the precipitation of metal oxides (e.g. Fe2O3 or Y2O3) or calcium hydroxyapatite Ca10(PO4)6(OH)2 with a small amount of CaPO4. The principle of the ion exchange mechanism relies on the formation of a PAH–inorganic layer followed by the coating with PAH/PSS multilayers. The latter are removed in a strong alkaline solution (pH > 12) leaving the inner PAH/anion layer for chemical reactions. The composite inorganic–polymer nanoshell exhibits higher mechanical stability and preserves its shape upon drying. This shell is versatile in the choice of methods to incorporate organic or bioorganic substances with permeability and release at an appropriate pH value. | ||
| Fig. 6 Schematic illustration of the preparation of inorganic/organic hollow capsules. ‘An−’ is F−, OH− or PO43−, ‘M+’ is Y3+, Fe2+/3+ or Ca2+ and ‘MAn’ is YF3, Fe3O4 or hydroxyapatite. In the lower row, scanning electron microscopy images of the YF3 capsules at different preparation stages. Reprinted with permission from reference D. G. Shchukin, G. B. Sukhorukov and H. Möhwald, Angew. Chem., Int. Ed., 2003, 42, 4472. | ||
Yttrium can be selectively recovered in polyelectrolyte nanoshells from aqueous solution containing much higher concentration of attendant metals (e.g. Na+, K+, Sn2+ ZrO2+ or Fe3+).140 The yield of Yttrium recovery can be lowered in the presence of Fe3+, Sn2+ or ZrO2+ due to the affinity to form stable complexes with fluoride anions, Na+ or K+ has no effect on the recovery yield. PAH in the polyelectrolyte nanoshell acts as an anion exchange resin, the positive charge of which can be neutralized by negative anions.141 For example, OH− anions introduced can participate in the chemical reaction with Mn2+, Co2+, Zn2+ or Fe3+ to form weakly crystallized ferrites (MnFe2O4, CoFe2O4, or ZnFe2O4) or magnetite with traces of single oxides (CoO, ZnO, Fe2O3) as minor components. The formation of these oxides can be explained by hydrolysis of inorganic salts in the presence of PAH, which increases the pH inside the capsule volume.142 In another example, PO43− anions inside polyelectrolyte capsules can be employed for formation of fluorescent rare earth oxides (e.g. La0.95Eu0.05PO4, Ce0.9Tb0.1PO4 or La0.4Ce0.45Tb0.15PO4).143 The latter are crystalline with high fluorescence yield and stable emission at low or neutral pH and do not require thermal annealing for crystallization.
In another example, the interior of the polyelectrolyte complex in a microenvironment, in addition to the nanoshell, provides conditions for inorganic crystallization, which is controlled by the composition of the outer shell.144 For example, nucleation and/or growth of CaCO3 can be controlled by the presence of PAH, and dextran sulfate: spherical particles are formed only in the capsule interior. In the bulk solution outside the nanoshell (no dextran sulfate) or in the absence of both capsules and dextran sulfate these particles are larger rhombohedral crystals. In the presence of only polymers no particles are observed under similar conditions. Inner polymeric structure allows adsorption of Ca ions to the negatively charged sulfate groups of PSS, viable nuclei are formed and crystal growth becomes dominant. The dimension of the polyelectrolyte complex is the only restriction to the crystal growth, which can be also influenced by noncomplexed calcium and carbonate ions in the eternal phase of an enclosed reservoir.
4 Acoustic emulsification in electric fields
To remind, in an acoustic field the interfacial zone between two phases undergoes disruption followed by the formation of droplets as explained in details in Fig. 3. Transient collapse of cavitation bubbles causes shock waves and liquid jets followed by turbulent flows and streams, which significantly increase the mass transfer and transport processes nearby the electrode. As a consequence the thickness of the diffusion layer decreases yielding higher reaction rates in mass and charge transfer. In this way, acoustic streaming, turbulence, jets and shock waves in the bubble clouds can be quantitatively and qualitatively examined by measuring the cyclic voltammograms.4.1. Mechanisms of ultrasonic processes at liquid interfaces in an electric field
 | ||
| Fig. 7 (A) Schematic illustration of the interaction of ultrasound (acoustic cavitation) at the interface of liquids with organic phase (liquid A) and aqueous phase (liquid B) in a bulk solution containing the surface of the electrodes. (B) Scheme of physico-chemical processes (formation of radicals, mass and charge transfer, reduction and oxidation) taking place between the acoustic cavitation zone (formed by an ultrasonic horn) and a surface of the electrode. | ||
At high frequency of ultrasound the limiting current increases with the square root of the ultrasonic power, as the absorption is a function of the fluid viscosity. At low frequency the ultrasonic energy is largely absorbed by cavitation being high at higher amplitudes of ultrasonic pressure. Acoustic streaming is not limited to a region, where ultrasonic energy is absorbed, as the flow of liquid driven by the radiation pressure can move away from this region. Consequently, the increase of electrochemical current can be observed even when the working electrode is located outside the point of high intensity of ultrasound.
The electrode surface bears an electrochemical potential, and a diffusion layer is determined by acoustic mechanisms and sonochemical reactions (Fig. 7B). Transient collapse of cavitation bubbles causes pyrolysis of water followed by formation of radicals (H˙, OH˙ and R˙) and their subsequent interaction with solvent molecules. Redox reactions become effective with higher rates nearby an electrode as a consequence of stronger mass transfer and a thinner diffusion layer. The latter two are higher in acoustic zones, where the ultrasound pressure field is more intense. The cavitation zone dissipates its energy with a shorter radius of the irradiating horn, but a longer distance and a larger angle (θ) along the direction of field propagation.152
4.2. Methods accessing acoustic cavitation active zones in electric field
Indirect measurements offer a greater scope of approaches, which involve a stimulated chemical reaction or the erosion effect of cavitation.155 In the first class, a typical sonochemical reaction is the liberation of chlorine from carbon tetrachloride dissolved in water. In the second class, erosion of a metal or a dielectric surface is directly proportional to the eroded surface area with a certain thickness.
In sonoelectrochemical methods most of the studies are performed at relatively large distances between the surfaces of the electrode and an ultrasonic horn. Fewer studies show that the area closely adjacent to the ultrasonic horn surface (<30 mm or <1/2 wavelength) can serve as a zone in assigning mass transfer parameters. At 20 kHz this area is a highly disturbed zone with large Sherwood (Sh) numbers and a single hydrodynamic behavior. Ultrasonically decreased diffusion layer thickness and increased limiting current156 depend on the solvent vapour pressure and ultrasonic intensity.157 The fluid dynamics and diffusion layer can be modelled by Finite Element Modelling (FEM) and reveal the proportionality between the limiting current and the diffusion coefficient.158
In the absence of electroactive species cavitational spikes are not detected, as a double layer charging peak does not appear at constant potentials. A double layer cannot be removed by cavitation due to the strong electrostatic attraction between the electrode surface and the ions of opposite charge in solution. The double layer charging is prevented, if a bubble partially blocks the surface followed by depletion in the current. Viscosity changes by ultrasound can be examined via AC or DC: the greater AC response in aqueous solutions indicates the higher energy collapse of bubbles.159 The decreased DC signal in aqueous solutions indicates the larger viscosity of the aqueous electrolyte solution.
Acoustic emulsification of two unlike liquids exhibits significant current spikes, which are attributed to the impact of droplets of a dispersed phase onto the electrode surface (e.g. heptane droplets).160 The bubble activity is examined in real time by studying the reduction of the ferricyanide anion in aqueous solution by either chronoamperometry or cyclic voltammetry. This approach is useful to determine conditions for shock wave or jet formation by ultrasound. The acoustic flow velocity is derived from the equations of a mass balance by analogy with the purely hydrodynamic flow.155,161,162 The values of the flow velocity are used to estimate the contribution of the acoustic cavitation to stirring and emulsification. Such a velocity value requires classical general correlations to describe convective mass transfer.
Another type of methods for measuring mass transfer involves visualization by Particle Image Velocimetry (PIV)164 or laser tomography.165 PIV is a non-intrusive optical method, in which acoustic flow motions are compared to computational results of fluid mechanical models.166 At low frequencies of ultrasound, PIV requires the seeding of fluorescent tracers inside the solution and filtering of the image. As a consequence the cavitation reaction zones are distinguished by measuring the average velocities of hydrodynamic flows. The latter are directly related to the bubble quantity: higher number of bubbles yields stronger hydrodynamic streams. Such velocities are higher at increased ultrasonic intensity and at shorter distances between the electrode and horn surfaces.
Ultrasonic treatment of liquid/liquid interfaces causes thinning of the diffusion layer at the surface of an electrode followed by the release of a dissolved material from the suspended phase within the diffusion layer.159 The nature of the cavitation interface is studied by nanosecond voltammetry as it allows steady-state measurements within a range of diffusion layer thicknesses. The bubble activity is examined by measuring the chronoamperometric currents in the reduction of ferricyanide anions in aqueous solution in the volume of different phases.
 | ||
| Fig. 8 Calculated volumes of the ultrasonically formed liquid jets near the electrode surface at different (A) current densities and (B) velocities of jets. The calculated data are fitted linearly (R2 = 1, in red) with the equations (A) y = −3.31 × 10−9 + 1.31 × 10−6x and (B) y = 2.73 × 10−10 + 1.13 × 10−7x. Calculated cavitation pressure (atm) from jets of bubbles within a volume (μL) versus (C) the current densities (mA cm−2) and (D) velocities of the jet (m s−1) in water for OH− species (as a model with a known diffusion coefficient). Fits are exponential decays (R2 = 0.99, in red) with equations (C) y = 398 + 384exp(−81040x) and (D) y = 82373–23150 × (1 − exp(−418410x) − 58318(1 − exp(−1930501x))). | ||
Overall, the pressure in jets is dissipated by one order of magnitude faster at higher velocity and within a smaller volume. As a consequence mass transfer enhancement becomes efficient within a broader range of the liquid volume (emulsion droplets). By this approach jet and shock wave impacts are distinguished with efficiency and accuracy comparable to sonoluminescence measurements. As another advantage, this method eliminates complicated procedures of liquid preparation, which normally involves long degassing/regassing cycles. Less time is needed for collection and analysis of data, which is laborious in sonoluminescence.
4.3. Electro-acoustical processes in cleaning and separation
Acoustic cavitation not only effectively promotes the convective motion, which is increased at the contact area, but also provides a large amount of energy for the dissolution of waste materials. Cleaning and removal of residues, degradation and disposal of organic substances (e.g. dyes, toxic compounds, etc.), encapsulation/release in droplets159 can be performed by ultrasound in a bulk solution and close to the electrode. In degradation ultrasound directs decomposition with selectivity being sensitive to stirring.155 Other processes include formation and modification of novel materials,172 electropolymerization,173 increase of product yields and acceleration of electrochemical reactions,174 electrodeposition, electroplating and electrochemical dissolution.175Heavy metals other than Pb can be determined through the physical adsorption of metal species onto the electrode surface or binding to a ligand as an adsorptive complex on the electrode. For example, copper can be extracted by ultrasonically induced changes of a solvent with a ligand dissolved in ethyl acetate in an aqueous phase through effective emulsification.187 Acorga P50 (2-hydroxy-5-nonyl benzaldoxine) can be used as a ligand with ethyl acetate as organic phase and aqueous copper solution as an aqueous phase. The removal of copper from the solution can be effectively performed by ultrasonically breaking the diffusion layer at the electrode during electrolysis.188 Ultrasound-assisted removal of copper reduces the energy cost to half as compared to silent electrolysis.
Sonoelectrochemical leaching of cobalt or lithium ions is more efficient in citric acid solution than in H2SO4 or HCl19 with more than 96% of Co and nearly 100% of Li recovery yield. High leaching efficiency is mainly ascribed to sonochemically formed H2O2 and can be achieved through the reduction of the high valence of Co to a lower state followed by chelation with the anion. The leaching process involves both a dissolution reaction and the chelation between a metal ion and an anion. In the presence of H2O2 and ultrasound the chelating reaction between citric acid and Co2+ is more favorable and faster, leading to a higher leaching efficiency of Co.189 To note, a chelating substance is a compound containing a ligand (frequently organic) bonded to a central metal atom at two or more points.
Recovery of rare metals from waste ash has been in the main research focus, as the consumption rates of these metals are continually increasing, while the supply of metals becomes more limited, because the resources are nonrenewable.193 Ultrasound can be used for the improved extraction of the chelant-assisted rare metals. As the metals partition within the crystalline lattice of the solid particles their extraction can be achieved mechano-chemically with the increased metal dissolution in the presence of chelants.194 Ultrasound enhances the transport towards the solution for almost all rare metals and assists their better dissolution with the chelant-loaded washing solution reducing concentration. Oil can be recovered from an aqueous emulsion by acoustic standing waves in a porous material containing cavities.195 A combination of ultrasound with the techniques employing electric fields yields flocculation and coalescence of micelles, which is used for oil recovery.196 Let us consider the mechanisms and principles of separation techniques, which use acoustic waves in combination with electric fields and also fluid flows.
In dielectrophoresis the electric field gradient acts on a polarized particle with the Lorentz force199 (Fig. 9A).200 If the frequency of the applied electric field is varied, the induced dipole moment in a particle forces the latter to move. If particles have a higher permeability than the surrounding fluid,201 particles move to a region of stronger electric field. This dielectrophoresis process is called positive DEP.202 The opposite occurs, if the particles have lower permeability than the fluid (Ep < Em), being repelled from the electrodes (negative DEP). For continuous fractionation positive DEP is not recommended, since particles are attracted toward the electrodes and the direct interaction may alter electrode characteristics. The negative DEP is used to form flow channels with flows, which are temporarily switched on or off. Functional DEP requires a non-uniform electric field, which is created by electrodes with different designs203 and insulating obstacles.204 The dielectrophoretic force scales with the volume of the particle yielding effective separation of larger species with strong deflection by the electrode at first and leaving smaller species unaffected in the flow.205 As an advantage, DEP forces do not depend on the polarity of the electric field but rather on the field gradient and can be used with either AC or DC or both, but not accessible in electrophoresis, which uses only DC.
 | ||
| Fig. 9 (A) Schematic illustration of the dielectrophoresis (DEP) of particles with the explanation of the principle of positive and negative DEP. (B) Schematic presentation of the principle of acoustophoresis, in which the standing acoustic waves are formed by a transducer (on the left) and reflected (on the right) producing pressure nodes and antinodes for the particle separation. The radiation acoustic forces (F) transport particles to different locations of nodes or antinodes due to the difference in the particle density, conductivity and compressibility (acoustic contrast factor). (C) A scheme, which depicts the separation mechanism from the mixture of different particles in a continuous sheath flow in applied multiple standing surface acoustic waves (SAW). Particles experience the acoustic driving force (Fs), which directs them to the pressure nodes or antinodes and forms separate lines (bands) of selected particles. (D) Fluorescence microscopy images of particle distribution in the microchannel of the microfluidics device with standing acoustic waves (SAW) with time t = 0 (a), 0.2 s (b), 0.4 s (c), 0.8 s (d), 1.6 s (e), and 2 s (f). Reprinted with permission from references H. Watarai, Annu. Rev. Anal. Chem., 2013, 6, 353 and Y. Ai and B. L. Marrone, Microfluidics, BioMEMS, and Medical Microsystems XII, 2014, p. 8976. | ||
In a standing acoustic pressure field ultrasound generates an acoustophoretic force F,206 which causes the migration of particles towards the pressure node or antinode (Fig. 9B). The value of the radiation acoustophoretic force depends on the physical properties of a particle with a characteristic acoustic contrast factor ϕ.206 If ϕ < 0, the particle moves to the pressure node and vice versa. Rigid particles and most cells have a negative ϕ-factor, while gaseous bubbles and lipids are collected at the anti-node.202 The undesirable adhesion of particles (e.g. lipids) to the channel walls can be solved by using protective sheath flows and a multi node standing wave.207 Particles, which are equally affected by acoustic radiation forces, can be separated by altering the density of the fluid. For example, the density of a fluid increases in the presence of caesium chloride yielding forces with different strengths resulting in a higher concentration of separated particles.
Other technologies in separation are introduced by techniques of physical screening (e.g. membranes or beds of filtration media, mechanical sieves), methods based on a gravitational force and centrifugal or magnetic fields. To recover a dispersed phase from a liquid emulsion, the type of the emulsion (e.g. oil-in-water or water-in-oil) and the viscosity, density and volume fraction should be considered in the applied method.209 Chemical methods involve additives in order to enhance the phase separation through the control over the molecular interactions within the system. Physical methods include conventional gravity settlers, but require large residence time, large physical space or the use of porous membranes to break the emulsion.210
5 Cleaning and separation of liquids by acoustic waves and ultrasonic membranes
5.1. Ultrasonic separation and recovery of oil droplets by multiple standing surface acoustic waves
The ability of ultrasound to separate and manipulate molecular species in a liquid suspension is the basis of various fractionation and filtration methods. Species dissolved in liquids respond to the resonant ultrasonic field, if there is a non-zero acoustic contrast between the dispersed phase and the suspending fluid. As one of the methods a one-dimensional sound field is used to assemble the species into thin parallel bands, which are separated by one-half acoustic wavelength spacing (Fig. 10A). The separation of species from their suspension occurs by placing closely spaced physical barriers between the bands followed by transportation of the species-free fluids and species-rich streams into separate exit flows. In another process species are transported in the opposite direction of the flowing fluid by using slowly moving pseudo-standing waves.211 | ||
| Fig. 10 (A) Schematic illustration of separation from a mixture of particles in liquids in an organic phase (liquid A) and an aqueous phase (liquid B) in an acoustic field with standing waves. On the left, the mixture of particles and liquids before exposure to the acoustic field. On the right, ultrasound is produced by a transducer and is reflected from the bottom yielding standing surface acoustic waves with a half wavelength. Particles are separated by acoustic radiation forces at the pressure node or antinode depending on the acoustic contrast factor of particles. Phase contrast images of (B) oil droplets in standing acoustic waves at 1.345 MHz in a quartz cuvette, (C and D) in a concentric cuvette at a modulated 1.67 kHz and 6 kHz frequencies in a cylindrical piezo transducer, (E) recovered oil droplets after 4 min of sonication with the control sample on the left. Phase contrast images are reprinted with permission from reference A. Garcia-Lopez and D. N. Sinha, J. Open Acous., 2008, 1, 66. | ||
Acoustic standing surface waves are generated in a fluid-filled cell and satisfy the path length being half the ultrasound wavelength.212 The displacement nodes appear at a spacing of λ/2 and the pressure nodes are formed at the positions of the displacement antinodes. In a two-phase emulsion system the droplets of one phase experience a time averaged primary axial acoustic force in a continuous dispersion of another phase (Fig. 10B). This force is generated by the interaction between droplets and the primary acoustic field (discussed earlier in the text). Droplets, which move closer to the pressure antinodes, experience a dominating attractive radiation force between two spheres in an acoustic field. This secondary force enhances the coalescence of the droplets and the latter grow with a larger diameter as the acoustic force is proportional to the particle volume. Simultaneously there is another radiation force, which acts in the direction perpendicular to the horizontal sound wave. Although the magnitude of this force is two orders smaller than the primary radiation force, it is sufficient to produce clumping of the droplets and to counter act the buoyancy force. At higher resonance frequency of the acoustic standing wave (1–2 MHz) oil droplets form well-defined separated lines in the cell (Fig. 10B).
The separation of oil droplets is reversible with a twice higher speed in the presence of additional acoustic waves of lower frequency (e.g. 49 kHz). If the acoustic cell has a circular geometry the standing waves of separated oil droplets appear as concentric rings (Fig. 10C). The coalescence of oil droplets in these rings increases, if the standing acoustic wave (2 MHz) is modulated with additional ultrasound of lower frequency (6 kHz) (Fig. 10D). The recovery of oil at the top of the liquid occurs faster in the acoustic field with or without modulation in the broad range of applied power (Fig. 10E).
Oil can be also recovered from aqueous solution by using a low-intensity resonant acoustic field within a rectangular chamber.195 As the oil droplets (1 to 15 μm) have a negative acoustic contrast factor, they are driven to the pressure antinodes of the standing acoustic field. Later these droplets undergo coalescence and/or wetting onto the internal surface of the chamber. In this way ultrasound transports the oil droplets to the walls and clarifies the water flowing out from the chamber. Some of the coalesced droplets, which are lighter than water, become entrained with the effluent. When the acoustic field is turned off the droplets, which are within the interior of the chamber, flow out leaving the larger droplets on the chamber walls. Oil droplets show a tendency to migrate towards the reflector and form a thin oily layer on the plastic sheet. In this way oil can be effectively collected in the area with an increased hydrophobicity and in the presence of a porous medium. The effect of the latter we consider next.
5.2. Cleaning of liquids by membranes
A porous medium (membrane) can be introduced by self-assembled colloids (e.g. glass beads) or a mesh (e.g. alumina or polymeric).4 The diameter distribution of oil droplets is not affected by the type of membrane, but the effluent, which has a higher concentration of smaller droplets and fewer larger droplets than the feed solution. This is in contrast to the system without internal porous medium. The instantaneous quantity of oil collection inside the chamber can be increased in the polymeric mesh (e.g. polyester) at a twice higher ultrasonic intensity. This is due to the different acoustic and wetting properties of the membrane, in which polyester has lower acoustic impedance. Consequently in a polyester mesh ultrasound has little internal reflections and scattering, enhancing the oil collection. The latter is more effective at higher wetting, i.e. increased hydrophobicity of a material. The better performance of the aluminum mesh compared to the glass is due to the higher porosity ratio (0.9 in aluminum and 0.4 in glass).Overall, the filtration efficiency strongly depends on both the emulsion flow rate and the path length and is sensitive to the residence time within the porous medium. The coalescence of oil droplets occurs much faster than their retention inside the membrane. The separation efficiency shows a similar tendency with electric fields (discussed earlier in the text) with little dependence on the internal surface area. The wettability of oil on the membrane increases the separation efficiency reducing the residence time within the porous structure, thus accelerating the cleaning.
5.3. Separation of liquids by membranes
In general, the porous medium is introduced by a mesh with different compositions and geometries213 in order to entrap impurities,214 extract and separate weak acids and bases, hydrocarbons, gas mixtures or amino acids.5 The separation of liquids increases with improved wettability and the synergy of hydrophobic/oleophilic properties by controlling the surface chemistry and roughness of a mesh.3 For example, a mesh with a polymeric coating, which is formed by an emulsification of polytetrafluoroethylene on the stainless-steel, allows effective spreading and penetration of an oil droplet within ms (Fig. 11A).3 This is due to the oleophilic properties of the coating with a nanostructured craterlike morphology and ball-like nm pores. At the same time, such a coating allows entrapment of water droplets due to the hydrophobic properties controlled by the pore dimensions, comparable with the surface of a lotus leaf. | ||
Fig. 11 (A) SEM images of the polymeric membrane film, which is prepared from a stainless steel mesh as a large-area view (image in the upper row) and with a higher magnification (image in the lower row), indicating nanostructured craters. The polymeric coating mesh film shows a special wettability with both super-hydrophobic and super-oleophilic characteristics: (a) shape of a water droplet on the mesh film and (b) a diesel oil droplet, which spreads on the coating mesh film, permeates by a quick flow through the film within only 240 ms. (B and C) Diagram of reversible formation of intermolecular hydrogen bonds between PNIPAAm chains and water molecules below and above the lower critical solution temperature (LCST) (upper row), which leads to switchable swelling or shrinking due to the intramolecular hydrogen bonding between C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H groups in PNIPAAm chains or dehydration. In the lower row in (B), a water droplet permeates through the mesh, while an oil droplet preserves its wetting at a temperature below LCST and in (C) a water droplet cannot permeate through the mesh, in contrast to an oil droplet at a temperature above the LCST. Reprinted with permission from references L. Feng, Z. Zhang, Z. Mai, Y. Ma, B. Liu, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 2012 and B. Xue, L. Gao, Y. Hou, Z. Liu and L. Jiang, Adv. Mater., 2013, 25, 273. O and N–H groups in PNIPAAm chains or dehydration. In the lower row in (B), a water droplet permeates through the mesh, while an oil droplet preserves its wetting at a temperature below LCST and in (C) a water droplet cannot permeate through the mesh, in contrast to an oil droplet at a temperature above the LCST. Reprinted with permission from references L. Feng, Z. Zhang, Z. Mai, Y. Ma, B. Liu, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 2012 and B. Xue, L. Gao, Y. Hou, Z. Liu and L. Jiang, Adv. Mater., 2013, 25, 273. | ||
The wettability of the oil/water liquids can be switched on/off if the surface of a polymeric mesh is composed of block copolymers3 (Fig. 11B and C). The switching property is controlled by the temperature-sensitive oelophobicity/oleophilicity of a polymer (e.g. poly(N-isopropylacrylamide), PNIPAAm), pH-sensitive poly(2-vinylpyridine)-polydimethylsiloxane (BCP) or electrochemically sensitive polypyrrole). The mesh can be opened to the water and closed to the oil, if the polymeric surface acquires two states of wettability (hydrophilicity/oleophobicity and hydrophobicity/oleophilicity) composed of both BCP and PNIPAAm. If the lower critical solution temperature (LCST) is below 15 °C, a water droplet is entrapped by the surface as the CO and N–H groups of PNIPAAm form intermolecular hydrogen bonds with water molecules (Fig. 11B). As a consequence the structure of PNIPAAm becomes more hydrated and swells. In contrast, at higher temperature (>35 °C) the PNIPAAm acquires a dehydrated state and shrinks, reducing the wettability of water (Fig. 11C). Consequently, the surface exhibits hydrophilic and oleophobic properties. In a shrunken state the surface area is still high, but the repellence of water is much stronger due to the dehydrated PNIPAAm and the hydrophobic BCP. The overall effect of hydrophilicity/oleophobicity and hydrophobicity/oleophilicity is due to the symmetric and reversible BCP-b-PNIPAAm self-assembly into lamellar nanostructures.
In another approach excellent antifouling capability for oil/water separation could be introduced by coating of a mesh with a thin graphene oxide layer as an alternative to the polymeric coating.215,216 Advanced oil/water separation could be performed by a combination of a hierarchical surface roughness and surface chemistry. This approach can allow for selective pore diameters from the nano-scale to the micro-scale. In this way the oil intrusion pressure can be increased with higher separation efficiency according to permeation theory. To note, the intrusion pressure is lower if a mesh material has a pore diameter of several tens of microns, limiting the practical application due to inefficient energy consumption.
5.4. Separation of liquids by ultrasonic membranes
The separation processes, which involve wetting and entrapment, can be effectively extended with the removal and transportation by an ultrasonic emulsion liquid membrane217 or a supported liquid membrane.218 In contrast to a mesh, in an emulsion liquid membrane ultrasound induces changes in the aqueous solution of sulfuric acid in the presence of additives, e.g. extractant (bis(2-ethylhexyl)phosphoric acid or D2EHPA) and a hydrophobic surfactant (sorbitan monooleate or Span 80 in organic phase (hexane).217 The removal of copper(II) ions is ultrasonically enhanced by a liquid surfactant membrane, which is formed through emulsification of the organic phase with Span 80 and aqueous solution with divalent Cu(II)–EDTA chelated anions as the tracers.217 The removal occurs within few minutes, not accessible by mechanical stirring.The ultrasonically formed fine w/o emulsion with nanoscale droplets (50 nm) mimics the membrane properties in the presence of a carrier in the aqueous phase. Cobalt(II) can be removed from aqueous solution through an enhanced counter transport mechanism, not accessible by a soaking procedure or treatment under vacuum.218 Higher rates of transport efficiency are explained by an ultrasonically increased pore radius and density of the support. The filling of these pores is increased by ultrasound due to the acting acoustic streams and jets.
Metal ions can be effectively removed by an ultrasonically formed bubble/protein soft membrane15 with different types of cysteine-rich proteins (e.g. egg white protein (EWP),219 bovine serum albumin (BSA)220 or human serum albumin (HSA).221 A protein coating layer bearing –COOH, –NH2, –SH and metal-binding functional groups facilitates effective complexation and chelation, physical or surface adsorption with copper ions. Higher copper adsorption can be obtained at elevated temperature and increased pH values. By this approach a higher concentration of copper is removed from a diluted feed solution of copper ions and lower bio-sorbent dosage.
Another type of ultrasonic membrane contains a silica gel for effective entrapment of large organic dye molecules within its porous structure.222 The formation of such an ultrasonic gel membrane requires Si(OC2H5)4 in water and an acid catalyst excluding a co-solvent (e.g. ethanol). Higher density, finer porosity with sphere-shaped pores and a smoother surface are the main advantages of this membrane over those formed by other conventional methods.
6 Concluding remarks and outlook
At a liquid/liquid interface ultrasound undergoes absorption, reflection, scattering or refraction in a similar fashion as light does. The absorption of ultrasound can be determined by its frequency, viscosity (η) and the relaxation time (τ) of a liquid phase. Reflection can be characterized by the acoustic impedance, Z, which is a product of the density (ρ) multiplied by the sound velocity (v) in liquids. In the acoustic field the liquid phase boundary undergoes disruption, which is proportional to the amplitude of ultrasound. The latter can be predicted by a liquid jet and determines the initial diameter of a separated emulsion droplet. With time the droplet diameter experiences the thermodynamic (Laplace and osmotic pressure) and acoustic forces (acoustic pressure, jets and shock waves) and can be decreased to a desired volume. Still little is understood about the relationship between the ultrasonic intensity and duration of sonication at the interface involving complex liquids. Indeed, these studies are limited to the oil/water interface with proteins or saccharides and metal ions of a narrow choice.Although the model of heterogeneous cavitation (sonochemistry) as the injected liquid droplet mechanism is valid, it is unclear if this model can be applied to complex liquids. Obviously, it requires an extension in order to reveal the dependence of the ultrasonic parameters on the properties of the liquid at the interface. On the other hand, processes of cleaning and separation are definitely advanced by ultrasound, but lack description by an appropriate physico-chemical model. In contrast, ultrasonic emulsification fits well to the mechanism of the initial droplet formation in the frame of the thermodynamic interplay between the parameters of enthalpy and entropy involving the Laplace and osmotic pressure. The latter can be better understood in terms of ultrasonic intensity, duration and dissolved gases and require more experimental data. Other parameters include viscosity, surface tension and the nature of additives in the mixture of unlike liquids and ions.
Cleaning of organic compounds by ultrasound can be explained by acoustic emulsification. In this mechanism, higher intensity and faster streams/flows can yield smaller droplets (intense cavitation active zone) and vice versa (calm cavitation zone). As a consequence with time the diameter distribution is controlled by irradiation time: longer treatment decreases the droplet size. Still the physical and chemical reasons of ultrasonic duration need explanation. The composition of a droplet shell and sonochemical cross-linking determine the colloidal stability of an ultrasonic emulsion. Despite the successful encapsulation of hydrophobic compounds (e.g. drugs, dyes or DNA, t-RNA) in the oil phase, other substances can be entrapped, encapsulated and undergo reactions inside the droplets. In our opinion, this field is promising and worth of further investigation especially in cleaning and separation as a new emerging branch of ultrasonic technology.
Undoubtedly phase transfer reactions are advanced by acoustic emulsification with higher yields and faster rates. Transesterification and degradation of compounds benefit from ultrasound due to the larger surface area and enhanced mass transfer. Acoustic standing waves and their combination with continuous fluid flows can significantly improve cleaning and separation allowing control and prediction. We believe that constrained fluid transport in acoustic fields (also with electric fields) can be extended to complex liquids with specific ions.
Liquid/liquid extraction by ultrasound lacks a comprehensive model, in contrast to emulsification. The synergy of acoustic and electric fields can quantitatively and qualitatively predict acoustic streams, turbulent flows and jets from cyclic voltammograms. The shape and peaks of the detected currents can indicate the bubble collapse with different symmetries (jets or shock waves). In a determined droplet volume they can allow the bubble pressure build up, and its values can distinguish a jet or shock wave scenario. This approach is promising due to less time and reduced labour, in contrast to the method involving sonoluminescence. Other ultrasonic processes such as mass transfer and the thinning of a diffusion layer can be also calculated by measuring the currents.
Ultrasonic membranes are a newly arising branch, which offers a versatility of methods for predicted and effective separation and recovery of liquids. As the number of studies in this field is small, we can assume that it is in its infancy, but definitely has a bright, promising future.
Acknowledgements
The financial support from the Max-Planck Society and the DFG RA2111 research grant are acknowledged. H.M. acknowledges fruitful discussions with T. Zemb, J. Duhamet and J. Theisen and support from the European Research Council under the ERC Grant Agreement No. 320915 “REE-CYCLE”: Rare Earth Element reCYCling with Low harmful Emissions. We also acknowledge support from DFG (G8-project SmartCoat) and the Fonds der Chemischen Industrie.References
- K. T. Chung and C. E. Cerniglia, Mutat. Res., 1992, 277, 201 CAS.
- A. Yaqub and H. Ajab, Rev. Chem. Eng., 2013, 29, 123 CAS.
- L. Feng, Z. Zhang, Z. Mai, Y. Ma, B. Liu, L. Jiang and D. Zhu, Angew. Chem., Int. Ed., 2004, 43, 2012 CrossRef CAS PubMed; B. Xue, L. Gao, Y. Hou, Z. Liu and L. Jiang, Adv. Mater., 2013, 25, 273 CrossRef PubMed.
- H. Matsumiya, T. Kageyama and M. Hiraide, Anal. Chim. Acta, 2004, 507, 205 CrossRef CAS.
- G. Schulz, Desalination, 1988, 68, 191 CrossRef CAS; C. Del Cerro and D. Boey, Chem. Ind., 1988, 21, 681 Search PubMed; M. P. Thien and T. A. Hatton, Sep. Sci. Technol., 1988, 23, 819 CrossRef; S. A. Perusich and R. C. Alkire, J. Electrochem. Soc., 1991, 138 CrossRef , 700 (part I) and 708 (part II).
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846 CrossRef CAS PubMed; D. F. Rivas, P. Cintas and H. J. G. E. Gardeniers, Chem. Commun., 2012, 48, 10935 RSC.
- H. Song, J. D. Tice and R. F. Ismagilov, Angew. Chem., 2003, 115, 792 ( Angew. Chem., Int. Ed. , 2003 , 42 , 768 ) CrossRef.
- A. Aota, M. Nonaka, A. Hibara and T. Kitamori, Angew. Chem., Int. Ed., 2007, 46, 878 CrossRef CAS PubMed.
- Z. Chu, Y. Feng and S. Seeger, Angew. Chem., Int. Ed., 2015, 54, 2328 CrossRef CAS PubMed.
- P. C. Chen and Z. K. Xu, Sci. Rep., 2013, 3, 2776 Search PubMed.
- C. Su, Y. Xu, W. Zhang, Y. Liu and J. Li, Appl. Surf. Sci., 2012, 258, 2319 CrossRef CAS; F. J. Wang, S. Lei, M. S. Xue, J. F. Ou and W. Li, Langmuir, 2014, 30, 1281 CrossRef PubMed; W. Zhang, Y. Zhu, X. Liu, D. Wang, J. Li, L. Jiang and J. Jin, Angew. Chem., Int. Ed., 2014, 53, 856 ( Angew. Chem. , 2014 , 126 , 875 ) CrossRef PubMed.
- T. Sun, L. Feng, X. Gao and L. Jiang, Acc. Chem. Res., 2005, 38, 644 CrossRef CAS PubMed; J. Lahann, Nat. Nanotechnol., 2008, 3, 320 CrossRef PubMed; X. Yao, Y. L. Song and L. Jiang, Adv. Mater., 2011, 23, 719 CrossRef PubMed.
- C. Bondy and K. Söllner, Trans. Faraday Soc., 1935, 31, 835 RSC.
- D. Y. C. Leung, X. Wu and M. K. H. Leung, Appl. Energy, 2010, 87, 1083 CrossRef CAS; I. B. Banković-Ilic, O. S. Stamenković and V. B. Veljković, Renewable Sustainable Energy Rev., 2012, 16, 3621 CrossRef.
- A. M. Nazari, P. W. Cox and K. E. Waters, Sep. Purif. Technol., 2014, 132, 218 CrossRef CAS.
- F. Petersson, A. Nilsson, H. Jönsson and T. Laurell, Anal. Chem., 2005, 77, 1216 CrossRef CAS PubMed.
- K. S. Suslick, Ultrasound, its physical, biological and chemical effects, VCH Science Publishers, Weinheim, Germany, 1988 Search PubMed; T. J. Mason and J. P. Lorimer, Sonochemistry: theory, applications and uses of ultrasound in chemistry, Ellis Horwood Ltd., Chichester, UK, 1988 Search PubMed; G. J. Price, Current trends in sonochemistry, Royal Society of Chemistry, Oxford, UK, 1993 Search PubMed; R. van Eldik and C. D. Hubbard, Chemistry under extreme or nonclassical conditions, John Wiley and Sons Inc., New York, 1997 Search PubMed.
- A. J. Saterlay, S. J. Wilkins, C. H. Goeting, J. S. Foord, R. G. Compton and F. Marken, J. Solid State Electrochem., 2000, 4, 383 CrossRef CAS; T. J. Mason and V. S. Bernal, Power ultrasound in electrochemistry: from versatile laboratory tool to engineering solution, An introduction to sonoelectrochemistry, John Wiley and Sons Inc., New York, 2012, ch. 1 Search PubMed.
- L. Li, L. Zhai, X. Zhang, J. Lu, R. Chen, F. Wu and K. Amine, J. Power Sources, 2014, 262, 380 CrossRef CAS.
- K. S. Suslick and M. W. Grinstaff, J. Am. Chem. Soc., 1990, 112, 7807 CrossRef CAS; A. Gedanken, Chem. – Eur. J., 2008, 14, 3840 CrossRef PubMed.
- M. Y. Koh and T. I. G. Mohd, Renewable Sustainable Energy Rev., 2011, 15, 2240 CrossRef CAS.
- T. Laurell, F. Petersson and A. Nilsson, Chem. Soc. Rev., 2007, 36, 492 RSC.
- M. R. Hoffmann, I. Hua and R. Hochemer, Ultrason. Sonochem., 1996, 3, 163 CrossRef.
- E. Joyce, S. S. Phull, J. P. Lorimer and T. J. Mason, Ultrason. Sonochem., 2003, 10, 315 CrossRef CAS PubMed.
- Acoustic pressure can be represented as PA = PAsin2πft, where PA is the maximum pressure amplitude and f is the frequency of the acoustic wave. The intensity of ultrasound, I, can be determined as I = PA2/2ρv, where ρ is the density of a liquid and v is the velocity of sound in this liquid.
- D. H. Trevena, Contemp. Phys., 1969, 10, 601 CrossRef CAS.
- M. K. Li and H. S. Fogler, J. Fluid Mech., 1978, 88, 499 CrossRef CAS.
- M. J. W. Povey, Ultrasonic techniques for fluids characterization, Academic Press, 1997, p. 214 Search PubMed.
- M. Eigen and K. Tamm, Z. Elektrochem., 1962, 66(2) Search PubMed , 93 (part I) and 107 (part II).
- E. B. Flint and K. S. Suslick, Science, 1991, 253, 1397 CrossRef CAS PubMed.
- B. P. Barber and S. J. Putterman, Nature, 1991, 352, 318 CrossRef CAS; L. A. Crum and T. J. Matula, Science, 1997, 276, 1348 CrossRef; V. Q. Vuong, A. J. Szeri and D. A. Young, Phys. Fluids, 1999, 11, 10 CrossRef; R. Pecha and B. Gompf, Phys. Rev. Lett., 2000, 84, 1328 CrossRef PubMed; M. P. Brenner, S. Hilgenfeldt and D. Lohse, Rev. Mod. Phys., 2002, 74, 425 CrossRef.
- L. A. Crum, J. Phys., 1979, 40, 285 Search PubMed; E. A. Neppiras, Phys. Lett., 1980, 61, 159 Search PubMed; T. G. Leighton, The acoustic bubble, Academic Press, 1994, p. 640 CrossRef CAS PubMed; T. J. Matula, Ultrasonics, 2000, 38, 559 CrossRef CAS PubMed.
- J. B. Jeffries, R. A. Copeland, K. S. Suslick and E. B. Flint, Science, 1992, 256, 248 Search PubMed; R. Löfstedt, B. P. Barber and S. J. Putterman, Phys. Fluids, 1993, 5, 2911 CrossRef CAS; B. P. Barber, K. Weninger, R. Löfstedt and S. J. Putterman, Phys. Rev. Lett., 1995, 74, 5276 CrossRef PubMed; A. J. Szeri, B. D. Storey, A. Pearson and J. R. Blake, Phys. Fluids, 2003, 15, 2576 CrossRef.
- S. A. Elder, J. Acoust. Soc. Am., 1959, 31, 54 CrossRef CAS; H. G. Flynn, Physics of acoustic cavitation in liquids, in Physical Acoustics, ed. W. P. Mason, Academic Press, New York, 1964, vol. 1, Part B, p. 57 CrossRef; P. Tho, R. Manasseh and A. Ooi, J. Fluid Mech., 2007, 576, 191 CrossRef; M. C. Schenker, M. J. B. M. Pourquié, D. G. Eskin and B. J. Boersma, Ultrason. Sonochem., 2013, 20, 502 CrossRef PubMed.
- P. Riesz, D. Berdahl and C. L. Christman, Environ. Health Perspect., 1985, 64, 233 CrossRef CAS PubMed; P. Riesz and T. Kondo, Free Radicals Biol. Med., 1992, 13, 247 CrossRef PubMed; J. L. Luche, Ultrason. Sonochem., 1997, 4, 211 CrossRef PubMed; G. Mark, A. Tauber, R. Laupert, H. P. Schuchmann, D. Schulz, A. Mues and C. von Sonntag, Ultrason. Sonochem., 1998, 5, 41 CrossRef PubMed; M. Ashokkumar, L. A. Crum, C. A. Frensley, F. Grieser, T. J. Matula, W. B. McNamara III and K. S. Suslick, J. Phys. Chem. A, 2000, 104, 8462 CrossRef; Y. T. Didenko and K. S. Suslick, Nature, 2002, 418, 394 CrossRef PubMed.
- K. S. Suslick, D. A. Hammerton and R. E. Cline, J. Am. Chem. Soc., 1986, 108, 5641 CrossRef CAS; K. S. Suslick, Y. Didenko, M. M. Fang, T. Hyeon, K. J. Kolbeck, W. B. McNamara III, M. M. Mdleleni and M. Wong, Philos. Trans. R. Soc., A, 1999, 357, 335 CrossRef; K. S. Suslick, Kirk-Othmer Encyclopedia of Chemical Technology, Sonochemistry, John Wiley and Sons Inc., New York, 1998, vol. 26, p. 516 CrossRef PubMed; D. J. Flannigan and K. S. Suslick, Phys. Rev. Lett., 2007, 99, 134301 CrossRef PubMed; H. Xu, N. C. Eddingsaas and K. S. Suslick, J. Am. Chem. Soc., 2009, 131, 6060 CrossRef PubMed.
- M. Ashokkumar and F. Grieser, Phys. Chem. Chem. Phys., 2007, 9, 5631 RSC; D. G. Shchukin and H. Möhwald, Phys. Chem. Chem. Phys., 2006, 8, 3496–3506 RSC; D. V. Radziuk, W. Zhang, D. Shchukin and H. Möhwald, Small, 2010, 6, 545 CrossRef CAS PubMed.
- T. J. Mason, E. Joyce, S. S. Phull and J. P. Lorimer, Ultrason. Sonochem., 2003, 10, 319 CrossRef CAS PubMed; GE Water & Process Technologies, Handbook of Industrial Water Treatment, General Electric Company (GE), Trevose, 2008 CrossRef PubMed; N. N. Mahamuni and Y. G. Adewuyi, Ultrason. Sonochem., 2010, 17, 990 CrossRef PubMed; P. Rajasekhar, L. Fan, T. Nguyen and F. A. Roddick, Water Res., 2012, 46, 4319 CrossRef PubMed.
- S. Gantert and D. Möller, Chem. Eng. Technol., 2012, 35, 576 CrossRef CAS.
- J. P. Lorimer and T. J. Mason, Chem. Soc. Rev., 1987, 16, 239 RSC.
- Y. Mizukoshi, T. Shuto, N. Masahashi and S. Tanabe, Ultrason. Sonochem., 2009, 16, 525 CrossRef CAS PubMed.
- L. Stegpniak, U. Kegpa and E. Stanczyk-Mazanek, Desalination, 2008, 223, 180 CrossRef CAS.
- K. Okitsu, M. Iwatani, B. Nanzai, R. Nishimura and Y. Maeda, Ultrason. Sonochem., 2009, 16, 387 CrossRef CAS PubMed.
- S. Merouani, O. Hamdaoui, F. Saoudi, M. Chiha and C. Pétrier, J. Hazard. Mater., 2010, 175, 593 CrossRef CAS PubMed.
- X. Liu, Z. Zhang, W. Hu, Q. Le, L. Bao, J. Cui and J. Jiang, Ultrason. Sonochem., 2015, 26, 73 CrossRef CAS PubMed.
- N. Gondrexon, V. Renaudin, P. Boldo, Y. Gonthier, A. Bernis and C. Petrier, J. Chem. Eng., 1997, 66, 21 CrossRef CAS.
- D. Peters, Ultrason. Sonochem., 2001, 8, 221 CrossRef CAS PubMed.
- J. P. Canselier, H. Delmas, A. M. Wilhelm and B. Ablsrnail, J. Dispersion Sci. Technol., 2002, 23, 333 CrossRef CAS.
- M. K. Li and H. S. Fogler, J. Fluid Mech., 1978, 88, 499 CrossRef CAS.
- The droplet volume is equal to the cylinder volume of the broken jet with a diameter d and a length lmax as ¼πd2lmax.
- C. Bondy and K. Söllner, Trans. Faraday Soc., 1935, 31, 835 RSC.
- R. W. Boyle and G. B. Taylor, Phys. Rev., 1926, 27, 518 Search PubMed.
- Acoustic energy, which is transmitted per sq. cm through a liquid, depends on cavitation as
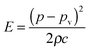 , where p and pv are hydrostatic and vapor pressure in the liquid, ρ is the liquid density and c is the sound velocity.
, where p and pv are hydrostatic and vapor pressure in the liquid, ρ is the liquid density and c is the sound velocity. - K. Makino, T. Mizorogi, S. Ando, T. Tsukamoto and H. Ohshima, Colloids Surf., B, 2001, 22, 251 CrossRef CAS.
- S. Avivi Levi and A. Gedanken, J. Biochem., 2002, 366, 705 CrossRef PubMed.
- X. Teng, D. G. Shchukin and H. Moehwald, Adv. Funct. Mater., 2007, 17, 1273 CrossRef CAS.
- Y. Han, D. Radziuk, D. Shchukin and H. Moehwald, Macromol. Rapid Commun., 2008, 29, 1203 CrossRef CAS.
- Y. Han, D. Radziuk, D. Shchukin and H. Moehwald, J. Mater. Chem., 2008, 18, 5162 RSC.
- In the frame of the acoustic emulsification model (Fig. 3) let us explain why droplets acquire larger or smaller diameter in acoustic zones with different intensity. The value of lmax is directly proportional to the amplitude of ultrasound, A, which can be deduced as
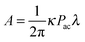 with k – compressibility of the medium, Pac – acoustic pressure and λ – wavelength from PA = AZ2πυ with Z – acoustic impedance of the medium as Z = ρv, ρ – density and υ – velocity of sound) and ν – frequency of ultrasound and
with k – compressibility of the medium, Pac – acoustic pressure and λ – wavelength from PA = AZ2πυ with Z – acoustic impedance of the medium as Z = ρv, ρ – density and υ – velocity of sound) and ν – frequency of ultrasound and 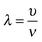 with
with 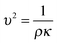 . The ultrasound wavelength in water is 7.5 cm and in silicone oil it is 5.78 cm assuming the velocity values in water νw = 1500 m s−1 and in oil νs = 1156 m s−1 and a frequency 20 kHz. The values of compressibility in water kw = 0.45 GPa−1 and in oil ks = 0.6 GPa−1. The A values in water and in silicone oil are about 34 μm and 35 μm, which are very close to the experimental values of proteinaceous microspheres at low acoustic intensity. Theoretically the droplet diameter is directly proportional to the maximum length of a jet, which is proportional to the amplitude A. In high acoustic pressure field (zone C) the intensity is high due to the dominance of the shock waves, which leads to energetically high emulsification and small diameter of droplets. In contrast, strong acoustic jets, flows and streams are developed in zones (‘A’ and ‘E’), with low intensity, which are weaker in the intermediate region of the acoustic field (zones B and D). In ‘A’ and ‘E’ zones longer jets form larger droplets, while shorter jets in stronger acoustic pressure regions (B and D) yield smaller bimodal microdroplets, in agreement with the model (Fig. 3).
. The ultrasound wavelength in water is 7.5 cm and in silicone oil it is 5.78 cm assuming the velocity values in water νw = 1500 m s−1 and in oil νs = 1156 m s−1 and a frequency 20 kHz. The values of compressibility in water kw = 0.45 GPa−1 and in oil ks = 0.6 GPa−1. The A values in water and in silicone oil are about 34 μm and 35 μm, which are very close to the experimental values of proteinaceous microspheres at low acoustic intensity. Theoretically the droplet diameter is directly proportional to the maximum length of a jet, which is proportional to the amplitude A. In high acoustic pressure field (zone C) the intensity is high due to the dominance of the shock waves, which leads to energetically high emulsification and small diameter of droplets. In contrast, strong acoustic jets, flows and streams are developed in zones (‘A’ and ‘E’), with low intensity, which are weaker in the intermediate region of the acoustic field (zones B and D). In ‘A’ and ‘E’ zones longer jets form larger droplets, while shorter jets in stronger acoustic pressure regions (B and D) yield smaller bimodal microdroplets, in agreement with the model (Fig. 3). - M. W. Grinstaff, P. Soon-Shiong, M. Wong, P. A. Sandford, K. S. Suslick and N. P. Desai, US Pat., 5,498,421, 1997 Search PubMed.
- U. Shimanovich, D. Eliaz, A. Aizer, I. Vayman, S. Michaeli, Y. Shav-Tal and A. Gedanken, ChemBioChem, 2011, 12, 1678 CrossRef CAS PubMed.
- U. Shimanovich, V. Volkov, D. Eliaz, A. Aizer, S. Michaeli and A. Gedanken, Small, 2011, 7, 1068 CrossRef CAS PubMed.
- M. Wong and K. S. Suslick, Abstr. Pap. Am. Chem. Soc., 1995, 210, 587 Search PubMed.
- U. Shimanovich, D. Matas, A. Cavaco-Paulo, S. Michaeli and A. Gedanken, Chem. – Eur. J., 2010, 16, 2108 CrossRef PubMed.
- T. Shiomi, T. Tsunoda, A. Kawai, H. Chiku, F. Mizukami and K. Sakaguchi, Chem. Commun., 2005, 5325 RSC.
- F. J. J. Toublan, S. Boppart and K. S. Suslick, J. Am. Chem. Soc., 2006, 128, 3472 CrossRef CAS PubMed.
- Y. Han, S. Li, X. Wang, X. Cao, L. Jia and J. Li, Colloid Polym. Sci., 2005, 284, 203 CAS.
- U. Shimanovich, I. D. Tkacz, D. Eliaz, A. Cavaco-Paulo, S. Michaeli and A. Gedanken, Adv. Funct. Mater., 2011, 21, 3659 CrossRef CAS.
- N. Skirtenko, T. Tzanov, A. Gedanken and S. Rahimipour, Chem. – Eur. J., 2010, 16, 562 CrossRef CAS PubMed; E. Colombo, F. Cavalieri and M. Ashokkumar, ACS Appl. Mater. Interfaces, 2015, 7, 12972 Search PubMed.
- T. N. Borodina, D. O. Grigoriev, M. A. Carillo, J. Hartmann, H. Moehwald and D. G. Shchukin, ACS Appl. Mater. Interfaces, 2014, 6, 6570 CAS.
- K. Xu and P. Yao, Langmuir, 2009, 25, 9714 CrossRef CAS PubMed.
- T. Borodina, D. Grigoriev, E. Markvicheva, H. Moehwald and D. G. Shchukin, Adv. Eng. Mater., 2011, 13, B123 CrossRef.
- M. A. Bradley, S. W. Prescott, H. A. S. Schoonbrood, K. Landfester and F. Grieser, Macromolecules, 2005, 38, 6346 CrossRef CAS.
- K. Fontenot and F. J. Schork, Ind. Eng. Chem. Res., 1993, 32, 373 CrossRef CAS; J. Rensen, D. Bosman, J. Magnaudet, C. D. Ohl, A. Prosperetti, R. Tögel, M. Versluis and D. Lohse, Phys. Rev. Lett., 2001, 86, 4819 CrossRef PubMed.
- E. S. Rajagopal, Emulsion Science, Academic Press, London, 1968 Search PubMed.
- O. Behrend, K. Ax and H. Schubert, Ultrason. Sonochem., 2000, 7, 77 CrossRef CAS PubMed.
- S. A. Neduzhii, Sov. Phys. Acoust., 1963, 9, 195 Search PubMed.
- V. S. Rodriguez, J. Delgado, C. A. Silebi and M. S. El-Aasser, Ind. Eng. Chem. Res., 1989, 28, 1 CrossRef.
- M. Willert, R. Rothe, K. Landfester and M. Antonietti, Chem. Mater., 2001, 13, 4681 CrossRef CAS.
- F. Tiarks, K. Landfester and M. Antonietti, Langmuir, 2001, 17, 5775 CrossRef CAS.
- D. M. El-Sherif and M. A. Wheatley, J. Biomed. Mater. Res., Part A, 2003, 66, 347 CrossRef PubMed.
- G. B. Sukhorukov, A. A. Antipov, E. Donath and H. Möhwald, Macromol. Rapid Commun., 2001, 22, 44 CrossRef CAS; A. A. Antipov, G. B. Sukhorukov, S. Leporatti, I. L. Radtchenko, E. Donath and H. Möhwald, Colloids Surf., A, 2002, 198, 535 CrossRef.
- G. Ibarz, L. Dähne, E. Donath and H. Möhwald, Adv. Mater., 2001, 13, 1324 CrossRef CAS; A. A. Antipov, G. B. Sukhorukov and H. Möhwald, Langmuir, 2003, 19, 2444 CrossRef; R. Georgieva, S. Moya, M. Hin, R. Mitlöhner, E. Donath, H. Kiesewetter, H. Möhwald and H. Bäumler, Biomacromolecules, 2002, 3, 517 CrossRef PubMed.
- X. Teng, D. G. Shchukin and H. Möhwald, Langmuir, 2008, 24, 383 CrossRef CAS PubMed.
- P. R. Gogate, Adv. Environ. Res., 2002, 6, 335 CrossRef CAS; Y. G. Adewuyi, Ind. Eng. Chem. Res., 2001, 40, 4681 CrossRef.
- N. R. Paliwal and A. K. Mungray, Polym. Degrad. Stab., 2013, 98, 2094 CrossRef CAS.
- P. Chand, V. R. Chintareddy, J. G. Verkade and D. Grewell, Energy Fuels, 2010, 24, 2010 CrossRef CAS.
- J. B. Bhasarkar, S. Chakma and V. S. Moholkar, Ultrason. Sonochem., 2015, 24, 98 CrossRef CAS PubMed.
- F. G. Antes, L. O. Diehl, J. S. F. Pereira, R. C. L. Guimarães, R. A. Guarnieri, B. M. S. Ferreira, V. L. Dressler and E. M. M. Flores, Ultrason. Sonochem., 2015, 25, 70 CrossRef CAS PubMed.
- K. Nakabayashi, H. Yanagi and M. Atobe, RSC Adv., 2014, 4, 57608 RSC.
- K. Nakabayashi, F. Amemiya, T. Fuchigami, K. Machida, S. Takeda, K. Tamamitsu and M. Atobe, Chem. Commun., 2011, 47, 5765 RSC.
- A. P. Vyas, J. L. Verma and N. Subrahmanyam, Fuel, 2010, 89, 1 CrossRef CAS; V. B. Veljković, J. M. Avramović and O. S. Stamenković, Renewable Sustainable Energy Rev., 2012, 16, 1193 CrossRef; A. S. Badday, A. Z. Abdullah, K. T. Lee and M. S. Khayoon, Renewable Sustainable Energy Rev., 2012, 16, 4574 CrossRef.
- W. Guo, H. Li, G. Ji and G. Zhang, Bioresour. Technol., 2012, 125, 332 CrossRef CAS PubMed.
- M. Takase, W. Feng, W. Wang, X. Gu, Y. Zhu, T. Li, L. Yang and X. Wu, Fuel Process. Technol., 2014, 123, 19 CrossRef CAS.
- S. Nii, S. Kikumoto and H. Tokuyama, Ultrason. Sonochem., 2009, 16, 145 CrossRef CAS PubMed.
- H. D. Hanh, N. T. Dong, K. Okitsu, R. Nishimura and Y. Maeda, Renewable Energy, 2009, 34, 780 CrossRef CAS.
- M. H. Entezari and A. Keshavarzi, Ultrason. Sonochem., 2001, 8, 213 CrossRef CAS PubMed.
- J. B. Bhasarkar, S. Chakma and V. S. Moholkar, Ultrason. Sonochem., 2015, 24, 98 CrossRef CAS PubMed.
- D. Zhao, H. Ren, J. Wang, Y. Yung and Y. Zhao, Energy Fuels, 2007, 21, 2543 CrossRef CAS.
- Z. Shayegan, M. Razzaghi, A. Niaei, D. Salari, M. T. S. Tabar and A. N. Akbari, Korean J. Chem. Eng., 2013, 9, 1751 CrossRef.
- M. Riccaboni, E. La Porta, A. Martorana and R. Attanasio, Tetrahedron, 2010, 66, 4032 CrossRef CAS.
- J. Friend and L. Y. Yeo, Rev. Mod. Phys., 2011, 83, 647 CrossRef.
- D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153 RSC.
- G. Taylor, Proc. R. Soc. A, 1950, 201, 192 CrossRef.
- J. Mir, J. Acoust. Soc. Am., 1980, 67, 201 CrossRef.
- C. Goodridge, W. Shi, H. Hentschel and D. Lathrop, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1997, 56, 472 CrossRef CAS.
- The α value is determined as
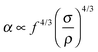 for a low-viscosity fluid such as water, acetone and ethanol.
for a low-viscosity fluid such as water, acetone and ethanol. - R. Liu, J. Yang, M. Z. Pindera, M. Athavale and P. Grodzinski, Lab Chip, 2002, 2, 151 RSC.
- J. Lee, T. Tuziuti, K. Yasui, S. Kentish, F. Grieser, M. Ashokkumar and Y. Iida, J. Phys. Chem. C, 2007, 111, 19015 CAS.
- T. Franke, A. R. Abate, D. A. Weitz and A. Wixforth, Lab Chip, 2009, 9, 2625 RSC; Z. Wang and J. Zhe, Lab Chip, 2011, 11, 1280 RSC.
- K. Kulkarni, J. Friend, L. Yeo and P. Perlmutter, Lab Chip, 2009, 9, 754 RSC.
- T. G. Leighton, Prog. Biophys. Mol. Biol., 2007, 93, 3 CrossRef CAS PubMed; J. Nilsson, M. Evander, B. Hammarstrom and T. Laurell, Anal. Chim. Acta, 2009, 649, 141 CrossRef PubMed.
- A. Aota, K. Mawataria and T. Kitamori, Lab Chip, 2009, 9, 2470 RSC.
- D. Di Carlo, Lab Chip, 2009, 9, 3038 RSC.
- In general, segmented flow chemistry, i.e. microfluidics, allows to control the multiphase of liquid/liquid interfaces by the physical forces in the microchannels. The interfacial parameters can be analyzed by dimensionless Bond (Bo) numbers as the gravity-to-tension ratio,
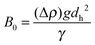 with Δρ – the density difference, g – gravitational acceleration (9.8 m s−2), γ – interfacial tension between two phases and dh – equivalent diameter. The driving force in liquids is a pressure, which is lower in the downstream part of the fluid due to the difference in the viscosities. When two fluids with different viscosity are brought into contact, the ΔPF pressure difference between the two phases is a function of the contact length and the flow velocity. The interface boundary within a point is determined by the balance between the ΔPL Laplace pressure (interfacial tension between two phases) and the ΔPF pressure difference. The ΔPL is derived by the Young–Laplace equation as
with Δρ – the density difference, g – gravitational acceleration (9.8 m s−2), γ – interfacial tension between two phases and dh – equivalent diameter. The driving force in liquids is a pressure, which is lower in the downstream part of the fluid due to the difference in the viscosities. When two fluids with different viscosity are brought into contact, the ΔPF pressure difference between the two phases is a function of the contact length and the flow velocity. The interface boundary within a point is determined by the balance between the ΔPL Laplace pressure (interfacial tension between two phases) and the ΔPF pressure difference. The ΔPL is derived by the Young–Laplace equation as 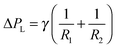 , where R1 and R2 are the curvature radii of the liquid–liquid interface in directions vertical and parallel to the liquid stream. For parallel multiphase microflows the equation is simplified as
, where R1 and R2 are the curvature radii of the liquid–liquid interface in directions vertical and parallel to the liquid stream. For parallel multiphase microflows the equation is simplified as 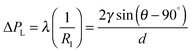 , where q and d are the dynamic contact angle and depth of the microchannels, with
, where q and d are the dynamic contact angle and depth of the microchannels, with 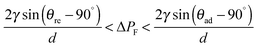 . The interface boundary is shifted towards the organic phase, if ΔPF is higher than the maximum value of ΔPL and vice versa. In the presence of dissolved gas the compression of the gaseous phase and the vapor pressure are to be considered.
. The interface boundary is shifted towards the organic phase, if ΔPF is higher than the maximum value of ΔPL and vice versa. In the presence of dissolved gas the compression of the gaseous phase and the vapor pressure are to be considered. - The primary acoustic radiation force, which dominates the motion of suspended particles, is derived as
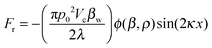 , where p0 – the pressure amplitude, Vc – the volume of the particle, βw – the compressibility of water, λ – the ultrasonic wavelength, k – the wavenumber, x – the direction of propagation and ϕ – the acoustic contrast factor or ϕ – factor, which is determined as
, where p0 – the pressure amplitude, Vc – the volume of the particle, βw – the compressibility of water, λ – the ultrasonic wavelength, k – the wavenumber, x – the direction of propagation and ϕ – the acoustic contrast factor or ϕ – factor, which is determined as 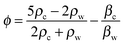 with ρc and ρw – densities of the particle and the medium, and βc and βw – the corresponding compressibility.
with ρc and ρw – densities of the particle and the medium, and βc and βw – the corresponding compressibility. - T. Kitamori, M. Tokeshi, A. Hibara and K. Sato, Anal. Chem., 2004, 76, 52A CrossRef CAS.
- A. Smirnova, K. Mawatari, A. Hibara, M. A. Proskurnin and T. Kitamori, Anal. Chim. Acta, 2006, 558, 69 CrossRef CAS.
- T. Maruyama, J. Uchida, T. Ohkawa, T. Futami, K. Katayama, K. Nishizawa, K. Sotowa, F. Kubota, N. Kamiya and M. Goto, Lab Chip, 2003, 3, 308 RSC.
- S. A. Bowden, P. B. Monaghan, R. Wilson, J. Parnell and J. M. Cooper, Lab Chip, 2006, 6, 740 RSC.
- S. Ohira and K. Toda, Lab Chip, 2005, 5, 1374 RSC.
- K. Mawatari, A. Aota, Y. Kikutani, N. Hanaoka and T. Kitamori, Proceedings of conference on microreaction technology, 2008 Search PubMed.
- C. Cocito, G. Caetano and C. Delfini, Food Chem., 1995, 5, 311 CrossRef CAS; D. H. Vila, F. J. H. Mira, R. B. Lucena and M. A. F. Recamales, Talanta, 1999, 50, 413 CrossRef; E. Gomez-Plaza, R. Gil-Munoz, J. Carreno-Espın, J. A. Fernandez-Lopez and A. Martınez-Cutillas, Eur. Food Res. Technol., 1999, 209, 257 CrossRef.
- I. Caldeira, R. Pereira, M. C. Clımaco, A. P. Belchior and R. B. de Sousa, Anal. Chim. Acta, 2004, 513, 125 CrossRef CAS.
- R. M. Pena, J. Barciela, C. Herrero and S. Garcıa-Martın, Talanta, 2005, 67, 129 CrossRef CAS PubMed.
- E. Alissandrakis, D. Daferera, P. A. Tarantilis, M. Polissiou and P. C. Harizanis, Food Chem., 2003, 82, 575 CrossRef CAS.
- Q. Tu, J. Qian and W. Frech, J. Anal. At. Spectrom., 2000, 15, 1361 RSC.
- K. Vilkhu, R. Mawson, L. Simons and D. Bates, Innovative Food Sci. Emerging Technol., 2008, 9, 161 CrossRef CAS.
- G. Cravotto, L. Boffa, S. Mantegna, P. Perego, M. Avogadro and P. Cintas, Ultrason. Sonochem., 2008, 15, 898 CrossRef CAS PubMed.
- G. Rocco, C. Toledo, I. Ahumada, B. Sepúlveda, A. Cañete and P. Richter, J. Chromatogr. A, 2008, 1193, 32 CrossRef CAS PubMed.
- L. Rusnáková, V. Andruch, I. S. Balogh and J. Škrlíková, Talanta, 2011, 85, 541 CrossRef CAS PubMed; M. Rezaee, Y. Yamini and M. Faraji, J. Chromatogr. A, 2010, 1217, 2342 CrossRef PubMed; Y. S. Su and J. F. Jen, J. Chromatogr. A, 2010, 1217, 5043 CrossRef PubMed; H. Xu, Y. Liao and J. Yao, J. Chromatogr. A, 2007, 1167, 1 CrossRef PubMed.
- F. Petersson, A. Nilsson, C. Holm, H. Johnsson and T. Laurell, Lab Chip, 2005, 5, 20 RSC.
- M. Murillo, A. Gonzalez, A. Ramırez and N. Guillen, At. Spectrosc., 1994, 15, 90 Search PubMed; T. B. Wang, X. J. Jia and J. Wu, J. Pharm. Biomed. Anal., 2003, 33, 639 CrossRef CAS PubMed.
- J. H. Lehr and J. Keeley, Water encyclopedia, Domestic, municipal and industrial water supply and waste disposal, John Wiley and Sons Inc., Hoboken, New Jersey, 2005 Search PubMed.
- D. Kundu, C. Hazra, A. Chatterjee, A. Chaudhari and S. Mishra, RSC Adv., 2014, 4, 24991 RSC.
- E. Donath, G. B. Sukhorukov, F. Caruso, S. Davis and H. Möhwald, Angew. Chem., 1998, 110, 2324 ( Angew. Chem., Int. Ed. , 1998 , 37 , 2202 ) CrossRef CAS; G. B. Sukhorukov, E. Donath, S. Davis, H. Lichtenfeld, F. Caruso, V. I. Popov and H. Möhwald, Polym. Adv. Technol., 1998, 9, 759 CrossRef.
- A. Voigt, E. Donath and H. Moehwald, Macromol. Mater. Eng., 2000, 282, 13 CrossRef CAS.
- G. B. Sukhorukov, E. Donath, S. Davis, H. Lichtenfeld, F. Caruso, V. I. Popov and H. Möhwald, Polym. Adv. Technol., 1998, 9, 759 CrossRef CAS.
- D. G. Shchukin, G. B. Sukhorukov and H. Mohwald, Angew. Chem., Int. Ed., 2003, 42, 4472 CrossRef CAS PubMed.
- D. G. Shchukin and G. B. Sukhorukov, Langmuir, 2003, 19, 4427 CrossRef CAS.
- D. G. Shchukin and G. B. Sukhorukov, Colloid Polym. Sci., 2003, 281, 1201 CAS.
- I. L. Radtchenko, M. Giersig and G. B. Sukhorukov, Langmuir, 2002, 18, 8204 CrossRef CAS.
- D. G. Shchukin, G. B. Sukhorukov and H. Mohwald, J. Phys. Chem. B, 2004, 108, 50 CrossRef.
- G. B. Sukhorukov, A. S. Susha, S. Davis, S. Leporatti, E. Donath, J. Hartmann and H. Möhwald, J. Colloid Interface Sci., 2002, 247, 251 CrossRef CAS PubMed.
- The radiation force, Frad, the driving force of the acoustic streaming, increases with the applied acoustic power as
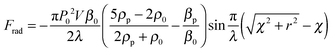 , where P0 is the amplitude of the driving acoustic pressure, V is the volume of the particle (impurity), β0 and βp is the compressibility of liquid and solid, being defined as
, where P0 is the amplitude of the driving acoustic pressure, V is the volume of the particle (impurity), β0 and βp is the compressibility of liquid and solid, being defined as 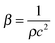 with c as the speed of sound, λ is the wavelength of the sound, ρ0 and ρp are the density of liquid and solid, χ is the distance from the electrode surface, r is the radius of the horn surface.
with c as the speed of sound, λ is the wavelength of the sound, ρ0 and ρp are the density of liquid and solid, χ is the distance from the electrode surface, r is the radius of the horn surface. - J. Klima, C. Bernard and C. Degrand, J. Electroanal. Chem., 1995, 399, 147 CrossRef.
- T. Matula, Philos. Trans. R. Soc., A, 1999, 357, 225 CrossRef CAS.
- T. G. Leighton, The acoustic bubble, Academic Press, London, 1994, Sec. 4.3.1, pp. 312–329 Search PubMed.
- P. R. Birkin, D. G. Offin, C. J. B. Vian, T. G. Leighton and A. O. Maksimov, J. Acoust. Soc. Am., 2011, 130, 3297 CrossRef PubMed.
- At the first several milliseconds the current decays as t−1/2, comparable to a Faradaic current with a diffusion controlled mass transfer according to the Cottrell's equation:
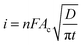 or
or 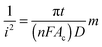 , where n – number of electrons, F – the Faraday constant, A – the electrode surface area, c – the bulk concentration of electroactive compound, D – the diffusion coefficient. After the jet impingement the current decreases for about 10 ms according to the Cottrell equation followed by an increase of the diffusion layer thickness as δ = (πDt)1/2.
, where n – number of electrons, F – the Faraday constant, A – the electrode surface area, c – the bulk concentration of electroactive compound, D – the diffusion coefficient. After the jet impingement the current decreases for about 10 ms according to the Cottrell equation followed by an increase of the diffusion layer thickness as δ = (πDt)1/2. - M. Atobe, M. Sashira and T. Nonaka, Ultrason. Sonochem., 2000, 7, 103 CrossRef CAS PubMed.
- The cavitation zone can be described as a function of the radius of the irradiating horn, a distance to the cavitation area and an acoustic field, which forms an angle along the propagation
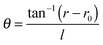 where r0 is the tip radius of the stepped horn, l is the distance from the top of the horn to the irradiation zone, and r is the sectional radius of the sonicated zone.
where r0 is the tip radius of the stepped horn, l is the distance from the top of the horn to the irradiation zone, and r is the sectional radius of the sonicated zone. - Y. Iida, K. Yasui, T. Tuziuti and M. Sivakumar, Microchem. J., 2005, 80, 159 CrossRef CAS.
- M. Romdhane, A. Gadri, F. Contamine, C. Gourdon and G. Casamatta, Ultrason. Sonochem., 1997, 4, 235 CrossRef CAS PubMed.
- Y. Iida, K. Yasui, T. Tuziuti and M. Sivakumar, Microchem. J., 2005, 80, 159 CrossRef CAS; F. Trabelsi, H. Ait-lyazidi, J. Berlan, P. L. Fabre, H. Delmas and A. M. Wilhelm, Ultrason. Sonochem., 1996, 3, S125 CrossRef; B. G. Pollet, J. Y. Hihn, M. L. Doche, J. P. Lorimer, A. Mandroyan and T. J. Mason, J. Electrochem. Soc., 2007, 154, E131 CrossRef; E. Maisonhaute, P. C. White and R. G. Compton, J. Phys. Chem. B, 2001, 105, 12087 CrossRef; J. Klima, Ultrasonics, 2011, 51, 202 CrossRef PubMed; B. G. Pollet, Power ultrasound in electrochemistry, John Wiley and Sons, Inc., Chichester, UK, 2012 Search PubMed.
- The limiting current is defined as Ilim = C(d,v)AD2/3C*Pw1/2, where d is the distance between electrode and horn surfaces, A is the area of the electrode, v is the kinematic viscosity, D is the diffusion coefficient, Pw is the ultrasonic power.
- P. R. Birkin, H. M. Hirsimaki, J. G. Frey and T. G. Leighton, Electrochem. Commun., 2006, 8, 1603 CrossRef CAS.
- I. E. Henley, A. C. Fisher, R. G. Compton and C. E. Banks, J. Phys. Chem. B, 2005, 109, 7843 CrossRef CAS PubMed.
- C. E. Banks, N. V. Rees and R. G. Compton, J. Phys. Chem. B, 2002, 106, 5810 CrossRef CAS.
- E. L. Coopert and L. A. Coury Jr., J. Electrochem. Soc., 1998, 145, 1801 CrossRef.
- F. Coeuret and A. Storck, Eléments de Génie Électrochimique, Tec-Doc Lavoisier, Paris, France, 1984–1993 Search PubMed.
- In a laminar flow over the plane surface, which is normal to the surface of the electrode, the velocity is uniform over the entire electrode at larger distances. The equivalent flow velocity can be estimated as
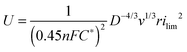 . By calculating the flow velocities a nondimensional hydrodynamic parameter such as Reynolds number, Re, is determined as
. By calculating the flow velocities a nondimensional hydrodynamic parameter such as Reynolds number, Re, is determined as 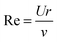 , where U is the flow velocity (m s−1), r is the electrode radius (m) and v is the kinematic viscosity (m2 s−1). The value of Re describes the mass transfer by convection, which is described by the Sherwood number. The latter is determined as Sh = 0.45Re1/2Sc1/3 or Sh = ilimr/n(FD)C* and Sc = v/D, where Sc is the dimensionless Schmidt number. These calculations confirm the dependence of Ilim on the square root of the ultrasonic intensity and the inverse square root of the distance between the electrode and horn surface.
, where U is the flow velocity (m s−1), r is the electrode radius (m) and v is the kinematic viscosity (m2 s−1). The value of Re describes the mass transfer by convection, which is described by the Sherwood number. The latter is determined as Sh = 0.45Re1/2Sc1/3 or Sh = ilimr/n(FD)C* and Sc = v/D, where Sc is the dimensionless Schmidt number. These calculations confirm the dependence of Ilim on the square root of the ultrasonic intensity and the inverse square root of the distance between the electrode and horn surface. - C. Klima, C. Bernard and J. Degrand, J. Electroanal. Chem., 1994, 367, 297 CrossRef.
- M. Barthès, G. Mazue, D. Bonnet, R. Viennet, J. Y. Hihn and Y. Bailly, Ultrasonics, 2015, 59, 72 CrossRef PubMed.
- A. Mandroyan, R. Viennet, Y. Bailly, M. L. Doche and J. Y. Hihn, Ultrason. Sonochem., 2009, 16, 88 CrossRef CAS PubMed; A. Mandroyan, M. L. Doche, J. Y. Hihn, R. Viennet, Y. Bailly and L. Simonin, Ultrason. Sonochem., 2009, 16, 97 CrossRef PubMed.
- M. Raffel, C. E. Willert, S. T. Wereley and J. Kompenhans, Particle Image Velocimetry, A Practical Guide, Springer-Verlag, Berlin Heidelberg, 2nd edn, 2007, p. 448 Search PubMed.
- The liquid jets can be quantified by measuring the current density i (A m−2 or mA cm−2 as more practical units), according to the following equation:
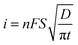 , where n is the number of electrons, F is the Faraday constant (96485S A mol−1), S is the surface area of the electrode (m2), D is the diffusion constant (m2 s−1), t is the time (s). As an example we consider OH− species as a model for diffusion assuming non-equilibrium conditions during acoustic cavitation within 2 s and a known diffusion coefficient being 5273 × 10−9 m2 s−1. The surface S is defined as
, where n is the number of electrons, F is the Faraday constant (96485S A mol−1), S is the surface area of the electrode (m2), D is the diffusion constant (m2 s−1), t is the time (s). As an example we consider OH− species as a model for diffusion assuming non-equilibrium conditions during acoustic cavitation within 2 s and a known diffusion coefficient being 5273 × 10−9 m2 s−1. The surface S is defined as 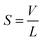 , where V is the volume (m3) and L is the length (m). The velocity of a liquid jet is derived as
, where V is the volume (m3) and L is the length (m). The velocity of a liquid jet is derived as 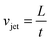 , where L is the length of a jet (m) and t is the time of its propagation (s). By replacing S with L defined through vjet and t the current density can be calculated as
, where L is the length of a jet (m) and t is the time of its propagation (s). By replacing S with L defined through vjet and t the current density can be calculated as 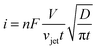 and the volume of a jet as
and the volume of a jet as 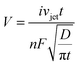 .
. - E. Samson, J. Marchand and K. A. Snyder, Mater. Struct., 2003, 36, 156 CrossRef CAS.
- The pressure, which is dissipated in a droplet volume near the electrode, is derived from the classical equation PV = νRT assuming adiabatic conditions of a nearly ideal gas (i.e. no heat transfer into the bulk solution). For 1 mol of a substance (e.g. OH−) at room temperature (i.e.T = 273 K) the pressure is determined within the droplet volume (from Fig. 8A and B) as P = RT/V with R being a gas constant (8.31 J K−1 mol−1).
- D. Radziuk, H. Möhwald and K. S. Suslick, Phys. Chem. Chem. Phys., 2014, 16, 3534 RSC.
- W. B. McNamara, Y. T. Didenko and K. S. Suslick, J. Phys. Chem. B, 2003, 107, 7303 CrossRef CAS.
- A. Gedanken, Ultrason. Sonochem., 2004, 11, 47 CrossRef CAS PubMed.
- R. Asami, M. Atobe and T. Fuchigami, J. Am. Chem. Soc., 2005, 127, 13160 CrossRef CAS PubMed.
- P. Cognet, A. M. Wilhelm, H. Delmas, H. Aıt Lyazidi and P. L. Fabre, Ultrason. Sonochem., 2000, 7, 163–175 CrossRef CAS PubMed.
- M. L. Doche, J. Y. Hihn, F. Touyeras, J. P. Lorimer, T. J. Mason and M. Plattes, Ultrason. Sonochem., 2001, 8, 291 CrossRef CAS PubMed; H. Ashassi-Sorkhabi and N. Ghalebsaz-Jeddi, Ultrason. Sonochem., 2006, 13, 180 CrossRef PubMed.
- J. Berlan, F. Trabelsi, H. Delmas, A. M. Wilhelm and J. F. Petrignani, Ultrason. Sonochem., 1994, 1, S97 CrossRef CAS; C. Petrier, A. Jeunet, J. L. Luche and G. Reverdy, J. Am. Chem. Soc., 1992, 114, 3148 CrossRef.
- G. Zhao, S. Shen, M. Li, M. Wu, T. Cao and D. Li, Chemosphere, 2008, 73, 1407 CrossRef CAS PubMed.
- X. Wang, J. Jia and Y. Wang, Ultrason. Sonochem., 2010, 17, 515 CrossRef CAS PubMed.
- D. J. Walton, L. D. Burke and M. M. Murphy, Electrochim. Acta, 1996, 41, 2747 CrossRef CAS.
- C. O. Neill, F. R. Hawkes, D. L. Hawkes, N. D. Lourenco, H. M. Pinheiro and W. Delee, J. Chem. Technol. Biotechnol., 1999, 74, 1009 CrossRef.
- D. Chatterjee, V. R. Patnam, A. Sikdar, P. Joshi, R. Misra and N. N. Rao, J. Hazard. Mater., 2008, 156, 435 CrossRef CAS PubMed.
- M. Rivera, M. Pazos and M. A. Sanrom, J. Chem. Technol. Biotechnol., 2009, 84, 1118 CrossRef CAS.
- M. D. Esclapez, V. Sáez, D. Milán-Yáñez, I. Tudela, O. Louisnard and J. González-García, Ultrason. Sonochem., 2010, 17, 1010 CrossRef CAS PubMed.
- W. Bourgeois, J. E. Burgess and R. M. Stuets, J. Chem. Technol. Biotechnol., 2001, 76, 337 CrossRef CAS.
- A. Yaqub, H. Ajab, M. H. Isa, H. Jusoh, M. Junaid and R. Farooq, J. New Mater. Electrochem. Syst., 2012, 15, 289 CAS.
- P. Tomcik, C. E. Banks and R. G. Compton, Electroanalysis, 2003, 15, 1661 CrossRef CAS.
- C. E. Banks, O. V. Klymenko and R. G. Compton, Phys. Chem. Chem. Phys., 2003, 5, 1652 RSC.
- R. Farooqa, Y. Wanga, F. Lina, S. F. Shaukatb, J. Donaldsonc and A. J. Chouhdary, Water Res., 2002, 36, 3165 CrossRef.
- L. Li, J. Ge and F. Wu, J. Hazard. Mater., 2010, 176, 288 CrossRef CAS PubMed.
- F. Rosalbino, D. Maccio, E. Angelini, A. Saccone and S. Delfino, J. Alloys Compd., 2005, 403, 275 CrossRef CAS.
- M. Fleischmann, D. Pletcher and A. Rafinski, J. Appl. Electrochem., 1971, 1, 1 CrossRef CAS; J. C. Farmer, F. T. Wang, R. A. Hawleyfedder, P. R. Lewis, L. J. Summers and L. Foiles, J. Electrochem. Soc., 1992, 139, 654 CrossRef; R. G. Hickman, J. C. Farmer and F. T. Wang, ACS Symp. Ser., 1993, 518, 430 CrossRef; A. J. Saterlay, F. Marken, J. S. Foord and R. G. Compton, Talanta, 2000, 53, 403 CrossRef PubMed.
- D. Peters, J. Mater. Chem., 1996, 6, 1605 RSC.
- H. Hasegawa, I. M. M. Rahman, Y. Egawa, H. Sawai, Z. A. Begum, T. Maki and S. Mizutani, Water, Air, Soil Pollut., 2014, 225, 2112 CrossRef.
- H. Hasegawa, I. M. M. Rahman, Y. Egawa, H. Sawai, Z. A. Begum, T. Maki and S. Mizutani, Microchem. J., 2013, 106, 289 CrossRef CAS.
- G. D. Pangu and D. L. Feke, Chem. Eng. Sci., 2004, 59, 3183 CrossRef CAS.
- L. A. Crum, J. Acoust. Soc. Am., 1972, 50, 157 CrossRef.
- J. C. Giddings, Unified Separation Chemistry, Johns and Wiley Ltd, New York, 1991 Search PubMed.
- The chemical potential of the system (μ) is the sum of external field effects (μext) and internal molecular interactions (μi), which is defined as μ = μext + μi = μext + μ0 + RTlnC, where μ0 is the internal standard chemical potential, R is the gas constant, T is the absolute temperature and C is the concentration of molecules. The force causes the relative displacement of molecules according to
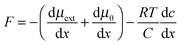 . The first term in parentheses is the force from the gradient of the external field, the second term is the force from the gradient at the liquid/liquid interface and the third term is the entropic force due to diffusion or Brownian motion. In the presence of particles this equation includes the microaction force
. The first term in parentheses is the force from the gradient of the external field, the second term is the force from the gradient at the liquid/liquid interface and the third term is the entropic force due to diffusion or Brownian motion. In the presence of particles this equation includes the microaction force 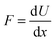 with U as a total potential of the particles.
with U as a total potential of the particles. - The Lorentz force depends on the complex permittivity
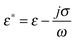 with ε dielectric constant, j = √−1, conductivity σ and angular frequency ω as ω = 2πν with ν the frequency of the applied field.
with ε dielectric constant, j = √−1, conductivity σ and angular frequency ω as ω = 2πν with ν the frequency of the applied field. - H. Watarai, Annu. Rev. Anal. Chem., 2013, 6, 353 CrossRef CAS PubMed.
- The real component of the Clausius–Mossotti factor is positive,
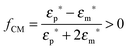 .
. - A. Lenshof and T. Laurell, Chem. Soc. Rev., 2010, 39, 1203 RSC.
- L. M. Barrett, A. J. Skulan, A. K. Singh, E. B. Cummings and G. J. Fiechtner, Anal. Chem., 2005, 77, 6798 CrossRef CAS PubMed.
- E. B. Cummings and A. K. Singh, Anal. Chem., 2003, 75, 4724 CrossRef CAS PubMed.
- U. Kim, J. R. Qian, S. A. Kenrick, P. S. Daugherty and H. T. Soh, Anal. Chem., 2008, 80, 8656 CrossRef CAS PubMed.
- The value of the radiation acoustophoretic force depends on the physical properties of a particle and is defined as
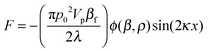 , where
, where 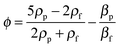 , where p0 is the acoustic pressure amplitude, Vp – volume of the particle, βp and βf and ρp and ρf are compressibility and density of the particle and the fluid, ϕ is the acoustic contrast factor, which governs the direction of particle movement.
, where p0 is the acoustic pressure amplitude, Vp – volume of the particle, βp and βf and ρp and ρf are compressibility and density of the particle and the fluid, ϕ is the acoustic contrast factor, which governs the direction of particle movement. - C. Grenvall, P. Augustsson, J. R. Folkenberg and T. Laurell, Anal. Chem., 2009, 81, 6195 CrossRef CAS PubMed.
- Y. Ai and B. L. Marrone, Microfluidics, BioMEMS, Medical Mcirosystems XII, 2014, p. 8976 Search PubMed.
- K. J. Lissant, Demulsi5cation: Industrial Applications, Marcel Dekker Inc., New York, 1983 Search PubMed; L. L. Schramm, Emulsions: Fundamentals and Applications in Petroleum Industry, American Chemical Society, Washington, 1992 Search PubMed.
- M. Cheryan and N. Rajagopalan, J. Membr. Sci., 1998, 151, 13 CrossRef CAS.
- T. L. Tolt and D. L. Feke, J. Acoust. Soc. Am., 1992, 91, 3152 CrossRef.
- A. Garcia-Lopez and D. N. Sinha, J. Open Acoust., 2008, 1, 66 CrossRef.
- M. Hiraide and K. Hasegawa, Fresenius' J. Anal. Chem., 1999, 363, 261 CrossRef CAS.
- S. Gupta and D. L. Feke, J. Acoust. Soc. Am., 1998, 104, 2189 CrossRef CAS; S. Gupta and D. L. Feke, AIChE J., 1998, 44, 1005 CrossRef; S. Gupta, Acoustically driven collection of suspended particles within porous media, PhD dissertation, Case Western Reserve University, Cleveland, OH, 1997 CrossRef PubMed; M. T. Grossner, A. E. Penrod, J. M. Belovich and D. L. Feke, Ultrasonics, 2003, 41, 65 CrossRef PubMed.
- H. Li, Y. Huang, Y. Mao, W. L. Xu and H. J. Ploehn, Chem. Commun., 2014, 50, 9849 RSC.
- M. Yu, Y. Huang, H. Li, L. Wang, Y. Qiao, C. Tang, C. Jung, Y. Yoon, S. Li and M. Yu, Adv. Mater. Interfaces, 2015, 2, 1400433 Search PubMed.
- M. Chiha, O. Hamdaoui, F. Ahmedchekkat and C. Petrier, Ultrason. Sonochem., 2010, 17, 318 CrossRef CAS PubMed; R. S. Juang and K. H. Lin, Colloids Surf., A, 2004, 238, 43 CrossRef.
- N. M. Kocherginsky, Q. Yang and L. Seelam, Sep. Purif. Technol., 2007, 53, 171 CrossRef CAS; G. León, G. Martínez, M. A. Guzmán, J. I. Moreno, B. Miguel and J. A. Fernández-López, Ultrason. Sonochem., 2013, 20, 650 CrossRef PubMed.
- S. Avivi Levi, Y. Nitzan, R. Dror and A. Gedanken, J. Am. Chem. Soc., 2003, 125, 15712 CrossRef PubMed.
- F. Cavalieri, M. Ashokkumar, F. Grieser and F. Caruso, Langmuir, 2008, 24, 10078 CrossRef CAS PubMed.
- F. L. Tchuenbou-Magaia, I. T. Norton and P. W. Cox, J. Cell. Plast., 2011, 47, 217 CAS.
- R. Litran, E. Blanco, M. Ramirezdelsolar and L. Esquivias, J. Sol-Gel Sci. Technol., 1997, 8, 985 CAS.





























| This journal is © the Owner Societies 2016 |