The origin of specificity and insight into recognition between an aminoacyl carrier protein and its partner ligase†
Received
27th May 2015
, Accepted 8th June 2015
First published on 12th June 2015
Abstract
Acyl carrier proteins (ACPs) are among the most promiscuous proteins in terms of protein–protein interactions and it is quite puzzling how ACPs select the correct partner between many possible upstream and downstream binding proteins. To address this question, we performed molecular dynamics simulations on dimeric Bradyrhizobium japonicum Gly:CP ligase 1 to inspect the origin of its selectivity towards the three types of carrier proteins, namely holoCP, apoCP, and holoCP–Gly, which only differ in the attached prosthetic group. In line with experiments, MM-GBSA analysis revealed that the ligase preferentially binds the holoCP form to both subunits with the binding free energies of −20.7 and −19.1 kcal mol−1, while the apoCP form, without the prosthetic group, is also recognized, but the binding values of −9.2 and −3.6 kcal mol−1 suggest that there is no competition for the ligase binding as long as the holoCP is present. After the prosthetic group becomes glycylated, the holoCP–Gly dissociates from the ligase, as supported by its endergonic binding free energies of 2.9 and 20.9 kcal mol−1. Our results indicate that these affinity differences are influenced by three aspects: the form of the prosthetic group and the specific non-polar hydrophobic interactions, as well as charge complementarity dominantly manifested through Arg220–Glu53 ion pair within the binding region among proteins. A careful examination of the bonding patterns within the ligase active site elucidated the interactions with Arg258, Asp215 and Tyr132 as being predominant in stabilizing the prosthetic group, which are significantly diminished upon glycation, thus promoting complex dissociation.
1. Introduction
Acyl carrier proteins (ACPs) are involved in a number of pathways of primary and secondary cell metabolism. Some of them function as stand-alone proteins in biosynthetic pathways, while others form a domain of large multidomain fatty acid or polyketide synthases. Also, there are peptidyl carrier proteins (PCP) as well as aryl carrier proteins (ArCPs) of nonribosomal peptide synthetases (NRPSs), which produce a large number of natural bioactive peptides in nucleic acid-independent mechanisms. ACPs are expressed as apoACPs and are activated to holoACPs by the corresponding ACP synthase (ACPS), which catalyzes the attachment of a 4′-phosphopantetheine (Ppant) moiety to the conserved Ser residue. This makes the Ppant prosthetic group (Scheme 1) a common feature of all acyl carrier proteins, through which ACPs are capable of binding, sequestering and delivering attached intermediates to a downstream acceptor protein or protein domain in numerous metabolic pathways. ACPs provide acyl groups for both lipid A1 and lipoic acid synthesis,2 and they participate in quorum sensing, bioluminescence and toxin activation.3–6 Moreover, together with PCPs, ACPs also take part in the polyketide and non-ribosomal peptide syntheses, which produce important secondary metabolites, such as lipopeptide antibiotic daptomycin and iron carrying siderophore enterobactin.7,8 Considering a large number of pathways in which ACPs participate, it is apparent that there are dozens of various partner proteins that interact with them. Thus, it remains a challenge to understand how ACPs both select and discriminate between appropriate partner proteins or partner domains, since they are among most promiscuous proteins in terms of protein–protein interactions in the cell.9
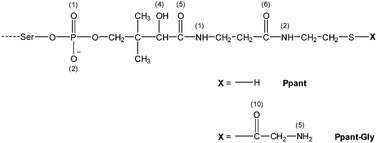 |
| | Scheme 1 Chemical structure of the Ppant prosthetic group and its aminoacylated derivative Ppant–Gly, both chemically bound to the Ser residue of the carrier protein. The numbering of atoms relevant for the discussion is indicated in round brackets. | |
It is presumed that the overall acidic nature of ACPs is the basis for interaction with their partner enzymes.10 Indeed, complementary basic regions, which form electrostatic interactions with the acidic helix II of ACP, have been identified in several enzymes in Escherichia coli fatty acid synthase II (FAS II).11 Thus, helix II of ACP is named as the “recognition helix” and it serves as a universal interaction site on ACP.12 The role of helix II became first apparent from the crystal structure of ACP complexed with holo-acyl carrier protein synthase (AcpS) from Bacillus subtilis.13 Moreover, the importance of these acidic residues has been further confirmed through extensive site-directed mutagenesis.14 In order to investigate the binding interactions between carrier protein and one of its protein partners, we choose a recently described complex formed between aminoacyl carrier protein from Bradyrhizobium japonicum (Bj CP) and its B. japonicum glycine:[carrier protein] partner ligase 1 (Bj Gly:CP ligase 1).15 Bj CP binds to Bj Gly:CP ligase 1 and attaches the glycyl moiety, derived from glycyl-adenilate formed in the active site of ligase, to the terminal –SH group of its Ppant moiety (Scheme 1). Subsequently, Bj CP, carrying the glycyl fragment, dissociates from Bj Gly:CP ligase 1 and delivers glycine to a downstream partner protein in a yet uncharacterized metabolic pathway.15 Recently published results15 have undoubtedly shown that, when both holo- and apoCP forms are present, biologically relevant recognition is achieved only between holoCP and Bj Gly:CP ligase 1, the latter interacting with moderate affinity (KD = 21.5 μM), as determined through a set of pull down experiments, as well as kinetic analysis and isothermal titration calorimetry (ITC) measurements. In this work, we were interested in elucidating the distinct features of the carrier protein in its apo and holo forms that govern the preferential binding of holoCP to Bj Gly:CP ligase 1, and rationalize the differing affinities of the ligase towards three ACP forms. Furthermore, since Bj Gly:CP ligase 1 is a dimer capable of binding one carrier protein per subunit (Fig. 1), we sought to find if subunits exhibit different bindings towards the same form of the carrier protein. Although the previously published co-crystal structure of holoCP and Bj Gly:CP ligase 115 provided global information about the interacting interfaces of these two proteins, the questions mentioned above remained to be answered.
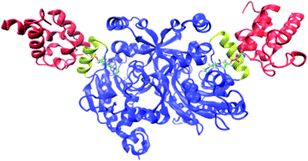 |
| | Fig. 1 The structure of holoCP – Bj Gly![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CP ligase 1 2:1 complex. Both holoCPs are given in red, while Bj Gly:CP ligase 1 is shown in blue with CP-binding helices given in yellow. Ppant prosthetic groups are given in the licorice representation. :CP ligase 1 2:1 complex. Both holoCPs are given in red, while Bj Gly:CP ligase 1 is shown in blue with CP-binding helices given in yellow. Ppant prosthetic groups are given in the licorice representation. | |
2. Methods
2.1 System preparation
The initial models for the molecular dynamics (MD) simulations were prepared using the three-dimensional structures determined by the X-ray diffraction. Two crystal structures of the enzyme B. japonicum Gly:CP ligase 1 in a complex with analogue of glycyl-adenylate bound to the cognate carrier protein (PDB ID code 4H2T, resolution 2.44 Å) and without it (PDB ID code 3MF1, resolution 2.20 Å) were both considered in setting up the initial models of the ligase interacting with its cognate carrier proteins. HoloCP–Gly bound to the ligase was modeled from the structure of B. japonicum glycine:[carrier protein] ligase complexed with the glycylated carrier protein (PDB ID code 4H2V, resolution 2.00 Å), while a complex of apoCP and Bj Gly:CP ligase 1 was modeled by deleting 4-phosphopantethein (Ppant) prosthetic group attached on an invariant serine residue of holoCP complexed with B. japonicum Gly:CP ligase 1 (PDB ID code 4H2T, resolution 2.44 Å). In all cases, the missing residues were built using the I-TASSER server.16–18 Altogether we prepared five molecular models for MD simulations (Table 1), which are all heteromeric complexes involving a dimeric ligase complexed with either two respective carrier proteins or two isolated prosthetic groups, symmetrically occupying both of its subunits (Fig. 1).
Table 1 Molecular models used in the molecular dynamics simulations
| Model number |
Molecular model |
| 1 |
apoCP:Bj Gly:CP ligase 1:apoCP |
| 2 |
holoCP:Bj Gly:CP ligase 1:holoCP |
| 3 |
holoCP–Gly:Bj Gly:CP ligase 1:holoCP–Gly |
| 4 |
Ppant:Bj Gly:CP ligase 1:Ppant |
| 5 |
Ppant–Gly:Bj Gly:CP ligase 1:Ppant–Gly |
2.2 Molecular dynamics simulations
The missing hydrogen atoms in the prepared models were added using the tleap module in AMBER12.19 The geometries of both the 4′-phosphopantetheine prosthetic group bound to the serine residue and an analogue of glycyl-adenylate were optimized using the HF/6-31G* level of theory in the Gaussian 09 program,20 after which the atomic partial charges were obtained by fitting the electrostatic potentials using the RESP fitting technique21 available in AMBER12. Prepared models (Table 1) were employed as the initial structures for the MD simulations. The general AMBER force field GAFF22 and the AMBER ff12SB force field23 were used for the substrate and the protein complexes, respectively. All structures were subsequently solvated in a truncated octahedral box of TIP3P water molecules spanning a 10 Å thick buffer of solvent around each protein. We carried out energy minimizations and MD simulations using the PMEMD module. Production runs, each 60 ns in length, were performed on GPU using the pmemd.CUDA engine.24–26 Protein geometry optimization was conducted in four cycles with different constraints. In the first cycle (1500 steps), water molecules and the substrate were relaxed, while the protein and zinc cation were constrained using a harmonic potential with a force constant of 32 kcal (mol Å2)−1. In the second (2500 steps), third (1500 steps), and fourth cycle (5000 steps), the same force constant was applied to the zinc cation, and the value of 10 kcal (mol Å2)−1 was imposed to constrain the protein backbone. The energy minimization procedure, consisting of 470 steps of the steepest descent followed by the conjugate gradient algorithm for the remaining optimization steps, was the same in all cycles. Optimized systems were gradually heated from 0 to 300 K and equilibrated during 30 ps using NVT conditions, and subjected to productive, unconstrained MD simulations at constant temperature (300 K) and pressure (1 atm). The temperature was held constant using a Langevin thermostat with a collision frequency of 1 ps−1. For the first 6 ns of the productive MD run, a time step of 1 fs was used and for the rest of MD simulations the time step was 2 fs. Bonds involving hydrogen atoms were constrained using the SHAKE algorithm.27 Structures were sampled every 1 ps over the first 6 ns and every 0.5 ps for the remaining 60 ns. The Particle Mesh Ewald method28 was applied to calculate long-range electrostatic interactions. The nonbonded interactions were truncated at 10.0 Å.
2.3 MM-GBSA free energy calculations
The binding free energies for the Bj Gly:CP ligase 1–Bj CP complexes, ΔGbind, were calculated according to the following:| | | ΔGbind = 〈Gcomplex〉 − 〈Gligase〉 − 〈Gcarrier_protein〉 | (1) |
where the symbol 〈〉 denotes the average value over 100 snapshots collected from the last 2 ns along each converged MD trajectory (Fig. S1, ESI†). The free energy of a system, Gcomplex/ligase/carrier_protein, can be approximated by three terms: the energy of the system in the gas phase, solvation free energy, and entropic contribution:
| | |
G
complex/ligase/carrier protein = EMM + Gsolv − TΔSMM
| (2) |
where
EMM, the gas-phase molecular mechanical energy, is calculated as the sum of
Einternal,
EvdW, and
Eelec contributions.
Gsolv, the solvation free energy is the sum of polar (
Gpolar) and nonpolar (
Gnonpolar) components, where the former was calculated by solving the finite-difference Generalized Born equation, while the latter was determined on the basis of the solvent-accessible surface area (SASA) using the following equation:
employing the recommended empirical parameters
γ = 0.0054 kcal mol
−1 Å
−2 and
β = 0.92 kcal mol
−1.
29 The solute conformational entropy (
SMM) was estimated by the normal-mode analysis. Free energy calculations were performed using the python script, MMPBSA.py
30 provided in AMBER12. We have used the modified GB model “OBC” with the parameters endowed by Onufriev and coworker (igb = 2).
31 The exterior and solute dielectric constants were set to 80 and 1, respectively.
32 According to the thermodynamic cycle depicted in
Fig. 2, we performed calculations on apoCP, holoCP, and holoCP–Gly proteins bound to Bj Gly:CP ligase 1 in order to clarify the differences in their binding free energies relative to each ligase subunit (
Fig. 1).
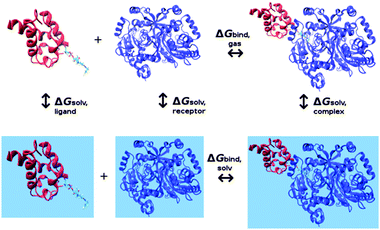 |
| | Fig. 2 Thermodynamic cycle used in MM-GBSA calculations. Blue surfaces represent calculations conducted in the aqueous solution. | |
MM-GBSA analysis has been carried out on six complexes, which differ in the choice of the form of the considered carrier protein (holoCP, apoCP, or holoCP–Gly) and the ligase subunit they are bound to (A or B), the latter assigned according to the PDB structures. As an illustrative example, both alternatives for the holoCP system are depicted in Fig. 3. Additionally, four model complexes involving isolated Ppant and Ppant–Gly prosthetic groups, each placed in both ligase subunits, were also analyzed to assess their influence on the overall binding.
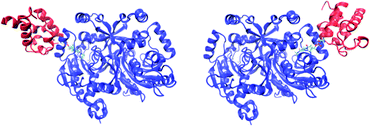 |
| | Fig. 3 The structures of holoCP carrier protein bound to subunits A (left) and B (right) of Bj Gly:CP ligase 1. | |
2.4 Computational alanine scanning of protein–protein interfaces
Experiments have shown that the basic CP-binding helix (residues R220-Q232) of Bj Gly:CP ligase 1 determines macromolecular recognition with acidic carrier protein.15 Wild-type Bj Gly:CP ligase 1 recognizes and binds the cognate Bj carrier protein with a moderate affinity of KD = 21.5 μM, while the enthalpy change with Bj Gly:CP ligase 1 and non-cognate carrier protein from A. tumefaciens (At) was not measurable. After the construction of the hybrid Bj ligase, where the CP-binding helix was replaced with the corresponding region from A. tumefaciens Ala:CP ligase, the selectivity of such a hybrid ligase was fully changed in favor of At CP, while Bj CP was no longer a partner to the ligase. In addition, upon deleting the CP-binding helix region, interactions with either cognate or heterologous carrier proteins were lost.15 Such strong experimental evidence that the CP-binding helix of Bj Gly:CP ligase 1 is principally responsible for the selection among number of possible carrier protein partners prompted us to use the in silico alanine scanning approach to probe the individual contribution of each residue within the mentioned region.33,34
It has been demonstrated that protein–protein interactions are critically dependent on just a few residues termed as “hot spots” at the interface, which exert a predominant contribution to the binding free energy,35,36 and which, if mutated, could disrupt the interaction. On the other hand, the majority of interface residues have a minimal effect on the binding upon alanine mutation. Alanine scanning mutagenesis investigates both the structural and energetic characteristics of protein–protein interactions by mutating individual amino acids to alanine and then scoring the impact of each mutation on the overall binding free energy (ΔΔGbind) among proteins. Alanine has a propensity for forming alpha helices but it can also occur in beta sheets. Since mutations to the alanine residue retain the β-carbon, thus wild-type flexibility, it follows that the polypeptide backbone conformations and, above all, secondary structure composition stay unchanged. For this work, residues are classified as hot spots if their mutation to alanine resulted in 10-fold or higher change in the ΔΔGbind.37
2.5 Sequence analysis
Sequences of aa:CP ligases were retrieved from the NCBI Protein database. The database was searched by PSI-BLAST, using an interactively built profile in order to retrieve all relevant sequences deposited in the database. Altogether, 806 sequences were retrieved after the BLAST search. To remove the redundancy of the initial dataset, sequences exceeding 90% identity to any other sequence were eliminated. The final sequence dataset contained 180 sequences which were aligned by ClustaX 2.1 and analysed.
2.6 Data analysis
Structures (RMSD, hydrogen bonds, distances) of the protein complexes were analyzed using the ptraj module38 available in the AmberTools12 and VMD,39 the latter also employed for the visualization together with the Pymol software.40 Graphs were plotted using the Xmgrace41 program. The analysis of the hydrogen-bonding occupancies along the MD trajectory was performed by employing the hydrogen bond donor–acceptor distance of 3.5 Å and the donor–hydrogen–acceptor angle of 135° as the cutoff values.
3. Results and discussion
3.1 Total binding free energies
We employed the MM-GBSA method to estimate the absolute binding free energies for the protein complexes and to obtain detailed information about energetic contributions that govern protein–protein interactions (Table 2). Looking first at the overall binding free energies, it turns out that both subunits of the ligase strongly favor binding to the holoCP form, as evidenced by ΔGbind values of −20.7 and −19.1 kcal mol−1, respectively, providing a convincing explanation why, in the crystal structures, both Bj Gly:CP ligase 1 subunits are occupied with the holoCP form. Partitioning of the overall interaction energies demonstrate that the non-polar parts (vdW + ESURF) prevail over the combined polar electrostatic contributions (EEL + EGB), thus representing the driving force for the binding, although a single most dominant promoting contribution is exerted by the gas-phase electrostatic component (EEL). On the other hand, the binding of the apoCP form is also favorable for both subunits, but the calculated free energies drop significantly and show notable asymmetry, −9.2 kcal mol−1 for the subunit A and −3.6 kcal mol−1 for the subunit B. We attribute this to a decrease in the favorable non-polar contributions relative to the holoCP forms. More precisely, a non-polar contribution for the formation of holoCP:Bj Gly:CP ligase 1 complex is approximately −120 kcal mol−1, while for apoCP it assumes around −95 kcal mol−1. The overall thermodynamic parameters suggest that there is no competition between holoCP and apoCP for the binding to ligase, meaning that when both forms are present, the ligase will exclusively bind the former system. These results are perfectly in line with the experimental data by Močibob et al.,15 which demonstrated that the acylation of holoCP protein to holoCP–Gly is not, in any way, affected by the presence of the apoCP form, even by adding a large excess of the latter. What is particularly interesting is the fact that, upon the glycation of the Ppant prosthetic group, the binding free energies for the holoCP–Gly form assume positive values, 2.9 kcal mol−1 for the subunit A, and 20.9 kcal mol−1, being largely positive for the subunit B. Although the origin of such a large difference in binding energies for each subunit is presently unclear, these results convincingly suggest that the enzymatic attachment of the glycyl moiety to the carrier protein will strongly promote the dissociation of both holoCP–Gly proteins from the Bj Gly:CP ligase 1, indicating that the departure of both CPs is not concerted. Let us also mention that, although the reliability of the correlation between in silico and experimental binding free energies is enhanced when the entropy is included, one should bear in mind that the calculation of the entropic contribution (TΔS) is still prone to errors and may introduce significant uncertainty in the results.42,43 In line with these observations, we noticed that our calculated results for the binding of the holoCP form (ΔGbind = −20.7 and −19.1 kcal mol−1) seem to be slightly overestimated in comparison with the experimental result of −6.4 kcal mol−1, as calculated from the measured value of KD = 21.5 μM, but the trend between the three forms of the carrier protein should be qualitatively reliable,44,45 which is the focus here. Future quantitative improvement might include the multiple trajectory protocol,46 which requires separate MD simulations for the unbound partners. This approach is computationally significantly more demanding and is beyond the scope here.
Table 2 Calculated binding free energies (ΔGbind) and their componentsa for ACP and prosthetic group interaction with the ligase obtained by the MM–GBSA approach (in kcal mol−1)
| System |
vdW |
EEL |
EGB |
ESURF |
ΔHgas |
ΔHsolv |
ΔHbind |
TΔS |
ΔGbind |
|
vdW = van der Waals contribution from MM; EEL = electrostatic energy as calculated by the MM force field; EGB = electrostatic contribution to the solvation free energy calculated by GB; ESURF = nonpolar contribution to the solvation free energy calculated by an empirical model.
|
| holoCP:ligase (subunit A) |
−107.2 |
−378.5 |
420.1 |
−12.9 |
−485.7 |
407.2 |
−78.5 |
−57.8 |
−20.7 |
| holoCP:ligase (subunit B) |
−104.5 |
−364.4 |
412.3 |
−12.4 |
−468.9 |
399.9 |
−69.0 |
−49.9 |
−19.1 |
| apoCP:ligase (subunit A) |
−70.8 |
−245.3 |
288.1 |
−8.4 |
−316.2 |
279.7 |
−36.5 |
−27.2 |
−9.2 |
| apoCP:ligase (subunit B) |
−84.4 |
−228.0 |
271.0 |
−9.3 |
−312.4 |
261.6 |
−50.7 |
−47.1 |
−3.6 |
| holoCP–Gly:ligase (subunit A) |
−82.3 |
−207.2 |
240.8 |
−13.2 |
−289.5 |
227.6 |
−61.9 |
−64.8 |
2.9 |
| holoCP–Gly:ligase (subunit B) |
−67.2 |
−163.4 |
200.6 |
−11.8 |
−230.6 |
188.8 |
−41.8 |
−62.9 |
20.9 |
|
Ppant:ligase (subunit A) |
−36.9 |
−157.5 |
160.6 |
−5.9 |
−194.3 |
154.6 |
−39.7 |
−27.5 |
−12.2 |
|
Ppant:ligase (subunit B) |
−38.3 |
−170.5 |
176.0 |
−5.9 |
−208.8 |
170.1 |
−38.7 |
−29.3 |
−10.4 |
|
Ppant–Gly:ligase (subunit A) |
−41.7 |
−122.2 |
132.2 |
−6.2 |
−163.9 |
126.1 |
−37.9 |
−32.7 |
−5.2 |
|
Ppant–Gly:ligase (subunit B) |
−37.6 |
−81.8 |
107.1 |
−5.6 |
−119.5 |
101.5 |
−17.9 |
−20.7 |
2.8 |
3.2 Binding analysis at the protein–protein interface
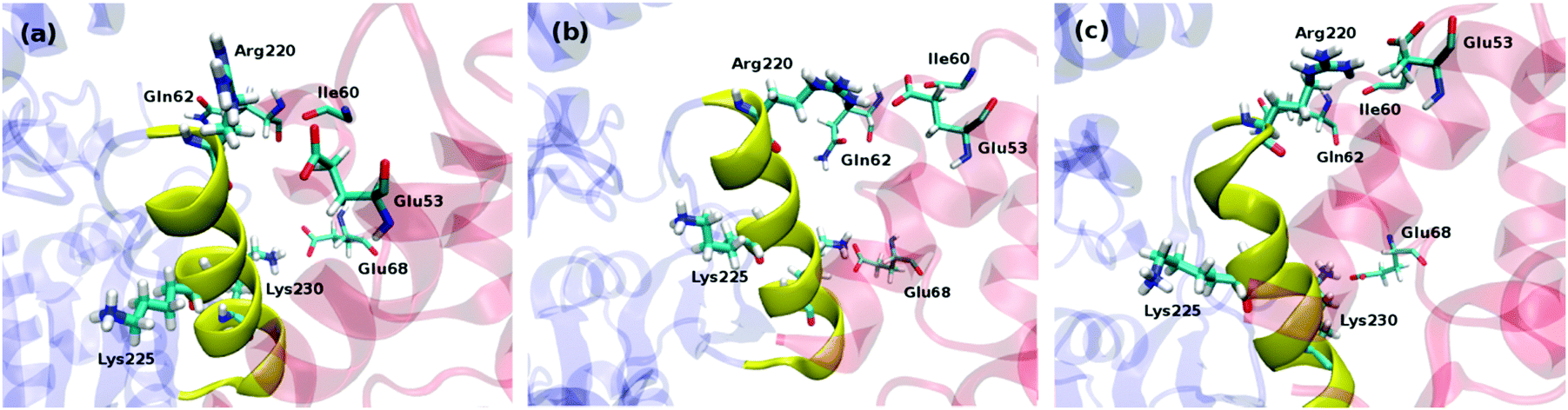
To get a further insight into the protein–protein binding interface and the resulting interactions, we employed in silico alanine scanning mutagenesis. All non-alanine and non-glycine residues in the CP-binding helix in both ligase subunits were mutated to alanine, including Arg220, Val221, Gln223, Met224, Lys225, Val227, Ser228, Gln229, Lys230, Gln231, and Gln232, assessing the influence of these point mutations on the total interaction energies with the holoCP. Calculations have revealed (Table S1, ESI,†Fig. 5) that almost all mutations identified these sites as “hot spot” residues, since they reduce the overall binding free energies by more than 1.36 kcal mol−1, which is equivalent to a 10-fold decrease in the KD value, the only exceptions provided by Val221, Gln223, Lys225, and Gln229 in subunit A and Val221 in subunit B. Interestingly, the K230A mutation in subunit B even increased the overall binding energy relative to the wild-type enzyme by 0.8 kcal mol−1. Common to both subunits is the large influence of the Arg220 residue, which, upon mutation, reduces the interaction in subunits A and B by 10.2 and 14.2 kcal mol−1, respectively, suggesting the significant role of Arg220 in the recognition and binding (see later). This large influence is attributed to a big drop in the electrostatic and solvation components of the overall ΔGbind value (Table S1, ESI†), which is a direct consequence of replacing a charged residue (Arg) with the uncharged apolar alternative (Ala). These high values are matched by a double-digit influence of the Lys230 and Gln231 in the subunit A, while the second-largest effect in the subunit B is exerted by Met224, being 7.5 kcal mol−1. Let us also mention that Lys230 and Gln231 are placed at the C-terminus of the CP-binding helix and both form hydrogen bonds with the side-chain of Glu68, and the backbone oxygen atoms of Val36 and Asp37 of the carrier protein, while Arg220 is placed at the N-terminus and creates hydrogen bonds with the side-chain of Glu53, and backbone carbonyl oxygen atoms of the Ile60 and Gln62 of the carrier protein (Fig. 4).
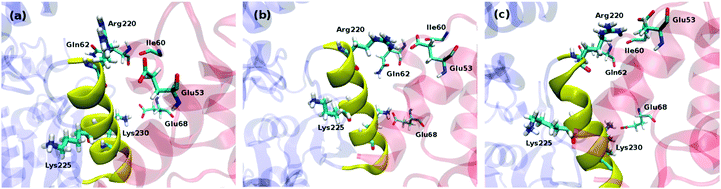 |
| | Fig. 4 Binding interfaces between ligase and various forms of carrier protein, namely apoCP (a), holoCP (b), and holoCP–Gly (c) in the ligase subunit A. CP-binding helix on ligase is given in yellow, while Bj Gly:CP ligase 1 and carrier proteins are given in transparent blue and red, respectively. | |
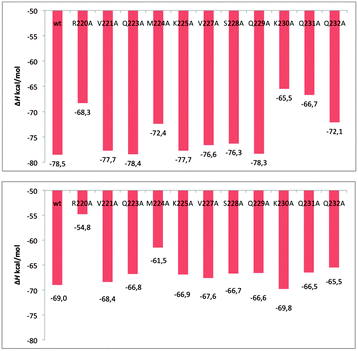 |
| | Fig. 5 The enthalpy changes calculated for sequentially mutated interface residue to alanine of complexes formed between holo carrier protein and Bj Gly:CP ligase 1 in its subunits A (top) and B (bottom). | |
Apart from inspecting the influence of each residue in the CP-binding helix on the binding energy through point-mutations to alanine, a very useful insight into the alterations in the environment around these residues imposed by the presence of the ACPs is offered by monitoring the differences in the pKa values of the ionisable residues47 in the mentioned ligase region. A closer look reveals that CP-binding helix is basic in nature consisting of thirteen amino acids in the RVGQMKAVSQKQQ sequence, with ionizable residues being Arg220, Lys225 and Lys230. Two of them, Arg220 and Lys230, are interacting with carrier proteins, while Lys225 is pointing to the interior of the ligase and is not directly participating in the interaction (Fig. 4). Their pKa values (Table 3) were obtained through the PROPKA3.0 server48 on 100 distinct snapshot structures of each complex attained during the last 5 ns of MD simulations with the 0.05 ns intervals. The reported pKa values were determined by taking the average value over all snapshots (Fig. S2, ESI†).49 If one considers the data for the “isolated” unbound ligase, obtained on the geometry from the crystal structure (3MF1.pdb), the values for Arg220 and Lys230 assume the values 13.0 and 9.6, respectively, suggesting the predominance of their cationic forms. The latter are only slightly modified relative to their aqueous solution values of 12.6 and 10.6, respectively. The presence of both holoCP and holoCP–Gly keeps the pKa value for Arg220 high at 12.6 and 12.7, respectively, being a consequence of the favorable interactions with the Glu53, Ile60 and Gln62 on the ACP (Fig. 4), in line with the high hydrogen bonding occupancy of these interactions (Table 4). This could explain a large negative effect on binding free energy upon R220A mutation (Fig. 4b), mentioned earlier, in comparison to all other mutated residues in both subunits. These contacts are significantly diminished in apoCP, which results in a lowering of the Arg220 pKa value to 12.2, thus contributing to the reduced affinity of the ligase towards the apoCP form. The changes in the pKa values of Lys230 are less obvious and, regardless of the form of the carrier protein, are clustered around 10.7 due to stabilizing interactions with Glu68, mirroring roughly the same occupancies in all three CP forms (Table 4). The pKa values for the Lys225 are also independent of the CP form and are grouped around 8.7, strongly indicating the hydrophobic nature of the ligase interior.
Table 3 PROPKA pKa values of the residues in the ligase CP-binding helix based on the average value from 100 snapshots of the last 5 ns of the MD trajectory. pKa,av correspond to the average value over both subunits
| Residue |
Unbound ligase (ID 3MF1) |
apoCP:ligase |
holoCP:ligase |
holoCP–Gly:ligase |
| Subunit A |
Subunit B |
pKa,av |
Subunit A |
Subunit B |
pKa,av |
Subunit A |
Subunit B |
pKa,av |
Subunit A |
Subunit B |
pKa,av |
| Arg220 |
13.0 |
(Missing residue) |
— |
12.3 |
12.0 |
12.2 |
11.6 |
13.6 |
12.6 |
12.6 |
12.7 |
12.7 |
| Lys225 |
9.3 |
(Missing residue) |
— |
8.9 |
8.7 |
8.8 |
8.7 |
8.3 |
8.5 |
8.0 |
9.1 |
8.6 |
| Lys230 |
10.3 |
8.8 |
9.6 |
11.0 |
10.6 |
10.8 |
10.6 |
10.5 |
10.6 |
10.7 |
10.8 |
10.8 |
Table 4 Hydrogen bonding analysis between ligase CP-binding helix (donors) and the matching carrier proteins residues (acceptors) at the binding interface based on the last 30 ns of the MD trajectory
| Donor |
Acceptor |
Occupancy in apoCP (%) |
Occupancy in holoCP (%) |
Occupancy in holoCP–Gly (%) |
| Residue |
Group |
Residue |
Group |
Subunit A |
Subunit B |
Subunit A |
Subunit B |
Subunit A |
Subunit B |
| Arg220 |
N–H1 |
Ile60 |
O |
45 |
77 |
64 |
73 |
18 |
— |
| Arg220 |
N–H2 |
Glu53 |
OE1 |
11 |
— |
23 |
21 |
26 |
34 |
| Arg220 |
N–H2 |
Glu53 |
OE2 |
10 |
— |
30 |
18 |
23 |
34 |
| Arg220 |
N–H2 |
Gln62 |
OE1 |
7 |
— |
2 |
11 |
— |
— |
| Lys230 |
N–H2 |
Glu68 |
OE1 |
31 |
— |
— |
20 |
24 |
15 |
| Lys230 |
N–H2 |
Glu68 |
OE2 |
22 |
— |
— |
15 |
21 |
8 |
As previously reported,15 the helix involved in the interaction with CPs is evolutionary not well preserved and ligases interact specifically with the cognate carrier protein. The results of in silico alanine scanning mutagenesis prompted us to re-analyze the conservation of particular residues (Fig. 6) in the context of their contribution to the interaction free energy. Curiously, all the residues with the most pronounced effect on CP binding are weakly conserved (Met224, Lys230), or not conserved (Arg220, Gln231, Gln232). The same is also true for other residues with an appreciable contribution to the free energy of CP binding (Lys225, Val227, Ser228). The only conserved residue within the ligase interacting region is Gln229, but it does not influence CP binding, in accordance with the results of in silico alanine scanning mutagenesis, because it is located on the opposite side of the interacting helix and does not contact CPs. Two conclusions can be drawn from the sequence divergence of the ligase helix involved in the interaction with CP: no specific residues are universally conserved to serve as the major anchoring points for CP binding across the whole ligase family, and CP recognition is likely species dependent. This is in line with previous experimental results which have shown that recognition of cognate CPs by A. tumefaciens and B. japonicum is mutually exclusive, and it can be easily inverted by a domain swap between the two ligases.15
 |
| | Fig. 6 Sequence conservation of the amino acid:[carrier protein] ligase residues engaged in aminoacyl carrier protein binding. 180 representative sequences were used to create multiple sequence alignment, eight of which are shown here. Different shades of gray represent 90, 70 and 50% conservation of identical or similar residues in the full sequence set. The sequence region corresponding to the helix interacting with the CP is boxed. The numbering on the top of the alignment corresponds to the sequence of the B. japonicum ligase. Abbreviations used and NCBI accession numbers of representative ligases: B. japonicum, Bradyrhizobium japonicum (NP_767597.1); A. tumefaciens, Agrobacterium tumefaciens (NP_355511.1); B. dolosa, Burkholderia dolosa (WP_006765793.1); M. extorquens, Methylobacterium extorquens (WP_015952737.1), T. paurometabola, Tsukamurella paurometabola (WP_013126227.1); S. cattleya, Streptomyces cattleya (WP_014144993.1); B. cereus, Bacillus cereus (WP_000811431.1); C. botulinum, Clostridium botulinum (WP_003385573.1). | |
3.3 Binding modes of the Ppant and Ppant–Gly prosthetic groups
To get a better insight into the observed exclusive binding preference of Bj Gly:CP ligase 1 towards holoCP over holoCP–Gly, we examined the interaction network formed between both Ppant and Ppant–Gly and their protein surrounding within the ligase active site. The important hydrogen-bonding interactions with key residues in the last 30 ns of MD trajectories are presented in Table 5, which suggest that the overall interaction network is more comprehensive and more persistent for Ppant than for Ppant–Gly (Fig. S3–S8, ESI†). Within 5 Å of the each atom of the Ppant prosthetic group in holoCP, we identified the five most dominant hydrogen bonding networks that promote the binding, where Ppant acts as a hydrogen bond acceptor to the side chains of Arg258, Gln232, and Tyr132 through its O1, O2 and O6 atoms, respectively, and as a hydrogen bond donor to the side chain of Asp215 through its N(2)–H group (Scheme 1, Fig. S8, ESI†). Inspection of the evolution of these hydrogen bonds along the trajectory (Fig. S3–S7, ESI†) reveals that, after the systems equilibrates, during the last 20–30 ns of the simulations all five of these interaction distances cluster below 2.5 Å in their donor–acceptor separation, which qualifies them as medium strong hydrogen bonds.50,51 Analogously, if we consider the same distances in the Ppant–Gly group of the holoCP–Gly form, these interacting partners exhibit significantly larger geometric separations and lower occupancies, which result in the diminished stabilization that, subsequently, works in the direction of dissociating the ACP with such prosthetic group from the ligase. It appears that the binding interface with Ppant is most dominated through interactions established by its anionic phosphate group, where both negatively charged oxygen atoms are stabilized by the Arg258 side chain, occupying on average 73% and 80% of snapshots in both subunits, the latter being reduced to 46% and 51% in Ppant–Gly (Table 5). The next in line are interactions with Asp215 and Tyr132 side chains which drop from 39% and 63% occupancies in holoCP, respectively, to 8% and 12% in holoCP–Gly, in the same order. Interestingly, we did not observe any stabilizing interactions originating from the hydrogen bonding exerted in the terminal –SH fragment of Ppant or, even surprisingly, in the analogous –S–C(O)–CH2–NH2 fragment of the glycated Ppant–Gly. This provides another evidence that glycation of the prosthetic group in holoCP–Gly does not stabilize the complex and promotes dissociation.
Table 5 Hydrogen bonding analysis between the ligase side-chains and the prosthetic group of the matching carrier protein within the ligase active site based on the last 30 ns of the MD trajectory
| Donor |
Acceptor |
Occupancy in holoCP (%) |
Occupancy in holoCP–Gly (%) |
| Residue |
Group |
Residue |
Group |
Subunit A |
Subunit B |
Subunit A |
Subunit B |
| Arg258 |
N–H2 |
Ppant
|
O1 |
74 |
72 |
25 |
66 |
| Arg258 |
N–H1 |
Ppant
|
O2 |
92 |
68 |
52 |
50 |
|
Ppant
|
N2–H |
Asp215 |
O1 |
10 |
58 |
0 |
16 |
| Tyr132 |
O–H |
Ppant
|
O6 |
41 |
85 |
0 |
23 |
|
Ppant
|
O4–H |
His260 |
N |
2 |
10 |
0 |
0 |
| Gln232 |
N–H |
Ppant
|
O2 |
60 |
0 |
1 |
15 |
| Gln229 |
N–H |
Ppant
|
O5 |
7 |
0 |
81 |
84 |
3.4 Contribution of prosthetic groups to the binding
In order to estimate the energy contribution of each form of the prosthetic group (Ppant or Ppant–Gly) to the overall protein–protein binding energies, we have modeled two additional complexes 4 and 5 (Table 1) by deleting protein parts of the corresponding carrier proteins in 2 and 3, and keeping the matching prosthetic group with the methyl group on its linker oxygen atom inside the ligase for additional 60 ns of MD simulations. It turned out (Table 2) that non-polar contribution (vdW + ESURF) is favorable but roughly the same for both prosthetic groups, while electrostatic contribution (EEL + EGB) is more affirmative for Ppant than for Ppant–Gly. Interestingly, the latter assume positive values, 4.3 and 17.7 kcal mol−1 on average for PPant and PPant–Gly, respectively, which strongly point to the overall non-polar hydrophobic nature of the ligase active site. However, the most interesting are the total ΔGbind, which are appreciably more favorable for PPant (−11.3 kcal mol−1 on average) than for PPant–Gly (−1.2 kcal mol−1 on average), thus mirroring the situation with the entire CPs. In the context of the total protein–protein binding affinities, the result for PPant suggests that a significant part of the ligase specificity towards holoCP originates from the stabilization of this form of the prosthetic group within its active site, which is a noteworthy outcome. However, this is not an exclusive component determining biological recognition, and one must not neglect differences in the protein part of each CP, as evident, for example, from a set of co-crystallization experiments involving Bj Gly:CP ligase 1 and coenzyme A, an in vivo precursor of the Ppant group, which resulted in the unproductive binding.52 All of this strongly indicates that the overall recognition between studied proteins is jointly affected by differences in the non-covalent interactions within binding areas and those in the ligase active site, and that each modification of carrier proteins, even in their prosthetic groups, results in distinct forms suitable for the specific binding with the cognate protein partner.
4. Conclusion
In this work we utilized molecular dynamics simulations, MM–GBSA binding free energy calculations, in silico alanine scanning mutagenesis, pKa analysis, and multiple sequence alignment to model the biological interactions of three forms of the aminoacyl carrier protein, namely holoCP, apoCP, and holoCP–Gly, with their partner protein Bj Gly:CP ligase 1. The results revealed that the ligase preferentially binds the holoCP form to both of its subunits with the binding free energies of −20.7 and −19.1 kcal mol−1. On the other hand, the apoCP form without the Ppant prosthetic group is also recognized by the ligase, ΔGbind assuming −9.2 and −3.6 kcal mol−1. The observed large difference between binding affinities of these two forms indicates the lack of competition between apo- and holo-forms for the ligase binding. After the prosthetic group is glycylated, the holoCP–Gly form dissociates from the complex towards its downstream partner protein, as Ppant–Gly is no longer engaged in interactions established between unmodified Ppant and its protein surrounding. This is in agreement with the observed endergonic binding free energies being 2.9 and 20.9 kcal mol−1. It is particularly interesting to see that such a small modification of the prosthetic group results in such a significant change in the binding affinities. Earlier experiments have shown that Bj Gly:CP ligase 1 does not aminoacylate the carrier protein prototype, holoACP of the fatty acid synthesis pathway (FAS II), despite the presence of the Ppant prosthetic group.53 Taken all together, this indicates that the attachment of the Ppant prosthetic group is an obligatory event that precedes the formation of the biologically active CP:Bj Gly:CP ligase 1 complex, but the prosthetic group per se is not the element which exclusively predetermines association. The latter was further revealed by analyzing complexes involving ligase and prosthetic groups alone, which confirmed more favorable binding affinities for Ppant than for Ppant–Gly. However, the productive protein–protein communication is also guided by the electrostatic complementarity manifested through the Arg220–Glu53 ion pair and the overall non-polar interplay of the interacting residues within the protein–protein binding interface. Evolutionary analysis revealed that CP recognition is species dependent and there are no specific residues that are universally conserved and that serve as the anchoring points for carrier protein binding across the whole ligase family. Therefore, each structural modification of the carrier protein results in its altered propensity towards one of the many heterologous protein partners and, moreover, prevents the false or nonproductive contact between numerous non-partner enzymes.
Acknowledgements
We would like to thank the Zagreb University Computing Centre (SRCE) for generously granting computational resources on the ISABELLA cluster (isabella.srce.hr) and the CRO–NGI infrastructure (http://www.cro-ngi.hr). A.M. wishes to thank the Croatian Science Foundation for a doctoral stipend through the Career Development Project for Young Researchers (Contract No. I-3376-2014). I.G.S. acknowledges support from the Croatian Science Foundation (Grant 09.01/293) and the European Community's Seventh Framework Programme (FP7RegPot/IntegraLife, 315997). R.V. gratefully acknowledges the European Commission for a Marie Curie FP7 Career Integration Grant (Contract No. PCIG12-GA-2012-334493).
References
- M. S. Anderson and C. R. Raetz, J. Biol. Chem., 1987, 267, 5159 Search PubMed
 .
.
- S. W. Jordan and J. E. Cronan, Jr., J. Biol. Chem., 1997, 272, 17903 CrossRef CAS PubMed .
- M. S. Anderson, C. E. Bulawa and C. R. Raetz, J. Biol. Chem., 1985, 260, 15536 CAS .
- A. L. Schaefer, D. L. Val, B. L. Hanzelka, J. E. Cronan, Jr. and E. P. Greenberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 9505 CrossRef CAS .
- J. P. Issartel, V. Koronakis and C. Hughes, Nature, 1991, 351, 759 CrossRef CAS PubMed .
- D. M. Byers and E. A. Meighen, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 6085 CrossRef CAS .
- A. Koglin and C. T. Walsh, Nat. Prod. Rep., 2009, 26, 987 RSC .
- R. S. Gokhale, R. Sankaranarayanan and D. Mohanty, Curr. Opin. Struct. Biol., 2007, 17, 736 CrossRef CAS PubMed .
- G. Butland, J. M. Peregrin-Alvarez, J. Li, W. Yang, X. Yang, V. Canadien, A. Starostine, D. Richards, B. Beattie, N. Krogan, M. Davey, J. Parkinson, J. Greenblatt and A. Emil, Nature, 2005, 433, 531 CrossRef CAS PubMed .
- T. A. Gould, H. P. Schweizer and M. E. Churchill, Mol. Microbiol., 2004, 53, 1135 CrossRef CAS PubMed .
- Y. M. Zhang, M. S. Rao, R. J. Heath, A. C. Price, A. J. Olson, C. O. Rock and S. W. White, J. Biol. Chem., 2001, 276, 8231 CrossRef CAS PubMed .
- Y. M. Zhang, B. Wu, J. Zheng and C. O. Rock, J. Biol. Chem., 2003, 278, 52935 CrossRef CAS PubMed .
- K. D. Parris, L. Lin, A. Tama, R. Mathewa, J. Hixonb, M. Stahla, C. C. Fritzb, J. Seehraa and W. S Somersa, Structure, 2000, 8, 883 CrossRef CAS PubMed .
- H. Gong, A. Murphy, C. R. McMaster and D. M. Byers, J. Biol. Chem., 2007, 282, 4494 CrossRef CAS PubMed .
- M. Močibob, N. Ivić, M. Luić and I. Weygand-Đurašević, Structure, 2013, 21, 614 CrossRef PubMed .
- J. Yang, R. Yan, A. Roy, D. Xu, J. Poisson and Y. Zhang, Nat. Methods, 2015, 12, 7 CrossRef CAS PubMed .
- A. Roy, A. Kucukural and Y. Zhang, Nat. Protoc., 2010, 5, 725 CrossRef CAS PubMed .
- Y. Zhang, BMC Bioinf., 2008, 9, 40 CrossRef PubMed .
-
D. A. Case, T. A. Darden, T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Merz, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Goetz, I. Kolossvai, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M.-J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, O. Farkas, J. J. Dannenberg, S. Dapprich, A. D. Daniels, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed .
- C. I. Bayly, P. Cieplak and P. A. Kollman, J. Phys. Chem., 1993, 97, 10269 CrossRef CAS .
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed .
- V. Hornak, R. Abel, A. Okur, B. Strockbine, A. Roitberg and C. Simmerling, Proteins: Struct., Funct., Bioinf., 2006, 65, 712 CrossRef CAS PubMed .
- A. W. Gotz, M. J. Williamson, D. Xu, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2012, 9, 1542 CrossRef PubMed .
- S. Le Grand, A. W. Gotz and R. C. Walker, Comput. Phys. Commun., 2013, 184, 374 CrossRef CAS .
- R. Salomon-Ferrer, A. W. Goetz, D. Poole, S. Le Grand and R. C. Walker, J. Chem. Theory Comput., 2013, 9, 3878 CrossRef CAS .
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, J. Comput. Phys., 1997, 23, 327 CrossRef .
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS .
- D. Sitkoff, K. A. Sharp and B. Honig, J. Phys. Chem., 1994, 98, 1978 CrossRef CAS .
- B. R. Miller III, T. D. McGee, Jr., J. M. Swails, N. Homeyer, H. Gohlke and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 3314 CrossRef .
- A. Onufriev, D. Bashford and D. A. Case, Proteins, 2004, 55, 383 CrossRef CAS PubMed .
- D. Bashford and D. A. Case, Annu. Rev. Phys. Chem., 2000, 51, 129 CrossRef CAS PubMed .
- I. Massova and P. A. Kollman, J. Am. Chem. Soc., 1999, 121, 8133 CrossRef CAS .
- S. Huo, I. Massova and P. A. Kollman, J. Comput. Chem., 2002, 23, 15 CrossRef CAS PubMed .
- T. Clackson and J. A. Wells, Science, 1995, 267, 383 CrossRef CAS PubMed .
- Y. Ofran and B. Rost, PLoS Comput. Biol., 2007, 3, e119 CrossRef PubMed .
- W. L. DeLano, Curr. Opin. Struct. Biol., 2002, 12, 14 CrossRef CAS PubMed .
- D. R. Roe and T. E. Cheatham, J. Chem. Theory Comput., 2013, 9, 3084 CrossRef CAS .
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33 CrossRef CAS PubMed .
- The PyMOL Molecular Graphics System, Version 1.2r3pre, Schrödinger, LLC.
-
P. J. Turner, Center for Coastal and Land-Margin Research Oregon Graduate Institute of Science and Technology Beaverton, Oregon .
- T. Hou, J. Wang, Y. Li and W. Wang, J. Chem. Inf. Model., 2011, 51, 69 CrossRef CAS PubMed .
- G. Rastelli, A. D. Rio, G. Degliesposti and M. Sgobba, J. Comput. Chem., 2010, 31, 797–810 CAS .
- H. Sun, Y. Li, S. Tian, L. Xu and T. Hou, Phys. Chem. Chem. Phys., 2014, 16, 16719 RSC .
- X. Kong, P. Pan, D. Li, S. Tian, Y. Li and T. Hou, Phys. Chem. Chem. Phys., 2015, 17, 6098 RSC .
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef CAS .
- P. J. Kundrotas and E. Alexov, Biophys. J., 2006, 9, 1724 CrossRef PubMed .
- M. H. M. Olsson, C. R. Sondergard, M. Rostkowski and J. H. Jensen, J. Chem. Theory Comput., 2010, 7, 525 CrossRef .
- H. W. van Vlijmen, M. Schaefer and M. Karplus, Proteins, 1998, 33, 145 CrossRef CAS .
- P. Gilli, L. Pretto, V. Bertolasi and G. Gilli, Acc. Chem. Res., 2009, 42, 33 CrossRef CAS PubMed .
- C. Laurence and M. Berthelot, Perspect. Drug Discovery Des., 2000, 18, 39 CrossRef CAS .
- M. Močibob, N. Ivić, D. Šubašić, M. Luić and I. Weygand-Đurašević, Croat. Chem. Acta, 2011, 84, 149 CrossRef .
- M. Močibob, N. Ivić, S. Bilokapić, T. Maier, M. Luić, N. Ban and I. Weygand-Đurašević, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14585 CrossRef PubMed .
Footnote |
| † Electronic supplementary information (ESI) available: The time evolution of the protein–protein RMSD values and selected hydrogen bond donor–acceptor distances along the MD trajectories, in silico alanine scanning energy contributions, pKa snapshot analysis of the selected residues. See DOI: 10.1039/c5cp03066h |
|
| This journal is © the Owner Societies 2015 |

 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *a
*a