
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Doped golden fullerene cages†
Francesca
Baletto
*a and
Riccardo
Ferrando
b
aPhysics Department, King's College London, Strand, WC2R 2LS, London, UK. E-mail: francesca.baletto@kcl.ac.uk
bPhysics Department, Univ. of Genova, Italy
First published on 15th April 2015
Abstract
A first-principles investigation of the effect of the doping of golden cages of 32 atoms is proposed. It is shown that Ag and Cu doping affects the geometrical stability of the icosahedral fullerene Au32 cage, where Ag-doping leads to a new, low symmetric, and prolate motif while Cu-doping leads to a lump, incomplete decahedral shape. Most significantly, the HOMO–LUMO gap depends strongly on the cluster geometry while its dependence on the cluster chemical composition seems to be weaker.
1 Introduction
As building blocks in many and various nanotechnological applications, metallic nanoparticles and nanoalloys are still continuing to receive significant attention. Among all the transition metals, gold is attracting extensive interest due to its unexpected catalytic properties combined with its plasmonic nature. The electronic properties of small nanoobjects depend on their geometry, which can differ remarkably from bulk structures because nanoparticles can break translational symmetry, thus allowing the stabilization of non-crystalline morphologies,1 including low symmetry structures. At small sizes, the minimization of the surface energy contribution together with the electronic shell closure are the major driving forces in the geometrical stabilization of pure metals. Thus, the prediction of the icosahedral “golden” fullerene Au32, the first all-gold fullerenic species,2 followed by Au42,3,4 Au505 and Au72,6 and other cagelike structures,7 came surprisingly and spurred considerable attention. Au32 shows a hollow structure with the same icosahedral symmetry as the C60 Buckminsterfullerene – from here the name – but where the Au atoms are at the centre of 5- and 6-rings instead. The remarkable stability of the golden fullerene Au32 has been explained in terms of its spherical aromaticity, as 32 electrons in the 6s level fulfill Hirsh's criterion for σ and π molecular orbitals.8 The first experimental evidence of metallic hollow cages came quickly after the theoretical prediction using photoelectron spectroscopy9,10 although at near room temperature it has been shown that Au32 preferentially adopts a compact motif due to thermal vibrations.11–14 Hollow cages have been predicted also for Au clusters adsorbed on MgO(100).15,16The surge of interest in hollow nanostructures, together with the wide range of possibilities offered by nanoalloys with respect to elemental clusters,17 has led recently to the study of bimetallic cages.18–21 However, these studies are often limited to the investigation of a single metal atom encapsulated into a golden cage,22–27 whereas a systematic study of the stability of golden cages after doping with other metallic species is still missing.
In this paper, we perform density functional theory (DFT) calculations to systematically study the effects of changes in composition on the Au32 fullerene, by doping it with silver or copper atoms. We observe that Ag-doping leads to the formation of a new low symmetry motif, always in very close competition with the fullerene – within 0.1–0.2 eV – for all compositions. On the other hand, doping by 50 at% of Cu leads to the preferential formation of compact shapes with decahedral symmetry. The dependence of the HOMO–LUMO (HL) gap on the geometry and on the doping is also discussed. On that regard, we have shown that the gap depends little on the chemical composition, while there is a huge difference between lump/compact and hollow/open geometries, with the latter having a gap at least 0.6 eV wider.
AuAg and AuCu have been chosen because they may be expected to show different behaviours, as follows from examining their bulk phase diagrams. In bulk crystals, AuAg forms solid solutions in the full composition range, while in AuCu three ordered phases are formed (Au0.5Cu0.5 (fcc, L10), Au0.25Cu0.75, and Au0.75Cu0.25 (fcc, L12)).28 In AuAg, Ag segregation is expected in bulk crystal surfaces, due to the lower surface energy of Ag. However, this trend can somewhat change in small clusters because charge transfer effects can lead Au atoms to occupy low-coordination sites, such as vertexes.29,30 In AuCu, some preference for Au surface segregation follows already both from the size effect – which drives the larger atom to the cluster surface – and from surface energy considerations. Recently, atomistic calculations confirm this trend.31,32 In both AuAg and AuCu, a high degree of intermixing is expected in the interior of nanoparticles.
2 Computational method
The energy stability of pure Au, Ag and Cu nanoparticles and AuAg, AuCu nanoalloys has been estimated throughout ab initio simulations using the Quantum Espresso package,33 a density functional theory based plane wave code. In these first-principles simulations, the exchange–correlation (XC) potential is described self-consistently within the generalized gradient approximation (GGA) throughout the Perdew–Burke–Ernzerhof (PBE). The Rabe–Rappe–Kaxiras–Joannopoulos ultrasoft pseudopotential has been used to model valence electron–nuclei interactions. The energy cut-off for the plane wave basis set has been set to 40 Ry with a charge density cut-off of 360 Ry. The Au electronic configuration considered is [Xe]4f145d106s1, featuring 5s and 5p semi-core states that strongly overlap the valence 5d orbitals. All the calculations have been performed at the Γ point only in a sufficient large simulation cubic box of at least 30 bohr. To improve convergence a Gaussian smearing has been introduced and the equivalent electronic temperature is 2 mRy. This has been checked to give the same qualitative and quantitative results at a lower electronic temperature of 0.5 mRy for pure Au clusters. Further, it was shown that an electronic temperature of 2 mRy is low enough for capturing even magnetic properties of small Pt and Pt-doped nanoclusters.34,35 In all cases, a DFT local optimisation, with no symmetry constraints, has been performed at the first-principles level within the Broyden–Fletcher–Goldfarb–Shann (BFGS) procedure, until the atomic forces have been converged to better than 4 meV Å−1.In order to estimate the HOMO–LUMO band gap (HL gap) a ΔSCF method has been used. This technique allows us to calculate the ionization potential (IP) and the electron affinity (EA) as the difference of two ground state configurations, one corresponding to the neutral system and the other obtained eliminating one electron from, or adding one to it (for IP and EA, respectively). For the treatment of the charged systems, where a positive uniform background is added, a Markov–Payne correction36 has been applied in order to improve the convergence with the cubic box size. The ΔSCF method has been shown to be particularly accurate for small and finite systems as the correlation effects on quasi-particle energy levels are generally captured by charge relaxation following the addition or the removal of an electron.37 Very recently, Baroni and coworkers38 have used the ΔSCF method within the Quantum Espresso distribution to calculate the IP and EA values of pure Au clusters in the size range between 2 and 32 atoms and the same authors have shown that the approach gives a similar trend with respect to the most accurate GW calculations and experiments. Here, our calculations are aimed at determining whether there is any clear effect of the doping on the HL gap.
3 Results and discussion
3.1 Structures of pure clusters
The structures that have been locally relaxed by DFT have been chosen in the following way. We have considered pure clusters first and we have generated a wide database of compact structures by means of basin hopping optimization runs in which the interaction was modelled by an empirical atomistic potential derived within the second-moment tight-binding approximation.39 This has allowed us to single out the two significant lump geometries, depicted in the top row of Fig. 1.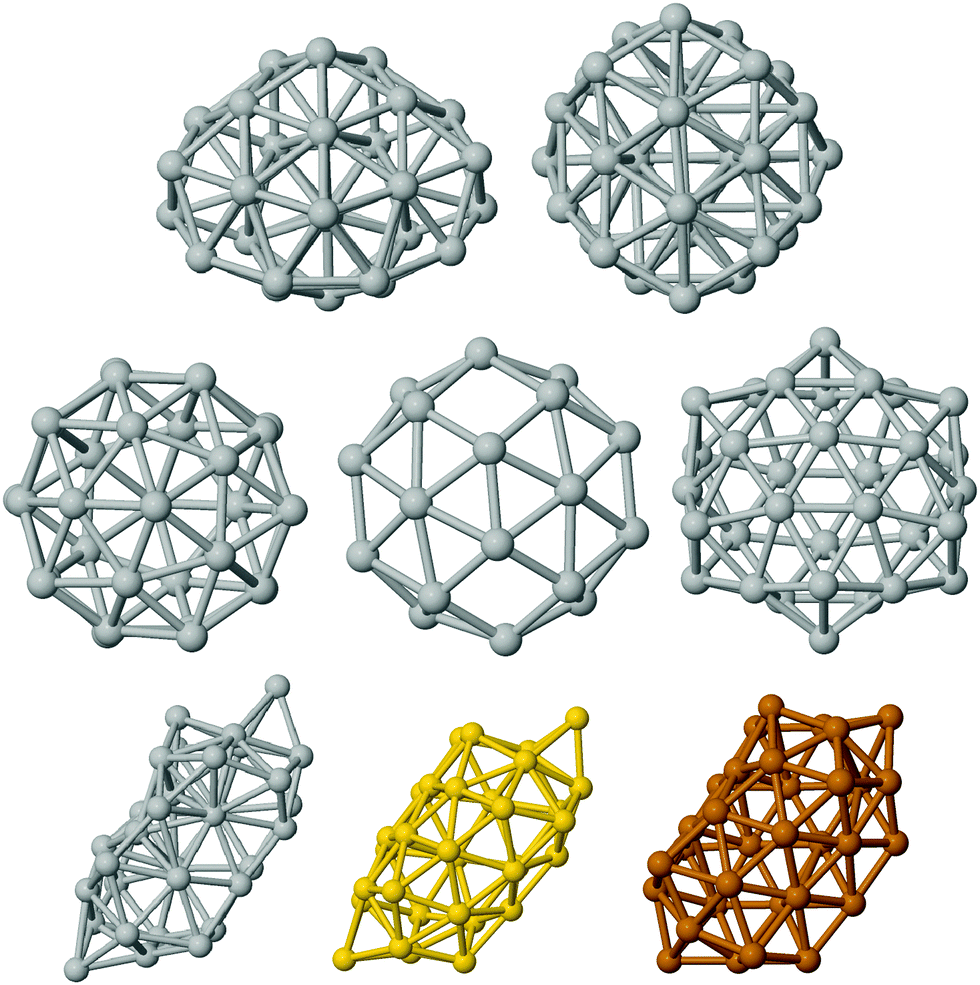 | ||
| Fig. 1 Different structures of 32 atoms for pure noble metals. Upper and central rows refer to silver. At the top fulfilled and compact isomers, the incomplete decahedron (hDh), and a poly-icosahedron (polyIh). Central row: hollow isomers, an empty pancake – poly-icosahedron – of 38 atoms, an empty truncated octahedron of 38 atoms, and the icosahedral fullerenic cage. Bottom row: the new low symmetry tubular, named worm, for silver, gold and copper. For Ag and Cu-“Worm” the atoms with more than 6 nearest neighbours are highlighted by black arrows. | ||
The first is a fragment of the Marks decahedron of 75 atoms (half-Dh, hDh), whereas the second is a poly-icosahedral arrangement. Hollow structures of pure clusters have been obtained by eliminating inner atoms from compact structures at larger sizes extracted from our structural database.15 This has allowed us to single out three significant cage motifs which are reported in the mid panel of Fig. 1. From left to right, we report the cage obtained by emptying a poly-icosahedral (polyIh) structure of 38 atoms, the cage obtained by emptying a truncated octahedron of 38 atoms, and the cage corresponding to the external shell of the anti-Mackay icosahedron of 45 atoms. The latter cage has icosahedral symmetry and it is usually called golden fullerene. In the bottom row of Fig. 1 we report the last open geometry. This is a new, low-symmetry shape, referred to as worm shape in the following, which has been obtained by the ionic relaxation of an empty decahedral shape. As Ag, Au and Cu worms have shown slightly different relaxations, thus they are all reported in the bottom row of Fig. 1. The longest length is about 13.1 Å, 13.2 Å, and 11.3 Å in Ag, Au and Cu, respectively. The empty region has a diameter of about 4.5 Å, 5 Å, and 5.5 Å in Ag, Cu, and Au, respectively. The number of nearest neighbour (NN) pairs varies depending on the metal, from 100 in Ag, to 97 in Cu, and to 92 in Au. This should be compared with the number of nearest neighbour pairs in a fullerene cage, which is 90, and in a compact poly-icosahedral shape, which is 114. The coordinates of the new worm shape for the pure metals are reported in the ESI.†
We have analysed the worm structure by the common-neighbour analysis.40 The CNA analysis gives a signature of three integer numbers (r,s,t) for each NN pair: r is the number of common nearest neighbors of the atoms of the pair, s is the number of nearest-neighbour bonds among these common nearest neighbours, and t is the length of the longest chain which can be formed with these common bonds. The CNA reveals that the worm structure has only surface signatures (in prevalence (200) and (300) signatures, which characterize (100) and (111) facets, respectively). In the case of silver, there is one atom that cannot be classified as a surface atom, leading to a small percentage of (433) CNA signatures. Anyway, there is still an empty region in the Ag worm, which thus preserves its hollow and elongated nature. In addition, this is the only silver isomer with a maximum pair distance longer than 10 Å and a minimum distance of 4.5 Å between atoms at the opposite sites with respect to the central axis. This is highlighted in Fig. 2 where the pair distribution function is plotted for four Ag isomers.
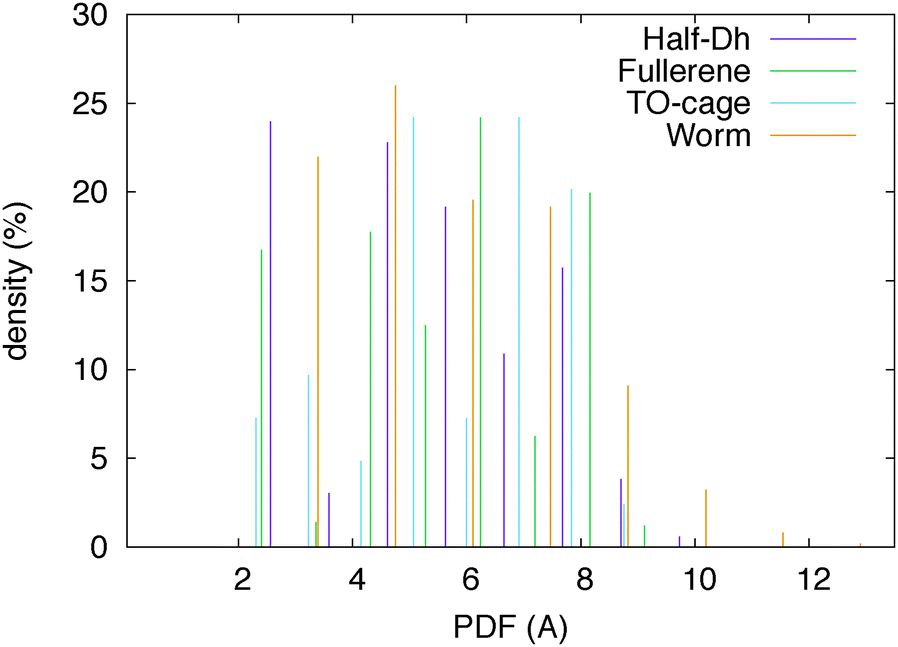 | ||
| Fig. 2 Pair distribution function of four Ag32 isomers. The worm shape is hollow and elongated in one direction. | ||
In agreement with other DFT studies, the best Au isomer is the icosahedral fullerene cage. This structure corresponds to the external shell of the anti-Mackay icosahedron of 45 atoms.41 The second isomer is the tubular worm at 0.6 eV above. All the other geometries – including the empty truncated octahedron and the empty pancake – are at least at 0.8 eV above. The two lump shapes result to be slightly distorted after the ionic relaxation, loosing their symmetry and they are considerably higher in energy above 1.8 eV. It should be noted that the relative stability of the ionic forms is slightly different compared to the neutral case, where the cationic worm is almost degenerate to the fullerene Au32+. But the anionic fullerene is still 0.2 eV better than the negatively charged worm. It should be noted that the third isomer for Au32− is the empty pancake being higher by 0.4 eV. Further, all but the incomplete Dh are within 0.9 eV.
Not surprisingly, the lowest energy isomer for Cu32 is found to have a compact decahedral shape, obtained cutting asymmetrically a larger Marks decahedron of 75 atoms in such a way that the 5-fold symmetry axis is displaced towards the surface. This isomer is in close competition with another lump poly-icosahedral configuration, just a few hundredths of eV above. On the other hand, any hollow geometry is lying at least 2 eV above – for the fullerene, while the empty TO and the empty pancake are at 3.5 and 3.3 eV, respectively. Charging the system does not change the overall picture, where hollow geometries remain very unfavourable. On the other hand, one should notice that Cu32− prefers to adopt a polyIh shape instead of an incomplete Dh, but the Cu32+ hDh gain is more stable than the polyIh by roughly 0.4 eV.
Ag32 shows a very interesting behaviour, indeed. First of all, all the considered structural motifs are within 1.46 eV. Four shapes are so close in energy as less as 0.25 eV. Among them, the new low symmetry tubular shape, named worm, has been identified. The worm and the incomplete Dh are isoenergetic within a few meV. The third isomer is the poly-icosahedral geometry at just 0.04 eV and the fullerene cage is at 0.24 eV. The worst two isomers, lying at 1.46 and 1.33 eV, are the empty pancake and the empty truncated octahedron, respectively.
It should be noted that Ag32+ has a different energy ladder, where the positively charged worm is now at 0.55 eV while the polyIh is at 0.4 eV above the incomplete Dh. The fullerene Ag32+ becomes really unfavorable being higher 1 eV than the best isomer. As observed for copper, Ag32− as a preference for the polyIh, followed by the worm-Ag32−. The incomplete Dh negatively charged is the third isomer at 0.35 eV above and followed by the fullerene at 0.6 eV. The charged empty cages are truly unfavourable from an energetic point of view.
3.2 Nanoalloy structures
Let us now describe the results obtained for the binary clusters. Starting from the geometries of pure clusters, we have generated binary nanoalloys by substituting atoms of different species one-by-one in many different configurations, in order to select the most stable ones. We note that our exploration of the possible chemical patterns and structures might be not exhaustive, but it is at least a thorough exploration of some significant low-energy motifs, for binary clusters. For the Au-containing nanoalloys, we have selected four of the seven motifs of Fig. 1: the fullerene cage, the worm, the incomplete decahedron and the poly-icosahedron (polyIh). As discussed before, those shapes result to be the best or the second isomer for the pure cases. The empty shapes obtained by a truncated octahedron and the poly-icosahedral pancake of 38 atoms are strongly unfavourable for the pure clusters, so that they are not considered further. Regarding the chemical ordering adopted by nanoalloys, we opt for a comparison between fully segregated and fully mixed chemical patterns, avoiding in that way a systematic search of the best chemical pattern for all the chemical compositions. As widely shown in the literature, Au atoms have a tendency to stay at the surface, and ideally at vertex sites, while smaller Cu atoms prefer to occupy core sites.31,32 The bulk phase diagram of AuCu shows three ordered phases at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3, 1:1, and 3:1 compositions, while AuAg presents solid solutions for all compositions. For a very small amount of doping as one, two and four Cu or Ag atoms, we have done a quite systematic search which confirms that trend with a preference for Ag atoms to form many mixed bonds.
:3, 1:1, and 3:1 compositions, while AuAg presents solid solutions for all compositions. For a very small amount of doping as one, two and four Cu or Ag atoms, we have done a quite systematic search which confirms that trend with a preference for Ag atoms to form many mixed bonds.
The structural relaxation of AuAg and AuCu cages has been summarised in Fig. 3 where the atomic binding energy,
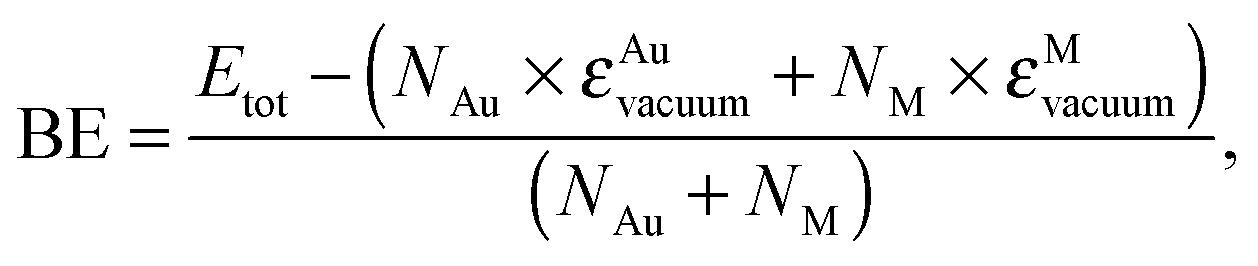 | (1) |
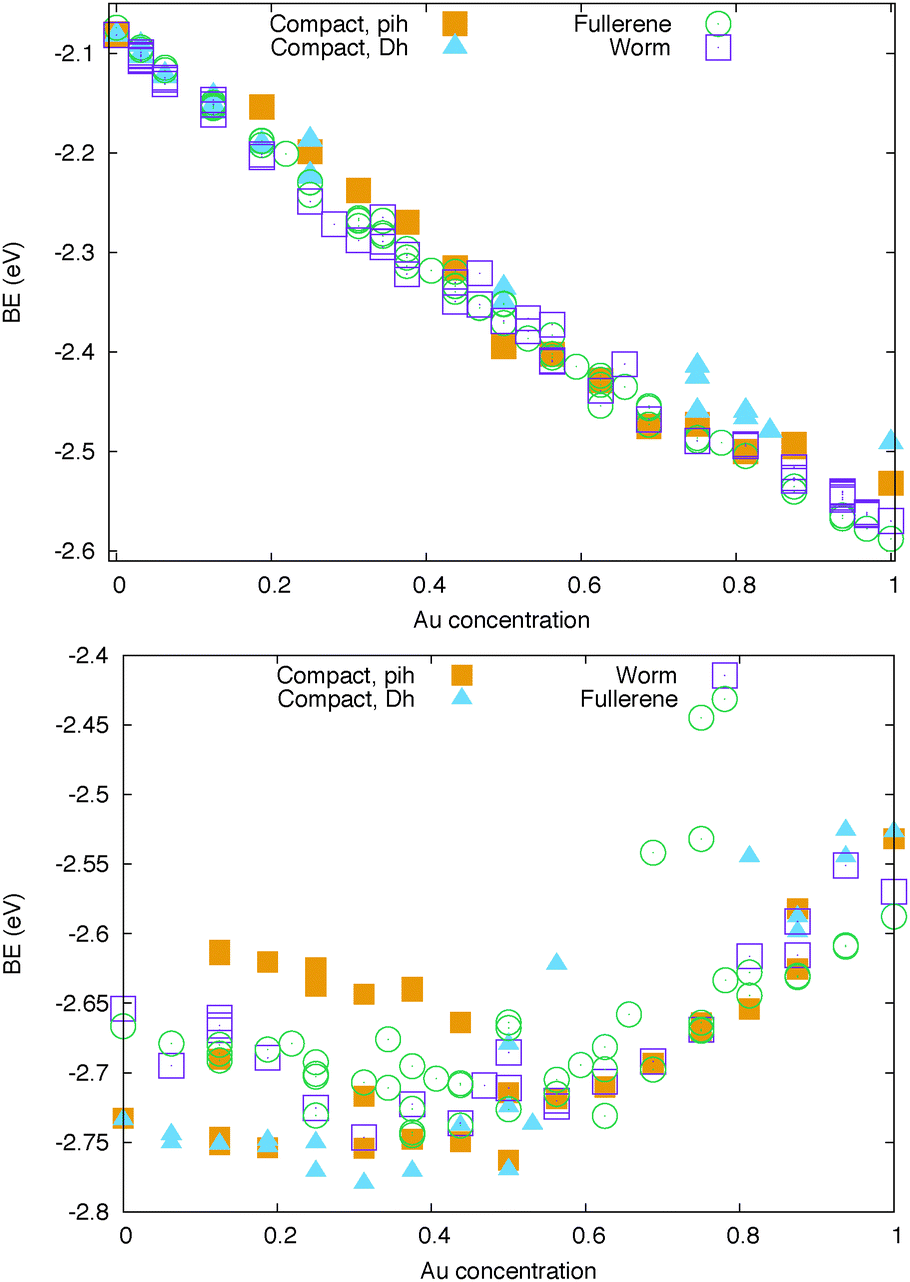 | ||
| Fig. 3 Binding energy per atom (eV) of Au-based nanocages at 32 atoms for Ag (top) and Cu (bottom) doping. Empty symbols stand for hollow geometries, while full symbols represent compact shapes. | ||
On the other hand, the specific chemical ordering pattern makes a difference for AuAg clusters. The BE shows a clear linear dependence on the Au-concentration. Ag-doped fullerene cages prefer to adopt the worm shape, with the exception of very high Au concentrations, as 80% and above. Anyway, at all concentrations, the fullerene cage and the worm shape are in very close competition and, thus, different chemical ordering patterns determine the lowest energy isomer corresponding to the different compositions. The best chemical ordering in AuAg is the one that maximizes the number of mixed pairs, as already suggested by Lopez and coworkers in ref. 43 for clusters of 13 atoms. Whenever it is possible, the best isomer is characterised by a complete intermixing while all segregated patterns, such as the Janus-like, are strongly unfavorable. At very low Au concentration, gold atoms prefer to occupy vertex positions while at low Ag concentration, silver atoms prefer to form only one Ag–Ag bond and to lie on the same facet. In hollow shapes, this tendency to intermixing often leads to an Au-line decoration pattern. For example, the best chemical pattern in the Au12Ag20 fullerene cage is the one where Au atoms formed a zig-zag line – resulting in only 48 mixed nearest neighbour pairs – depicted in Fig. 4, while the full mixing – characterised by 60 mixed pairs – lies 0.27 eV above.
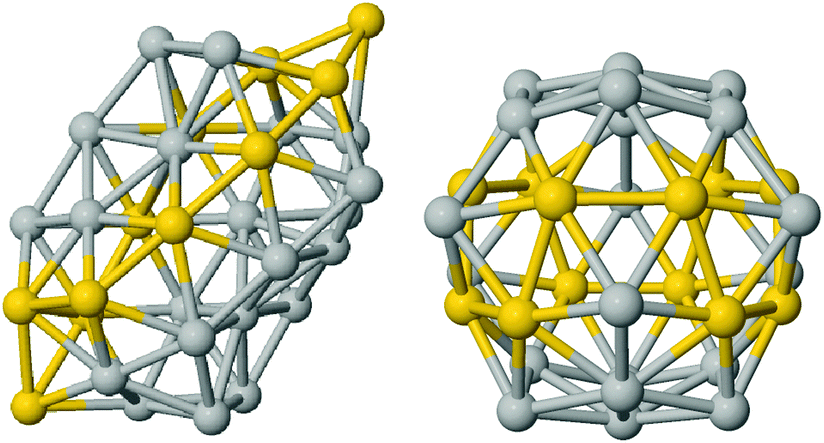 | ||
| Fig. 4 Golden line decoration on silver cages, worm (left) and fullerene (right). Silver are in grey and gold in yellow. | ||
The linear decoration pattern of Au seems however to be present only in cages that have a real empty space inside, see Fig. 4. This follows from considering the Ag tetrahedron of 20 atoms, which is a structure with no internal atoms, but without a notable empty inner space. In this Ag20 tetrahedron, we have substituted 4 Ag atoms with Au atoms, finding that placing the Au atoms at vertices is more favourable than placing them on a line on the same edge, by 0.27 eV. About the anionic and cationic AuAg32, we should notice that the Au24Ag8 icosahedral cage becomes unfavorable with respect to the worm or the polyIh by 0.3 eV.
3.3 Electronic structure properties
As noble metal clusters may play a role as plasmonic objects, their optical properties are of primary importance. Here we analyze the behaviour of the HL gap. The energy gap between the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) is a significant parameter in terms of the electronic stability and for optical properties of metallic clusters. We have applied a ΔSCF method to calculate the HL-gap, which is reported in Fig. 5, for the pure cases and for different chemical compositions, namely gold at 25%, 50% and 75%. Using the ΔSCF method within standard DFT calculations, we found that the HL-gap of pure clusters is considerably large for hollow symmetric cages, as the empty truncated octahedron and the fullerene cage, as high as 3.9–4.1 eV. This is in agreement with recent GW calculations proposed by Umari and coworkers.38 There, they have shown that the quasi-particle gap is 4.5 eV for Au20 and 3.9 eV for Au32.38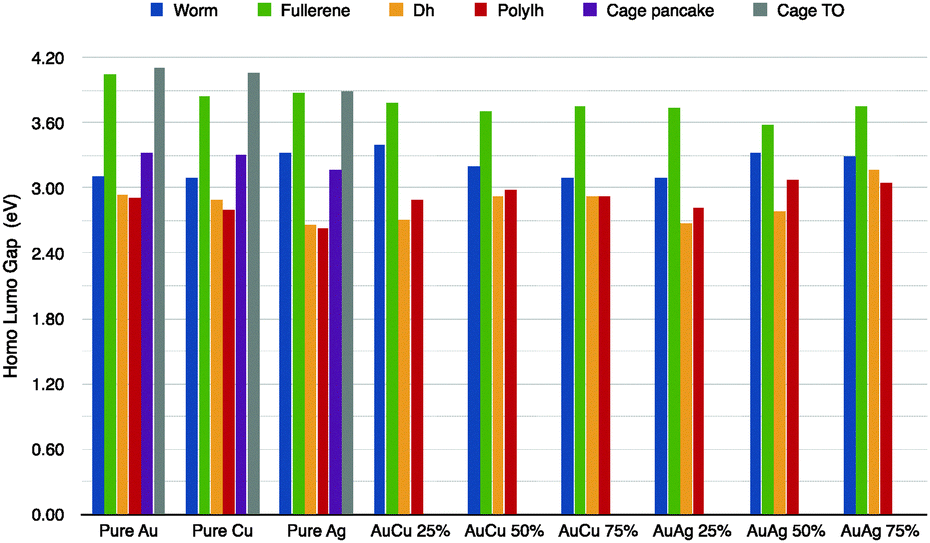 | ||
| Fig. 5 HL gap in eV calculated using the ΔSCF method for different geometries and for pure and mixed systems. The first three group of the histrogram refers to the pure case. The 4th to 6th group to the AuCu and the last to AgAu. In the nanoalloys, the Au concentration increases from 25%, to 50% and then 75%. Different colours refer to the various geometries considered. Hollow cages: worm (blue), fullerene (green), empty pancake (purple), and empty TO (grey). Lump shapes: hDh (yellow) and polyIh (red). | ||
In the golden fullerene, a large band gap should be expected due to its spherical aromaticity (2(N + 1)2) as discussed in ref. 44. A low symmetry cage as the “worm” shows a HL-gap of 3.1–3.3 eV, while lump, compact shapes the gap is from 2.6 to 2.9 eV. In the case of nanoalloys, we have limited our analysis to AuCu and AuAg clusters with an Au concentration of 25%, 50% and 75% and we have considered only the best chemical pattern for each geometrical motifs. We should notice that doping has a very little effect on the gap, while it depends strongly on the geometry, where open motifs have a consistently wider gap than the decahedral or poly-icosahedral ones. A doped Au–fullerene cage shows a gap between 3.6–3.8 eV for both AuCu and AuAg. The worm gap is slightly more affected by the doping and the gap is between 3.1 and 3.7 eV. On the other hand, incomplete Dh has a 2.7–3.1 eV gap.
Further, we would like to discuss the values of the first ionization potential, as reported in Table 1 for the elemental noble metal clusters and in Fig. 6 for the binary cases. The bulk values are 7.7 eV, 7.6 eV and 9.2 eV, respectively, for Cu, Ag and Au. In the case of pure metals, it is clear that the IP is dramatically lower than the corresponding bulk value. The ionization potential depends little on the geometrical shape, with a minimum for the hDh, 5.2 for Ag, 5.4 eV for Cu and 6.5 for Au, and a peak for the truncated octahedral cage, 6.1 for Ag, 6.4 for Cu 7.3 eV for Au. For the nanoalloys, the first IP increases as the gold concentration increases, with a maximum for the fullerenic cage – between 6.4–6.7 eV – and a minimum for the incomplete decahedron at 5.6 eV for both Au8Cu24 and Au8Ag24. As far as we can see, the specific chemical ordering pattern should affect little the optical properties of these small nanoalloys, in qualitative agreement with time-dependent DFT calculations.45
| Worm | Fullerene | hDh | polyIh | |
|---|---|---|---|---|
| Pure Au | ||||
| IP | 6.48 | 7.02 | 6.52 | 6.80 |
| EA | 3.37 | 2.98 | 3.58 | 3.89 |
| Pure Cu | ||||
| IP | 5.74 | 6.30 | 5.43 | 5.77 |
| EA | 2.65 | 2.45 | 2.54 | 2.97 |
| Pure Ag | ||||
| IP | 5.74 | 6.03 | 5.19 | 5.54 |
| EA | 2.41 | 2.15 | 2.53 | 2.91 |
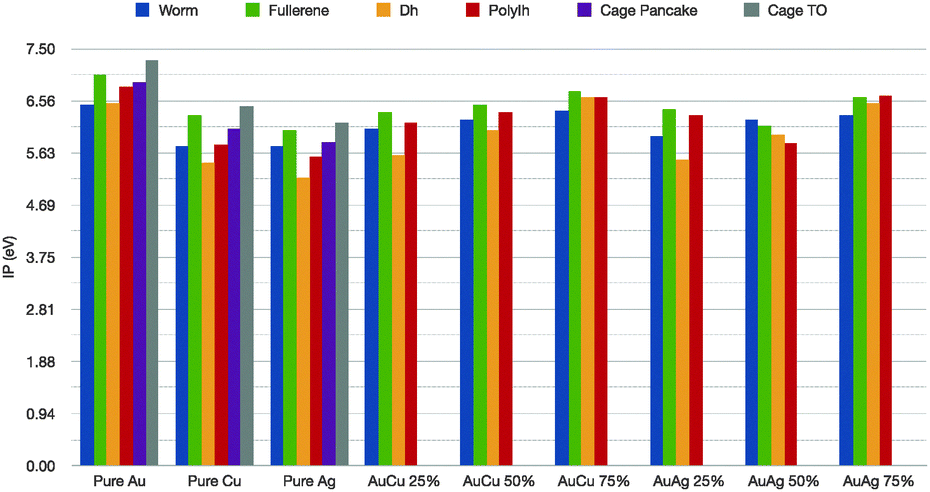 | ||
| Fig. 6 First ionization potential in eV for pure and mixed clusters, colour scheme as in Fig. 5. | ||
4 Conclusions
A new and low symmetry hollow/worm structure has been found by means of density-functional-theory simulations for pure Ag and AuAg at 32 atoms. At all compositions, the worm and the fullerene cages are in close competition. Generally speaking, fully intermixed structures are preferred, however, often gold forms line decoration of the silver cage. We have shown that a rich-Au composition AuCu fullerene is still the best isomer, but as one moves towards Cu-rich compositions the lump decahedral shape is energetically more favourable for that alloy with an Au-rich shell and copper inside.We have shown that the HL gap seems to depend prevalently on the geometry more than on the chemical composition. These results are somehow expected taking into account the Jellium model. Recently, it has been shown that this should be independent of the chemical composition.46 We hope that our work stimulate further theoretical investigations and experiments, because those hollow bimetallic isomers with a large gap might be considered as a new state of matter, and classified as super-cages.
Acknowledgements
The authors acknowledge support from the Cost Action MP0903 NANOALLOY. FB thanks the financial support by UK research council EPSRC, under Grants No. EP/GO03146/1 and No. EP/J010812/1 and the Faculty and Departmental computational facilities at King's College London managed by A. Comisso.References
- F. Baletto and R. Ferrando, Rev. Mod. Phys., 2005, 371, 77 Search PubMed.
- P. Johansson, D. Sundholm and J. Vaara, Angew. Chem., 2004, 43, 2678 CrossRef PubMed.
- Y. Gao and X. Zeng, J. Am. Chem. Soc., 2005, 127, 3698 CrossRef CAS PubMed.
- H. Ning, et al. , J. Phys. Chem. Solids, 2014, 75, 696 CrossRef CAS PubMed.
- D. Tian, et al. , J. Phys. Chem. A, 2007, 111, 411 CrossRef CAS PubMed.
- A. Karttunen, et al. , Chem. Commun., 2008, 465 RSC.
- X. Gu, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 205401 CrossRef.
- A. Hirsch, Z. Chen and H. Jiao, Angew. Chem., Int. Ed., 2001, 40, 2834 CrossRef CAS.
- M. Ji, et al. , Angew. Chem., 2005, 44, 7119 CrossRef CAS PubMed.
- S. Bulusu, et al. , Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326 CrossRef CAS PubMed.
- H. De, et al. , Catal. Today, 2012, 198, 106 CrossRef CAS PubMed.
- D. Tian, et al. , Comput. Mater. Sci., 2011, 50, 2359 CrossRef CAS PubMed.
- N. Shao, et al. , J. Am. Chem. Soc., 2010, 132, 6596 CrossRef CAS PubMed.
- N. Shao, et al. , J. Phys. Chem. C, 2014, 118, 6887 CAS.
- R. Ferrando, et al. , Phys. Rev. Lett., 2009, 102, 216102 CrossRef CAS.
- R. Ferrando, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 045418 CrossRef.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS PubMed.
- Priyanka and K. Dharamvir, Phys. Chem. Chem. Phys., 2013, 15, 12340 RSC.
- R. Fournier and S. Afzal-Hussain, J. Chem. Phys., 2013, 138, 054303 CrossRef PubMed.
- M. Zhang, et al. , Chin. Phys. B, 2012, 21, 056102 CrossRef.
- Q. Deng, et al. , Comput. Mater. Sci., 2011, 976, 183 CAS.
- D. Manna, et al. , J. Phys. Chem. C, 2013, 117, 18777 CAS.
- Z. Lee, et al. , Nanoscale, 2013, 5, 9620 RSC.
- Y. Pei, et al. , ACS Nano, 2011, 5, 1441 CrossRef CAS PubMed.
- Y. Gao, et al. , Sci. Rep., 2014, 4, 5886 Search PubMed.
- H.-W. Yang, et al. , J. Phys. Chem. A, 2013, 117, 2672 CrossRef CAS PubMed.
- H.-F. Li, et al. , Phys. Chem. Chem. Phys., 2014, 16, 244 RSC.
- R. Hultgren, P. D. Desai, D. T. Hawkins, M. Gleiser and K. K. Kelley, Values of the Thermodynamic Properties of Binary Alloys, American Society for Metals, Berkeley, 1981 Search PubMed.
- F. Y. Chen and R. L. Johnston, Acta Mater., 2008, 56, 2374 CrossRef CAS PubMed.
- M. Cerbelaud, et al. , Phys. Chem. Chem. Phys., 2011, 13, 10232 RSC.
- S. Darby, et al. , J. Chem. Phys., 2002, 116, 1536 CrossRef CAS PubMed.
- T. J. Toai, G. Rossi and R. Ferrando, Faraday Discuss., 2008, 138, 49 RSC.
- P. Giannozzi, et al. , J. Phys.: Condens. Matter, 2009, 21, 395502 CrossRef PubMed.
- C. Di Paola and F. Baletto, Proc. for CMMSE XI, 2011, p. 451.
- C. Di Paola and F. Baletto, Eur. Phys. J. D, 2013, 67, 49 CrossRef.
- G. Makov and M. C. Payne, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 51, 4014 CrossRef CAS.
- L. A. Curtiss, P. C. Redfern, K. Raghavachari and J. A. Pople, J. Chem. Phys., 1998, 109, 42 CrossRef CAS PubMed.
- J. Xian, S. Baroni and P. Umari, J. Chem. Phys., 2014, 140, 124101 CrossRef PubMed.
- G. Rossi and R. Ferrando, Chem. Phys. Lett., 2006, 17 CrossRef CAS PubMed.
- F. Baletto, C. Mottet and R. Ferrando, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 155408 CrossRef.
- R. Ferrando, J. Phys.: Condens. Matter, 2015, 27, 013003 CrossRef PubMed.
- D. D. C. Rodrigues, et al. , Chem. Phys., 2008, 340, 91 CrossRef PubMed.
- X. Lopez-Lozano, et al. , J. Phys. Chem. C, 2013, 117, 3602 Search PubMed.
- J. Wang, et al. , J. Phys. Chem. A, 2005, 109, 9265 CrossRef CAS PubMed.
- G. Barcaro, et al. , J. Phys. Chem. C, 2011, 115, 24085 CAS.
- D. Cricchio, et al. , Phys. Rev. A: At., Mol., Opt. Phys., 2012, 86, 013201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cp01061f |
| This journal is © the Owner Societies 2015 |