
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ PM-IRRAS of a glassy carbon electrode/deep eutectic solvent interface
Luciana
Vieira
ab,
Robert
Schennach
c and
Bernhard
Gollas
*ab
aCEST Competence Centre for Electrochemical Surface Technology GmbH, Viktor-Kaplan-Strasse 2, 2700 Wiener Neustadt, Austria
bInstitute for Chemistry and Technology of Materials, Graz University of Technology, Stremayrgasse 9, 8010 Graz, Austria. E-mail: bernhard.gollas@tugraz.at; Tel: +43 316 87332338
cInstitute of Solid State Physics, Graz University of Technology, Petersgasse 16/2, 8010 Graz, Austria
First published on 16th April 2015
Abstract
The interface of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar choline chloride/ethylene glycol deep eutectic solvent with a glassy carbon electrode has been investigated by polarization modulation reflection-absorption spectroscopy (PM-IRRAS). Temporal spectral changes at open circuit potential show the experiments to be surface sensitive and indicate slow adsorption of electrolyte molecules on the electrode surface. In situ spectroelectrochemical PM-IRRAS measurements reveal characteristic potential-dependent changes of band intensities and wavenumber-shifts in the surface spectra. The potential dependent spectral changes are discussed in terms of adsorption, reduction, desorption and reorientation of choline cations at the interface. Analogies are drawn to the ionic layer structure proposed for the architecture of electrode/ionic liquid interfaces. The results show that in situ PM-IRRAS is generally applicable to glassy carbon electrodes and to electrode interfaces with deep eutectic solvents.
:2 molar choline chloride/ethylene glycol deep eutectic solvent with a glassy carbon electrode has been investigated by polarization modulation reflection-absorption spectroscopy (PM-IRRAS). Temporal spectral changes at open circuit potential show the experiments to be surface sensitive and indicate slow adsorption of electrolyte molecules on the electrode surface. In situ spectroelectrochemical PM-IRRAS measurements reveal characteristic potential-dependent changes of band intensities and wavenumber-shifts in the surface spectra. The potential dependent spectral changes are discussed in terms of adsorption, reduction, desorption and reorientation of choline cations at the interface. Analogies are drawn to the ionic layer structure proposed for the architecture of electrode/ionic liquid interfaces. The results show that in situ PM-IRRAS is generally applicable to glassy carbon electrodes and to electrode interfaces with deep eutectic solvents.
1. Introduction
Deep eutectic solvents (DESs) consist of a mixture of a quaternary ammonium halide (NR4+X−) with a hydrogen bond donor (RZ; Z = CONH2, COOH, OH).1 These compounds have many of the physical–chemical properties of classical room temperature ionic liquids (RTILs), such as negligible vapour pressure, good ionic conductivity and thermal stability.2 Moreover, they are air- and water-stable, easy to prepare and low in cost, which make them an attractive alternative to some highly reactive RTILs.3One of the most attractive characteristics of RTILs and DESs is their wide electrochemical window, which can span more than 4 Volts and allows the electrodeposition of metals that cannot be electroplated from aqueous solutions.4,5 However, the electrochemical processes taking place at the electrode/RTIL interface are quite different from those in aqueous solutions.6 The Gouy–Chapman–Stern model of the electric double layer (EDL) for metal interfaces with aqueous electrolyte solutions7 cannot be directly transferred to RTILs. Mainly because the former has been formulated to explain diluted electrolytes, in which the ions are treated as point charges, whereas RTILs consist of neat ionic fluids.8,9
In situ measurements such as X-ray reflectometry10 and atomic force microscopy11 have been used to investigate the near-surface structure of the electrode/RTIL interface. Multi-layers of alternating ions with well ordered compact layers of RTILs have been proposed to explain such interfaces.12 Consequently, mass transport and charge transfer kinetics of electrode reactions can be significantly affected leading to unexpected electrochemical outcomes.13–15
Several surface sensitive spectroscopic techniques have been used to investigate RTIL/electrode interfaces, including sum frequency generation (SFG),16–19 surface-enhanced Raman spectroscopy (SERS),20,21 surface-enhanced infrared absorption spectroscopy (SEIRAS),22,23 subtractive normalized interfacial Fourier transform infrared spectroscopy (SNIFTIRS)24 and infrared reflection absorption spectroscopy (IRRAS).25,26 Spectroscopic and theoretical investigations on electrified electrode/ionic liquid interfaces suggest that the electrode surface acts as an electronic and structural template, where cations adsorb at potentials negative of the potential of zero charge (PZC) and that at potentials positive of the PZC the cations are replaced by anions.6,27–29 However, this potential-dependent re-arrangement of cations and anions is not that simple and adsorption–desorption hysteresis19,22 as well as ultra-slow restructuring30,31 have also been reported.
Infrared reflection absorption spectroscopy (IRRAS or RAIRS) is a powerful technique to interrogate metal–liquid interfaces at a molecular level.32,33 However, difficulties may arise from the strong IR absorption of the electrolyte in external reflection geometry, which usually masks the IR signal of the surface species. Due to the difficulty of acquiring a background spectrum of the solution under exactly the same conditions (for a solid–liquid interface), a background correction can be made by either potential or polarization modulation.34,35
In potential modulation, the background is an IRRAS spectrum recorded at a given reference potential. This technique has been used for the spectroelectrochemical investigation of the ionic liquid [EMIM][BF4] (1-ethyl-3-methylimidazolium tetrafluoroborate) at charged gold electrodes.24 Variation of the band intensity of anions and cations in the IR spectra with the applied potential indicated an overall change of the interfacial species. Moreover, this technique also allowed to predict the orientation of the EMIM+ cations at the surface, which allegedly had a vertical orientation towards the gold surface.
IRRAS has already been used to study ionic liquid/metal interfaces, namely [EMIM] [Tf2N] (1-ethyl-3-methylimidazolium bis-(trifluoromethylsulfonyl)imide) on Ag.25,26 The temperature effect was investigated and the thin-film IRRAS spectra were compared to those of the bulk liquids.26 Differences for the IR bands of bulk and thin film spectra arising from the substrate–IL interactions at open circuit potential were reported.25
Polarization modulation is based on the surface selection rule,36 which state that the p-polarized radiation is absorbed by surface and liquid phase species, whereas the s-polarized light is only absorbed by the species in the liquid phase. Thus, the s-spectrum can be used as a background.37 The difference between the p-and s-polarized spectra divided by the sum of the two yields an IR spectrum of the surface species. This technique is called polarization modulation infrared reflection absorption spectroscopy (PM-IRRAS).
PM-IRRAS is a distinguished technique to study adsorption38 and has been successfully employed to study the molecular structure of self-assembled monolayers, biomimetic membranes and Langmuir Blodgett films at charged metal electrodes by Lipkowski's group.39–46 To date, however, no PM-IRRAS study of electrode/ionic liquid interfaces has been reported in the literature.
We have extensively studied the electrochemistry of choline chloride (ChCl)/ethylene glycol (EG) deep eutectic electrolytes containing ZnCl2, mainly at glassy carbon (GC) electrodes.47,48 Unusual voltammetric behaviour such as cathodic peaks for Zn electrodeposition in the reverse anodic scan, have been observed for this DES47–49 on glassy carbon and also in other RTILs.50,51 Strong adsorption of the electrolyte and potential-dependent blocking of the active sites of the electrode surface may account for these effects.47 Here, we present a PM-IRRAS study of the adsorption and potential dependent changes of ChCl/EG on a glassy carbon electrode. This is the first report on in situ PM-IRRAS of an electrode/DES interface and of in situ PM-IRRAS on glassy carbon electrodes.
2. Experimental
Materials
Ethylene glycol (99.8%, anhydrous, Sigma Aldrich) was used as received. Solutions of 1:2 molar ratio of ChCl and EG, referred to as 12CE, were prepared as described elsewhere.47 The electrolyte was added to the spectroelectrochemical (SEC) cell under dry N2 atmosphere.
ATR-FTIR
Infrared spectra of the bulk electrolyte were measured on a Bruker Alpha FTIR spectrometer equipped with an ALPHAs platinum attenuated total reflectance (ATR) single reflection diamond. The spectra were recorded in the ATR module with a spectral range between 4000 and 400 cm−1, averaged 200 times at a resolution of 4 cm−1.Spectroelectrochemical cell
A PM-IRRAS cell suitable for work with ionic liquids was designed and constructed in house (Fig. 1). The cell was custom-made of stainless steel and can be sealed with o-rings. Ports with polytetrafluoroethylene (PTFE) tubes connected to 3-port valves permit gas purging, filling with electrolyte (3 mL) or complete sealing of the cell. The working electrode consisted of a 14 mm diameter glassy carbon disk inserted into a threaded shroud, which was used to accurately control the distance between the electrode and the zinc selenide optical window via a micrometer screw. The zinc selenide window is sealed with an o-ring and a polyether ether ketone (PEEK) cell body, which also serves as the electrolyte compartment. Zinc wires were used as quasi-reference and counter electrodes to make these measurements better comparable to previous studies.47,48 They were connected via insulated wires and an external pin connector to the potentiostat. The temperature of the cell could be controlled internally by a ceramic heating element. The experiments described here were all performed at room temperature.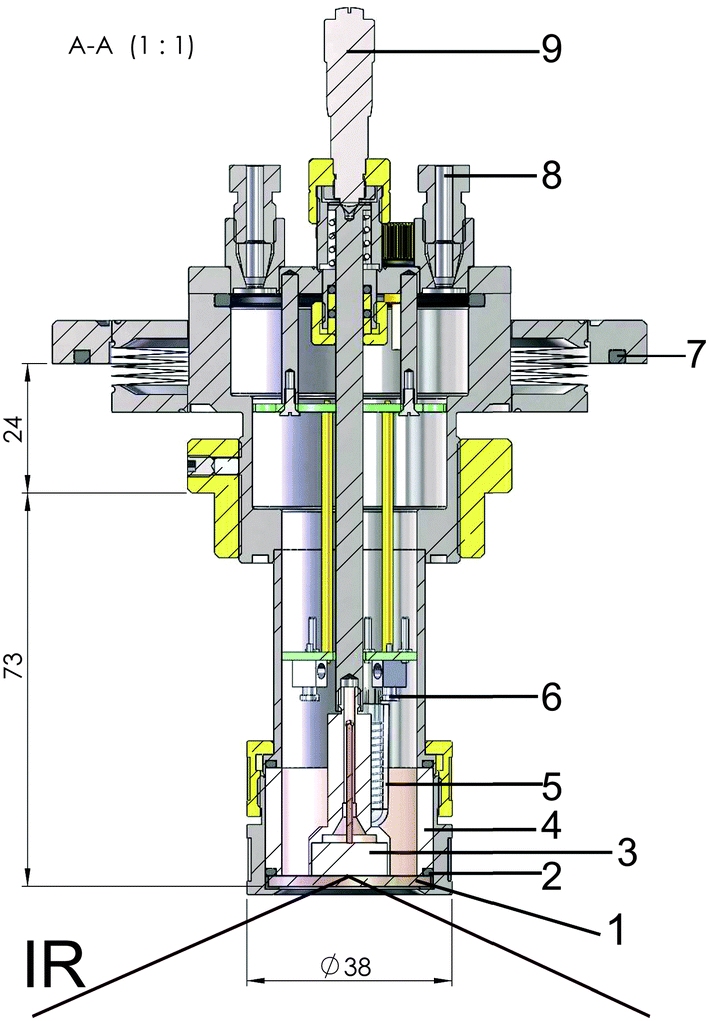 | ||
| Fig. 1 Sketch of the spectroelectrochemical cell designed for PM-IRRAS experiments with ionic liquids. (1) ZnSe window, (2) o-ring, (3) GC working electrode, (4) PEEK cell body (5) thermo element, (6) contact for a counter electrode and quasi reference electrode wires, (7) o-ring for sealing of the spectrometer, (8) hollow steel cell body with threaded inlet and outlet ports (PTFE tubes) for connection to gas flow and electrolyte filling, (9) micrometer screw. | ||
In situ PM-IRRAS spectroelectrochemistry
Electrochemical in situ experiments were carried out by hooking up the spectroelectrochemical cell to a Metrohm Eco Chemie Autolab PGSTAT100 potentiostat/galvanostat, controlled by NOVA 1.06 software.Chronoamperometry and simultaneous PM-IRRAS were synchronized manually. Electrochemical experiments were performed in external reflection geometry (10 μm thick layer of electrolyte). An uncompensated solution resistance (iR drop) of 190 Ω was estimated using the current interrupt and the positive feedback tools the NOVA software of the potentiostat. The electrochemical experiments were performed with an automatic iR drop compensation.
PM-IRRAS measurements were carried out with a Bruker IFS66 vacuum Fourier transform infrared spectrometer. The light beam was incident at 70° with respect to the surface normal onto the sample and the light was detected by a liquid nitrogen-cooled mercury cadmium telluride detector. Two wire-grid polarizers were placed in the incident beam light path, one mounted onto a rotatable mount and the other on a motor mount controlled by Opus Spectroscopy software (Bruker). A script was written to rotate the polarizer through 90° and to collect the spectrum sequentially with s- and p-polarized radiation. The intensity of the glow bar is constant over several hours. In addition, the geometry of the two polarisers ensures a constant intensity in s and p. A detailed description of the method has been published by Schennach et al.37 All spectra were collected at 4 cm−1 spectral resolution using 200 scans and are presented without smoothing.
The PM-IRRAS geometry was checked with respect to the optical constants of the ZnSe window and the electrolyte used, in order to avoid total reflection. The refractive index (η) of ZnSe and 12CE are 2.63235 and 1.4652,53 respectively. According to Snell's Law, total reflection at the ZnSe/12CE interface would take place at incident angles above 33.69°, probing only the ZnSe/12CE interface. Hence, the measurements were carried out with an incident angle at the vacuum/ZnSe interface of 70°, which results in an incident angle of 21° at the ZnSe/12CE interface and an incident angle of 40° at the 12CE/GC interface giving maximum intensity. This incident angle with respect to the normal on the GC surface clearly is not ideal, but could not be further increased due to restrictions of the reflection unit/cell geometry in combination with the high refractive index of 12CE.
For interpretation of the IR results the surface selection rule was used.36 This rule is valid for all conducting samples. Nevertheless, a surface selection rule for non-conducting samples is also known.54 Additionally, the electrode must be highly reflective to the IR radiation. Although this would be better achieved with metals such as gold, suitable IR reflectivity has also been reported for glassy carbon.33
For each measurement, a p- and a s-polarized spectrum has been obtained. The ratio of the difference over the sum of the two spectra, calculated according to eqn (1), results in the PM-IRRAS spectrum, which yields information about the species on the surface.37 It is important to remark that within the electrolyte layer the intensity of s- and p-polarized light might not be the same. Therefore, the vibrations of species in the bulk electrolyte might not cancel out completely. For this reason, we have additionally normalized the spectra with respect to the initial spectrum at t1 for time dependent and to the spectrum at E1 for potential dependent measurements. As shown in the time-resolved measurements below, however, we have clear evidence that mainly surface-near species are detected.
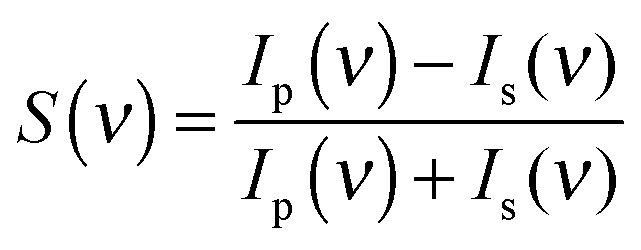 | (1) |
3. Results and discussion
FTIR characterization of the DES
The compounds of interest were first investigated by ATR-FTIR spectroscopy (Fig. 2) in order to compare and understand the characteristics of the vibrations of 12CE on a GC surface.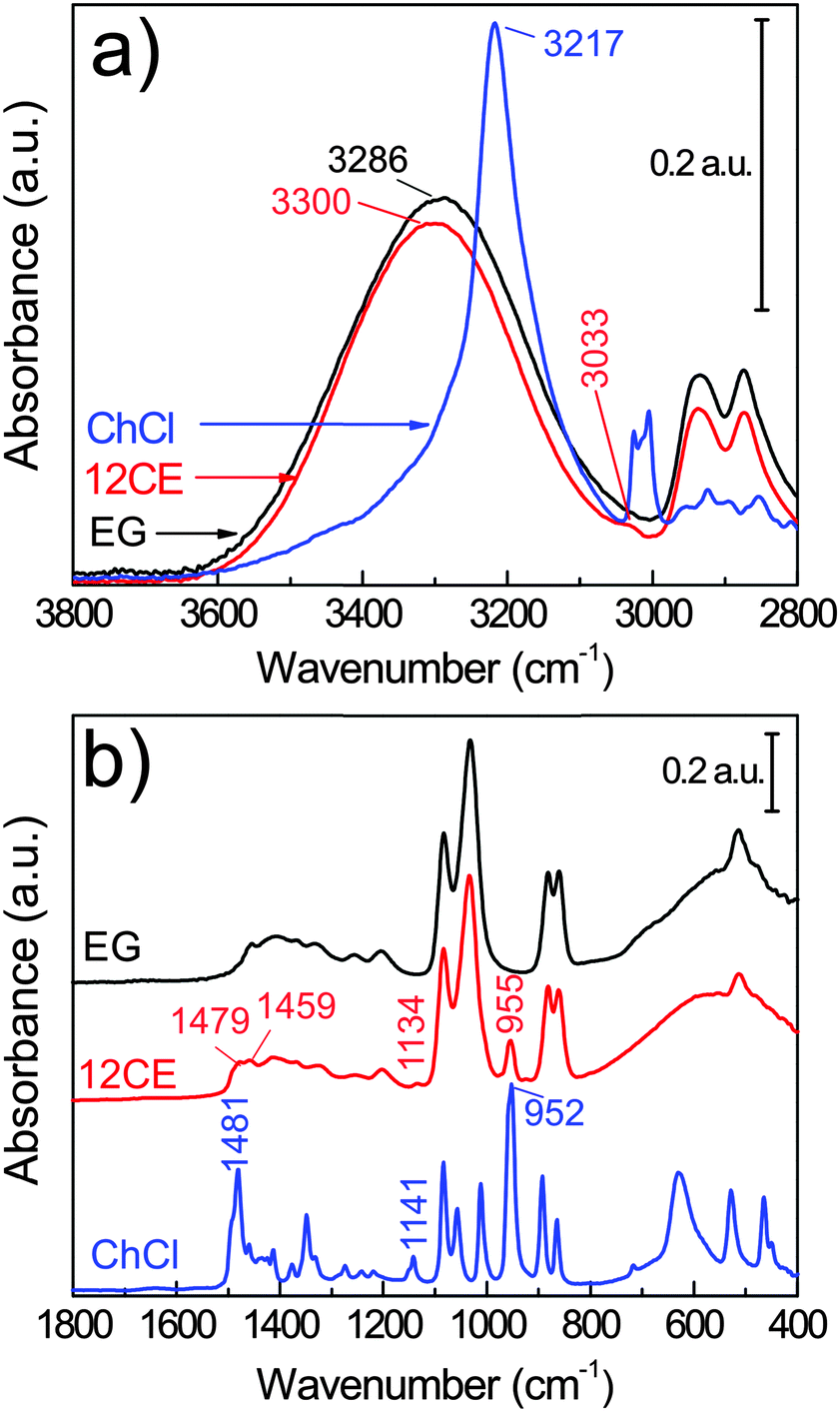 | ||
| Fig. 2 Bulk FTIR spectra of choline chloride (ChCl), ethylene glycol (EG) and their eutectic 1:2 molar mixture 12CE. | ||
The vibrational spectra of ethylene glycol55,56 and of choline chloride57,58 are well described in the literature. However, there are only a few spectroscopic investigations on the interaction of these two components in a deep eutectic solvent.59,60
Deep eutectic solvents are formed due to the strong interaction between a hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA). The alcohol moiety of the HBD, ethylene glycol, delocalizes the charge of the HBA choline chloride through interactions with the chloride ion,61 according to Scheme 1. However, this illustration is insufficient to explain the various types of H-bond interactions in 12CE.62 The spectrum of 12CE is dominated by ethylene glycol, which shows a very broad band at 3286 cm−1 for ν(O–H) due to extensive intermolecular OH(EG)–O(EG) bonding. The OH(EG)–Cl− interaction upon mixing of 2 mole of EG with 1 mole of ChCl results in a decrease of the extent of OH(EG)–O(EG) bonding. This is reflected in the spectrum by a slight decrease of band intensity and band width as well as a small shift to 3300 cm−1 (Fig. 2a). The broad nature of these bands makes unambiguous assignments difficult, but it has been suggested that the OH stretching region reflects the presence of multiple types of hydrogen bonds in the form of OH(EG)–Cl−, OH(Ch+)–Cl− and OH(EG)–O(EG),62 similar to observations reported for DESs formed by combination of choline chloride with urea58 and lactic acid63 as HBDs.
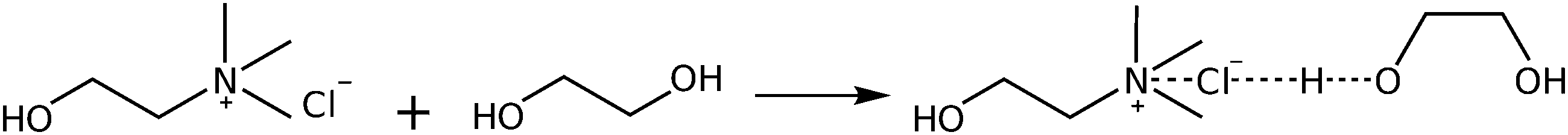 | ||
| Scheme 1 Interaction of choline chloride (HBA) and ethylene glycol (HBD) to form the 12CE DES. | ||
The OH stretching mode associated with the choline chloride OH(Ch+)–Cl− hydrogen bond at 3217 cm−1 seems to have disappeared. Nevertheless, characteristic absorptions originating from the (CH3)3N+ group of ChCl are observed at 3033 and 1477 cm−1.64 The bands at 955 and 923 cm−1 (too small to be visible in Fig. 2) are attributed to the asymmetric and symmetric stretching modes of C–N, respectively.65
PM-IRRAS characterization of 12CE on glassy carbon
PM-IRRAS spectra of a glassy carbon surface immersed into ethylene glycol and 12CE electrolyte are presented in Fig. 3. In the PM-IRRAS spectrum of EG significant differences compared to the bulk-IR can be observed. In the wavenumber region of the O–H stretching mode (3600–3200 cm−1) we observe two overlapping bands with two maxima at 3430 and 3260 cm−1. In the FTIR of the bulk electrolyte, only one band is observed, at 3286 cm−1. In this measurement, the absorption by species with a dipole moment perpendicular to the surface is intensified and thus the presence of two overlapping bands may originate from the vibrational modes of two different OH groups, one adsorbed on the electrode surface and a second at the other end of the EG molecule.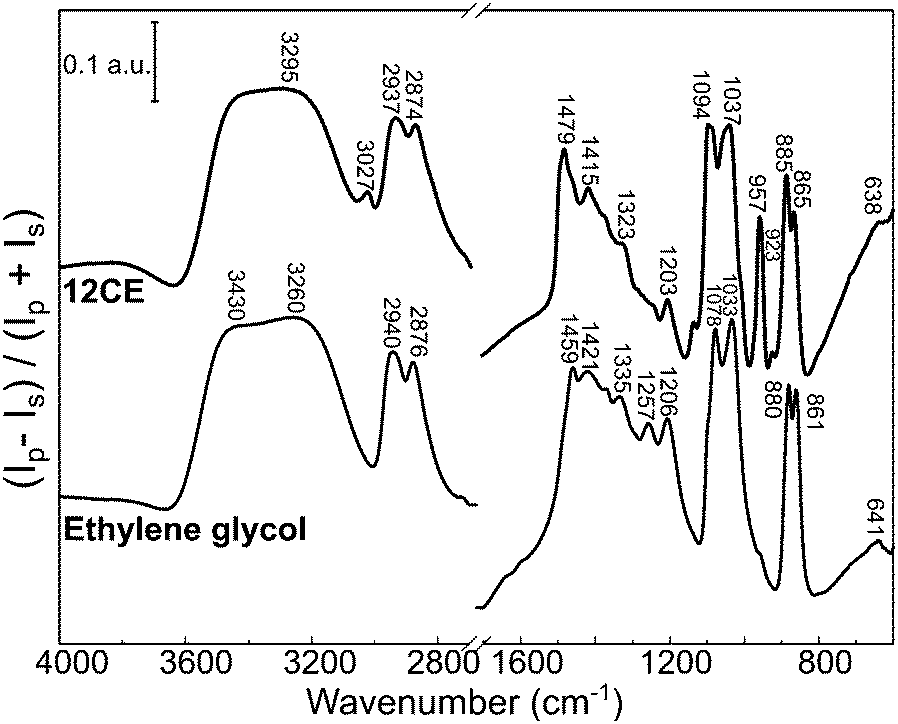 | ||
| Fig. 3 PM-IRRAS surface spectrum of the deep eutectic solvent 12CE and ethylene glycol on glassy carbon. | ||
Similar changes in the vibrational spectrum of ethylene glycol caused by adsorption on a metal surface have been reported for EG on platinum,66 palladium,67 rhodium68 and silver69,70 surfaces using high resolution electron energy loss spectroscopy (HREELS). It was alleged that ethylene glycol interacts with Pt, Pd and Rh through both oxygen atoms (Fig. 4b).66–68 Monodentate –OCH2CH2OH species have been observed by in situ IRRAS studies of EG on Mo(110)71 and Rh(100)72 surfaces. Monodentate species would allow adjacent O–H groups to interact with each other giving rise to a second ν(O–H) band at lower frequency.71 A similar statement can be made for the νs(C–O) and the νas(C–O) mode observed at 1083 cm−1 and at 1032 cm−1 respectively in the bulk. If EG was adsorbed exclusively in a bidentate orientation on the surface, the latter mode should not be observed, because it would be effectively screened out by the image dipole of the conducting surface.71 However, in the surface spectra of both EG and 12CE bands in both wavenumber regions are observed (Fig. 3). This means that a significant fraction of the species are adsorbed in a monodentate orientation giving rise to the νs(C–O) and the νas(C–O) modes. In this orientation the latter mode is allowed. Our results thus indicate that EG has its C–C bond axis predominantly tilted vertically to the glassy carbon surface (Fig. 4a).
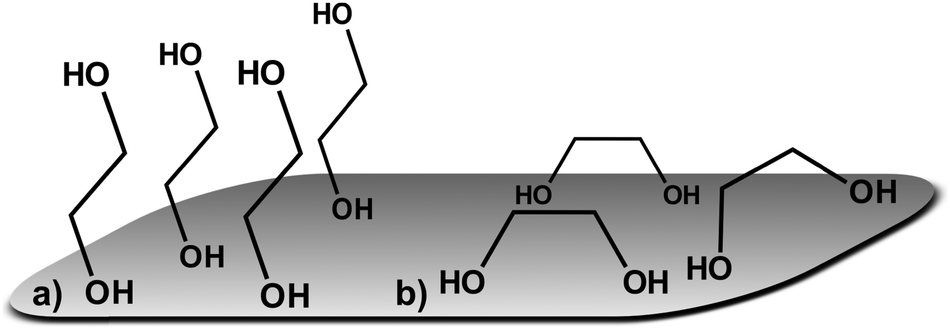 | ||
| Fig. 4 Possible orientations for ethylene glycol on glassy carbon: (a) monodentate and (b) bidentate. | ||
The spectrum of 12CE shows a very intense absorption for OH at 3295 cm−1. It also seems to be composed of two overlapping bands, similarly to the spectrum of pure EG. The bands associated with the ammonium function are sharper and more intense compared to the spectrum of the bulk 12CE. The asymmetrical bending of (CH3)3N+ at 1479 cm−1 and the asymmetrical stretching of C–N at 957 cm−1 show a significantly stronger absorption on GC. In none of the spectra of the two liquids a band associated with water at around 1650 cm−1 was observed, which confirms the water content to be very low.
A comparison of the vibrational modes of EG and 12CE in the bulk and on a GC surface is shown in Table 1. The bands were assigned according to the literature for EG and ChCl.55–57 It should be noted that most of the C–O vibrations as well as the C–N modes of ChCl show a significant shift or/and increase in intensity when measured on the GC substrate. The presence of choline chloride results in competition for active sites on the surface, which is evident by the increase of the absorption band intensities related to this group in the PM-IRRAS spectrum. The higher intensity of the ammonium group in the surface spectrum of 12CE, compared to the bulk IR spectrum, suggests that the choline molecules adsorb through this functional group on the glassy carbon surface at open circuit potential.
| EG | EG on GC | 12CE | 12CE on GC | Assignment |
|---|---|---|---|---|
|
a Frequencies in cm−1.
b ν = stretching, δ = bending; γ = wagging; τ = twisting; ρ = rocking, as = asymmetric; s = symmetric.
|
||||
| 3430 | 3290 | 3295 | ν(OH) | |
| 3286 | 3260 | |||
| 2935 | 2940 | 2936 | 2937 | ν as(C–H) |
| 2874 | 2876 | 2872 | 2874 | ν s(C–H) |
| 1477 | 1479 | δ as[N+(CH3)3] | ||
| 1454 | 1459 | 1459 | δ(CH2) | |
| 1408 | 1421 | 1413 | 1415 | δ(C–O–H) |
| 1368 | 1370 | 1369 | 1367 | γ(CH2) |
| 1334 | 1334 | 1325 | 1323 | γ(CH2) |
| 1256 | 1257 | 1253 | τ(CH2) | |
| 1204 | 1206 | 1202 | 1203 | τ(CH2) |
| 1134 | 1133 | C–N | ||
| 1083 | 1078 | 1083 | 1083 | ν s(C–O) |
| 1032 | 1033 | 1032 | 1036 | ν as(C–O) |
| 955 | 953 | ν as[C–N+(CH3)3] | ||
| 925 | 923 | ν[O–CCN] | ||
| 881 | 880 | 881 | 882 | ρ(CH2) |
| 859 | 861 | 861 | 862 | ν(C–C) |
| 641 | 638 | τ(C–CO) | ||
| 512 | 512 | γ(C–CO) | ||
Time-resolved measurements
PM-IRRAS spectra of a glassy carbon electrode at open circuit potential (OCP) in EG and 12CE were recorded as a function of time at regular intervals. The measurement was started immediately after filling of the cell with electrolyte and the subsequent approach of the working electrode to the ZnSe window. The temporal evolution of band intensities clearly demonstrates the sensitivity of the experiment for the glassy carbon/12CE interface. No temporal changes are observed in control experiments of the ZnSe/12CE interface. The initial time zero (t0) indicates the first contact of the electrolyte with the GC electrode and the first measurement started 5 minutes later at time t1. The spectra here are presented as the ratio of the spectral intensity at sampling time tx over that at the first measurement at t1. Fig. 5a and b show the surface spectra of ethylene glycol as a function of time, monitored from t1 = 5 minutes up to 300 minutes.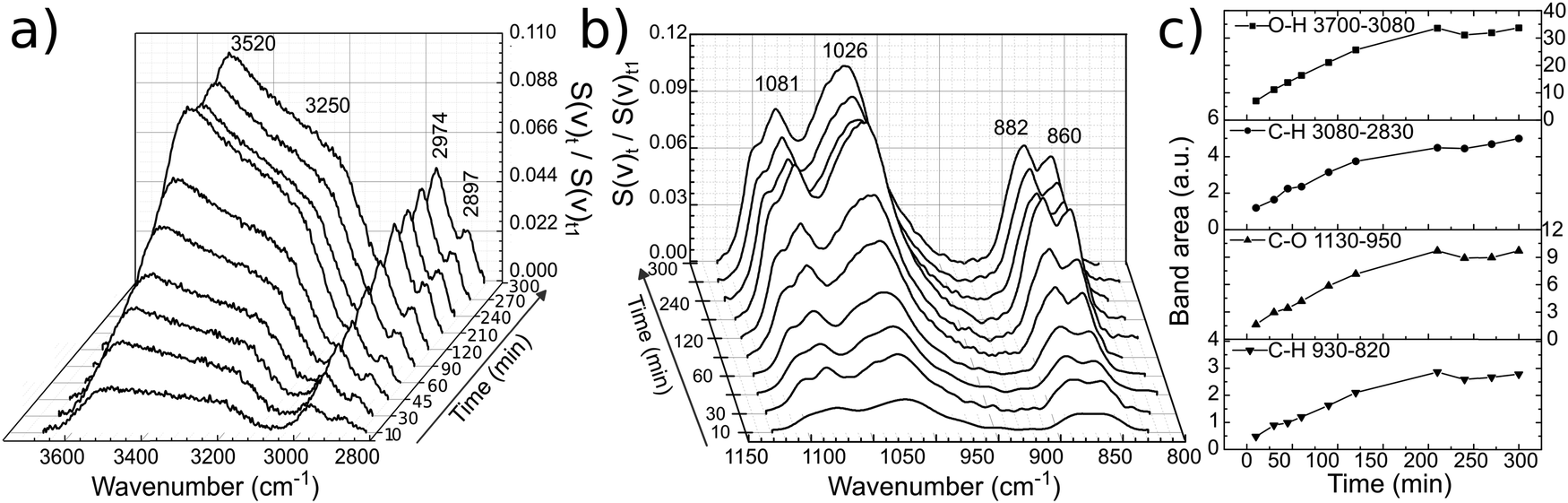 | ||
| Fig. 5 Temporal evolution of spectral bands of ethylene glycol on glassy carbon (a), (b) and their respective band area versus time (c). The lines between data points in (c) serve only as guide to the eye. | ||
The absorption bands, particularly those related to the O–H and C–O groups, increase significantly within this period of time, e.g. the ν(O–H) at 3700–3100 cm−1 and ν(C–O) at 1130–950 cm−1. Similarly, the ρ(CH2) and ν(C–C) skeleton vibrations of ethylene glycol at 930–820 cm−1 show a stronger IR absorption with time. The increase of the ν(O–H), ν(C–O) and C–H bands can be followed by plotting the integrated band intensity versus time (Fig. 5c).
The O–H stretching at 3520 cm−1 turns much more intense compared to that at 3250 cm−1 and continuously increases with time indicating self-assembly of EG molecules on the surface. This results in an increase of the surface population and consequently of the IR signal. The layering of the molecules at OCP proceeds very slowly and reaches a plateau after 200 minutes. It is important to mention that PM-IRRAS is also sensitive to the molecules that are in the proximity of the electrode surface (ca. 1 μm, or 1/4 of the wavelength of the incident radiation).35 Hence, the absorption increase may be the result of an increasing transition dipole moment of the molecules in the vicinity of the electrode surface caused by an increase in population and/or a more vertical orientation of the molecules. Although there might be condensation of water on the detector, from the liquid nitrogen cooling, the O–H absorption by water molecules is annulled through the subtraction of the s- from p-polarized spectrum, since the absorption will be the same in both polarized components.
The EG was removed and 12CE electrolyte was inserted into the cell. For this second experiment it is important to remark that the GC was pre-wetted with EG. A slow increase in band intensities similar to that of EG was observed for the DES 12CE (Fig. 6). The intensity increase of the asymmetric stretching mode of (CH3)3N+ at 950 cm−1 indicates that the choline ammonium group competes with the OH group for adsorption sites on the electrode surface. However, the intensities of the two overlapping OH bands at about 3400 and 3200 cm−1 are similar, in contrast to the spectrum of EG, where the OH group thought to be in contact with the surface shows a higher intensity.
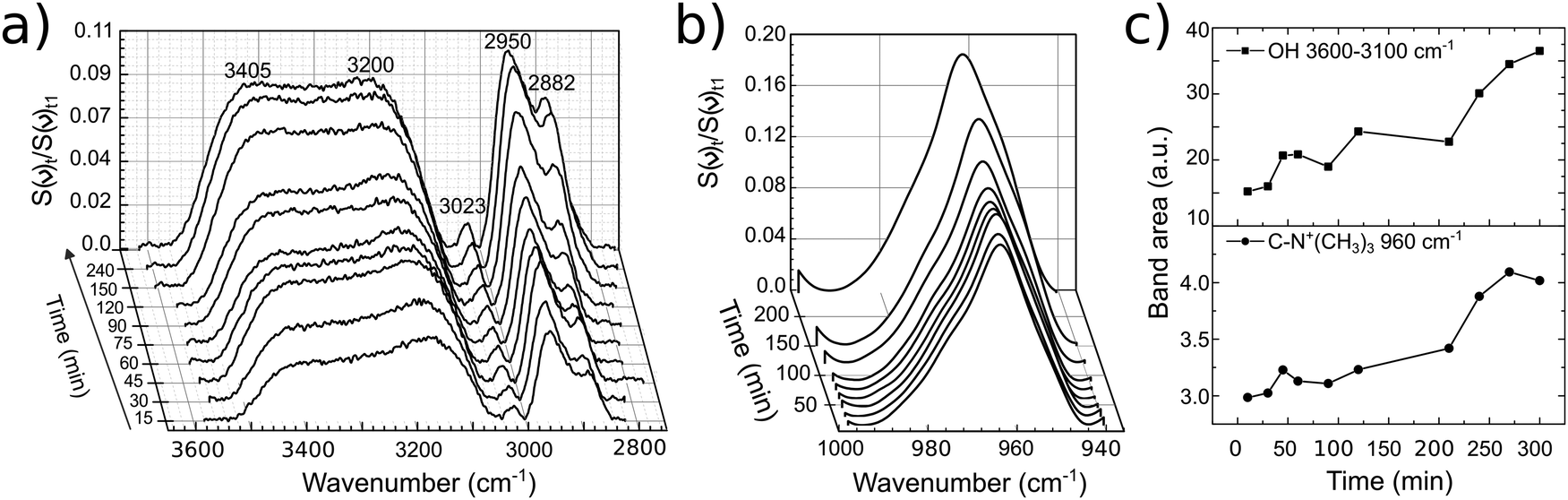 | ||
| Fig. 6 Temporal evolution of bands of 12CE on GC (a), (b) with their respective band area versus time (c). The lines between data points in (c) serve only as guide to the eye. | ||
Layering of organic solvents73,74 and ionic liquids75–78 near solid surfaces at OCP have been detected by in situ AFM of metal/liquid interfaces. Molecular ordering at solid surfaces has been found a common phenomenon for organic solvents and ionic liquids. However, the adsorption strength of ionic liquids can be much higher than that of organic solvents, as indicated by the AFM rupture force for such ionic layers.79 Moreover, it has been shown that the layering of ionic liquids is promoted by applying an external potential to the electrode, which can also change the order of cations and anions and their orientation on the surface.12
Spectroelectrochemistry
In situ PM-IRRA spectra were recorded during a chronoamperometric multi-step sequence. The sequence of potential steps together with the current responses and the corresponding PM-IRRA spectra are presented in Fig. 7 and 8, respectively.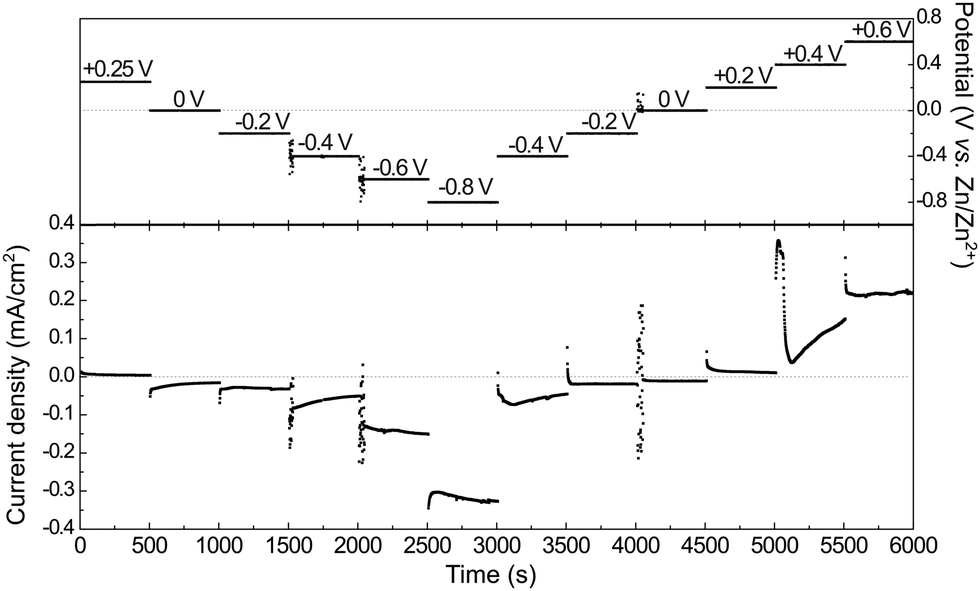 | ||
| Fig. 7 Electrode potential and current density versus time of 12CE in the spectroelectrochemical cell during in situ PM-IRRAS experiments. | ||
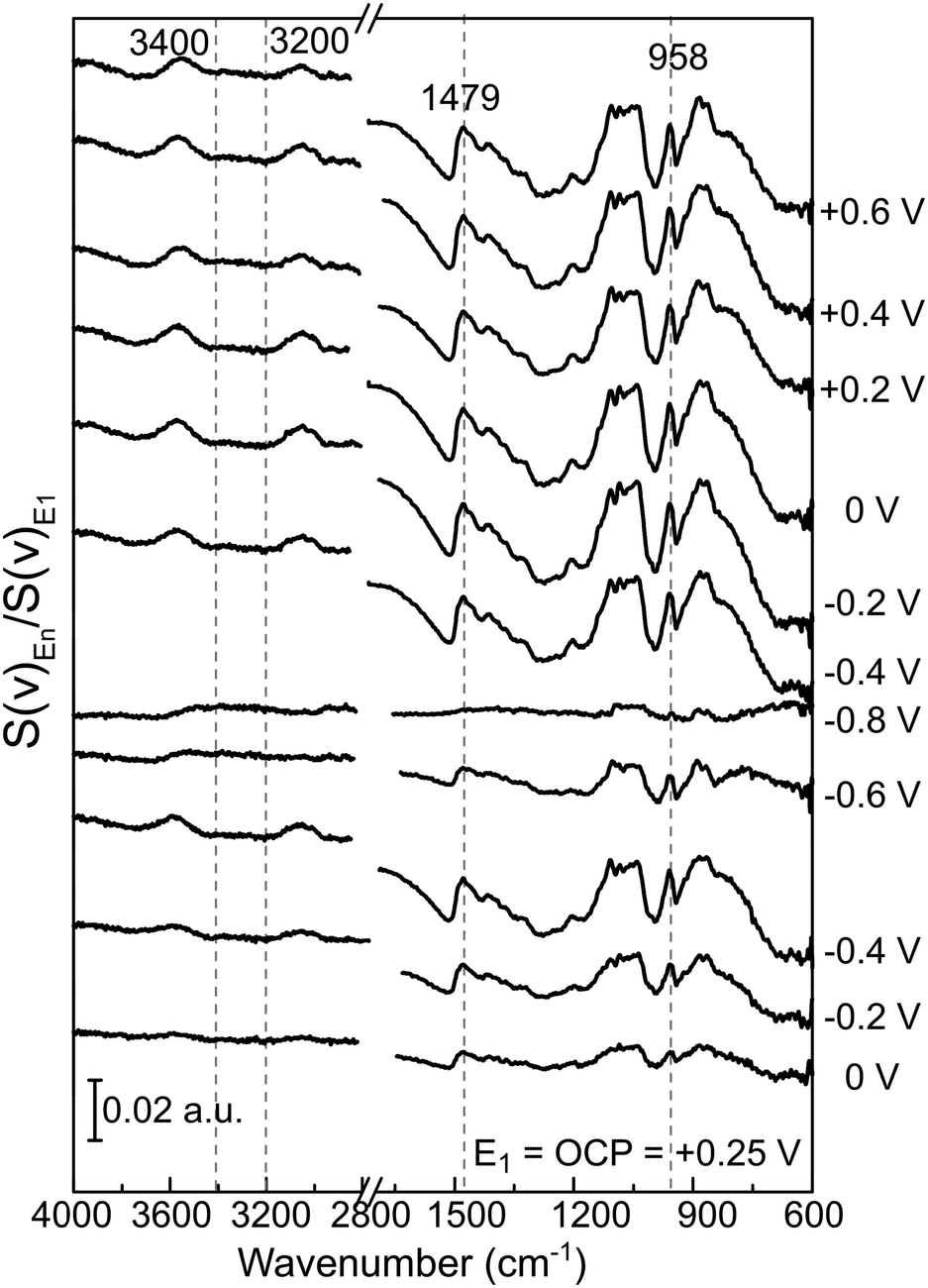 | ||
| Fig. 8 PM-IRRAS surface spectra of GC/12CE recorded at the indicated potentials. All spectra are differentially normalized to the PM-IRRAS spectrum at the same value as OCP E1 = Eocp = +0.25 V. | ||
The spectrum recorded at the first potential step (+0.25 V vs. Zn/Zn2+) was chosen as reference. Since this potential is measured at open circuit, no faradaic reactions should take place here. All following spectra recorded at different applied potentials (En) have been normalized (by division) to this reference (E1). Therefore, bands pointing up indicate an increase in absorption, whereas those pointing down have a lower IR absorption compared to the reference spectrum. From +0.25 V, the potential was stepped in the negative direction until −0.8 V in intervals of 0.2 V and then back positively till +0.6 V.
During the first potential step at +0.25 V vs. Zn/Zn2+ (about 10 minutes after filling the SEC cell with 12CE), the molecules presumably have a random orientation at the GC surface. Thus initially, ammonium as well as hydroxyl groups might be in contact with the electrode. By stepping the potential negatively to 0 V, adsorption of choline molecules at the GC becomes evident by an increase in the intensity of the bands in the fingerprint region (1500–800 cm−1), particularly those attributed to the vibrations of the quaternary ammonium group at 1479 and 959 cm−1. This increase in absorption intensity is accentuated as the potential is stepped more negative to −0.2 and −0.4 V vs. Zn/Zn2+. A corresponding increase in the cathodic charging current can be observed at −0.4 V, likely due to a stronger adsorption of choline cations.
Stepping the potential to −0.6 and −0.8 V causes a decrease of the PM-IRRAS signal, compared to that at −0.4 V. For these potential steps, a significant increase in current density is also observed, indicating reduction of the electrolyte (ChCl, EG).47 At −0.6 V reduction of the electrolyte might already occur on some active sites. At −0.8 V, electrolyte reduction takes place on the whole electrode surface. Consequently, the adsorbed layer is desorbed and a decrease of the band intensities is observed in the whole PM-IRRAS spectrum. No evidence for massive formation of hydrogen gas is observed though. In that case the layer cavity would be completely filled with the gas, thus the IR absorbance would be much lower than during the initial state of the experiment. Instead, the PM-IRRAS spectrum at −0.8 V is similar to the one at OCP (+0.25 V), where the molecules are randomly oriented and more statistically distributed. Presumably, disordered layers of Ch+ and EG molecules are now separated from the surface by adsorbed hydrogen, which does not absorb in the IR.
Primary cathodic reduction products of the choline cations probably are choline radicals. These radicals could decompose to trimethylamine and ethanol, dimethylaminoethanol and/or acetaldehyde.80 The latter three could be further reduced to form alkoxy species (RO−).47,48 Alternatively, electrochemical grafting of the generated radicals on the GC surface could occur.81 The IR bands of these radicals and decomposition products appear at wavenumbers similar to those of the functional groups of the choline, and are thus difficult to identify.
The evolution of the bands with applied potential can be better visualized in the spectra with a corrected baseline as presented in Fig. 9. The baseline was corrected using the spline interpolation as described by Zamlynny et al.35 The spectra have been divided into three regions: the ν(O–H) region from 3600 to 2700 cm−1, the δ[(CH3)3N+] region from 1500 to 1300 cm−1 and the ν(C–N) region from 1000 to 800 cm−1. The band areas have been integrated with respect to the local baseline and are presented in Fig. 10.
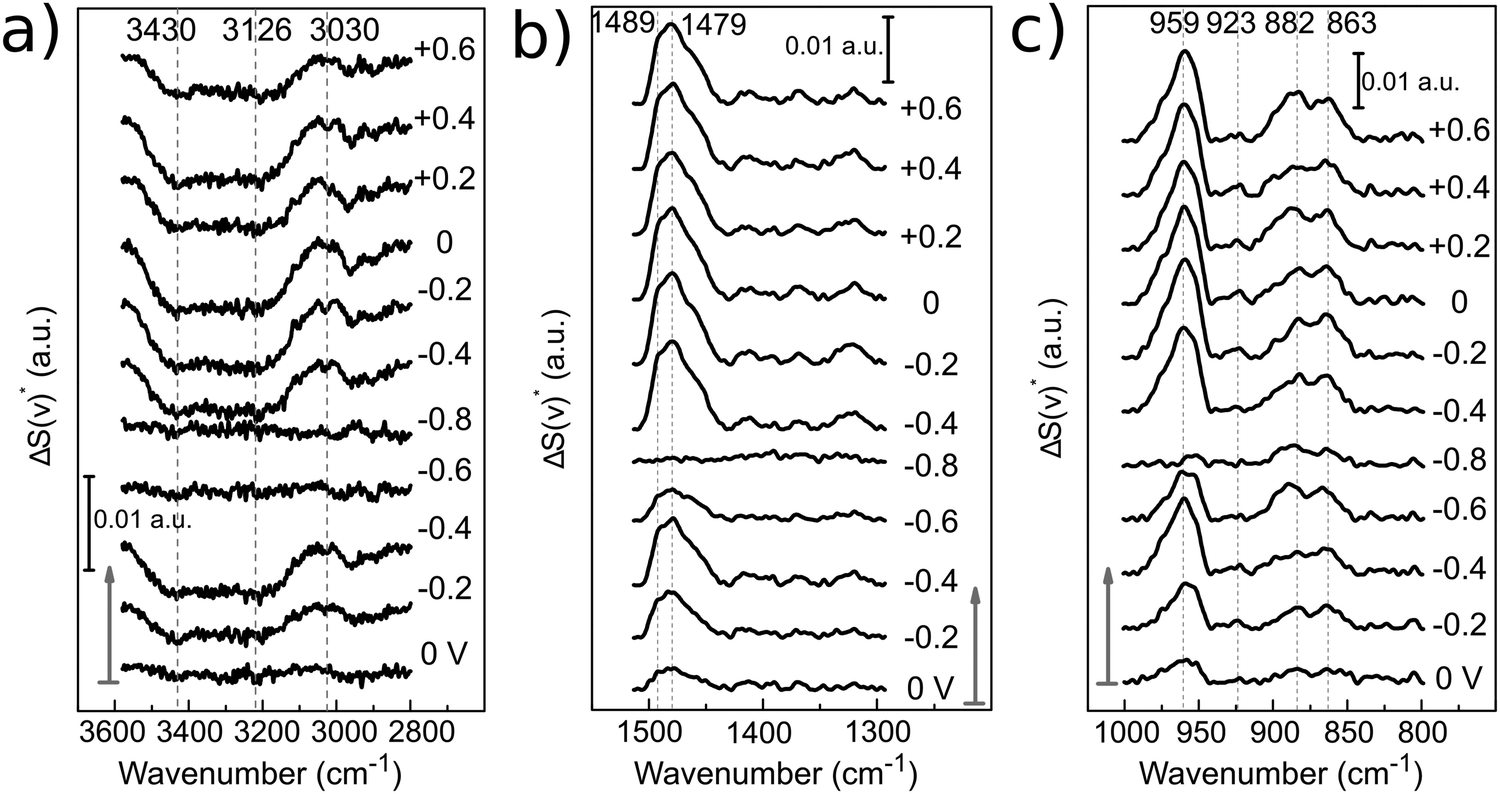 | ||
| Fig. 9 Baseline corrected PM-IRRAS surface spectra [ΔS*(ν)] of 12CE at different applied potentials. Arrows indicate the start of the experiment. All spectra are normalized to the PM-IRRAS spectrum at open circuit potential (+0.25 V). The spectral regions correspond to: (a) O–H and C–H stretching, (b) C–H bending and (c) C–N stretching. | ||
 | ||
| Fig. 10 PM-IRRAS band intensities of (a) ν(O–H), (b) δas[N+(CH3)3] and (c) νas[C–N+(CH3)3] of 12CE on GC as a function of electrode potential. The bands were integrated in the corrected baseline spectra in Fig. 9 with respect to zero. Arrows indicate potential step direction from the start of the experiment. | ||
In the ν(O–H) region (3600–3000 cm−1), the O–H stretching band intensity decreased upon decreasing the potential from 0 to −0.4 V. This indicates that the O−H groups of Ch+ and EG gradually desorb, compared to the initial state and are probably replaced by the choline ammonium group. Decreasing the potential further to −0.6 and −0.8 V vs. Zn/Zn2+ increased the intensity and it became more similar to the initial state. This can be correlated to a closer approach and reduction of the OH groups. Stepping the potential positively again did not result in an increase of the absorption back to the initial value.
The asymmetrical C–H methylene bending modes at 1489 and 1479 cm−1 and C–N stretching of the ammonium group at 959 cm−1 show a common trend, because they belong to the same functional group. Stepping the potential negatively from 0 to −0.4 V, the increase in band intensities indicate adsorption of the positively charged choline groups on the GC electrode. From −0.6 to −0.8 V Ch+ ions are presumably replaced by adsorbed hydrogen, therefore a decrease in absorption is observed. Stepping from −0.8 to −0.4 V causes re-adsorption of the Ch+, which is evident by the strong increase of band intensities for these two vibrational modes. From −0.4 to 0 V the Ch+ remains adsorbed on the surface.
Stepping positively from 0 V up to +0.6 V caused the band area of δas[N+(CH3)3] and νas[C–N+(CH3)3] to decrease slightly. Adsorption of the ammonium group at a positively charged electrode seems rather unlikely. The anions in the deep eutectic electrolyte presumably replace the cations at positive potentials. In the 12CE, these anions would be either Cl− or RO− species, where the latter are generated through the reduction of the Ch+ (or EG) at E ≤ −0.6 V. In Fig. 7, an anodic current density of 0.22 mA cm−2 is observed at +0.6 V indicating oxidation of cathodic reduction products of the electrolyte. Simultaneously, there is a significant absorption increase of the bands in the 1200–1000 cm−1 region (νas and νs of C–O, Fig. 8). However, integration of these C–O bands is difficult due to the overlap with νs(C–N) at 1133 cm−1. The increase in this region, however, could also be caused by the oxidation of the glassy carbon, generating –O–H, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and –COOH groups on the surface.82
O and –COOH groups on the surface.82
The binding strength of the Ch+ to the GC surface can be evaluated by the position of the δas and νas N+(CH3)3 band centers as a function of the applied potential (Fig. 11). Variations on the vibration frequency of a band may indicate changes in adsorption geometry, surface coverage or the electrostatic properties of the molecules on the electrode surface.83 Band-shifts to lower wavenumbers (red-shift) indicate that the bond became weaker. On the contrary, shifts to higher wavenumbers (blue-shift) suggest a strengthening of the chemical bond. If the Ch+ group is adsorbed to the electrode surface at negative potentials, it is expected that the induction of charge from the electrode will make the bond stronger, causing a blue-shift. Indeed, for the δas[N+(CH3)3], a blue-shift of 6 cm−1 is observed, from 1478 cm−1 to 1484 cm−1 upon stepping the potential from 0 to −0.2 V. At −0.8 V, the frequency shifts to 1467 cm−1, indicating the desorption of the Ch+. Similar behaviour is observed for the νas[N+(CH3)3], firstly a red-shift from 958 cm−1 at 0 V to 962 cm−1 at −0.6 V due to direct adsorption of Ch+ then a blue shift to 952 cm−1 at −0.8 V due to its desorption. Switching the potential to positive values caused the peak centres to return to 1479 cm−1 and 959 cm−1, probably due to the induction of charge from Cl−.
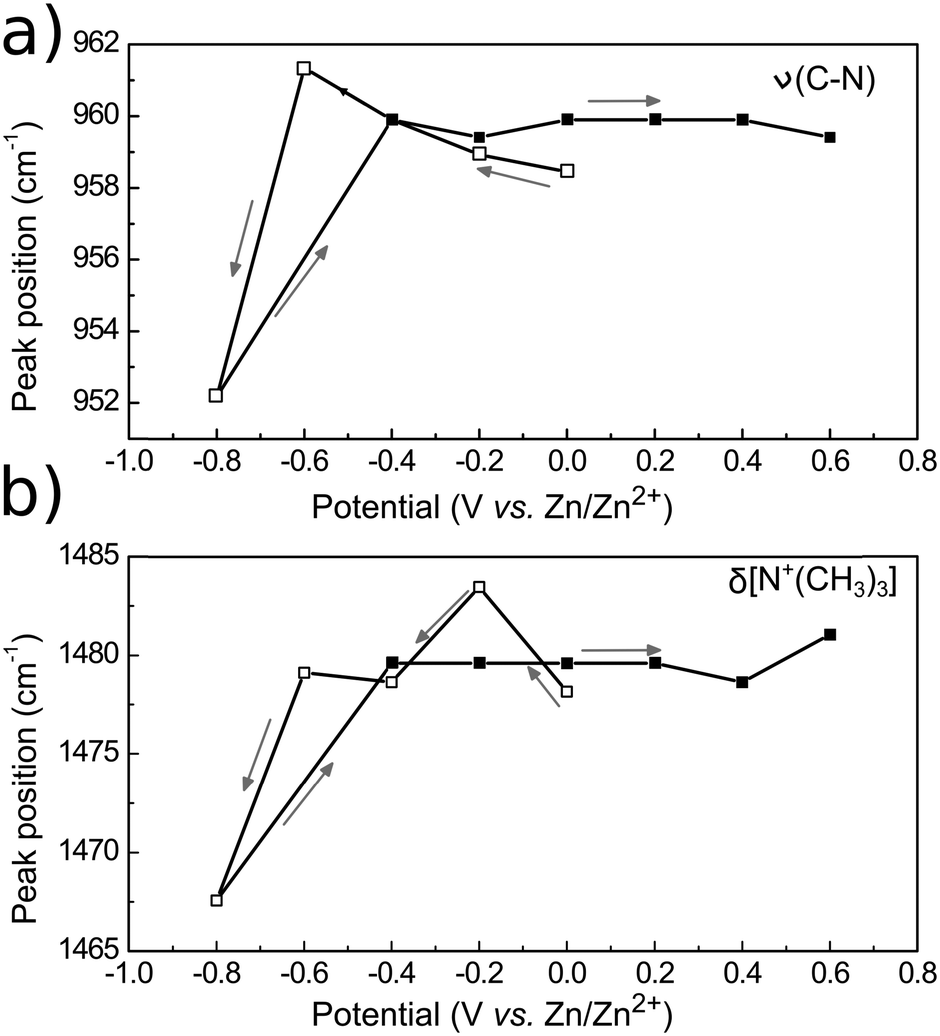 | ||
| Fig. 11 Frequency shift as a function of the electrode potential for the vibrational modes νas[C–N+(CH3)3] (a) and δas[N+(CH3)3] (b). Empty squares indicate forward potential steps (from 0 to −0.8 V vs. Zn2+) while filled squares indicate the steps in a positive direction (from −0.4 to +0.6 V vs. Zn2+). Arrows point in the direction of the potential steps and the lines between the data points serve only as guide to the eye. | ||
Our in situ PM-IRRAS results show that electrolyte components adsorb on the glassy carbon electrode. Analogously to the results reported for other electrode/RTIL interfaces, our results also demonstrate that the layer architecture and binding strength of the adsorbate depend on the applied potential. These findings could help in explaining potential dependent blocking effects we observed during zinc electrodeposition from 12CE on glassy carbon.47 Although our observations are consistent with complimentary studies on electrode/RTIL interfaces by others, we are aware that our interpretation is preliminary. Density functional theory calculations and molecular dynamic simulations are currently in progress for this system and will contribute to a clearer and more quantitative picture of these interfacial processes.
4. Conclusions
The glassy carbon electrode/electrolyte interface has been studied in situ by PM-IRRAS for the first time. This is a rather new approach also for spectroelectrochemical investigations of ionic liquids. To date, PM-IRRA spectroelectrochemistry has not been reported for ionic liquids or deep eutectic solvents.The glassy carbon electrode interface with ethylene glycol and 12CE gave a good response to PM-IRRAS and most of the vibrational modes of the liquids could be identified. The increase of the band intensities with time at open circuit potential indicates self-assembly of EG and 12CE at the surface and demonstrates the surface sensitivity of the external reflection geometry.
The spectroelectrochemical study of 12CE on glassy carbon electrodes showed characteristic potential-dependent changes of the surface spectra. They indicate that decreasing the potential from open circuit to −0.6 V vs. Zn/Zn2+ causes mostly choline cations to adsorb on the surface. From 0 to −0.6 V the adsorbed Ch+ molecules become more tilted toward the electrode surface, consequently decreasing the resultant dipole moment and the PM-IRRAS signal. At −0.8 V, the Ch+ molecules are reduced, and as a result hydrogen is likely to adsorb on the GC surface.
From −0.6 to +0.4 V vs. Zn/Zn2+ Ch+ molecules re-adsorb on the GC surface, and an increasing IR absorbance is observed for the δ and ν vibrational modes of the choline N+(CH3)3 group. At E ≥ +0.4 V a re-arrangement of the interfacial species takes place and Ch+ is probably replaced by Cl−. In this potential range, the bands attributed to the vibrational modes of Ch+ increase due to a change in molecular orientation, which becomes more vertical with respect to the surface in order to maximize the electrostatic interaction with the primary adlayer of Cl−.
Acknowledgements
The authors acknowledge funding by the COMET program of the Austrian Research Advancement Agency (FFG) and the government of Lower Austria. This work was additionally supported by Atotech Deutschland GmbH, Akzo Nobel Surface Chemistry AB and Andritz AG. The authors are indebted to Martin Kornschober for designing and building the spectroelectrochemical cell.References
- E. L. Smith, A. P. Abbott and K. S. Ryder, Chem. Rev., 2014, 114, 11060–11082 CrossRef CAS PubMed.
- Q. Zhang, K. D. O. Vigier, S. Royer and F. Jerome, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC.
- M. H. Chakrabarti, F. S. Mjalli, I. M. AlNashef, M. A. Hashim, M. A. Hussain, L. Bahadori and C. T. J. Low, Renewable Sustainable Energy Rev., 2014, 30, 254–270 CrossRef CAS.
- Electrodeposition from Ionic Liquids, ed. F. Endres, A. Abbott and D. MacFarlane, Wiley-VCH, Weinheim, 2008 Search PubMed.
- Electrochemical aspects of ionic liquids, ed. H. Ohno, Wiley Interscience, 2005 Search PubMed.
- M. V. Fedorov and A. A. Kornyshev, Chem. Rev., 2014, 114, 2978–3036 CrossRef CAS PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical methods: fundamentals and applications, Wiley, New York, 2001 Search PubMed.
- A. Kornyshev, J. Phys. Chem. B, 2007, 111, 5545–5557 CrossRef CAS PubMed.
- K. B. Oldham, J. Electroanal. Chem., 2008, 613, 131–138 CrossRef CAS.
- M. Mezger, H. Schröder, H. Reichert, S. Schramm, J. S. Okasinski, S. Schöder, V. Honkimäki, M. Deutsch, B. M. Ocko, J. Ralston, M. Rohwerder, M. Stratmann and H. Dosch, Science, 2008, 322, 424–428 CrossRef CAS PubMed.
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855–6863 CAS.
- R. Atkin, N. Borisenko, M. Drüschler, F. Endres, R. Hayes, B. Huber and B. Roling, J. Mol. Liq., 2014, 192, 44–54 CrossRef CAS.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238–2264 CrossRef CAS PubMed.
- L. E. Barrosse-Antle, A. M. Bond, R. G. Compton, A. M. O'Mahony, E. I. Rogers and D. S. Silvester, Chem. – Asian J., 2010, 5, 202–230 CrossRef CAS PubMed.
- F. Endres, O. Hofft, N. Borisenko, L. H. Gasparotto, A. Prowald, R. Al-Salman, T. Carstens, R. Atkin, A. Bund and S. Z. El Abedin, Phys. Chem. Chem. Phys., 2010, 12, 1724–1732 RSC.
- S. Baldelli, Acc. Chem. Res., 2008, 41, 421–431 CrossRef CAS PubMed.
- S. Baldelli, J. Phys. Chem. Lett., 2013, 4, 244–252 CrossRef CAS PubMed.
- B. Bozzini, A. Bund, B. Busson, C. Humbert, A. Ispas, C. Mele and A. Tadjeddine, Electrochem. Commun., 2010, 12, 56–60 CrossRef CAS.
- W. Zhou, S. Inoue, T. Iwahashi, K. Kanai, K. Seki, T. Miyamae, D. Kim, Y. Katayama and Y. Ouchi, Electrochem. Commun., 2010, 12, 672–675 CrossRef CAS.
- V. O. Santos, M. B. Alves, M. S. Carvalho, P. A. Z. Suarez and J. C. Rubim, J. Phys. Chem. B, 2006, 110, 20379–20385 CrossRef CAS PubMed.
- Y. Yuan, T. Niu, M. Xu, J. Yao and R. Gu, J. Raman Spectrosc., 2010, 41, 516–523 CrossRef CAS.
- K. Motobayashi, K. Minami, N. Nishi, T. Sakka and M. Osawa, J. Phys. Chem. Lett., 2013, 4, 3110–3114 CrossRef CAS.
- Y. Yang, L. Zhang, M. Osawa and W. Cai, J. Phys. Chem. Lett., 2013, 4, 1582–1586 CrossRef CAS PubMed.
- N. Nanbu, Y. Sasaki and F. Kitamura, Electrochem. Commun., 2003, 5, 383–387 CrossRef CAS.
- O. Hoefft, S. Bahr and V. Kempter, Anal. Sci., 2008, 24, 1273–1277 CrossRef CAS.
- O. Hoefft, S. Bahr and V. Kempter, Langmuir, 2008, 24, 11562–11566 CrossRef CAS PubMed.
- Y. Su, Y. Fu, Y. Wei, J. Yan and B. Mao, ChemPhysChem, 2010, 11, 2764–2778 CrossRef CAS PubMed.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106, 046102 CrossRef PubMed.
- K. Kirchner, T. Kirchner, V. Ivaništšev and M. V. Fedorov, Electrochim. Acta, 2013, 110, 762–771 CrossRef CAS.
- S. Makino, Y. Kitazumi, N. Nishi and T. Kakiuchi, Electrochem. Commun., 2011, 13, 1365–1368 CrossRef CAS.
- N. Nishi, Y. Hirano, T. Motokawa and T. Kakiuchi, Phys. Chem. Chem. Phys., 2013, 15, 11615–11619 RSC.
- W. Iwasita and F. Nart, Prog. Surf. Sci., 1997, 55, 271–340 CrossRef.
- S. P. Best, S. J. Borg and K. A. Vincent, in Spectroelectrochemistry, ed. W. Kaim and A. Klein, The Royal Society of Chemistry, 2008, pp. 1–30 Search PubMed.
- B. Beden and C. Lamy, in Spectroelectrochemistry: Theory and Practice, ed. R. J. Gale, Plenum Press, New York, 1988, pp. 189–261 Search PubMed.
- V. Zamlynny and J. Lipkowski, Advances in Electrochemical Science and Engineering, in Diffraction and Spectroscopic Methods in Electrochemistry, ed. R. C. Alkire, D. M. Kolb, J. Lipkowski and P. N. Ross, Wiley-VCH, Weinheim, 2008, vol. 9, pp. 315–376 Search PubMed.
- R. G. Greenler, J. Chem. Phys., 1966, 44, 310–315 CrossRef CAS.
- R. Schennach, C. Hirschmugl, E. Gilli and W. T. Tysoe, Appl. Spectrosc., 2009, 63, 369–372 CrossRef CAS PubMed.
- A. H. Kycia, Z. Su, C. L. Brosseau and J. Lipkowski, in Vibrational Spectroscopy at Electrified Interfaces, ed. A. Wieckowski, C. Korzeniewski and B. Braunschweig, John Wiley & Sons, Hoboken, 2013, pp. 345–417 Search PubMed.
- I. Burgess, M. Li, S. L. Horswell, G. Szymanski, J. Lipkowski, J. Majewski and S. Satija, Biophys. J., 2004, 86, 1763–1776 CrossRef CAS PubMed.
- I. Burgess, M. Li, S. L. Horswell, G. Szymanski, J. Lipkowski, S. Satija and J. Majewski, Colloids Surf., B, 2005, 40, 117–122 CrossRef CAS PubMed.
- J. Lipkowski, Phys. Chem. Chem. Phys., 2010, 12, 13874–13887 RSC.
- C. L. Brosseau, J. Leitch, X. Bin, M. Chen, S. G. Roscoe and J. Lipkowski, Langmuir, 2008, 24, 13058–13067 CrossRef CAS PubMed.
- I. Zawisza, X. Bin and J. Lipkowski, Langmuir, 2007, 23, 5180–5194 CrossRef CAS PubMed.
- X. Bin, S. L. Horswell and J. Lipkowski, Biophys. J., 2005, 89, 592–604 CrossRef CAS PubMed.
- X. Bin and J. Lipkowski, J. Phys. Chem. B, 2006, 110, 26430–26441 CrossRef CAS PubMed.
- S. L. Horswell, V. Zamlynny, H. Li, A. R. Merrill and J. Lipkowski, Faraday Discuss., 2002, 121, 405–422 RSC.
- L. Vieira, A. H. Whitehead and B. Gollas, J. Electrochem. Soc., 2014, 161, D7–D13 CrossRef CAS.
- A. H. Whitehead, M. Polzler and B. Gollas, J. Electrochem. Soc., 2010, 157, D328–D334 CrossRef CAS.
- N. M. Pereira, P. M. V. Fernandes, C. M. Pereira and A. Fernando Silva, J. Electrochem. Soc., 2012, 159, D501–D506 CrossRef CAS.
- T. J. Simons, A. A. J. Torriero, P. C. Howlett, D. R. MacFarlane and M. Forsyth, Electrochem. Commun., 2012, 18, 119–122 CrossRef CAS.
- M. Tulodziecki, J.-M. Tarascon, P. L. Taberna and C. Guéry, Electrochim. Acta, 2014, 134, 55–66 CrossRef CAS.
- K. Shahbaz, F. S. G. Bagh, F. S. Mjalli, I. M. AlNashef and M. A. Hashim, Fluid Phase Equilib., 2013, 354, 304 CrossRef CAS.
- R. B. Leron, A. N. Soriano and M. Li, J. Taiwan Inst. Chem. Eng., 2012, 43, 551–557 CrossRef CAS.
- V. P. Tolstoy, I. Chernyshova and V. A. Skryshevsky, Handbook of Infrared Spectroscopy of Ultrathin Films, Wiley, 2003 Search PubMed.
- K. Krishnan and R. Krishnan, Proc. – Indian Acad. Sci., Sect. A, 1966, 64, 111–122 CAS.
- W. Sawodny, K. Niedenzu and J. W. Dawson, Spectrochim. Acta, Part A, 1967, 23, 799–806 CrossRef CAS.
- K. M. Harmon, S. L. Madeira, M. J. Jacks, G. F. Avci and A. C. Thiel, J. Mol. Struct., 1985, 128, 305–314 CrossRef CAS.
- S. L. Perkins, P. Painter and C. M. Colina, J. Phys. Chem. B, 2013, 117, 10250–10260 CrossRef CAS PubMed.
- D. Yue, Y. Jing, J. Sun, X. Wang and Y. Jia, J. Mol. Liq., 2011, 158, 124–130 CrossRef CAS.
- H. Wang, Y. Jing, X. Wang, Y. Yao and Y. Jia, J. Mol. Liq., 2012, 170, 20–24 CrossRef CAS.
- A. Abbott, D. Boothby, G. Capper, R. Rasheed and D. Davies, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed.
- S. L. Perkins, P. Painter and C. M. Colina, J. Chem. Eng. Data, 2014, 59, 3652–3662 CrossRef CAS.
- M. Francisco, A. Bruinhorst and M. Kroon, Angew. Chem., Int. Ed., 2013, 52, 3074–3085 CrossRef CAS PubMed.
- B. H. Stuart, Infrared Spectroscopy: Fundamentals and Applications, Wiley, 2004 Search PubMed.
- P. Larkin, Infrared and Raman Spectroscopy; Principles and Spectral Interpretation, Elsevier Science, 2011 Search PubMed.
- O. Skoplyak, M. A. Barteau and J. G. Chen, Surf. Sci., 2008, 602, 3578–3587 CrossRef CAS.
- M. B. Griffin, E. L. Jorgensen and J. W. Medlin, Surf. Sci., 2010, 604, 1558–1564 CrossRef CAS.
- N. F. Brown and M. A. Barteau, J. Phys. Chem., 1994, 98, 12737–12745 CrossRef CAS.
- A. J. Capote and R. J. Madix, Surf. Sci., 1989, 214, 276–288 CrossRef CAS.
- A. J. Capote and R. J. Madix, J. Am. Chem. Soc., 1989, 111, 3570–3577 CrossRef CAS.
- K. T. Queeney, C. R. Arumainayagam, M. K. Weldon, C. M. Friend and M. Q. Blumberg, J. Am. Chem. Soc., 1996, 118, 3896–3904 CrossRef CAS.
- M. M. M. Jansen, B. E. Nieuwenhuys and H. Niemantsverdriet, ChemSusChem, 2009, 2, 883–886 CrossRef CAS PubMed.
- S. J. O'Shea, M. E. Welland and J. B. Pethica, Chem. Phys. Lett., 1994, 223, 336–340 CrossRef.
- R. Y. H. Lim and S. J. O'Shea, Langmuir, 2004, 20, 4916–4919 CrossRef CAS PubMed.
- T. Carstens, R. Gustus, O. Hoefft, N. Borisenko, F. Endres, H. Li, R. J. Wood, A. J. Page and R. Atkin, J. Phys. Chem. C, 2014, 118, 10833–10843 CAS.
- T. Carstens, R. Hayes, S. Z. E. Abedin, B. Corr, G. B. Webber, N. Borisenko, R. Atkin and F. Endres, Electrochim. Acta, 2012, 82, 48–59 CrossRef CAS.
- D. Wakeham, R. Hayes, G. G. Warr and R. Atkin, J. Phys. Chem. B, 2009, 113, 5961–5966 CrossRef CAS PubMed.
- R. Atkin, S. Z. E. Abedin, R. Hayes, L. H. S. Gasparotto, N. Borisenko and F. Endres, J. Phys. Chem. C, 2009, 113, 13266–13272 CAS.
- F. Endres, N. Borisenko, S. Z. El Abedin, R. Hayes and R. Atkin, Faraday Discuss., 2012, 154, 221–233 RSC.
- K. Haerens, E. Matthijs, K. Binnemans and B. Van der Bruggen, Green Chem., 2009, 11, 1357–1365 RSC.
- D. Belanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995–4048 RSC.
- B. E. Conway, in Electrochemical Supercapacitors, ed. B. E. Conway, Springer, US, 1999, pp. 183–220 Search PubMed.
- M. Roeefzaad, D. Thanh Pham and K. Wandelt, in Vibrational Spectroscopy at Electrified Interfaces, ed. A. Wieckowski, C. Korzeniewski and B. Braunschweig, John Wiley & Sons, Inc, 2013, pp. 307–326 Search PubMed.
| This journal is © the Owner Societies 2015 |