
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
On the origin and variation of colors in lobster carapace†
Shamima
Begum
a,
Michele
Cianci
 b,
Bo
Durbeej
c,
Olle
Falklöf
c,
Alfons
Hädener‡
a,
John R.
Helliwell
*a,
Madeleine
Helliwell
a,
Andrew C.
Regan
a and
C.
Ian F. Watt
a
b,
Bo
Durbeej
c,
Olle
Falklöf
c,
Alfons
Hädener‡
a,
John R.
Helliwell
*a,
Madeleine
Helliwell
a,
Andrew C.
Regan
a and
C.
Ian F. Watt
a
aSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: john.helliwell@manchester.ac.uk
bEMBL, PETRA III, DESY, Notkestrasse 85, 22603 Hamburg, Germany
cDivision of Computational Physics, IFM, Linköping University, SE-58183 Linköping, Sweden
First published on 13th March 2015
Abstract
The chemical basis of the blue-black to pink-orange color change on cooking of lobster, due to thermal denaturation of an astaxanthin–protein complex, α-crustacyanin, in the lobster carapace, has so far been elusive. Here, we investigate the relaxation of the astaxanthin pigment from its bound enolate form to its neutral hydroxyketone form, as origin of the spectral shift, by analyzing the response of UV-vis spectra of a water-soluble 3-hydroxy-4-oxo-β-ionone model of astaxanthin to increases in pH, and by performing extensive quantum chemical calculations over a wide range of chemical conditions. The enolization of astaxanthin is consistent with the X-ray diffraction data of β-crustacyanin (PDB code: 1GKA) whose crystals possess the distinct blue color. We find that enolate formation is possible within the protein environment and associated with a large bathochromic shift, thus offering a cogent explanation for the blue-black color and the response to thermal denaturation and revealing the chemistry of astaxanthin upon complex formation.
1. Introduction
In the last decade the carotenoid astaxanthin (3,3′-dihydroxy-β-carotene-4,4′-dione) has gained interest in the nutraceutical and cosmetic industries due to its antioxidant activity with potential applications against cardiovascular problems, different types of cancer and some diseases of the immunological system.1 Very recently, for instance, astaxanthin preparations have reached the mass market as anti-aging skincare products with the commercial name of ASTALIFT (© FUJIFILM Corporation). The red color displayed by isolated astaxanthin or its preparations, 1 (Scheme 1), often differs from that of the in vivo material,2 as represented by the familiar blue-black of the shell of live lobsters which reverts on cooking to the pink-orange color of dilute astaxanthin solutions. The dark color arises from an astaxanthin–protein complex, α-crustacyanin,3 whose atomic structure, despite recent progress,4,5 is as yet unknown. The crystal structure of a blue crystal of a dissociation product, β-crustacyanin,6 prompted renewed interest in the origins of the large bathochromic shift of the UV-vis absorption.7 Compared with uncomplexed 1, shifts are 100 and 160 nm for β-crustacyanin and α-crustacyanin, respectively.6 | ||
| Scheme 1 The structure of astaxanthin (1) and acid–base (HA and B) induced tautomerism of astaxanthin producing the enolate 1− and the ene-diol tautomer 1ed. Initial slow deprotonation of 1 at C3 followed by rapid proton transfer from O3 to O4 yield the enolate 1− capable of extended formal delocalization of the negative charge through the polyene chain to O4′. Reprotonation at O3 yields the ene-diol tautomer 1ed. Further similar chemistry leads to 1ded. | ||
The crystal structure of β-crustacyanin reveals two protein sub-units linked by a pair of astaxanthin molecules,6 modeled therein as α-hydroxyketone tautomers 1. Terminal oxygenated rings are in close association, at one end of each astaxanthin, with the nitrogen of a histidine of one protein and, at the other end, with a water molecule and the acidic phenolic hydroxyl of a tyrosine residue of the second protein. These rings are almost coplanar with the polyene chains, which are slightly bowed with a closest approach of about 7 Å.
Two distinct themes have developed in explanation for the coloration. The first, and most thoroughly investigated, focuses on physical arrangements of the astaxanthins in the protein complexes. The angles of twist about the C6–C7 (and C6′–C7′) bonds of the astaxanthins are much reduced from those found in the 6-s-cis conformation of uncomplexed astaxanthin (ca. 50°), to a 6-s-trans conformation with end rings coplanar with the polyene chains in the protein bound form, permitting enhanced overlap along the conjugated array in the complexed forms. Exciton coupling between the paired astaxanthins in β-crustacyanin has also been considered. Quantum chemical calculations and experiment, however, suggest that neither conformational change nor exciton coupling can account for more than 30% of the observed shift, with the larger contribution arising from co-planarization of the end rings.8–10 Kuhn and Sörensen suggested in 1938 a second theme,11 which remained largely neglected, involving a reversible ionization of the astaxanthins upon complexation. The consequences of deprotonation of an α-hydroxycyclohexenone residue, and subsequent proton transfers, are shown in Scheme 1. However, Buchwald and Jencks12 stated that the proteins involved did not contain a site sufficiently basic to retain a proton.
The viability of enolate formation, however, also depends on the acidity of astaxanthin and although this remains unquantified, we note that much simpler compounds, also capable of forming an α-hydroxylated enolate in which the negative charge is distributed between two oxygen atoms, including 2,3-dihydroxy-2-propenal (triose reductone) and ascorbic acid,13,14 are relatively strong organic acids (pKa(1) = 5.2 and 4.1, respectively) with second ionizations (pKa(2) ≈ 13) also accessible in water. A priori rejection of the possibility of enolate formation within the protein thus seems premature. While solutions of astaxanthin in a strongly basic organic medium are known to develop deep blue colors,15 quantitative studies in aqueous media require disentangling the effects of aggregation16,17 from those of ionization. We have therefore examined the behavior of 3-hydroxy-4-oxo-β-ionone, 2, C13H18O3, as a water-soluble model that retains the trimethylated α-hydroxycyclohexenone of astaxanthin but replaces the rest of the molecule with a simpler α-β-unsaturated ketone at the 6-position of the ring. This permits formal conjugation of the ring carbonyl (at C4) with that on the side chain at C9 through the C6–C7 bond, which occupies the same steric environment as the corresponding bond in astaxanthin. This compound, readily synthesized from β-ionone,18,19 is shown by NMR spectroscopy to exist as its ketonic tautomeric form in both methanol and chloroform solution. Its enolization and enolate chemistry, expected to mimic that of astaxanthin (Scheme 1), is shown in Scheme 2. Our UV-vis experiments on an astaxanthin model compound and quantum chemical calculations support a mechanism by which the colors of crustacyanin protein complexes arise because astaxanthin is bound in its 1− form.
 | ||
| Scheme 2 The structures of 3-hydroxy-4-oxo-β-ionone (2) and its ene-diol tautomer 2ed, and their relationships to possible anionic deprotonation products. Deprotonation of 2 at C3 followed by proton transfer from O3 to O4 yield the enolate 2−, with a sharing of the negative charge by conjugation between O3 and O9. Reprotonation at O3 yields the ene-diol tautomer 2ed. A dianion, 2-dianion, is accessible through deprotonation of the enolic hydroxyl group of 2−. | ||
2. Experimental
2.1 Preparation of compounds
3-Hydroxy-4-oxo-β-ionone (systematically 6-hydroxy-2,4,4-trimethyl-3-[(1E)-3-oxo-1-buten-1-yl]-2-cyclohexen-1-one), 2, was prepared by the published methods from β-ionone,18,19 and obtained as off-white crystals from hexane whose spectroscopic and physical properties were in accord with literature reports. 2,3-Dehydro-3-hydroxy-4-oxo-β-ionone (systematically 6-hydroxy-2,4,4-trimethyl-3-[(1E)-3-oxo-1-buten-1-yl]-2,4-cyclohexadien-1-one), 3, was prepared by dissolving 2 (0.011 g) in dry methanol (5 ml) in a 50 ml wide-neck RB flask. Aqueous NaOH solution (20 ml of 0.2 M) was added to provide an immediate deep orange colored solution, which was shaken vigorously to ensure full aeration. The open flask was then allowed to stand at ambient temperature (21 °C) with periodic vigorous shaking for 24 hours during which time the orange color faded noticeably. Glacial acetic acid was then added by microdropper until the residual color was discharged (pH < 8). The colorless solution was then evaporated to dryness under vacuum maintaining T < 30 °C, the oily residue was taken up in chloroform, and filtered through a cotton wool wad. Examination by TLC (silica, eluting with 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 vol/vol ethyl acetate:hexane) showed that the solution contained traces of residual unchanged 2 and a single major, more mobile product, which was isolated by preparative TLC on silica and crystallized from ether–hexane as off-white needles. For details, see the ESI.†
:50 vol/vol ethyl acetate:hexane) showed that the solution contained traces of residual unchanged 2 and a single major, more mobile product, which was isolated by preparative TLC on silica and crystallized from ether–hexane as off-white needles. For details, see the ESI.†
2.2 General methods
NMR spectra were recorded on Brüker 300, 400 or 500 AMX spectrometers. Infrared spectra were recorded on a Perkin-Elmer Spectrum FTIR and were run as liquid films or solids in an ATR attachment. UV-vis investigations were carried out on a Cary-50 BIO spectrometer fitted with a thermostated block set to 30 °C. Mass spectra were recorded on a Hewlett Packard HP5791A MSD. For details, see the ESI.†2.3 Quantum chemical calculations
UV-vis spectra and pKa values were determined in aqueous and methanol solutions based on primarily density functional theory (DFT) calculations. Bulk solvation effects were accounted for using the CPCM polarizable conductor calculation model.20 Geometry optimizations were throughout carried out in a water or methanol solvent using the B3LYP hybrid density functional in combination with a Pople style basis set, either 6-31G(d,p) that does not include diffuse functions, or 6-31+G(d,p) that includes diffuse functions on second-row atoms. Complementary geometry optimizations were for benchmark purposes also carried out using three alternative density functionals (BP86, BLYP and M06-2X21), as well as Møller–Plesset second-order perturbation theory (MP2) and the ab initio approximate coupled-cluster singles and doubles method (CC2).22,23 Some of these optimizations were done in the gas phase and with the cc-pVDZ basis set. Based on optimized ground-state geometries, absorption maxima were then obtained as vertical excitation energies calculated using time-dependent DFT (TD-DFT).24–26 The TD-DFT calculations were performed with seven different density functionals covering a range of exchange–correlation potentials. Some of these functionals – including M06-HF,27 LC-ωPBE28,29 and LC-BLYP30 – have partly been designed to offer a better description of excited states than standard functionals. To assess the performance of TD-DFT, complementary excited-state calculations were also performed with CC2 and complete active space second-order perturbation theory (CASPT2).31 While CC2 is one of the least costly correlated ab initio methods available today, CASPT2 is widely regarded as the gold standard in the field.The DFT and MP2 calculations were performed with the GAUSSIAN 09 program.32 The CC2 and CASPT2 calculations, in turn, were performed with the TURBOMOLE 6.333,34 and MOLCAS 7.635 programs, respectively. Further details on the quantum chemical calculations are given in the ESI.†
2.4 Crystallographic refinement
CNS solve V1.336 was used to refine the β-crustacyanin structure (PDB code: 1GKA and r1gkasf6) in its complex with the two astaxanthin molecules in the hydroxyketone form and the enolate form. For details, see the ESI.†3. Results and discussion
3.1 Spectroscopic studies of the behavior of 3-hydroxy-4-oxo-β-ionone
The UV-vis spectra of dilute methanolic solutions of 2 exhibit a λmax at 272 nm (εmax = 5.1 × 103 dm3 mol−1 cm−1), with a low-intensity shoulder extending out to 394 nm. Solutions in pure water show only a small shift (λmax = 275 nm), and no change in either extinction coefficient or shape of the absorption peak. When dilute aqueous solutions are prepared by addition of small aliquots (20 μl) of more concentrated methanolic solutions to water, the spectra, after the mixing period (ca. 20 seconds), are stable and show no changes comparable to those observed by Giovannetti, et al.17 for similar aqueous dilutions of astaxanthin. Thus, we conclude that the smaller hydroxyketone 2 does not participate in aggregate formation at the concentrations (ca. 10−4 M) used in these experiments.Spectra of the aqueous solutions were also unchanged over long periods after acidification with dilute hydrochloric acid. Addition of aqueous NaOH solution, with cells maintaining a nitrogen atmosphere over the solution, enabled preparation and monitoring of basic solutions of 2 in water containing 0.7% (vol/vol) methanol. A typical sequence of spectra, following addition of 2 to a 0.2 M aqueous NaOH solution, is shown in Fig. 1a.
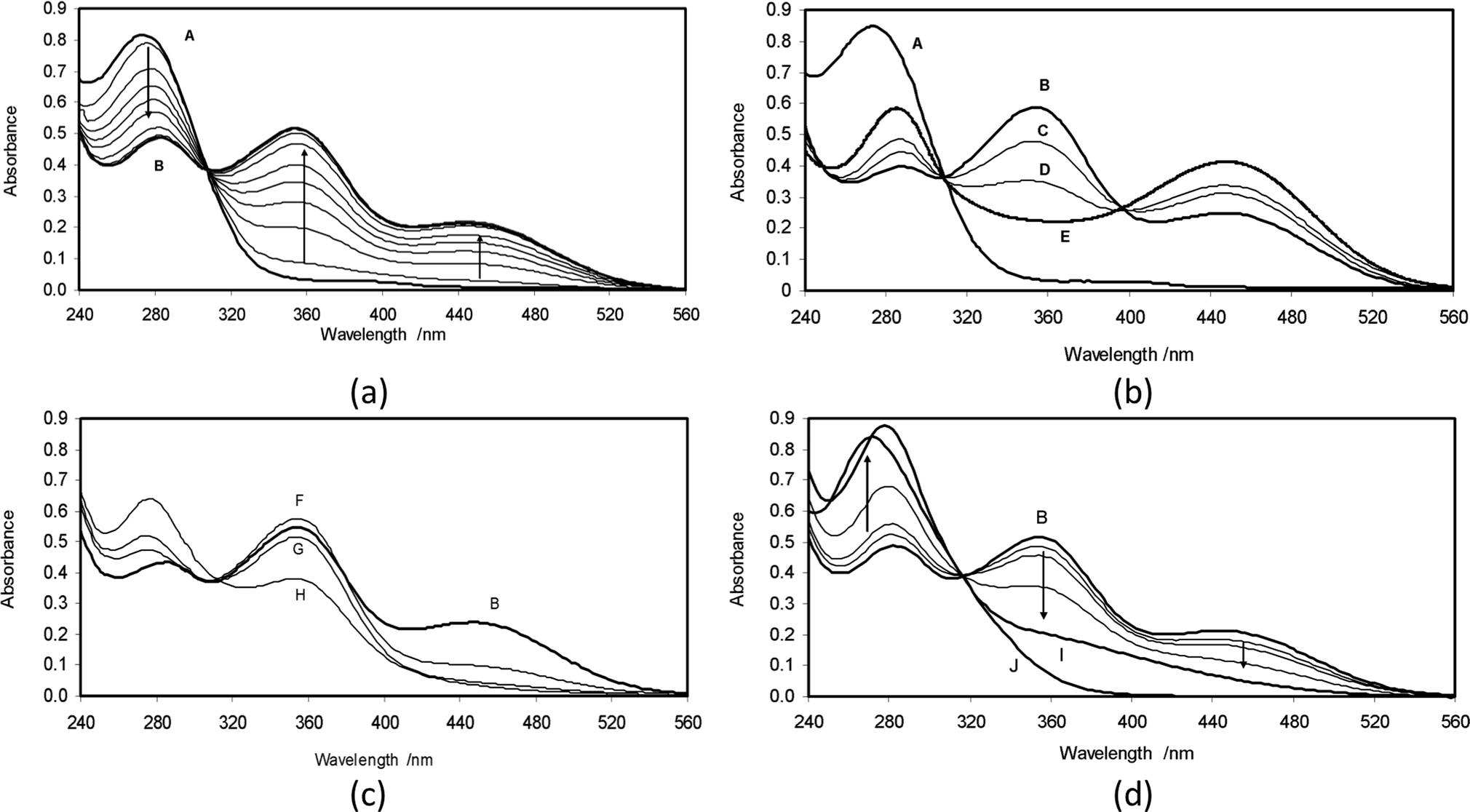 | ||
| Fig. 1 Solution spectra of 3-hydroxy-4-oxo-β-ionone (2): (a) evolution of UV-vis spectra on addition of 2 to a deoxygenated 0.2 M aqueous NaOH solution at 30 °C. Trace A is the initial spectrum of 2 with λmax at 275 nm. Over about 45 minutes, this spectrum equilibrates to trace B; (b) comparison of UV-vis spectra at 30 °C of 2 non-equilibrated (trace A) and equilibrated (trace B = pH 13.2) in 0.2 M aqueous NaOH with those in more concentrated NaOH solutions (trace C = pH 13.3, trace D = pH 13.7, trace E = pH 14.5); (c) comparison of UV-vis spectra at 30 °C of 2 equilibrated in 0.2 M aqueous NaOH (trace B = pH 13.2) with those arising when solutions are acidified (trace F = pH 12.6, trace G = pH 12.1, trace H = pH 10.4); (d) evolution of UV-vis spectra at 30 °C of 2 equilibrated in 0.2 M aqueous NaOH (trace B = pH 13.2) following opening of the cell to air (after 65 minutes, trace I), followed by acidification (pH 4, trace J). | ||
In Fig. 1a, trace A is the initial spectrum of 2 with λmax at 275 nm. Over about 45 minutes, this spectrum evolves to trace B, by decay of the peak at 275 nm and development of new maxima at 278, 356 and 454 nm (the longest-wavelength new maximum thus presents a bathochromic shift of no less than 180 nm relative to the parent material). The sequence displays a tight isosbestic point at 308 nm, and sections at any of the new maxima show that the absorbance changes are first order, with a rate constant kobs = 9.7 × 10−4 s−1. Spectra at this stage were stable for at least one hour, with slow decays associated with leakage of atmospheric oxygen into the cell (see further below). At this stage also, acidification of the solution to pH 4 by addition of the calculated amount of acetic acid quickly regenerated, with no detectable intermediate stages, the original spectrum (trace A). Furthermore, chromatography of an extract confirmed the recovery of 2 and showed only minor traces of other materials.
The spectroscopic changes, A → B (Fig. 1a), are consistent with the formation of either of the anions 2− and 2-dianion, or indeed of a rapidly equilibrating mixture thereof. Properly distinguishing between these possibilities led to examining spectra after equilibration in buffered basic solutions over as wide a pH range as possible. To avoid prolonged equilibration times and reproducibility issues due to slow infusion of atmospheric oxygen into the media, the solutions were equilibrated in 0.2 M NaOH, (spectrum B in Fig. 1a), and then the spectra were examined after adjustment of pH in both acidic and basic directions.
The results of increasing the basicity of the medium are shown in Fig. 1b. While the original trace A is included for comparison, the starting point for the chemistry here is trace B, corresponding to equilibrated 2− and 2-dianion species in a 0.2 M NaOH solution at pH 13.2. Traces C and D arose from sequential additions of highly concentrated NaOH, increasing the basicity firstly to pH 13.3 and then to pH 13.7. In both cases, spectra were corrected for small volume changes associated with the additions. In these spectra, the peaks at 278 and 454 nm increase with increasing basicity, whereas the peak at 356 nm decreases, with a new isosbestic point appearing at 395 nm. These changes are consistent with a situation in which, in trace B, the peak at 356 nm is associated with 2− and the peaks at 278 and 454 nm with the 2-dianion species. In a further experiment, 2 was equilibrated directly with 2 M NaOH (pH 14.5), which produced a spectrum that after scaling to match the 395 nm isosbestic point is shown as trace E in Fig. 1b. Despite the large variations in ionic strength, trace E falls into the pattern shown by traces B, C and D. A near superimposable trace was again produced when 1.5 M NaOH was used, indicating that at pH 14.5, 2 is almost fully converted to its 2-dianion form.
The results of decreasing the basicity of the medium are shown in Fig. 1c. Again, the starting point is the equilibrated state of 2− and 2-dianion species (trace B). Addition of measured amounts of neat trifluoroethanol, pKa = 12.4,37 then provided solutions with some buffering at pH > 12. In trace F (pH = 12.6), the original peak at 454 nm associated with 2-dianion decreased substantially, while the original peak at 356 nm associated with 2− increased slightly. Trace G (pH = 12.1) shows some small depletion of 2− and almost complete loss of the 2-dianion. For a more acidic medium yet, a portion of solid sodium bicarbonate, calculated to produce a solution buffered at pH 10.4, was added to a freshly equilibrated solution of 2 in 0.2 M aqueous NaOH. After small scaling of the spectrum to match the isosbestic point, this yielded trace H, in which it is clear that the 356 nm peak has dropped to about half its original value and there has been substantial growth of the 272 nm peak associated with 2 itself. Further acidifications continue the trend of reversion to the spectrum of 2.
The spectra and responses to pH changes presented in Fig. 1a–c are fully consistent with the scheme of deprotonations and proton transfers shown above in Scheme 2, where: (i) deprotonation of 2 at C3 followed by proton transfer from O3 to O4 yield 2−, with a sharing of the negative charge by conjugation between O3 and O9; (ii) reprotonation at O3 yields the ene-diol tautomer 2ed; and (iii) deprotonation of the enolic hydroxyl group of 2− produces 2-dianion. The spectra also permit initial estimates of the dissociation constants Ka(1) and Ka(2) defined in Scheme 2. From Fig. 1c, a comparison of traces H and F suggests that at pH 10.4, there is a ca. 50% conversion of 2 to 2−, with little further conversion to the 2-dianion. In that case, application of the Henderson–Hasselbach equation to the equal concentrations of 2 and 2− gives pKa(1) ≈ 10.4. Similar inspection of Fig. 1b, comparing traces B and E, suggests that at pH 13.2, the equilibrated solution contains near equal amounts of 2− and 2-dianion, with little residual un-ionized 2, so that pKa(2) ≈ 13.2. These values of the dissociation constants might be compared with the corresponding values of 4.12 (pKa(1)) and 11.51 (pKa(2)) for ascorbic acid.38
The preceding analysis providing estimates of Ka(1) and Ka(2) does not take into account the possibility that 2 can also be ionized to form its alkoxide, by deprotonation of the secondary alcohol (Scheme 3a), a process expected to have little effect on the UV-vis spectrum of 2. However, since this process involves equilibration of an oxygen acid, the associated relaxation in either acid or base in aqueous medium should be fast39 compared to that for the deprotonation leading to 2−, irrespective of thermodynamic acidities. The value of 10.4 for pKa(1) extracted above is not then simply that for dissociation of 2 as a carbon acid. Rather, any appreciable alkoxide formation would call for a correction according to the equation included in Scheme 3a, whereby the apparent ionization constant will be an underestimate of the true value, i.e., pKa(C3–H) < pKa(1).
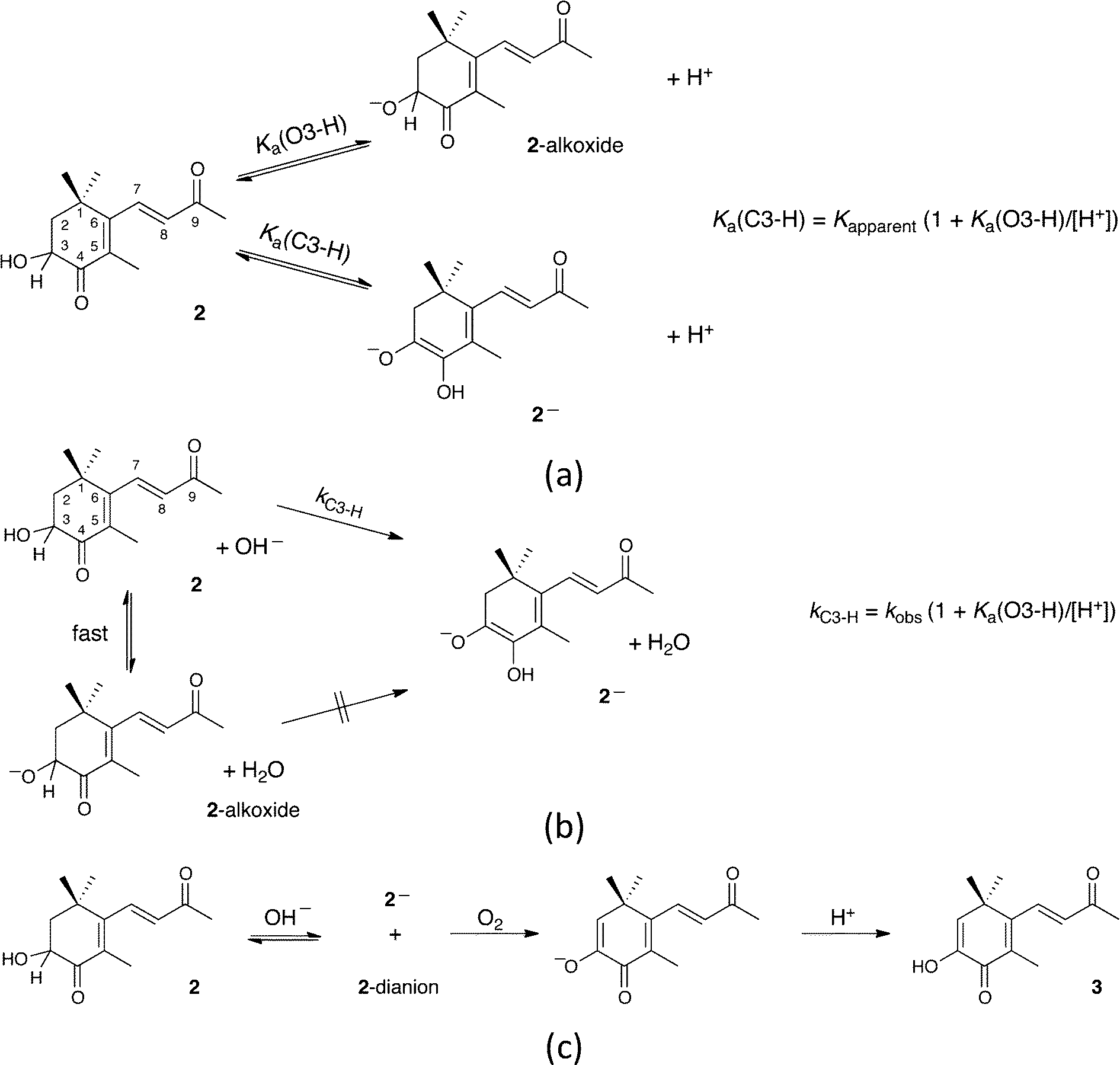 | ||
| Scheme 3 Equilibria of 3-hydroxy-4-oxo-β-ionone (2): (a) competing O- and C-dissociations; (b) the effect of fast equilibration with alkoxide on the observed rate constant for enolate formation; (c) the action of oxygen on basic solutions. | ||
For simple secondary aliphatic alcohols (16 < pKa(O3–H) < 16.5), extents of alkoxide formation would be small, even in strongly basic aqueous solutions, and the discrepancy between apparent and actual carbon acid dissociation constants would be undetectably small. The secondary alcohol in 2, however, is in a polar environment, and empirical additivity relationships (based on a very sparse data set) have yielded a relationship40 between acidity and Taft's polar substituent parameters (σ*) which, using σ* = 2.0 for –COCH = CH2,41 gives pKa(O3–H) ≈ 13.1 for 2. Even with this value, the difference between apparent and actual carbon acid dissociation constants would be small.
However, the uncertainty in the estimate of pKa(O3–H) is large, but while we have no direct experimental measurement, the kinetics of formation of 2− indicates that the value may be much higher than 13.1. This is also found by the quantum chemical calculations further described below. Earlier, we noted that the formation of 2− and 2-dianion mixture in 0.2 M NaOH show first-order behavior with kobs = 9.7 × 10−4 s−1. Assuming that the reaction is also first order in hydroxide, the second-order rate constant for the reaction is then 4.9 × 10−3 M−1 s−1. Comparison with the second-order rate constants for deprotonation (by hydroxide) at 25 °C of more familiar carbon acids such as nitromethane (pKa = 10.2) and acetylacetone (pKa = 8.9) of 2.76 × 10 M−1 s−1 (ref. 42) and 3.98 × 104 M−1 s−1,43 respectively, makes it very clear that carbon deprotonation of 2 is >103 slower than for a carbon acid of similar acidity. Nothing obvious in the structure of 2 accounts for the low reactivity, but if, under the conditions, 2 existed largely as its alkoxide and did not rearrange or react with hydroxide to yield 2−, the rate constant found here would need to be scaled upwards as shown in Scheme 3b to yield a true value, kC3–H, for the second-order rate constant for carbon deprotonation of 2 by hydroxide. If the correction factor kC3–H/kobs is indeed ca. 103, it is possible to work out an estimate of 10.0 for the true pKa(C3–H) value of 2 (cf. with the above estimate of 10.4 for the pKa(1) value of 2).
3.2 The oxidation chemistry
As noted earlier, the basic solutions of 2 are extremely sensitive to oxygen. In view of possible parallels with the reported reactions of astaxanthin with oxygen in basic medium,44 we have examined this behavior (Fig. 1d). Trace B evolves, after opening to air following zero-order kinetics over 65 minutes, to yield spectrum I. Further changes then become extremely slow with spectrum I persisting indefinitely if the cell is again closed. Qualitatively, zero-order rates are proportional to hydroxide concentration so that 2-dianion is implicated as the reactive species. After acidification to pH 4, there is an immediate change to yield spectrum J.To identify the oxidation products, a large-scale reaction was carried out. Chromatography showed formation of a single new product. After acidification of the solution, a crystalline product was isolated, molecular formula C13H16O3, thus a simple oxidation product of 2. Revealingly, proton NMR spectroscopy confirmed loss of the ABX signal, associated with the C2 and C3 hydrogens of 2 and its replacement by a single one-proton signal at δ 6.04. Geminal methyl groups now show a single signal indicating equivalent environments. The spectrum also shows a broad 1H signal at δ 6.28, assigned to a hydrogen bonded OH, whose presence was confirmed by IR spectroscopy. The analytical data are consistent with this oxidation product being the enol 2,3-dehydro-3-hydroxy-4-oxo-β-ionone (3 in Scheme 3c), bearing the same relationship to 2 as astacene bears to astaxanthin (with which it often co-occurs1). The UV-vis spectra of 3 in neutral and basic aqueous solution were superimposable on traces J and I in Fig. 1d. Responses of spectra to changes in pH were immediate, and the behavior in borate buffers gave pKa = 9.9. The chemistry is summarized in Scheme 3c in which we suggest that atmospheric oxygen reacts with 2-dianion, yielding the enolate of 3 and probably hydrogen peroxide, as its anion.
3.3 Quantum chemical calculations on the model system
Since similar aqueous solution experiments with astaxanthin itself are not possible, due to aggregate formation and the low solubility of astaxanthin in water, we have performed complementary TD-DFT calculations with seven different density functionals, first establishing the ability of these to accurately reproduce the UV-vis spectroscopic behavior of our model compound 2 (see Table S1 of the ESI†). Notably, the methods to be used for the full astaxanthin system below (M06-HF, LC-ωPBE and LC-BLYP) reproduce the experimental λmax of 2 with an accuracy of ∼10 nm (∼0.15 eV) for the hydroxyketone and di-anion forms (2 and 2-dianion, Scheme 2), and are in qualitative accord with all the experimental UV-vis spectra in that they predict a substantial red shift in the absorption upon formation of the mono-anion from the parent hydroxyketone. Notice, however, that the calculations give larger shifts than that observed experimentally.In light of this discrepancy, we have quite exhaustively investigated whether factors that may have a negative impact on the accuracy of TD-DFT excitation energies are at play. This investigation, which is fully described in Sections 4.1–4.5 of the ESI† and presented in Tables S1 and S3–S9, includes a comparison of TD-DFT results with CC2 and CASPT2 ab initio results, an assessment of how molecular geometries and choice of basis set influence the calculated λmax, and additional benchmark calculations on the oxidation product of 2 (2,3-dehydro-3-hydroxy-4-oxo-β-ionone) and on 2,3-dihydroxy-2-propenal, which contains the minimum carbon framework in which conjugation between a carbonyl group and an ene-diol may occur. Overall, this investigation shows that TD-DFT calculations constitute a reliable tool for obtaining a qualitatively accurate assessment of how acid–base chemistry influences the UV-vis spectroscopic behavior of astaxanthin and models thereof, and strongly indicates that more elaborate ab initio calculations would support the same conclusions.
Describing the solvent using a hybrid cluster-continuum approach,45 we also calculated the pKa dissociation constants of 2 (Table S2, ESI†). Since most of these pKa values are available from experiment, the calculations serve both as a test of the experimental predictions and a validation of the computational accuracy of DFT for the present systems, with implications also for the excited-state calculations discussed below.
Firstly, it can be seen that the experimentally measured pKa of 10.4 for deprotonation of the hydroxyketone at C3 (pKa(C3–H) in Table S2 (ESI†) and Scheme 3; pKa(1) Scheme 2) to form the mono-anion is reproduced very well, and that the calculated estimates (ranging between 7.7 and 10.6) are not particularly sensitive to the number of explicit water molecules included in the modeling. Given that we propose a mechanism for the bathochromic shift in crustacyanin based on formation of the mono-anion of astaxanthin, it is also notable that this pKa is fairly small. From calculations on the ketone form lacking the C3 hydroxyl group (4-oxo-β-ionone), it is predicted that the smallness of pKa(C3–H) for the hydroxyketone is due precisely to the C3 hydroxyl group, because without this group, the calculated pKa(C3–H) values amount to ∼20 or even higher.
The calculations further predict that pKa(C3–H) for the hydroxyketone is considerably smaller than the pKa for deprotonation of the C3 hydroxyl group to form the alkoxide of the hydroxyketone (pKa(O3–H) in Table S2, ESI†). Thus, alkoxide formation should not impact on mono-anion formation. Although the estimates of pKa(O3–H) vary somewhat with the number of water molecules, the difference between pKa(O3–H) and pKa(C3–H) appears to converge to a value of around 10 in the calculations.
In addition to pKa values of the hydroxyketone, we have also calculated the pKa for deprotonation of the C4 hydroxyl group of the mono-anion (pKa(O4–H) in Table S2 (ESI†); pKa(2) in Scheme 2) to form the di-anion. This pKa has also been determined experimentally, and pleasingly, the calculated values approach (23.6 → 17.9 → 14.5) the experimental estimate (13.2) to within close to 1 pKa unit as the number of water molecules is increased.
3.4 Quantum chemical calculations on different forms of astaxanthin
The UV-vis λmax of the parent keto form of astaxanthin (1, Scheme 1) was calculated using TD-DFT,24–26 employing three different density functionals and two different basis sets. These results were then compared with the corresponding results obtained for the mono-enol, di-enol, mono-anion and di-anion forms (1ed, 1ded, 1−, and 1-dianion, Scheme 1). In a preceding step discussed in more detail in Section 4.1 of ESI,† the choice of density functionals for these calculations was established from analogous calculations on the 3-hydroxy-4-oxo-β-ionone model system (2, Scheme 2) and its enolic and anionic forms (2ed, 2−, and 2-dianion, Scheme 2).Starting with the keto form of astaxanthin, it can be seen from Table 1 that all three density functionals (M06-HF, LC-ωPBE and LC-BLYP) give, in the absence of effects linked to aggregation phenomena, very similar λmax for the water and methanol solvents, and that the experimental value (476 nm) pertaining to a water–methanol mixture is reproduced with ∼20 nm (∼0.1 eV) accuracy. It can also be seen that the relatively small 6-31G(d,p) basis set gives similar λmax as the larger aug-cc-pVDZ basis set, which reflects the pronounced valence character of the strongly absorbing “Bu-like” excited state. Indeed, the diffuse functions included in the aug-cc-pVDZ basis set red shifts the λmax by no more than 13 nm (0.07 eV).
| Method/basis setb | Solvent | Keto | Mono-enol | Di-enol | Mono-anion | Di-anion |
|---|---|---|---|---|---|---|
| a All calculations based on geometries optimized using B3LYP/6-31G(d,p) in combination with a CPCM description of the water or methanol solvent. b Basis set I = 6-31G(d,p); basis set II = aug-cc-pVDZ. c Solvent effects obtained using the cc-pVDZ basis set and added to gas-phase data obtained with the target aug-cc-pVDZ basis set. d Experimental value in a water–methanol mixture. e ε max given in units of dm3 mol−1 cm−1. | ||||||
| M06-HF/I, λmax (ΔE, f) | H2O | 457 (2.71, 4.39) | 463 (2.68, 4.48) | 459 (2.70, 4.56) | 660 (1.88, 4.04) | 790 (1.57, 4.84) |
| LC-ωPBE/I, λmax (ΔE, f) | H2O | 473 (2.62, 4.53) | 482 (2.57, 4.64) | 477 (2.60, 4.73) | 658 (1.88, 4.43) | 779 (1.59, 5.16) |
| LC-BLYP/I, λmax (ΔE, f) | H2O | 463 (2.68, 4.59) | 470 (2.64, 4.70) | 465 (2.66, 4.79) | 643 (1.93, 4.45) | 754 (1.64, 5.19) |
| M06-HF/II,cλmax (ΔE, f) | H2O | 470 (2.64, 4.31) | 476 (2.60, 4.37) | 471 (2.63, 4.44) | 666 (1.86, 4.17) | 816 (1.52, 4.67) |
| LC-ωPBE/II,cλmax (ΔE, f) | H2O | 482 (2.57, 4.47) | 490 (2.53, 4.55) | 484 (2.56, 4.64) | 665 (1.86, 4.45) | 796 (1.56, 4.99) |
| LC-BLYP/II,cλmax (ΔE, f) | H2O | 474 (2.62, 4.52) | 481 (2.58, 4.61) | 475 (2.61, 4.69) | 651 (1.90, 4.50) | 774 (1.60, 5.02) |
| M06-HF/I, λmax (ΔE, f) | MeOH | 456 (2.72, 4.39) | 463 (2.68, 4.48) | 459 (2.70, 4.56) | 668 (1.86, 4.05) | 791 (1.57, 4.84) |
| LC-ωPBE/I, λmax (ΔE, f) | MeOH | 473 (2.62, 4.52) | 481 (2.58, 4.64) | 477 (2.60, 4.73) | 666 (1.86, 4.45) | 779 (1.59, 5.16) |
| LC-BLYP/I, λmax (ΔE, f) | MeOH | 463 (2.68, 4.59) | 470 (2.64, 4.70) | 465 (2.67, 4.79) | 651 (1.90, 4.46) | 754 (1.64, 5.19) |
| Experimentdλmax (ΔE, εmaxe) | Mixture | 476 (2.61, 10000) |
— | — | — | — |
Turning to the calculations on the other forms of astaxanthin also summarized in Table 1, but for which experimental data are not available, we first observe that neither single nor double enolization is predicted to significantly shift the λmax of the parent keto form, excluding tautomerism alone in the protein-bound astaxanthins as the origin of the much larger observed bathochromic shift. Notably, however, the calculations show that deprotonation of C3 followed by proton transfer between the oxygen atoms attached to C3 and C4 (or, equivalently, single enolization followed by deprotonation of the C3 hydroxyl group) to form the mono-anion, 1−, is associated with a red shift, lying uniformly between 177 and 212 nm (0.71–0.86 eV) for all methods, thus larger than the bathochromic shift in β-crustacyanin (λmax at 570 nm) and consistent with that in α-crustacyanin (λmax at 630 nm). For the dianion (1-dianion), the calculations yield a bathochromic shift of the order of 300 nm, bringing the absorption into the near-infrared (λmax ≈ 750–800 nm).
3.5 Crystallographic refinement
Re-examination of the crystal structure of β-crustacyanin6 indeed confirms that enolate (1−) and enolic (1ed) forms are as compatible with the crystal structure as the hydroxyketone form 1. Bond angles and bond distances for the end rings and polyene chain were derived from the published models of astaxanthin, thus maintaining the 6-s-trans conformation around the C6–C7 and C6′–C7′ bonds46,47 (see also the ESI†).4. Conclusions
The experiments and calculations that we have presented show that our model system 2 behaves kinetically and thermodynamically as a pseudo-acidic indicator,39 with slow carbon deprotonation yielding an extended enolate stable enough to exist in water at pH ≈ 10. The ability of the computational methods to reproduce experimental observations for the model system validates their application to astaxanthin itself, 1, its tautomers 1ed and 1ded, and the enolate 1−, thus confirming that enolate formation is associated with a 177–212 nm (0.71–0.86 eV) bathochromic shift. Since the conjugation in astaxanthin is much more extensive than in the model compound we have studied, its colored enolate should be even more stable and accessible at lower pH. Additionally, deprotonation within the protein would yield the extended enolate bound in a pre-organized site, retained by hydrogen bonding between O3 and the histidine at one end, and between O4’ and the water–tyrosine pair at the other (i.e., in Scheme 1, B = His and HA = HOH + Tyr). The blue-black to pink-orange color change on cooking of lobster would then be explained by thermal denaturation of α-crustacyanin releasing the astaxanthin from its bound enolate form, permitting relaxation to its neutral α-hydroxyketone form.For this explanation to be valid chromophore–protein interactions should favor the enolate form over the α-hydroxyketone form. Thus, it would be of interest to investigate how specific short-range chromophore–protein interactions and long-range bulk electrostatic effects stabilize the various forms of astaxanthin inside the binding pocket of β-crustacyanin. While a detailed analysis of such interactions is beyond the scope of this work, it has previously been suggested, based on quantum chemical calculations,48 that the most proximal protein residues do not interact strongly with the neutral α-hydroxyketone form, which at least does not go against the idea that the protein shifts the equilibrium between enolate and α-hydroxyketone toward the former. Furthermore, this study also showed that the absorption of the α-hydroxyketone form is rather insensitive to short-range chromophore–protein interactions, which suggests that a more severe perturbation of the chromophore is needed to explain the bathochromic shift in the protein. Finally, preliminary B3LYP calculations that we have performed indicate that the histidine shifts the equilibrium between enolate and α-hydroxyketone by at least 8 kJ mol−1 toward the former species.
Astaxanthin is not unique amongst carotenoids in its ability to form colored complexes with the crustacyanin protein, but the overwhelming majority of color-forming carotenoids (including actinioerythrol, violerythrin, iso-norastaxanthin, astacene, 19,19′-dinorastacene, 4-hydroxy-4′-oxo-β-β-carotene, adonirubin, 7,8-didehydroastaxanthin and 15,15′-didehydroastaxanthin) possess structures with a fully conjugated polyene chain,2,49 linking oxygenated terminal rings with relatively acidic enolic or enolizable sites. Binding of carotenoids as an extended enolate within the protein may thus be a mechanism of some generality for formation of these spectacularly colored complexes in nature.
Acknowledgements
We thank Professor P. Zagalsky for invaluable comments and discussion. This work was supported by Bildungs- und Kulturdepartement des Kantons Luzern, Switzerland (A.H.), the Swedish Research Council (B.D.), the Olle Engkvist Foundation (B.D.), the Nuffield Foundation (S.B.), the School of Chemistry, University of Manchester (J.R.H., M.H., A.R., I.W.) and the European Molecular Biology Laboratory (M.C.).References
- I. Higuera-Ciapara, L. Félix-Valenzuela and F. M. Goycoolea, Crit. Rev. Food Sci. Nutr., 2006, 46, 185–196 CrossRef CAS PubMed.
- G. Britton, R. J. Weesie, D. Askin, J. D. Warburton, L. Gallardo-Guerrero, F. J. Jansen, H. J. M. de Groot, J. Lugtenburg, J.-P. Cornard and J.-C. Merlin, Pure Appl. Chem., 1997, 69, 2075–2084 CrossRef CAS.
- C. D. Dellisanti, S. Spinelli, C. Cambillau, J. B. C. Findlay, P. F. Zagalsky, S. Finet and V. Receveur-Brechot, FEBS Lett., 2003, 544, 189–193 CrossRef CAS.
- N. E. Chayen, M. Cianci, J. G. Grossmann, J. Habash, J. R. Helliwell, G. A. Nneji, J. Raftery, P. J. Rizkallah and P. F. Zagalsky, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2003, 59, 2072–2082 Search PubMed.
- N. H. Rhys, M.-C. Wang, T. A. Jowitt, J. R. Helliwell, J. G. Grossmann and C. Baldock, J. Synchrotron Radiat., 2011, 18, 79–83 CAS.
- M. Cianci, P. J. Rizkallah, A. Olczak, J. Raftery, N. E. Chayen, P. F. Zagalsky and J. R. Helliwell, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9795–9800 CrossRef CAS PubMed.
- J. R. Helliwell, Crystallogr. Rev., 2010, 16, 231–242 CrossRef CAS.
- A. A. C. van Wijk, A. Spaans, N. Uzunbajakava, C. Otto, H. J. M. de Groot, J. Lugtenburg and F. Buda, J. Am. Chem. Soc., 2005, 127, 1438–1445 CrossRef CAS PubMed.
- A. Strambi and B. Durbeej, J. Phys. Chem. B, 2009, 113, 5311–5317 CrossRef CAS PubMed.
- N. Christensson, K. Žídek, N. C. M. Magdaong, A. M. LaFountain, H. A. Frank and D. Zigmantas, J. Phys. Chem. B, 2013, 117, 11209–11219 CrossRef CAS PubMed.
- R. Kuhn and N. A. Sörensen, Angew. Chem., 1938, 51, 465–466 CrossRef CAS.
- M. Buchwald and W. Jenks, Biochemistry, 1968, 7, 844–859 CrossRef CAS.
- Y. Abe, H. Horii, S. Taniguchi, K. Kamai and M. Takagi, Bull. Chem. Soc. Jpn., 1983, 56, 467–470 CrossRef CAS.
- M. M. T. Khan and A. E. Martell, J. Am. Chem. Soc., 1967, 89, 7104–7111 CrossRef CAS.
- V. R. Salares, N. M. Young, H. J. Bernstein and P. R. Carey, Biochim. Biophys. Acta, 1979, 576, 176–191 CrossRef CAS.
- C. Köpsel, H. Möltgen, H. Schuch, H. Auweter, K. Kleinermanns, H.-D. Martin and H. Bettermann, J. Mol. Struct., 2005, 750, 109–115 CrossRef PubMed.
- R. Giovannetti, L. Alibabaei and F. Pucciarelli, Spectrochim. Acta, Part A, 2009, 73, 157–162 CrossRef PubMed.
- E. Becher, R. Albrecht, K. Bernhard, H. G. W. Leuenberger, H. Mayer, R. K. Müller, W. Schüep and H. P. Wagner, Helv. Chim. Acta, 1981, 64, 2419–2435 CrossRef CAS.
- F. D. Negra, G. Santoni, M. Stivanello, M. Soukup and M. Fachini, PCT Int. Appl., WO2007072529, A2 20070628, 2007 Search PubMed.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- O. Christiansen, H. Koch and P. Jørgensen, Chem. Phys. Lett., 1995, 243, 409–418 CrossRef CAS.
- C. Hättig, J. Chem. Phys., 2003, 118, 7751–7761 CrossRef PubMed.
- M. A. L. Marques and E. K. U. Gross, Annu. Rev. Phys. Chem., 2004, 55, 427–455 CrossRef CAS PubMed.
- A. Dreuw and M. Head-Gordon, Chem. Rev., 2005, 105, 4009–4037 CrossRef CAS PubMed.
- M. E. Casida, J. Mal. Struct.: THEOCHEM, 2009, 914, 3–18 CrossRef CAS PubMed.
- Y. Zhao and D. G. Truhlar, J. Phys. Chem. A, 2006, 110, 13126–13130 CrossRef CAS PubMed.
- O. A. Vydrov and G. E. Scuseria, J. Chem. Phys., 2006, 125, 234109 CrossRef PubMed.
- O. A. Vydrov, J. Heyd, A. V. Krukau and G. E. Scuseria, J. Chem. Phys., 2006, 125, 074106 CrossRef PubMed.
- H. Iikura, T. Tsuneda, T. Yanai and K. Hirao, J. Chem. Phys., 2001, 115, 3540–3544 CrossRef CAS PubMed.
- K. Andersson, P.-Å. Malmqvist and B. O. Roos, J. Chem. Phys., 1992, 96, 1218–1226 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, USA, 2009 Search PubMed.
- TURBOMOLE V6.3 2011, a development of University of Karlsruhe and Forschungszentrum Karlsruhe GmbH, 1989–2007, TURBOMOLE GmbH, since 2007; available at: http://www.turbomole.com, accessed July 25, 2013.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- F. Aquilante, L. D. Vico, N. Ferré, G. Ghigo, P.-Å. Malmqvist, P. Neogrády, T. B. Pedersen, M. Pitoňák, M. Reiher, B. O. Roos, L. Serrano-Andrés, M. Urban, V. Veryazov and R. Lindh, J. Comput. Chem., 2010, 31, 224–247 CrossRef CAS PubMed.
- A. T. Brünger, Nat. Protoc., 2007, 2, 2728–2733 CrossRef PubMed.
- C. H. Arrowsmith, A. J. Kresge and Y. C. Tang, J. Am. Chem. Soc., 1991, 113, 179–182 CrossRef CAS.
- N. Kagayama, M. Sekiguchi, Y. Inada, H. D. Takagi and S. Funahashi, Inorg. Chem., 1994, 33, 1881–1885 CrossRef CAS.
- J. E. Crooks, Proton Transfer Reactions, Chapman and Hall, London, 1975 Search PubMed.
- S. Takahashi, L. A. Cohen, H. K. Miller and E. G. Peake, J. Org. Chem., 1971, 36, 1205–1209 CrossRef CAS.
- W. Adcock, J. Alste, S. Q. A. Rizvi and M. Aurangzeb, J. Am. Chem. Soc., 1976, 98, 1701–1711 CrossRef CAS.
- R. P. Bell and D. M. Goodall, Proc. R. Soc. London, Ser. A, 1966, 294, 273–297 CrossRef CAS.
- R. Brouillard and J.-E. Dubois, J. Org. Chem., 1974, 39, 1137–1142 CrossRef CAS.
- R. Kuhn and N. A. Sörensen, Ber. Dtsch. Chem. Ges. B, 1938, 71, 1879–1888 CrossRef.
- J. R. Pliego Jr and J. M. Riveros, J. Phys. Chem. A, 2002, 106, 7434–7439 CrossRef.
- G. Bartalucci, S. Fisher, J. R. Helliwell, M. Helliwell, S. Liaaen-Jensen, J. E. Warren and J. Wilkinson, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 238–247 CAS.
- G. Bartalucci, J. Coppin, S. Fisher, G. Hall, J. R. Helliwell, M. Helliwell and S. Liaaen-Jensen, Acta Crystallogr., Sect. B: Struct. Sci., 2007, 63, 328–337 CAS.
- J. Neugebauer, J. Veldstra and F. Buda, J. Phys. Chem. B, 2011, 115, 3216–3225 CrossRef CAS PubMed.
- W. L. Lee and P. F. Zagalsky, Biochem. J., 1966, 101, 9c–11c CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Complementary material for compound preparation and characterization and details and results of the quantum chemical calculations. See DOI: 10.1039/c4cp06124a |
| ‡ Pfaffenlohweg 29, CH-4125 Riehen, Switzerland. |
| This journal is © the Owner Societies 2015 |