
Native silica nanoparticles are powerful membrane disruptors
Hend I.
Alkhammash
ab,
Nan
Li
a,
Rémy
Berthier
a and
Maurits R. R.
de Planque
*a
aElectronics and Computer Science & Institute for Life Sciences, University of Southampton, Southampton, SO17 1BJ, UK. E-mail: mdp@ecs.soton.ac.uk
bDepartment of Physics, Faculty of Science, Taif University, Taif, Saudi Arabia
First published on 19th January 2015
Abstract
Silica nanoparticles are under development for intracellular drug delivery applications but can also have cytotoxic effects including cell membrane damage. In this study, we investigated the interactions of silica nanospheres of different size, surface chemistry and biocoating with membranes of phosphatidylcholine lipids. In liposome leakage assays many, but not all, of these nanoparticles induced dose-dependent dye leakage, indicative of membrane perturbation. It was found that 200 and 500 nm native-silica, aminated and carboxylated nanospheres induce near-total dye release from zwitterionic phosphatidylcholine liposomes at a particle/liposome ratio of ∼1, regardless of their surface chemistry, which we interpret as particle-supported bilayer formation following a global rearrangement of the vesicular membrane. In contrast, 50 nm diameter native-silica nanospheres did not induce total dye leakage below a particle/liposome ratio of ∼8, whereas amination or carboxylation, respectively, strongly reduced or prevented dye release. We postulate that for the smaller nanospheres, strong silica–bilayer interactions are manifested as bilayer engulfment of membrane-adsorbed particles, with localized lipid depletion eventually leading to collapse of the vesicular membrane. Protein coating of the particles considerably reduced dye leakage and lipid bilayer coating prevented dye release all together, while the inclusion of 33% anionic lipids in the liposomes reduced dye leakage for both native-silica and aminated surfaces. These results, which are compared with the effect of polystyrene nanoparticles and other engineered nanomaterials on lipid bilayers, and which are discussed in relation to nanosilica-induced cell membrane damage and cytotoxicity, indicate that a native-silica nanoparticle surface chemistry is a particularly strong membrane interaction motif.
Introduction
The spatial localization of biochemical reactions at a cellular and sub-cellular level is maintained by membranes that contain ion channels and transporters for tightly controlled uptake and export of ions and molecules, while larger biomolecules can traverse membranes by receptor-mediated endocytosis or exocytosis. In recent years, it has become evident that a wide variety of inorganic nanoparticles, without specific biomolecular receptor interactions, can also traverse membranes.1–3 These engineered nanomaterials (ENMs) have favorable properties for biomedical applications such as subcellular imaging, localized phototherapy and targeted drug delivery,4,5 with approximately 250 nanoparticle-based medicines already being approved or in clinical trials.6The exact mechanism by which ENMs translocate over membranes is not known and it may be different for different particles. ENMs tend to be observed in endosomes, indicating an activation of endocytosis mechanisms that does not require specific biomolecular interactions with receptor domains on the cell surface.1,2 However, passive uptake of ENMs, presumably by direct penetration of the membrane, has also been reported.1,7 An intimate contact between a nanoparticle and the membrane is required for cellular uptake of biomedical ENMs by endocytosis mechanisms or by direct translocation,5,8,9 but nanoparticle–membrane interactions may also have bioadverse effects such as an increase of the membrane's permeability to ions and metabolites or even a complete disintegration of the membrane.10,11
Most of our knowledge on ENM–membrane interactions derives from cell cultures, either in the form of microscopy images that show nanoparticle–membrane contact and particle endocytosis,1,2 or as biochemical assays that indirectly probe membrane damage.12–15 However, microscopy does not give information on membrane permeability and biochemical assays, which probe the presence of molecules in the extracellular medium which should have been retained in the cytosol (or vice versa), cannot determine whether the compromised membrane integrity that enabled release or uptake of these molecules is a primary effect of ENM–membrane interactions or a secondary effect of ENM-induced cellular metabolism dysfunction.12,13 Another concern is that ENMs tend to acquire a complex dynamic protein corona in biological solutions, which changes the surface properties of the particles.16–18
The physicochemical properties of ENMs, including size, shape, surface chemistry and surface morphology, determine the particle dispersion state in biological solutions and the biophysicochemical interactions that occur at the nano–bio interface.11 A systematic characterization of these interactions, which are still poorly understood, will facilitate the design of biomedical ENMs that can associate with and translocate across the membrane without compromising its barrier function.10,19 Research on natural membrane-targeting compounds such as protein and peptide toxins and amyloid particulates has historically employed lipid-only model membranes to investigate toxin–membrane interactions at a molecular level. These systems offer controlled conditions, i.e. a limited number of biomolecular components, and are amenable to biophysical methods that measure, for example, lipid order or membrane permeability.20–26
In recent years, the lipid bilayer methodology has started to be applied to the study of ENM–membrane interactions,27,28 primarily in the context of establishing structure–function relationships for nanotoxicology assessment.29,30 For example, electrical measurements of ENM-exposed suspended bilayers have shown that quantum dots, carbon nanotubes and polystyrene or silica nanospheres render bilayers permeable to ions, indicating a nanoparticle-induced perturbation of the bilayer structure.31–35 It has also been visualized with atomic force microscopy that cationic dendrimers and silica nanospheres can create holes in mica-supported bilayers,36 and fluorimetry measurements have demonstrated that titanium dioxide and silica nanospheres, cationic and anionic gold nanoparticles and protein-coated carbon nanotubes are all able to induce the release of fluorescent dyes from lipid vesicles.37–40 But because most studies only concern a small number of ENM species, structure–function relationships remain elusive. However, a cationic surface chemistry is commonly identified as a membrane-perturbing motif for ENMs.8,36,41–44
In the present study we have employed the liposome leakage assay24,45–48 to probe the interactions with zwitterionic and net anionic vesicular bilayers of amorphous silica nanospheres with a diameter of ≥50 nm, the mesoporous analogs of which are under development for targeted drug delivery.49–51 Silica nanospheres of various size and surface chemistry, including biocoating with proteins and lipid bilayers, were investigated. We observed distinct differences in the amount of nanoparticle-induced dye release, at a range of particle/liposome ratios, with a pronounced influence of nanoparticle surface chemistry for the smaller particles and of nanoparticle biocoating for all the particles. Furthermore, net anionic liposomes exhibited decreased particle-induced dye release, indicative of weaker particle–bilayer interactions, in comparison to zwitterionic liposomes. It is postulated that the 200 and 500 nm diameter particles induce total dye release from individual vesicles through the formation of a particle-supported bilayer with concomitant loss of the vesicle structure, while the 50 nm nanospheres gradually permeabilize the vesicular membrane through lipid depletion by bilayer engulfment of membrane-adsorbed nanospheres. For the smaller particles, which in terms of particle/liposome size ratio are most relevant for extrapolation to cell membranes, our data imply that a native-silica surface chemistry, rather than a cationic functionalization, presents the strongest nanosphere–membrane interaction motif.
Materials and methods
Materials
The lipids 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) were purchased from Avanti Polar Lipids (Alabaster, AL). Avidin, calcein and Sephadex-G50 were from Sigma Aldrich (St. Louis, MO), and sucrose, 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), sodium hydroxide, potassium chloride and 4-(1,1,3,3-tetramethylbutyl)phenyl-polyethylene glycol (Triton X-100) from Fisher Scientific (Loughborough, UK). Silica nanospheres with nominal diameters of 50, 100, 200 and 500 nm, either with a native, non-functionalized, silica surface or with an amine-, carboxyl- or avidin-functionalized surface were obtained from G. Kisker GbR (Steinfurt, Germany). The nanospheres were supplied as 25 or 50 mg mL−1 stock dispersions in surfactant-free (personal communication from manufacturer) deionized water. Polystyrene nanospheres with nominal diameters of 50, 100 and 200 nm, and carboxylated polystyrene particles of 200 nm diameter were from the same supplier. Mass concentrations of the particle dispersions were converted to number concentrations by assuming an ideal spherical geometry of the nanospheres and a density of 2.0 g cm−3 for silica or 1.05 g cm−3 for polystyrene particles.Dynamic light scattering
The hydrodynamic radius and the zeta potential of the silica nanospheres were characterized with a Zetasizer Nano ZS dynamic light scattering system with a 633 nm laser (Malvern Instruments, Malvern, UK) operating with a 173° backscatter angle for hydrodynamic radius measurements or with a 17° scatter angle for zeta potential determination. Nanosphere dispersions, diluted from deionized water stock solutions into a buffer solution of 100 mM KCl, 20 mM HEPES, pH 7.5 were measured in DTS1060 folded capillary cells (Malvern Instruments) at 25 °C. The functionalized and native-silica nanospheres with a nominal diameter of 50 nm were also investigated in phosphate-buffered KCl solutions of 2 mM ionic strength with pH values ranging from pH 2 to pH 14. Using Malvern Instruments software version 6.01, hydrodynamic radii were obtained by cumulants analysis of the correlation functions and the zeta potentials were derived from the measured electrophoretic mobilities with the Smoluchowski approximation.Liposome preparation
Calcein-loaded large unilamellar vesicles were prepared by hydration of a dry lipid film, vesicle extrusion and size exclusion chromatography. A dry film of 6.7 μmol total lipid was obtained by addition of a lipid–chloroform stock solution (either DOPC in chloroform or a mixture of DOPC and POPG at 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio in chloroform) to a glass vial, removal of chloroform by a stream of nitrogen gas and subsequent exposure to high vacuum. The lipid film was then hydrated with 1 mL of a 50 mM calcein solution, which had been adjusted to pH 7.5 with NaOH to ensure solubilization of the calcein crystals, which gave a dispersion of multilamellar vesicles. This dispersion was then passed 21 times through a polycarbonate membrane filter with a pore size of 400 nm (Whatman International Ltd, Maidstone, UK) with a mini-extruder from Avanti Polar Lipids, converting the multilamellar vesicles to large unilamellar vesicles. To separate the calcein-loaded vesicles from the free calcein molecules, a gel filtration step was performed. Approximately 750 μL of a suspension of Sephadex G50 beads in the leakage assay buffer (100 mM KCl, 500 mM sucrose, 20 mM HEPES, pH 7.5) was loaded in an empty 2 mL Zeba spin desalting column (Fisher Scientific) positioned in a 1.5 mL Eppendorf tube, which was centrifugated at 1000g for 3 minutes to remove excess buffer. Subsequently, 200 μL of the extruded liposome dispersion was added dropwise to the partially dried Sephadex bed and the column was centrifugated at 1000g for 45 s. The collected eluate was then applied to a second column with fresh partially dried Sephadex, and following centrifugation a new eluate was obtained. This process was repeated; when the sample had passed through four Sephadex columns, the final eluate (typically 40 μL of an orange-coloured dispersion) consisted of calcein-loaded large unilamellar vesicles without free calcein dye. These vesicles were stored at +4 °C and were used within two days.
:1 molar ratio in chloroform) to a glass vial, removal of chloroform by a stream of nitrogen gas and subsequent exposure to high vacuum. The lipid film was then hydrated with 1 mL of a 50 mM calcein solution, which had been adjusted to pH 7.5 with NaOH to ensure solubilization of the calcein crystals, which gave a dispersion of multilamellar vesicles. This dispersion was then passed 21 times through a polycarbonate membrane filter with a pore size of 400 nm (Whatman International Ltd, Maidstone, UK) with a mini-extruder from Avanti Polar Lipids, converting the multilamellar vesicles to large unilamellar vesicles. To separate the calcein-loaded vesicles from the free calcein molecules, a gel filtration step was performed. Approximately 750 μL of a suspension of Sephadex G50 beads in the leakage assay buffer (100 mM KCl, 500 mM sucrose, 20 mM HEPES, pH 7.5) was loaded in an empty 2 mL Zeba spin desalting column (Fisher Scientific) positioned in a 1.5 mL Eppendorf tube, which was centrifugated at 1000g for 3 minutes to remove excess buffer. Subsequently, 200 μL of the extruded liposome dispersion was added dropwise to the partially dried Sephadex bed and the column was centrifugated at 1000g for 45 s. The collected eluate was then applied to a second column with fresh partially dried Sephadex, and following centrifugation a new eluate was obtained. This process was repeated; when the sample had passed through four Sephadex columns, the final eluate (typically 40 μL of an orange-coloured dispersion) consisted of calcein-loaded large unilamellar vesicles without free calcein dye. These vesicles were stored at +4 °C and were used within two days.
Liposome leakage assay
A 4 mL quartz cuvette was filled with 2.5 mL of leakage assay buffer to which 1 μL of the suspension of calcein-loaded vesicles was added, except for the experiments with biocoated particles, where 0.5 μL was added. The cuvette was placed in a SLM 8100C fluorometer (Urbana, IL) where the assay solution was continuously stirred at room temperature. With an excitation wavelength of 490 nm and an emission wavelength set at 520 nm, a baseline fluorescence signal (intensity I0), was recorded. Subsequently, the appropriate amount of nanoparticles was added, typically as a ∼10 μL particle dispersion in deionized water, and any change in fluorescence intensity, a result of nanoparticle-induced dye release from the liposomes, was monitored for ∼10 min. Finally, 51 μL of a 100 mM solution of Triton X-100 was added to completely disrupt the vesicles and to determine the fluorescence intensity (Imax) for complete release of the self-quenching dye from the vesicles. For each time point of the fluorescence intensity curve (I(t)), the percentage of dye release from the vesicles was calculated as ((I(t) − I0)/(Imax − I0)) × 100. Multiple experiments were performed for selected nanoparticle–liposome combinations to verify the reproducibility of the measurements.Biocoating of nanoparticles
Silica nanospheres were coated with DOPC bilayers by a modification of the solvent-casting method described by Ross et al.52 Firstly, 40 μL of a 50 mg mL−1 dispersion of 200 nm silica nanospheres in deionized water was pipetted in a 2 mL glass vial, and the water was allowed to evaporate overnight. Subsequently, 100 μL of a 67 mM DOPC–chloroform solution was added to the dry particles, with 1–2 seconds of bath sonication to achieve a homogeneous slurry, from which the chloroform was allowed to evaporate overnight. The dry mixture of nanoparticles and lipids was then rehydrated with 1 mL of the leakage assay buffer, followed by vortex mixing and a 1–2 second bath sonication step to effectively suspend the lipid-coated particles. Finally, the particles were pelleted twice by 3 min centrifugation at 14000g and removal of the supernatant, which contains excess lipids, and the lipid-coated nanoparticles were resuspended in 40 μL of the leakage assay buffer. For protein coating, 40 μL of a 50 mg mL−1 dispersion of 100 nm silica nanospheres was mixed with 1 mL of a 1 mg mL−1 solution of avidin in phosphate buffered saline, followed by overnight incubation at room temperature. Next, the particles were pelleted twice by 3 min centrifugation at 14000g and removal of the supernatant, which contains non-bound protein, and the protein-coated nanoparticles were resuspended in 40 μL of the leakage assay buffer. Lipid and protein coating protocols were verified by the inclusion of fluorescently labeled lipid or avidin and by using larger silica particles, which can be observed by optical microscopy. Images of glass slide-deposited coated nanoparticles of 500 nm diameter showed co-localization of the fluorescence signal with the particles, while ∼100 μm diameter glass beads that had been coated using the above protocols showed a fluorescent outline indicative of full lipid or protein coverage.
Results
Silica nanosphere characterization
The silica nanosphere dispersions were characterized by dynamic light scattering and zeta potential measurements. Non-functionalized silica particles with nominal diameters of 50, 100, 200 and 500 nm were found to have hydrodynamic diameters of approximately 52, 107, 214 and 535 nm in a buffer of 20 mM HEPES, 100 mM KCl, pH 7.5, with polydispersity values ≤0.17 (see Table 1). Surface-functionalized, aminated or carboxylated, nanoparticles have similar hydrodynamic diameters (Table 1). It should be noted that throughout the manuscript we refer to the nominal particle diameters because these present, in approximation, the average diameters for the particles with different surface chemistries. At an ionic strength corresponding to the liposome leakage assay buffer, the zeta potentials for all the silica particles are approximately −30 mV (Table 1). The zeta potentials of the various particles at pH 7 and a low ionic strength (1 mM KCl) are all negative, with values ranging from −58 to −84 mV (data not shown), regardless of the surface functionalization. It is only at extreme pH values, i.e. pH < 5 and pH > 13, that the zeta potentials of non-functionalized, aminated and carboxylated nanospheres have significantly different values, and that the expected trend of larger zeta potentials for aminated particles and smaller zeta potentials for the carboxylated particles is observed, as depicted in Fig. 1 for 50 nm diameter nanospheres. This can be explained by a relatively low surface density of functionalized groups, with a large surface area of non-functionalized, overall negatively charged, silica. Negative zeta potentials in pH 7 buffer solution for non-modified, aminated and carboxylated silica nanospheres from the same supplier were also observed in a recent study, where these nanospheres were shown to accumulate distinctly different protein coronas.53| Nominal diameter (nm) | Surface functionalization | Z-average diameter (nm) | Polydispersity | Zeta potential (mV) |
|---|---|---|---|---|
| a Dispersed in 100 mM KCl, 20 mM HEPES, pH 7.5. | ||||
| Silica | ||||
| 50 | None | 51.7 | 0.171 | −22.0 |
| 100 | None | 106.5 | 0.050 | −27.6 |
| 200 | None | 213.9 | 0.021 | −30.5 |
| 500 | None | 534.5 | 0.109 | −34.9 |
| 50 | Carboxylated | 55.3 | 0.146 | −24.0 |
| 200 | Carboxylated | 196.8 | 0.002 | −33.7 |
| 500 | Carboxylated | 453.8 | 0.011 | −41.5 |
| 50 | Aminated | 46.9 | 0.015 | −32.7 |
| 200 | Aminated | 192.2 | 0.023 | −30.8 |
| 500 | Aminated | 516.1 | 0.072 | −32.0 |
| Polystyrene | ||||
| 50 | None | 46.4 | 0.018 | −54.6 |
| 100 | None | 100.5 | 0.014 | −44.8 |
| 200 | None | 203.0 | 0.008 | −46.0 |
| 200 | Carboxylated | 199.3 | 0.006 | −50.0 |
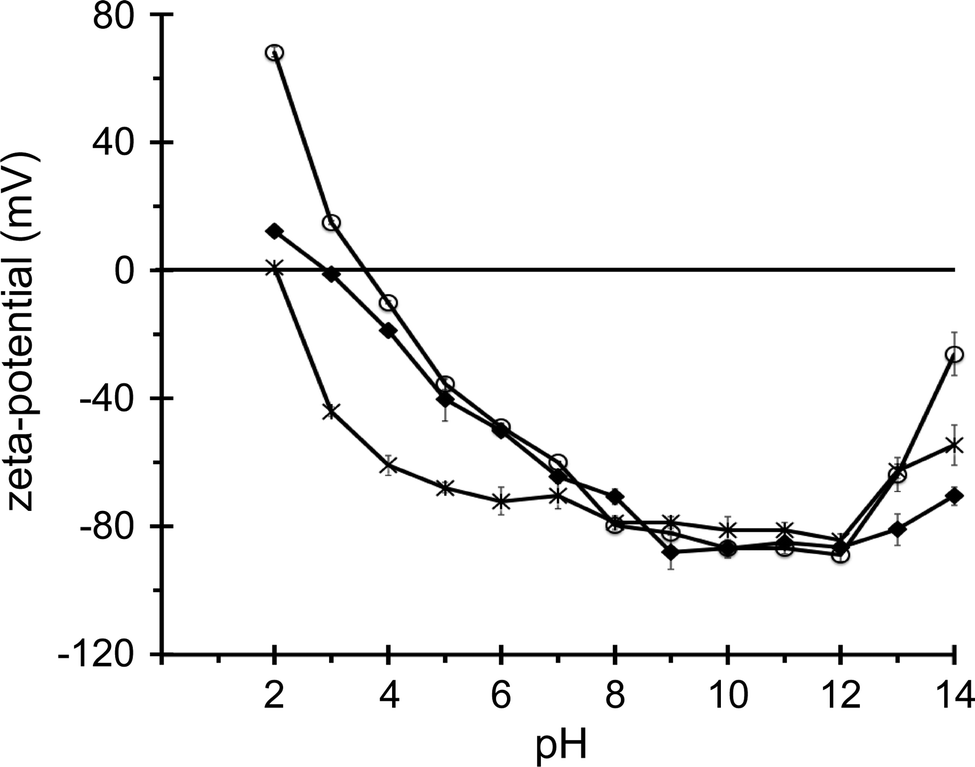 | ||
| Fig. 1 Zeta potential of 50 nm diameter silica nanospheres in KCl–phosphate buffers of different pH with an ionic strength of 2 mM. Zeta potential values for nanospheres with a native silica surface (◆), carboxylated surface (×) and aminated surface (○) are connected as a guide for the eye. | ||
Interaction with zwitterionic liposomes
Modulation of the permeability of lipid bilayers of unilamellar liposomes by the silica nanospheres was assessed with a calcein liposome leakage assay, as commonly employed for membrane-active biomolecules.24,45–48 Liposomes filled with a high concentration of the dye were diluted into a cuvette with buffer solution and after stabilization of the fluorescence signal, nanoparticles were added to the liposome dispersion. The fluorescence signal was monitored for 7–10 minutes, after which a detergent was added to solubilize the bilayers and determine the fluorescence signal corresponding to a total release of the dye from all the liposomes. The key property of the assay is that the fluorescence of the dye is quenched inside the vesicles, but upon release into the large volume of measurement buffer in the cuvette, the dye becomes diluted and will fluoresce. Thus, the fluorescence intensity is directly proportional to the amount of dye that has leaked out of the liposomes. When the fluorescence intensity of the liposome dispersion prior to nanoparticle addition is defined at 0% leakage, and the intensity after detergent-induced liposome rupture is defined as 100% leakage, the fluorescence intensity at any point after nanoparticle addition can be converted to a percentage of leaked dye. Nanoparticle–liposome interactions can be evaluated in terms of the extent of dye release, with substantial amounts of leaked dye corresponding to significant nanoparticle-induced bilayer perturbation.An example of fluorescence versus time data, for dye-loaded liposomes of zwitterionic DOPC lipids upon exposure to six different concentrations of 200 nm non-functionalized silica nanospheres, is shown in Fig. 2. The fluorescence intensity of the liposome–nanoparticle dispersions, expressed as the percentage of released dye, is clearly time- and dose-dependent. At the highest particle concentration of 20 pM, approximately 70% of the dye is released within the first 150 s following nanoparticle addition, after which the fluorescence signal gradually increases until it levels off, approximately 300 s after nanoparticle introduction, at a value corresponding to 91% dye release. At lower particle concentrations, the initial rate of dye release is consistently slower and the percentage of the dye that has leaked out of the liposomes after 415 s, at which point detergent was added, is consistently smaller (Fig. 2). At the lowest particle concentration of the 200 nm plain silica nanospheres, 0.6 pM, the amount of leaked dye slowly increases over time, reaching a value corresponding to 23% dye release after 415 s.
 | ||
| Fig. 2 Normalized fluorescence intensity of dispersions of calcein-loaded 400 nm diameter DOPC vesicles at 10 pM concentration in the presence of 200 nm non-functionalized silica nanospheres at concentrations in the range 0.6–20 pM. The fluorescence intensity before addition of nanoparticles to the liposome dispersion, indicated by the arrow, is defined at 0% dye leakage while the intensity after addition of liposome-disrupting Triton X-100 detergent, indicated by the asterisk, is defined at 100% dye leakage. | ||
Assuming that no lipid molecules were lost during the liposome preparation protocol, and considering that a 400 nm liposome of phosphatidylcholine lipids contains ∼1.4 million lipids, the liposome concentration in the measurement solution was approximately 10 pM. The near-total dye release observed at the highest particle concentration of 20 pM suggests that either each individual nanoparticle has triggered the release of all the dye from a single liposome, or that multiple particles are required to achieve complete dye release from a single liposome, after which the same particles would then have to associate with other liposomes and permeabilize the membranes of those liposomes as well. It is clear, however, that particle concentrations significantly below the estimated liposome concentration do not result in efficient dye release, which indicates that a single particle does not readily induce dye leakage from multiple liposomes.
We also measured the fluorescence intensity of nanoparticle–liposome mixtures at different concentrations of the DOPC liposomes but with a fixed estimated 1:1 molar ratio of liposomes and 200 nm diameter non-functionalized silica nanospheres. Fig. 3 shows that at lower liposome concentrations, the absolute fluorescence intensity after detergent-induced membrane rupture is reduced. This decrease in the maximum fluorescence signal is directly proportional to the liposome concentration and hence to the maximum concentration of free calcein dye. However, it is also observed that for lower liposome and nanoparticle concentrations the percentage of released dye decreases, which is evident from the increasingly large change in fluorescence intensity when the detergent is added at the end of each experiment (Fig. 3). For example, at a liposome (and nanoparticle) concentration of 5.0 pM the dye release percentage is ∼75% and at 1.2 pM it is ∼15%, whereas at the reference concentration of 10 pM, the leakage percentage is almost 100%. It thus appears that also at a constant (1:1) number ratio of liposomes and nanospheres the extent of dye release can vary significantly, with a more diluted system resulting in less bilayer perturbation, potentially because of a decreased likelihood of particle–liposome diffusional contact.
 | ||
| Fig. 3 Absolute fluorescence intensity for equimolar mixtures of calcein-loaded DOPC liposomes and non-functionalized 200 nm diameter silica nanoparticles at concentrations of (a) 10 pM nanospheres and 10 pM liposomes, (b) 5 pM, (c) 2.5 pM, (d) 1.2 pM and (e) 0.6 pM. Nanoparticles were added to the liposome dispersions at t ≈ 25 s and detergent was added at t ≈ 500 s. Note that a decrease in concentration at a fixed 1/1 nanoparticle/liposome ratio leads to a pronounced decrease in nanoparticle-induced dye leakage. | ||
Before addressing possible scenarios of nanoparticle–bilayer association, we first evaluated dye leakage from DOPC liposomes for different silica nanospheres. The general observation for all these ENMs is that fluorescence intensity increases with the duration of the liposome exposure to the particles, and that higher particle concentrations cause more dye to leak out of the liposomes. These data are summarized in Fig. 4, where the maximum dye release observed during the assay, i.e. the endpoint leakage value, is plotted against the number of particles and also against the total surface area of all the particles in the measurement solution. Fig. 4A shows the maximum percentage of dye leakage observed for the non-functionalized silica nanospheres with diameters of ∼50, 200 and 500 nm as a function of the number of particles, up to a particle concentration of 20 pM, which was the highest concentration investigated for the 500 nm particles. The effect of the non-functionalized silica nanospheres is size-dependent, with the larger particles causing more leakage at the same particle concentration. However, when the maximum leakage percentage is given as a function of the total surface area of the particles in the measurement solution (Fig. 4D), it appears that the 50 nm diameter particles are considerably more effective in causing dye release than the 200 and 500 nm particles. Interestingly, the latter two nanospheres induce approximately 90–100% dye release when the total particle surface area is close to the total outer surface area of the liposomes (estimated as 3.0 m2 L−1), whereas the 50 nm particles already cause 100% dye release at a 7-fold lower total particle surface area.
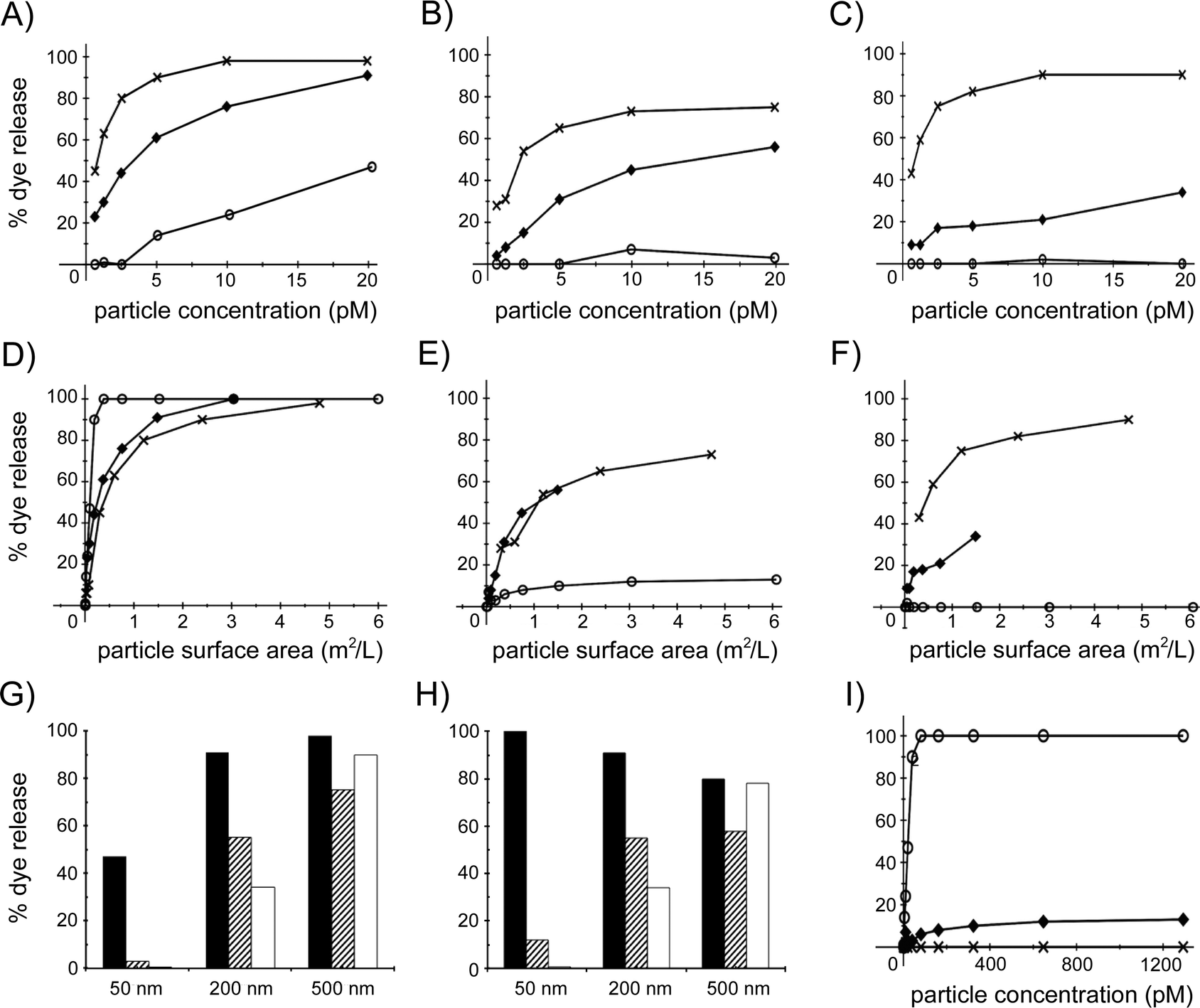 | ||
| Fig. 4 Endpoint dye leakage percentages for different concentrations of a range of silica nanospheres. The concentration of calcein-loaded DOPC liposomes is 10 pM for all experiments, corresponding to an outer surface area of ∼3.0 m2 L−1. (A–C) Dye leakage as a function of the concentration of non-functionalized (A), aminated (B) and carboxylated (C) silica nanospheres of 50 nm (○), 200 nm (◆) and 500 nm (×) diameter. (D–F) Dye leakage as a function of the surface area per volume of non-functionalized (D), aminated (E) and carboxylated (F) silica nanospheres of 50 nm (○), 200 nm (◆) and 500 nm (×) diameter. (G–H) Comparison of endpoint dye release percentages for 50 nm, 200 nm and 500 nm non-functionalized (black), aminated (hatched) and carboxylated (white) nanospheres at a concentration of 20 pM (G) or at a surface area of ∼1.4 m2 L−1 (H). (I) Endpoint dye leakage values for higher concentrations of 50 nm diameter non-functionalized (○), aminated (◆) and carboxylated (×) silica nanospheres. | ||
For the aminated and carboxylated silica nanospheres, the same trend is observed as with the non-functionalized nanospheres; at the same number of nanoparticles, the larger particles are able to induce a larger fraction of the dye to leak out of the liposomes. However, for both the aminated and the carboxylated particles of 200 and 500 nm, the amount of released dye is somewhat lower than for the non-functionalized analogs, whereas the 50 nm functionalized nanospheres release almost no dye, even at a concentration of 20 pM (Fig. 4B and C). This trend is also apparent when dye release is presented as a function of total particle surface area for the aminated and carboxylated nanospheres. At the same total particle surface area, 200 and 500 nm aminated particles appear equally effective at inducing liposome leakage, but not as effective as their non-functionalized analogs, and induce considerably more dye leakage than the 50 nm functionalized particles (Fig. 4E). The pattern is somewhat different for the carboxylated particles, of which, compared to their aminated analogs, the 500 nm particles cause considerably more, and the 200 nm particles somewhat less, dye release, and the 50 nm particles do not induce significant leakage (Fig. 4F).
The relationship between surface chemistry and dye leakage for the three particle sizes is summarized in Fig. 4G, where the percentage dye leakage is given for the highest particle concentration, 20 pM, in Fig. 4A–C. The three different 500 nm, 200 nm and 50 nm particles induce >75%, 35–90% and 0–47% dye leakage, respectively. The surface chemistry of the largest particles does not affect the leakage significantly, whereas for the 200 nm and the 50 nm particles, the non-functionalized silica particles release more dye than the functionalized analogs. Surprisingly, for the smallest nanospheres, only the non-functionalized analogs are able to induce a significant amount of dye leakage. At higher particle concentrations this remains the case, with non-functionalized, aminated and carboxylated 50 nm nanospheres at 1.3 nM concentration inducing, respectively, total, 12% and nil dye release (Fig. 4I). Hence, the ability of the silica nanospheres to perturb the membrane of the liposomes increases with particle size, especially for the carboxylated and aminated particles. These trends are somewhat different when the effect of the particles is compared at a total particle surface area of 1.4 m2 L−1 (Fig. 4H). Although surface chemistry again does not significantly modulate the effect of the largest particles and the non-functionalized 50 and 200 nm nanospheres again clearly induce more dye leakage than their carboxylated and aminated analogs, the non-functionalized particles now appear approximately equally effective at bilayer perturbation (80–100% leakage) for each particle size. These data indicate that the interactions between silica nanospheres and lipid bilayers are modulated by both particle size and particle surface chemistry.
At the start of a liposome leakage assay, the amount of leaked dye is low because there still is a considerable fraction of non-perturbed liposomes in the measurement solution. Typically, as can be seen for the time-leakage curves in Fig. 2, the leakage percentage initially increases linearly with time, after which, particularly at higher leakage percentages (>30–40%), the rate of dye release slows down. To gain further insight into the nanosphere–bilayer interactions, we derived the rate of dye release from the slope of the time-leakage curves for different concentrations of the various nanospheres for the first 25 seconds after particle addition to the liposome dispersion, and plotted this rate against the concentration of particles (Fig. 5A, C and E) and against the total particle surface area (Fig. 5B, D and F). At the same number of particles, the initial leakage rate follows the pattern 50 nm < 200 nm ≪ 500 nm diameter particles, for all three surface chemistries (Fig. 5A, C and E). However, for the same total particle surface area, the 200 and 500 nm nanospheres give similar initial leakage rates, with the 50 nm non-functionalized particles causing a considerably higher rate and both the functionalized 50 nm particles causing a significantly lower rate, close to zero percent dye leakage per second (Fig. 5B, D and F).
 | ||
| Fig. 5 Initial rate of dye release from 400 nm DOPC liposomes for non-functionalized (A, B), aminated (C, D) and carboxylated (E, F) silica nanospheres of 50 nm (○), 200 nm (◆) and 500 nm (×) diameter as a function of nanoparticle concentration (A, C, E) or of particle surface area per volume (B, D, F). The liposome concentration in all experiments is 10 pM, corresponding to an outer surface area of ∼3.0 m2 L−1. | ||
These leakage rates, which relate to the initial stage of liposome–nanoparticle interactions when the liposomal membranes are still largely uncompromised, confirm that 50 nm non-functionalized particles have a considerable larger effect than 50 nm carboxylated or aminated analogs, and also that the influence of surface functionalization for the 500 and 200 nm particles is less pronounced (Fig. 4G and H). Unlike the summary in Fig. 4H, which presents endpoint leakage values (i.e. most or all liposomes have been in contact with the nanospheres), the initial leakage rates at the same particle total surface area (Fig. 5B, D and F) suggest that the 200 and 500 nm particles interact with the 400 nm diameter DOPC liposomes in a very similar manner, which is distinctly different from the smaller 50 nm particles. As discussed below, we relate this to the relative dimensions of the particles and the liposomes.
Negatively charged liposomes and biocoated nanospheres
To investigate the role of electrostatic interactions between silica nanospheres and lipid bilayers, the fluorescence leakage assay was also performed with liposomes with a net negative charge. Fig. 6A shows that 200 nm diameter non-functionalized silica nanospheres cause calcein dye to leak out of DOPC/POPG (2/1 molar ratio) liposomes in a dose-dependent manner, as also observed for zwitterionic liposomes of pure DOPC (Fig. 2). However, at a particle concentration of 20 pM, where ∼95% dye release is observed for DOPC liposomes (Fig. 2 and 6C), only 28% of the dye leaks out of the PC:PG liposomes. A similar trend is apparent for 50 nm non-functionalized silica particles, which at a concentration of 5 pM induce ∼28% dye leakage from DOPC liposomes but only ∼3% dye leakage from PC:PG liposomes (Fig. 6D), indicating that the presence of the net negatively charged POPG lipid significantly reduces the extent of bilayer perturbation by non-functionalized silica nanospheres. The 200 nm diameter aminated nanospheres were found to induce 17% dye release (Fig. 6B), but the 50 nm diameter aminated particles did not cause any leakage at 20 pM concentration (data not shown), and even at a 100-fold higher concentration only resulted in ∼7% dye release from the PC:PG liposomes (Fig. 6E). Hence it appears that the perturbation of the bilayer by both non-functionalized and aminated 50 and 200 nm silica particles is significantly reduced when the vesicular membrane surface has become net anionic by the inclusion of 33% POPG lipids. | ||
| Fig. 6 Fluorescence intensity curves normalized to dye leakage percentages for dispersions of net anionic liposomes with a DOPC/POPG = 2/1 molar ratio. (A, B) 10 pM PC:PG liposomes with 4 pM (a), 8 pM (b) and 20 pM (c) non-functionalized (A) or aminated (B) silica nanospheres of 200 nm diameter. A comparison with 100% DOPC liposomes is given in (C, D): (C) 10 pM DOPC liposomes with 200 nm non-functionalized silica particles at 10 pM concentration (a) and 10 pM PC:PG liposomes with non-functionalized (b) and aminated (c) 200 nm particles at 10 pM concentration; (D) non-functionalized silica nanospheres of 50 nm diameter at 5 pM concentration in 5 pM vesicle dispersions of DOPC (a) and DOPC:POPG (b). Aminated 50 nm silica particles require a 100-fold higher concentration to achieve a similar extent of dye release, as shown in (E) for 16 pM non-functionalized particles (a) and 1600 pM aminated analogs (b) in 16 pM dispersions of PC:PG vesicles. | ||
The role of particle surface chemistry was further explored by coating silica nanospheres with proteins or with lipids. Non-functionalized, aminated and carboxylated 200 nm silica nanospheres were covered with a DOPC bilayer using the lipid–solvent casting protocol described in the Methods section. None of these bilayer-coated nanospheres caused dye release from DOPC liposomes at a concentration where the non-coated analogs induced significant dye leakage. This was also observed for bilayer-coated 50 nm diameter native-silica nanospheres (data not shown). To address the impact of a protein corona on nanosphere–liposome interactions, we used commercially available avidin-functionalized (covalent coupling) silica nanospheres of 100 nm diameter as well as 100 nm native-silica nanospheres that were coated with avidin (by physisorption) by exposure to a solution of 1 mg mL−1 protein. Under conditions were ∼75% dye leakage was observed for non-coated non-functionalized 100 nm silica nanospheres, the particles with covalently attached and the particles with physisorbed avidin, were found to induce, respectively, only 3% and 5% dye leakage from the DOPC vesicles (data not shown). Hence, these preliminary experiments indicate that silica nanospheres that are covered by a lipid bilayer do not compromise the structural integrity of DOPC vesicular membranes, whereas the bilayer perturbing properties of nanoparticles with a native-silica surface chemistry are significantly reduced, but not abolished, by biocoating with the avidin protein.
Finally, since polystyrene particles have been used in conjunction with silica nanoparticles in bionanoscience studies,17,54,55 we also investigated non-functionalized polystyrene particles with nominal diameters of 50, 100 and 200 nm, as well as 200 nm carboxylated analogs, the zeta potentials of which ranged from −45 to −55 mV (Table 1), with the liposome leakage assay. Non-functionalized 200 nm particles induced minor leakage, ∼4%, from DOPC vesicles at a particle concentration of 20 pM, while 200 nm carboxylated, 100 nm non-functionalized and 50 nm non-functionalized polystyrene nanospheres did not cause any dye release at particle concentrations of, respectively, 30, 300 and 3100 pM (data not shown). The nil or minor dye leakage from DOPC liposomes at high concentrations of these polystyrene particles stands in marked contrast to the bilayer perturbing potential of the non-functionalized silica nanospheres.
Discussion
Liposome leakage assays with engineered nanomaterials
To date, a small number of studies has employed the liposome leakage assay to probe interactions between inorganic or metallic ENMs and the membrane of large unilamellar vesicles. Goodman et al. used gold nanospheres with a core diameter of 2 nm with either an anionic or a cationic surface functionalization.37 With zwitterionic SOPC liposomes, these anionic and cationic gold ENMs induced, respectively, up to 15% and 5% dye leakage at a particle concentration of 220 nM. For net negatively charged SOPC/SOPS liposomes, this trend was reversed, with the anionic and cationic ENMs causing, respectively, ∼3% and ∼20% dye leakage, suggesting that electrostatic interactions between these ENMs and lipid bilayers contribute to bilayer perturbation. These effects were dose dependent, with 875 nM of the cationic gold particle inducing ∼50% dye leakage.37 Hirano et al. measured dye leakage from net negatively charged DOPC/DOPG liposomes in the presence of lysozyme-coated, i.e. cationic, single walled carbon nanotubes and observed a dose-dependent leakage, with a maximum leakage value of 30% at a particle concentration of 80 ng mL−1.38 These two studies do not elaborate on ENM/liposome ratios but Moghadam et al. explicitly address this point for 100 nm diameter zwitterionic DOPC liposomes exposed to a range of concentrations of functionalized gold and titanium dioxide particles of 8–21 nm diameter.39 With a cationic surface functionality, both types of ENM gave rise to near-total dye release when the particle concentration approximates the liposome concentration, whereas an anionic surface chemistry resulted in only ∼7% leakage. Lower ENM/liposome ratios resulted in a smaller percentage of dye release and a longer time to reach steady-state leakage.39 For the silica nanospheres investigated in the present study, we also observe dose-dependent dye leakage from DOPC liposomes, and for some particles total dye release is observed when the particle/liposome molar ratio is ∼1/1 (e.g.Fig. 2).However, Moghadam et al. report a time scale of hours to achieve 100% leakage at ∼1/1 ENM/liposome ratio,39 whereas in our experiments total dye release can occur in <10 minutes, which is similar to the time observed by Goodman et al. and Hirano et al. to reach steady-state leakage.37,38 We postulate that this is due to the 30-fold lower liposome (and ENM) concentration employed by Moghadam et al., which will decrease the rate of ENM–liposome diffusive contact. This interpretation is supported by our experiments with lower liposome concentrations: maintaining a nanosphere/liposome ratio of ∼1/1 while diluting the sample increases the amount of time to reach steady-state leakage and also reduces the percentage of nanosphere-induced dye release (Fig. 3). Based on their observation that ∼15 nm diameter cationic, but non anionic, ENMs with a gold or titanium dioxide core were able to induce near-total dye release, Moghadam et al. suggested that bilayer disruption is mediated by attractive electrostatic interactions between cationic ENM surface charges and the DOPC liposome surface, which has a small negative zeta potential of −12 mV.39 However, we observe that, at an approximately equimolar ENM/liposome ratio, 50 nm non-functionalized silica ENMs induce significant dye leakage (∼46%) from 400 nm diameter DOPC vesicles (Fig. 4G), even though the zeta potential of these nanospheres is −22 mV (Table 1).
Interaction of liposomes with planar and curved surfaces
ENMs cannot modulate the permeability or structural integrity of lipid bilayers without a prior ENM–bilayer association step. Although little is known about ENM–bilayer association motifs, there is an extensive literature on the association of liposomes with planar solid materials, particularly for silica. Unilamellar liposomes, typically of ∼30–200 nm diameter, can adsorb to silica surfaces, forming a solid-supported lipid bilayer when the adsorbed vesicles deform and rupture.56–58 Electrostatic interactions play a prominent role in vesicle–silica association. For example, it has been shown that vesicles of positively charged DOTAP lipids associate with the negatively charged silica surface and rupture as individual vesicles, whereas DOPC/DOPS (1:2) vesicles with a high net negative charge do not adsorb to silica.56 Vesicles of an intermediate charge, low positive (DOPC/DOTAP = 4/1), zwitterionic (DOPC) or low-to-medium negative (DOPC/DOPS = 4/1 or 2/1), do adsorb to a silica surface but are not sufficiently deformed to rupture as isolated vesicles, with the formation of a silica-supported lipid bilayer resulting from vesicle–vesicle interactions at a sufficiently high silica surface density of adsorbed vesicles.56 The role of the solid support is, however, not well understood. Solid-supported bilayers are readily formed by vesicle association, deformation and rupture on silica, glass and mica surfaces, but not on platinum, gold or titanium dioxide surfaces.57 For gold and titanium dioxide surfaces, it has been demonstrated that zwitterionic PC vesicles do adsorb, but do not deform to an extent that leads to vesicle rupture,59,60 indicating that vesicle–surface attractive forces are not as strong as in the case of silica. However, when a gold surface is functionalized with a high density, ∼75%, of cationic carboxyl or anionic amine groups in a matrix of hydroxyl-terminated thiols, zwitterionic PC vesicles are able to adsorb and to fuse into a supported bilayer.61
For our experiments with 50–500 nm silica nanospheres, it is also of interest to consider previous work on the formation of supported lipid bilayers on silica beads. It is well known that vesicle adsorption and supported bilayer formation are also possible on the curved surface of silica beads with micrometer diameters,62,63 but vesicle adsorption can also lead to the formation of supported bilayers on nanospheres, even if these are smaller than the vesicles. For example, Wunder and co-workers demonstrated that exposure of silica nanospheres with diameters ranging from 4 to 100 nm to DMPC liposomes of 50–200 nm diameter resulted in the formation of supported lipid bilayers on all nanoparticles.64–66 Mornet et al. visualized with cryotransmission electron microscopy that PC vesicles with a diameter of ∼40 nm formed supported lipid bilayers on ∼110 nm silica particles when the vesicles had a positive, a neutral or a low-negative charge (DOPC/DOPS = 4/1 or 3/1), whereas vesicles with a high negative charge (DOPC/DOPS = 1/1) adsorbed to the silica nanospheres and deformed, but did not rupture.67 Also, Wang and Liu reported recently that 100 nm DOPC liposomes formed supported bilayers on 20–50 nm diameter silica nanospheres, but not on 20–50 nm diameter titanium oxide, iron oxide or zinc oxide nanoparticles.68 Instead, surface-supported vesicles were observed at a pH where these surfaces were positively charged, with no vesicle association at higher pH values where these surfaces were negatively charged.68 These observations with ENMs are consistent with vesicle–surface association studies for planar materials,56,57,59,60 indicate a complex dependence on the physicochemical properties of metal oxide surfaces, and confirm an (exceptionally) strong interfacial attraction between PC vesicles and native silica, which has been quantified as an adhesion energy of up to 1 mJ m−2.69
In our experiments we observe time- and dose-dependent release of fluorescent molecules from DOPC vesicles that are exposed to silica nanospheres of various size and surface chemistry with a diameter of 50, 200 or 500 nm. The endpoint leakage percentages presented in Fig. 4G and H indicate that lipid bilayer perturbation is modulated by both particle size and by particle surface chemistry. Specifically, the 500 nm particles induce a similar amount of dye leakage, ∼75–100% at a particle concentration of 20 pM (which approximates the estimated liposome concentration of 10 pM) and ∼60–80% at a total particle surface area of 1.4 m2 L−1 (i.e. about half of the estimated total liposome area of 3.0 m2 L−1) regardless of nanosphere surface chemistry, whereas for the smaller 50 nm particles only the non-functionalized nanospheres cause significant dye leakage. This suggests that the 50 and 500 nm particles interact with the 400 nm diameter DOPC vesicles through different mechanisms. We propose that adsorption of a single 500 nm particle to a 400 nm vesicle induces vesicle rupture and the formation of a particle-supported DOPC bilayer,62–67 as observed by Wang et al. in a system of 100 nm DMPC vesicles and 100 nm silica particles.70 In this scenario, pronounced vesicle deformation, followed by vesicle rupture, occurs because a large proportion of the vesicle surface becomes associated with the silica surface,56–58 with each of the three surface chemistries employed in this study. Because bilayer-coated silica nanoparticles cannot induce dye leakage from liposomes (see below), maximum leakage would not be expected at particle/liposome ratios smaller than ∼1. In terms of the endpoint leakage for 200 nm particles (Fig. 4), the different surface chemistries all lead to significant dye leakage, in the order non-functionalized (∼90% leakage) > amine-functionalized (∼50%) > carboxyl-functionalized (∼30%), but in terms of the initial leakage rate at the same total particle surface area, the bilayer perturbing effect of the 200 nm particles, for each surface chemistry, is almost the same as that of the 500 nm particles (Fig. 5B, D and F). Hence we hypothesize that the 200 nm diameter particles also induce the formation of nanosphere-supported bilayers, but that this process is less effective than with the larger 500 nm particles because of a reduced particle–vesicle contact area, and is therefore influenced by particle surface chemistry, with both carboxyl- and amine-modification reducing the extent of endpoint dye leakage from the vesicles (Fig. 4G and H).
In contrast to the 200 and 500 nm nanoparticles, which induce near-total dye release at a particle/liposome ratio of ∼1/1, the non-functionalized 50 nm silica nanospheres only cause 100% dye release at concentrations ≥80 pM (Fig. 4I), which corresponds to a nanoparticle/vesicle ratio of ∼8/1. Given that for a 50 nm nanoparticle associated with a 400 nm vesicle, the relative surface area of the vesicle adsorbed to a silica surface will be significantly smaller than for a 200 nm or 500 nm particle, we propose that this can only result in local bilayer perturbation (as opposed to the global vesicle reorganization leading to a particle-supported bilayer). It should be noted that, to our knowledge, the only previous study on silica nanospheres with the conventional format of the liposome leakage assay as employed in the present work, reported that small, 14 nm diameter, non-functionalized silica particles at a very high particle concentration of 100 μg mL−1 (∼45 nM) only induced ∼5–20% dye release from 400 nm liposomes (∼15 pM concentration) of POPC/DOPE/DOPS/cholesterol.71 However, an initial increase in fluorescence signal was followed by a substantial intensity decrease,71 indicative of photobleaching. Various other recent studies with biomembrane model systems, outlined below, have established that silica nanospheres are indeed able to perturb, by surface association, the structure of a lipid bilayer or monolayer, and that the extent of perturbation depends, also for particles <200 nm in diameter, on nanosphere size.
Lipid bilayer perturbation by silica nanospheres
Vakurov et al. exposed an electrode-supported monolayer of DOPC lipids to dispersions of silica nanospheres ranging in diameter from 14 to 172 nm and established by electron microscopy and cyclic voltammetry that all nanoparticles associated with the monolayer surface and reduced the mobility of the DOPC lipids, with the smaller particles having a larger rigidifying effect, probably because these pack more efficiently on the lipid monolayer.72 For silica nanospheres of 4, 10 and 20 nm diameter, Wang et al. concluded from calorimetry measurements that surface association with 80 or 200 nm diameter DLPC or DOPC liposomes also leads to local gelation of the lipids.73 Liu et al. exposed an electrode-supported DPPTE-tethered lipid bilayer of DOPC to non-functionalized, carboxyl-functionalized and amine-functionalized silica nanoparticles of ∼50 nm diameter, all with a negative zeta potential, and determined with impedance measurements that all particles reduced the electrical resistance of the bilayer, indicative of bilayer defect formation (possibly by particle-mediated lipid removal), with the aminated particles acting at the shortest time scale.74 Interestingly, they also measured the hydrodynamic radius of the particles after incubation with the bilayer, and found that only the native-surface silica particles had aggregated,74 suggesting a distinct bilayer–nanosphere interaction mechanism for the non-functionalized analogs. With a more direct electrical method, we have shown previously that the same non-functionalized and amine-functionalized 50 and 500 nm diameter nanospheres as employed in the present study were able, at fM to pM concentration, to gradually increase the current flowing over voltage-clamped aperture-suspended lipid bilayers of DOPC or asolectin lipids, with short-lived current spikes indicative of (transient) nanosphere-induced bilayer defects.31 At 8 pM particle concentration, the 50 nm native-surface silica particles caused the largest increase in baseline current of an asolectin bilayer while the 500 nm aminated analogs induced the highest transient current spikes, which could correlate with bilayer translocation events.31Several interesting microscopy studies give some insight in the mechanism by which silica nanospheres interact with liposomal lipid bilayers. Le Bihan et al. visualized with cryo-electron microscopy that silica-shell particles of 15 and 20 nm adhere to the outer surface of 200 nm DOPC vesicles, but that particles of 30–190 nm diameter are internalized by these liposomes.75 They observed intermediate stages in this process, where the vesicular bilayer curves around the nanosphere, which is internalized, as a bilayer-covered particle, when the bilayer ‘neck’ ruptures.75 In agreement with this study, Hoffmann et al. inferred from cryo-electron microscopy images and neutron scattering data that, at a 12-fold excess of particles over vesicles, 16 nm diameter silica nanospheres adsorb to the outer surface of 100 nm diameter DOPC vesicles without significantly affecting the vesicle structure, although neutron spin-echo data indicated an increased flexibility of the vesicular bilayer, which could be related to nanoparticle-induced indentations of the vesicle surface.76 Fluorescence microscopy of giant unilamellar vesicles of DOPC, with a diameter of ∼10–20 μm, was used by Zhang et al. to investigate vesicle morphology following bilayer adsorption of silica nanospheres of 18, 78 or 182 nm diameter.77 They concluded that the two larger particles become wrapped by the vesicular bilayer, which eventually leads to collapse of the vesicle because of lipid depletion, whereas the 18 nm particles adsorb to the bilayer and cause a local decrease in lipid dynamics, the stress of which is released by detachment of a particle-covered bilayer patch, which can leave a micrometer-sized opening, potentially stabilized by the 18 nm particles, in the vesicular bilayer.77
With our liposome leakage assay we observe that 50 nm non-functionalized silica nanospheres are only able to induce 100% dye leakage from 400 nm DOPC liposomes when the nanosphere/liposome ratio is 8/1 or higher (Fig. 4I). Given that the adsorption of 8 nanospheres of 50 nm diameter to a 400 nm diameter liposome would result in a liposome surface coverage of only ∼4%, it appears unlikely that a local bilayer rigidification as reported for supported monolayers or bilayers,72,73 even when associated with bilayer defect formation,31,74 could lead to total release of the calcein dye within the 10 minute duration of a typical experiment. Instead, we postulate that the vesicle-adsorbed nanospheres become engulfed by the vesicular lipid bilayer, as described by Le Bihan et al. and Zhang et al.,75,77 and that with a sufficient number of engulfment events, which may be associated with an endocytosis-like particle uptake, the vesicle collapses, with total loss of any enclosed dye molecules, because of lipid depletion from its membrane. It can be expected that the threshold number of bilayer-engulfed nanoparticles that a vesicular bilayer can accommodate without rupturing depends on the size of both the particle and the liposome. Interestingly, Pera et al. showed, using particle–liposome dispersions in a 96-well plate, that for 240 nm diameter DOPC vesicles (∼2 pM concentration), 16 nm diameter non-functionalized silica nanospheres started to cause severe (∼80%) dye leakage at a ∼10/1 nanosphere/liposome ratio,40 which could follow the same scenario as proposed for our dispersion of 50 nm nanoparticles and 400 nm liposomes. However, 66 nm analogs caused maximum dye leakage (∼85%) from the 240 nm vesicles at a number ratio of ∼1/1,40 suggesting nanoparticle-induced supported bilayer formation as proposed above for our 500 and 200 nm particles. It should be noted, however, that in this adaptation of the liposome leakage assay,40 the nanoparticle–liposome dispersions could not be stirred, the recordings consisted of a small number of time points and primary data were not shown.
Effect of silica functionalization and biocoating
It is remarkable that amine or carboxyl functionalization of the 50 nm silica particles drastically reduces or abolishes, respectively, dye release from and hence bilayer perturbation of the 400 nm DOPC liposomes. Exposure of the vesicles at 10 pM concentration to 20 or 300 pM functionalized 50 nm particles did not result in significant dye leakage (Fig. 4G and H), and even at a much higher particle concentration of 1.3 nM the carboxylated analogs did not result in any dye release, whereas the aminated particles only caused 12% of the dye to leak out of the liposomes (Fig. 4I). Since for our particles the zeta potential in the measurement buffer is, given the accuracy of these measurements, similar to that of native silica (Table 1) and significant differences in zeta potential only become apparent at low or high pH (Fig. 1), the surface density of the functionalized groups is likely to be low in relation to the characteristic groups of a native silica surface. Puddu and Perry also reported a low amine surface density for aminated 500 nm silica nanospheres, but this extent of functionalization proved sufficient to drastically change peptide–particle association.78 The surface chemistry of hydrated amorphous silica consists of various silanol Si–OH (single, geminal, vicinal), siloxane Si–O–Si and ionic siloxide Si–O− moieties, with the relative surface density of these groups depending on the exact synthesis conditions.79,80 Our non-functionalized silica nanospheres, in the measurement buffer, can be expected to have surface densities of 3.7–4.5 silanol groups per nm2 and 0.2–1.0 siloxide groups per nm2,79,80 and should hence be net negatively charged, as confirmed by the measured zeta potentials of −22 to −35 mV in a buffer of 100 mM KCl, 20 mM HEPES, pH 7.5 (Table 1). The strong adsorption of PC bilayer surfaces to native silica is not fully understood,56–58,61 but is thought to involve a range of interaction forces: ion pairing and hydrogen bonding as well as ion–dipole, dipole–dipole and van der Waals interactions.80 Our results with amine- and carboxyl-functionalized 50 nm silica nanospheres suggest that these chemical modifications either weaken the silica–bilayer interactions by introducing unfavourable chemical groups and/or by removing favourable moieties from the silica surface. Although amination of ENMs is often related to an increase in bilayer or cell membrane perturbation,8,36,37,41,42 this may not apply to silica particles, which already possess a favourable surface chemistry for bilayer association.56–58,61For the 200 nm and 50 nm silica nanospheres with either a native silica or an aminated surface, bilayer perturbation is significantly reduced when the liposomes are net negatively charged by the inclusion of 33% POPG lipids (Fig. 6). This indicates that nanosphere–bilayer interactions are weakened by increased electrostatic repulsion, as observed in vesicle fusion studies,56,57 and that partial amination of the particle surface does not result in enhanced particle–liposome attraction through ion pairing between the particle amine moieties and the PG headgroups. For both the non-functionalized and the aminated 200 nm silica particles, introduction of 33% POPG lipids in the vesicular membrane results in an approximately 5-fold reduction in dye leakage with respect to 100% DOPC vesicles (Fig. 4A, B and 6CversusFig. 6A and B). However, for the 50 nm nanospheres this reduction is about 10-fold (Fig. 6D), which again suggests that the smaller particles induce dye release by a different mechanism than the larger analogs, with the 50 nm nanospheres requiring stronger interactions with the vesicle surface for bilayer disruption to occur. When, changing the nanoparticle surface rather than the liposome surface, 100 nm native-silica nanospheres were covered by the protein avidin their ability to induce dye release was strongly reduced, while DOPC bilayer-coated 50 nm and 200 nm native-silica and 200 nm aminated and carboxylated particles did not cause any dye release (data not shown). This indicates that bio-coated silica particles either do not (for lipid coating) or infrequently (for avidin coating) adhere to the zwitterionic liposomes or that they adhere without (significant) concomitant bilayer perturbation. For the native-surface and carboxylated polystyrene ENMs included in the present study, for which no or negligible dye leakage was observed, the latter scenario is more likely because there is evidence that polystyrene particles do associate with vesicular PC bilayers.73,81 In summary, all these observations emphasize the strong interaction forces between a native silica and a zwitterionic phosphatidylcholine bilayer surface.69
Cell membrane interactions of silica nanospheres
Although an understanding of the interactions between silica nanospheres and phospholipid liposomes is of considerable interest in itself, for example to tune the permeability of the vesicular bilayer, for intra-cellular drug delivery applications49–51 the interactions with cell membranes, rather than lipid-only bilayers, are critical. Several groups have investigated cellular uptake and toxicity of silica particles, using a variety of cell lines and cell culture media. For example, at relatively high nanoparticle concentrations of ∼50–500 μg mL−1, Napierska et al. established that for 16, 19, 60, 104 and 335 nm silica particles, a reduction in particle size is correlated with increased cytotoxic potential for a human endothelial cell line, while Foldbjerg et al. established that, for five out of six different cell types, 25–30 nm BSA-coated silica nanospheres were 2–4 times less toxic than non-functionalized analogs.82,83 At lower dose, for example the ≤10 μg mL−1 concentration of 50 or 400 nm silica nanospheres as employed by Chu et al. with four different cell lines, silica particles typically do not give rise to cytotoxic effects, and microscopy images are consistent with non-specific endocytosis of the nanoparticles, which in the study of Chu et al. were mostly present as membrane-bound vesicle-like organelles.84Interestingly, in a recent comprehensive study, Lesniak et al. demonstrated that 50 nm silica nanospheres, at 25 μg mL−1 concentration, strongly associate with the membranes of A549 lung epithelial cells, as evident from (1) cell morphology changes associated with membrane damage, (2) the presence of nanoparticle clusters in proximity to the plasma membrane, (3) an accumulation of membrane-associated proteins on isolated extracellular particles, and (4) a high number of internalized particles, mostly engulfed in vesicles and lysosomes but also free in the cytosol, which could be a result of the observed membrane damage.55 When the same particles were coated with a protein corona from fetal bovine serum prior to exposure to the cells, no membrane damage or membrane-associated particle clusters were observed, particle uptake was reduced and all internalized particles were present in endosomal vesicles, which the authors contributed to substantially weaker particle–membrane adhesion.55 In a follow-up study on the relation between nanoparticle–membrane adhesion and the extent of particle uptake, Lesniak et al. explicitly recommend that ENMs are not only characterized in terms of physicochemical properties such as size and surface charge but also in terms of their adhesion to relevant membranes.85 Given that our experiments as described in the present paper identify a strong particle–bilayer interaction for 50 nm non-functionalized silica nanospheres which is significantly reduced by protein coating, we conclude that the straightforward liposome leakage assay provides a convenient and quantitative method for the high-throughput preliminary assessment of ENM–membrane interactions.
Conclusions
The liposome leakage assay gives insight into the interaction between nanoparticles and vesicular lipid bilayers. For particles with a native-silica surface, nanospheres of 500 nm or 200 nm diameter induce near-total dye release from 400 nm zwitterionic DOPC vesicles when the number of particles in the dispersion approximates the number of liposomes, which we attribute to adhesion of the vesicular bilayer to the nanoparticle surface followed by an increase in particle–bilayer contact area with concomitant vesicle flattening, bilayer rupture and the formation of a particle-supported bilayer. In contrast, native-silica nanospheres of 50 nm diameter cause complete dye release only at an ∼8 fold excess of nanoparticles over liposomes, which can be attributed to particle engulfment by the vesicular bilayer with eventual collapse of the vesicle because of lipid depletion from the liposomal membrane. Both these nanoparticle–liposome interaction mechanisms involve adhesion of the vesicular bilayer to the native-silica particle surface, with the nanoparticle/liposome size ratio, i.e. the relative vesicle surface area in contact with a single nanoparticle, determining whether nanoparticle wrapping by the bilayer leads to a global vesicle deformation and vesicle rupture or only to localized bilayer indentation, possibly leading to pinch-off of the bilayer-coated particle.We explored the effect of chemical and biochemical functionalization of the native-silica particle surface and of the inclusion of 33% net anionic POPG lipids in the liposomes. It is a striking observation that all these modifications significantly reduced or even abolished dye release from the liposomes, which indicates that nanoparticle–bilayer interactions were weakened. For the POPG/DOPC liposomes this is most likely due to an increased electrostatic repulsion between the particles and the liposomes, whereas for the aminated and carboxylated particles, as well as for the protein- and bilayer-coated particles, this could be related to (partial) masking of the silanol and siloxide groups of native silica and an associated loss of hydrogen bonding, ion–dipole, dipole–dipole and/or van der Waals interactions. In this view, the common statement that ENMs become membrane-active by introducing a cationic surface chemistry8,36,41–44 does not apply to silica particles. Indeed, the particular surface chemistry of silicon oxide enables an exceptionally strong adhesion to phosphatidylcholine bilayers,56–58,61 which does not appear to be the case for related native metal oxides such as titanium oxide,59,68 for native gold,60 and based on our observations with 200 nm particles, for native or carboxylated polystyrene.
However, for drug delivery applications, mere adhesion of drug-loaded nanoparticles to the surface of the cell membrane is a sufficient requirement because membrane-associated particles up to ∼300 nm in size are taken up by non-specific endocytosis.2,3 Here, strong interactions with the membrane are unfavourable because these can lead to membrane damage, possibly through lipid wrapping of nanoparticles, and hence cytotoxic effects.55,85 In this context, the liposome leakage assay could present a suitable method for the preliminary assessment of the membrane disrupting potential of nanoparticles, particularly with further development of high-throughput formats such as the 96-well assay employed by Pera et al.40 Biomimetic applications such as vesicular protocells,86–88 on the other hand, could also benefit from an understanding of the interactions between nanoscale silica and liposomes. For instance, it would be of interest to explore whether, at a low particle/liposome ratio, enzyme-loaded mesoporous silica particles are taken up by protocells as bilayer-enclosed compartments with a distinct biochemical functionality, mimicking sub-cellular organelles.
Acknowledgements
H.I.A. thanks Taif University and the Royal Embassy of Saudi Arabia Cultural Bureau (reference TU065) for funding. M.d.P. acknowledges the financial support of the University of Southampton through its Adventure in Research (A/2008/11) and Medicine-PAS Interface grants. We are grateful to Nik Rogers and Anthony Lee for assistance with the liposome leakage assay, to Natalie Smithers for fluorimeter instructions and to Shuangfan Yang, Thuy Nguyen and Nicolas Green for the initial zeta potential measurements.References
- L. Treuel, X. Jiang and G. U. Nienhaus, J. R. Soc., Interface, 2013, 10, 20120939 CrossRef PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- K. Kettler, K. Veltman, D. van de Meent, A. van Wezel and A. J. Hendriks, Environ. Toxicol. Chem., 2014, 33, 481–492 CrossRef CAS PubMed.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- Y. Yan, G. K. Such, A. P. R. Johnston, J. P. Best and F. Caruso, ACS Nano, 2012, 6, 3663–3669 CrossRef CAS PubMed.
- M. L. Etheridge, S. A. Campbell, A. G. Erdman, C. L. Haynes, S. M. Wolf and J. McCullough, Nanomedicine, 2013, 9, 1–14 CrossRef CAS PubMed.
- K. Unfried, C. Albrecht, L. O. Klotz, A. von Mikecz, S. Grether-Beck and R. P. F. Schins, Nanotoxicology, 2007, 1, 52–71 CrossRef CAS.
- M. Zhu, G. Nie, H. Meng, T. Xia, A. Nel and Y. Zhao, Acc. Chem. Res., 2013, 46, 622–631 CrossRef CAS PubMed.
- H. Kettiger, A. Schipanski, P. Wick and J. Huwyler, Int. J. Nanomed., 2013, 8, 3255–3269 Search PubMed.
- A. Nel, T. Xia, H. Meng, X. Wang, S. Lin, Z. Ji and H. Zhang, Acc. Chem. Res., 2013, 46, 607–621 CrossRef CAS PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- B. J. Marquis, S. A. Love, K. L. Braun and C. L. Haynes, Analyst, 2009, 134, 425–439 RSC.
- S. A. Love, M. A. Maurer-Jones, J. W. Thompson, Y. S. Lin and C. L. Haynes, Annu. Rev. Anal. Chem., 2012, 5, 181–205 CrossRef CAS PubMed.
- X. Han, R. Gelein, N. Corson, P. Wade-Mercer, J. Jiang, P. Biswas, J. N. Finkelstein, A. Elder and G. Oberdörster, Toxicology, 2011, 287, 99–104 CrossRef CAS PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- M. Mahmoudi, I. Lynch, M. R. Ejtehadi, M. P. Monopoli, F. B. Bombelli and S. Laurent, Chem. Rev., 2011, 111, 5610–5637 CrossRef CAS PubMed.
- A. E. Nel, E. Nasser, H. Godwin, D. Avery, T. Bahadori, L. Bergeson, E. Beryt, J. C. Bonner, D. Boverhof, J. Carter, V. Castranova, J. R. DeShazo, S. M. Hussain, A. B. Kane, F. Klaessig, E. Kuempel, M. Lafranconi, R. Landsiedel, T. Malloy, M. B. Miller, J. Morris, K. Moss, G. Oberdörster, K. Pinkerton, R. C. Pleus, J. A. Shatkin, R. Thomas, T. Tolaymat, A. Wang and J. Wong, ACS Nano, 2013, 7, 6422–6433 CrossRef CAS PubMed.
- C. E. Dempsey, Biochim. Biophys. Acta, 1990, 1031, 143–161 CrossRef CAS.
- H. Duclohier, Mini-Rev. Med. Chem., 2002, 2, 331–342 CrossRef CAS.
- B. Bechinger, Mol. Membr. Biol., 2000, 17, 135–142 CrossRef CAS.
- A. Drechsler, G. Anderluh, R. S. Norton and F. Separovic, Biochim. Biophys. Acta, 2010, 1798, 244–251 CrossRef CAS PubMed.
- M.-A. Sani, E. Gagne, J. D. Gehman, T. C. Whitwell and F. Separovic, Eur. Biophys. J., 2014, 43, 445–450 CrossRef CAS PubMed.
- M. R. R. de Planque, V. Raussens, C. Antoranz Contera, D. T. S. Rijkers, R. M. J. Liskamp, J.-M. Ruysschaert, J. F. Ryan, F. Separovic and A. Watts, J. Mol. Biol., 2007, 368, 982–997 CrossRef CAS PubMed.
- A. Hirano, H. Yoshikawa, S. Matsushita, Y. Yamada and K. Shiraki, Langmuir, 2012, 28, 3887–3895 CrossRef CAS PubMed.
- R. Michel and M. Gradzielski, Int. J. Mol. Sci., 2012, 13, 11610–11642 CrossRef CAS PubMed.
- M. Schulz, A. Olubummo and W. H. Binder, Soft Matter, 2012, 8, 4849–4864 RSC.
- A. Negoda, Y. Liu, W. C. Hou, C. Corredor, B. Y. Moghadam, C. Musolff, L. Li, W. Walker, P. Westerhoff, A. J. Mason, P. Duxbury, J. D. Posner and R. M. Worden, Int. J. Biomed. Nanosci. Nanotechnol., 2013, 3, 52–83 CrossRef CAS.
- P. R. Leroueil, S. Hong, A. Mecke, J. R. Baker Jr, B. G. Orr and M. M. Banaszak Holl, Acc. Chem. Res., 2007, 40, 335–342 CrossRef CAS PubMed.
- M. R. R. de Planque, S. Aghdaei, T. Roose and H. Morgan, ACS Nano, 2011, 5, 3599–3606 CrossRef CAS PubMed.
- A. Negoda, K. J. Kim, E. D. Crandall and R. M. Worden, Biochim. Biophys. Acta, 2013, 1828, 2215–2222 CrossRef CAS PubMed.
- S. A. Klein, S. J. Wilk, T. J. Thornton and J. D. Posner, J. Phys.: Conf. Ser., 2008, 109, 012022 CrossRef.
- S. Ramachandran, G. L. Kumar, R. H. Blick and D. W. van der Weide, Appl. Phys. Lett., 2005, 86, 083901 CrossRef PubMed.
- C. Corredor, W. C. Hou, S. A. Klein, B. Y. Moghadam, M. Goryll, K. Doudrick, P. Westerhoff and J. D. Posner, Carbon, 2013, 60, 67–75 CrossRef CAS PubMed.
- P. R. Leroueil, S. A. Berry, K. Duthie, G. Han, V. M. Rotello, D. Q. McNerny, J. R. Baker Jr, B. G. Orr and M. M. Holl, Nano Lett., 2008, 8, 420–424 CrossRef CAS PubMed.
- C. M. Goodman, C. D. McCusker, T. Yilmaz and V. M. Rotello, Bioconjugate Chem., 2004, 15, 897–900 CrossRef CAS PubMed.
- A. Hirano, K. Uda, Y. Maeda, T. Akasaka and K. Shiraki, Langmuir, 2010, 26, 17256–17259 CrossRef CAS PubMed.
- B. Y. Moghadam, W. C. Hou, C. Corredor, P. Westerhoff and J. D. Posner, Langmuir, 2012, 28, 16318–16326 CrossRef CAS PubMed.
- H. Pera, J. M. Kleijn and F. A. M. Leermakers, Chem. Lett., 2012, 41, 1322–1324 CrossRef CAS.
- E. Fröhlich, Int. J. Nanomed., 2012, 7, 5577–5591 CrossRef PubMed.
- J. Chen, J. A. Hessler, K. Putchakayala, B. K. Panama, D. P. Khan, S. Hong, D. G. Mullen, S. C. Dimaggio, A. Som, G. N. Tew, A. N. Lopatin, J. R. Baker, M. M. Holl and B. G. Orr, J. Phys. Chem. B, 2009, 113, 11179–11185 CrossRef CAS PubMed.
- P. Ruenraroengsak, P. Novak, D. Berhanu, A. J. Thorley, E. Valsami-Jones, J. Gorelik, Y. E. Korchev and T. D. Tetley, Nanotoxicology, 2012, 6, 94–108 CrossRef CAS PubMed.
- S. Anguissola, D. Garry, A. Salvati, P. J. O'Brien and K. A. Dawson, PLoS One, 2014, 9, e108025 Search PubMed.
- B. A. Howell, Y. Kapoor and A. Chauhan, in Methods in Bioengineering: Alternatives to Animal Testing, ed. T. J. Maguire and E. Novik, Artech House, Boston, 2010, ch. 5, pp. 99–114 Search PubMed.
- A. S. Ladokhin, W. C. Wimley and S. H. White, Biophys. J., 1995, 69, 1964–1971 CrossRef CAS.
- S. Rex, Biophys. Chem., 1996, 58, 75–85 CrossRef CAS.
- A. M. Powl, J. M. East and A. G. Lee, Biochemistry, 2008, 47, 4317–4328 CrossRef CAS PubMed.
- A. Popat, S. B. Hartono, F. Stahr, J. Liu, S. Z. Qiao and G. Q. M. Lu, Nanoscale, 2011, 3, 2801–2818 RSC.
- D. Douroumis, I. Onyesom, M. Maniruzzaman and J. Mitchell, Crit. Rev. Biotechnol., 2013, 33, 229–245 CrossRef CAS PubMed.
- A. Baeza, M. Colilla and M. Vallet-Regí, Expert Opin. Drug Delivery, 2015, 12, 319–337 CrossRef PubMed.
- E. E. Ross, S.-W. Mok and S. R. Bugni, Langmuir, 2011, 27, 8634–8644 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- M. P. Monopoli, C. Aberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 7, 5845–5857 CrossRef PubMed.
- R. Richter, A. Mukhopadhyay and A. Brisson, Biophys. J., 2003, 85, 3035–3047 CrossRef CAS.
- R. P. Richter, R. Bérat and A. R. Brisson, Langmuir, 2006, 22, 3497–3505 CrossRef CAS PubMed.
- P. S. Cremer and S. G. Boxer, J. Phys. Chem. B, 1999, 103, 2554–2559 CrossRef CAS.
- E. Reimhult, F. Höök and B. Kasemo, J. Chem. Phys., 2002, 117, 7401–7404 CrossRef CAS PubMed.
- E. Reimhult, M. Zäch, F. Höök and B. Kasemo, Langmuir, 2006, 22, 3313–3319 CrossRef CAS PubMed.
- T. Cha, A. Guo and X.-Y. Zhu, Biophys. J., 2006, 90, 1270–1274 CrossRef CAS PubMed.
- T. M. Bayerl and M. Bloom, Biophys. J., 1990, 58, 357–362 CrossRef CAS.
- T. Buranda, J. Huang, G. V. Ramarao, L. K. Ista, R. S. Larson, T. L. Ward, L. A. Sklar and G. P. Lopez, Langmuir, 2003, 19, 1654–1663 CrossRef CAS.
- S. Savarala, S. Ahmed, M. A. Ilies and S. L. Wunder, ACS Nano, 2011, 5, 2619–2628 CrossRef CAS PubMed.
- S. Savarala, S. Ahmed, M. A. Ilies and S. L. Wunder, Langmuir, 2010, 26, 12081–12088 CrossRef CAS PubMed.
- S. Ahmed and S. L. Wunder, Langmuir, 2009, 25, 3682–3691 CrossRef CAS PubMed.
- S. Mornet, O. Lambert, E. Duguet and A. Brisson, Nano Lett., 2005, 5, 281–285 CrossRef CAS PubMed.
- F. Wang and J. Liu, Small, 2014, 10, 3927–3931 CrossRef CAS PubMed.
- T. H. Anderson, Y. Min, K. L. Weirich, H. Zeng, D. Fygenson and J. N. Israelachvili, Langmuir, 2009, 25, 6997–7005 CrossRef CAS PubMed.
- H. Wang, J. Drazenovic, Z. Luo, J. Zhang, H. Zhoua and S. L. Wunder, RSC Adv., 2012, 2, 11336–11348 RSC.
- Q. Mu, N. S. Hondow, L. Krzemiński, A. P. Brown, L. J. Jeuken and M. N. Routledge, Part. Fibre Toxicol., 2012, 9, 29 CrossRef CAS PubMed.
- A. Vakurov, R. Brydson and A. Nelson, Langmuir, 2012, 28, 1246–1255 CrossRef CAS PubMed.
- B. Wang, L. Zhang, S. C. Bae and S. Granick, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18171–18175 CrossRef CAS PubMed.
- Y. Liu, Z. Zhang, Q. Zhang, G. L. Baker and R. M. Worden, Biochim. Biophys. Acta, 2014, 1838, 429–437 CrossRef CAS PubMed.
- O. Le Bihan, P. Bonnafous, L. Marak, T. Bickel, S. Trépout, S. Mornet, F. De Haas, H. Talbot, J.-C. Taveau and O. Lambert, J. Struct. Biol., 2009, 168, 419–425 CrossRef CAS PubMed.
- I. Hoffmann, R. Michel, M. Sharp, O. Holderer, M.-S. Appavou, F. Polzer, B. Faragob and M. Gradzielski, Nanoscale, 2014, 6, 6945–6952 RSC.
- S. Zhang, A. Nelson and P. A. Beales, Langmuir, 2012, 28, 12831–12837 CrossRef CAS PubMed.
- V. Puddu and C. C. Perry, Langmuir, 2013, 30, 227–233 CrossRef PubMed.
- A. Rimola, D. Costa, M. Sodupe, J.-F. Lambert and P. Ugliengo, Chem. Soc. Rev., 2013, 113, 4216–4313 CrossRef CAS PubMed.
- S. V. Patwardhan, F. S. Emami, R. J. Berry, S. E. Jones, R. R. Naik, O. Deschaume, H. Heinz and C. C. Perry, J. Am. Chem. Soc., 2012, 134, 6244–6256 CrossRef CAS PubMed.
- L. Zhang and S. Granick, Nano Lett., 2006, 6, 694–698 CrossRef CAS PubMed.
- D. Napierska, L. C. J. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846–853 CrossRef CAS PubMed.
- R. Foldbjerg, J. Wang, C. Beer, K. Thorsen, D. S. Sutherland and H. Autrup, Chem.-Biol. Interact., 2013, 204, 28–38 CrossRef CAS PubMed.
- Z. Chu, Y. Huang, Q. Tao and Q. Li, Nanoscale, 2011, 3, 3291–3299 RSC.
- A. Lesniak, A. Salvati, M. J. Santos-Martinez, M. W. Radomski, K. A. Dawson and C. Åberg, J. Am. Chem. Soc., 2013, 135, 1438–1444 CrossRef CAS PubMed.
- P. Stano and P. L. Luisi, Curr. Opin. Biotechnol., 2013, 24, 633–638 CrossRef CAS PubMed.
- P. Stano, Biotechnol. J., 2011, 6, 850–859 CrossRef CAS PubMed.
- P. L. Luisi, Anat. Rec., 2002, 268, 208–214 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |