The reactions of N-methylformamide and N,N-dimethylformamide with OH and their photo-oxidation under atmospheric conditions: experimental and theoretical studies†
Received
11th December 2014
, Accepted 6th February 2015
First published on 17th February 2015
Abstract
The reactions of OH radicals with CH3NHCHO (N-methylformamide, MF) and (CH3)2NCHO (N,N-dimethylformamide, DMF) have been studied by experimental and computational methods. Rate coefficients were determined as a function of temperature (T = 260–295 K) and pressure (P = 30–600 mbar) by the flash photolysis/laser-induced fluorescence technique. OH radicals were produced by laser flash photolysis of 2,4-pentanedione or tert-butyl hydroperoxide under pseudo-first order conditions in an excess of the corresponding amide. The rate coefficients obtained show negative temperature dependences that can be parameterized as follows: kOH+MF = (1.3 ± 0.4) × 10−12 exp(3.7 kJ mol−1/(RT)) cm3 s−1 and kOH+DMF = (5.5 ± 1.7) × 10−13 exp(6.6 kJ mol−1/(RT)) cm3 s−1. The rate coefficient kOH+MF shows very weak positive pressure dependence whereas kOH+DMF was found to be independent of pressure. The Arrhenius equations given, within their uncertainty, are valid for the entire pressure range of our experiments. Furthermore, MF and DMF smog-chamber photo-oxidation experiments were monitored by proton-transfer-reaction time-of-flight mass spectrometry. Atmospheric MF photo-oxidation results in 65% CH3NCO (methylisocyanate), 16% (CHO)2NH, and NOx-dependent amounts of CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NH and CH3NHNO2 as primary products, while DMF photo-oxidation results in around 35% CH3N(CHO)2 as primary product and 65% meta-stable (CH3)2NC(O)OONO2 degrading to NOx-dependent amounts of CH3NCH2 (N-methylmethanimine), (CH3)2NNO (N-nitroso dimethylamine) and (CH3)2NNO2 (N-nitro dimethylamine). The potential for nitramine formation in MF photo-oxidation is comparable to that of methylamine whereas the potential to form nitrosamine and nitramine in DMF photo-oxidation is larger than for dimethylamine. Quantum chemistry supported atmospheric degradation mechanisms for MF and DMF are presented. Rate coefficients and initial branching ratios calculated with statistical rate theory based on molecular data from quantum chemical calculations at the CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ level of theory show satisfactory agreement with the experimental results. It turned out that adjustment of calculated threshold energies by 0.2 to 8.8 kJ mol−1 lead to agreement between experimental and predicted results.
NH and CH3NHNO2 as primary products, while DMF photo-oxidation results in around 35% CH3N(CHO)2 as primary product and 65% meta-stable (CH3)2NC(O)OONO2 degrading to NOx-dependent amounts of CH3NCH2 (N-methylmethanimine), (CH3)2NNO (N-nitroso dimethylamine) and (CH3)2NNO2 (N-nitro dimethylamine). The potential for nitramine formation in MF photo-oxidation is comparable to that of methylamine whereas the potential to form nitrosamine and nitramine in DMF photo-oxidation is larger than for dimethylamine. Quantum chemistry supported atmospheric degradation mechanisms for MF and DMF are presented. Rate coefficients and initial branching ratios calculated with statistical rate theory based on molecular data from quantum chemical calculations at the CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ level of theory show satisfactory agreement with the experimental results. It turned out that adjustment of calculated threshold energies by 0.2 to 8.8 kJ mol−1 lead to agreement between experimental and predicted results.
1. Introduction
N-methylformamide (MF) and N,N-dimethylformamide (DMF) are High Production Volume (HPV) chemicals on the US HPV Challenge Program Chemical List along with around 2800 other compounds.1 MF and DMF have also been reported as important products in the atmospheric degradation of dimethylamine and trimethylamine, respectively.2 The toxicology of dimethyl and monomethyl derivatives of acetamide and formamide was recently reviewed;3 DMF is handled under the OECD HPV Screening Information Data Set (SIDS) Program4 while the test plan for MF has not yet been implemented.1
Barnes et al.5 reviewed the scarce information on amides, presented new kinetic and product information, and outlined the photo-oxidation mechanisms for formamides and acetamides. Recently, Borduas et al.6 reported photo-oxidation products and kinetic data for OH reactions with five amides and presented additional mechanistic insight from quantum chemistry calculations. In short: the atmospheric gas phase chemistry of amides is not yet fully understood. The amide + OH and amide + NO3 reactions are relatively fast (rate coefficients in the order of 10−11 and 10−14 cm3 s−1, respectively),5 and the average global lifetimes of amides with respect to reactions with OH and NO3 radicals will be less than a few days; reactions with Cl atoms and O3 do not constitute important sinks for amides. The gas phase UV spectra of amides are structureless and show low absorption cross-sections (<10−20 cm2) above 270 nm.7 Consequently, tropospheric photolysis of amides is not an important loss process. Due to their solubility in water, uptake into droplets and subsequent deposition may be an important sink.
The present communication reports on experimental kinetic studies of MF and DMF reactions with OH radicals at atmospherically relevant temperatures and from product studies in photo-oxidation experiments of MF and DMF. The results are corroborated by high-level quantum chemical calculations and statistical rate theory.
2. Experimental methods
2.1. Laser photolysis experiments
The kinetic experiments were performed by using the flash photolysis/laser-induced fluorescence technique in a coolable slow-flow reactor with helium as bath gas. The experimental setup was described in detail elsewhere,8 so only a brief description will be given here.
The reaction cell has an inner volume of ca. 640 cm3 and is enclosed in a steel housing that can be evacuated to prevent ice formation on the windows and to improve thermal insulation. The gas flows were controlled by calibrated mass flow controllers, and accumulation of reaction products is avoided by choosing appropriate flow rates. OH radicals were produced by pulsed photolysis of 2,4-pentanedione9 (for OH + DMF) or tert-butyl hydroperoxide10 (t-BuOOH, for OH + MF) with a KrF excimer laser at 248 nm. The OH concentration-time profiles were monitored by recording the laser-induced fluorescence (LIF) in a wavelength range of (308 ± 7.5) nm after excitation at 281.9 nm. For excitation a frequency-doubled dye laser was used that was operated with rhodamine 6G and pumped with a Nd:YAG laser. The fluorescence signal was detected with a photomultiplier oriented perpendicularly to the antiparallel excimer and dye laser beams. A variation of the pulse energy of the photolysis laser between 60 and 300 mJ corresponding to fluences of 20–100 mJ cm−2 had no significant effect on the rate coefficient, which indicates that unwanted radical–radical reactions are unimportant under our experimental conditions.
A bath gas flow with a well-known concentration of the corresponding amide was produced by flowing helium through a saturator that consisted of a conventional gas washing bottle with a fritted disc. The washing bottle containing the pure amide was kept in a water bath at 294 K, and the possibility of evaporative cooling was checked by monitoring the temperature at the highest flow rates for 90 minutes. No change in temperature was observed. In general, one bottle (height of the liquid column: ca. 10 cm, helium flow: 5–50 sccm) was found to be sufficient for saturation under our experimental conditions. Test measurements with a second bottle in a two-stage arrangement gave identical results. The amide concentrations were calculated with the ideal gas law from the measured vapor pressures by assuming complete saturation.
To assess the measured values, the vapor pressure of DMF was calculated from Antoine equation, log(P/bar) = A − [B/(T/K + C)],11 with the constants from ref. 12: A = 3.93068, B = 1337.716, and C = −82.648 (determined in the temperature range 303–363 K).12,13 For T = 294 K, one obtains a vapor pressure of P = 3.99 mbar. For MF the Antoine constants in ref. 12, A = 4.99796, B = 2134.031, and C = −45.071, were obtained in the temperature range 370–472 K.12,14 Because this is much higher than the temperature in our saturator, we determined the vapor pressure of MF at 294 K in separate experiments. We used a 200 cm3 stainless steel vacuum line connected to a turbo pump and a capacitance manometer. The leak rate of this vacuum line was determined to be 0.002 mbar min−1. The MF was purified by first purging it with helium in a gas washing bottle for 3 hours and then thoroughly degassing it in repeated freeze–pump–thaw cycles. The gas washing bottle was connected to the vacuum line, and the pressure rise was measured until the change in pressure reached the leak rate determined before. In this way, we obtained a vapor pressure of P = 0.33 mbar at T = 294 K. This value is somewhat higher than the value of 0.266 mbar obtained with the above Antoine parameters. However, we note that extrapolations with the Antoine equation outside the temperature range, where the parameters were determined, have to be considered with great caution.
The mixtures of 2,4-pentanedione and t-BuOOH in helium were manometrically prepared in gas cylinders and allowed to homogenize for at least 16 hours before use.
2,4-Pentanedione (99%, Sigma Aldrich), t-BuOOH (80% in H2O, Sigma Aldrich), MF (99% Sigma Aldrich), and DMF (99% Alfa Aesar) were purified by repeated freeze–pump–thaw cycles, and the gas washing bottles containing the amides were purged before each experiment by a helium flow for at least 30 minutes. The purity of the helium was >99.999% (Air Liquide).
In our kinetic experiments pseudo-first order conditions were ensured by keeping the amide concentrations in large excess of the initial OH concentration. Typical amide concentrations were in the range 1014–1016 cm−3 whereas OH concentrations can be estimated from absorption cross-sections of our precursors as being typically in the order of 1011 cm−3. The decay of the OH concentration, of which an example is shown in Fig. 1, is then given by a first-order rate law, d[OH]/dt = −kpseudo[OH], where kpseudo = kbim[amide] + kd, with kbim being the bimolecular rate coefficient of the OH + amide reaction and kd characterizing other first-order loss processes of the OH radicals (diffusional loss, reaction with precursor etc.). If kd is assumed to be constant for a given temperature and pressure, the bimolecular rate coefficient kbim is given as the slope of kpseudo plotted vs. amide concentration. An example plot is given in Fig. 2. We note that in some experiments we observed biexponential decays. However, because the scattering of the low LIF intensities at longer times was considerable, we refrained from performing biexponential analyses in these cases and determined the rate coefficients kpseudo always from the initial, linear part of the semi-logarithmic plots.
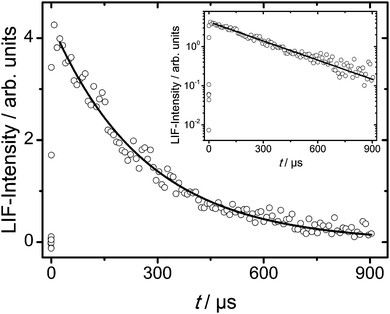 |
| | Fig. 1 LIF intensity–time profile and least-squares fit for MF + OH experiment at T = 295 K, P = 600 mbar, and [MF]0 = 1.4 × 1016 cm−3. | |
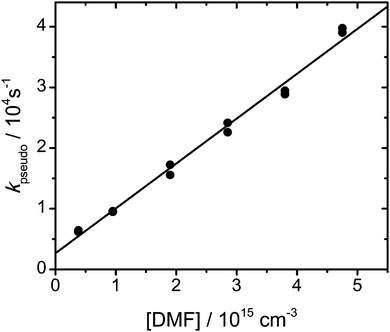 |
| | Fig. 2 Pseudo-first order plot for OH + DMA (T = 294 K, P = 100 mbar). | |
The experimental conditions for the OH + MF and OH + DMF experiments are summarized in Tables S1 and S2 of the ESI.† Due to the multitude of parameters that influence the uncertainty of the experiments, and which are difficult to quantify exactly, a maximum error of 30% for the rate coefficients was estimated. We note, however, that the statistical error is smaller (see below).
2.2. Photo-oxidation studies
OH + amide reaction product studies were conducted in a 480 l Teflon coated reaction chamber made of glass and surrounded by 18 UV/Vis lamps (λ ≥ 300 nm) to simulate sunlight.15 Experiments were performed in dry synthetic air at an absolute pressure of 970 mbar and temperatures in the range between 302 K and 306 K. OH radicals were produced by photolysis of isopropyl nitrate. The initial reaction conditions were 250 ppbV of the reagent amide, 100 ppb of NO and 250 ppbV of isopropyl nitrite. The reagent mixture was irradiated for 15 minutes. During photo-oxidation, NO was continuously added to the chamber at a rate of 6.8 ppb min−1. Synthetic air was continuously added to the chamber at a flow rate of 3.26 slpm to replenish instrumental sampling flows. A large amount of NO was injected into the chamber 10 minutes after the lamps had been turned off for identification of rapidly decaying thermally unstable acylperoxynitrate compounds. Duplicate experiments were carried out for each reagent.
The chemicals and gases used in the experiments and their stated purities were as follows: N-methylformamide (99%, Sigma-Aldrich), N,N-dimethylformamide (99.8%, Sigma-Aldrich), synthetic air (5.0, Messer Austria), nitrogen oxide in nitrogen (401 mg m−3 NO; Linde). Isopropyl nitrite was synthesized from sodium nitrite in water, isopropanol and sulfuric acid.
Analytical instrumentation coupled to the chamber included a high-resolution proton-transfer-reaction time-of-flight mass spectrometer (PTR-TOF 8000, Ionicon Analytik GmbH)16 for amide and amide photo-oxidation product measurements, a chemiluminescence NO-detection instrument (CLD770 AL ppt, ECO PHYSICS) combined with a photolytic converter (PLC 760 MH, ECO PHYSICS) for NO2 detection, a UV photometric ozone analyzer (49i, Thermo Scientific) and a temperature/relative humidity sensor (UFT75-AT, MELTEC). The PTR-ToF-MS was interfaced to the chamber via a PEEK capillary tube (temperature: 50 °C; flow: 0.15 slpm). The PTR-ToF-MS was operated at a reduced electric field of 52 Td and 85 Td, respectively, during the duplicate experiments. Chemical ionization reagent ions were H3O+ and H3O+(H2O), respectively, for the two PTR-ToF-MS operating modes.
For PTR-ToF-MS data analysis, we used the “PTR-TOF Data Analyzer” software.17 Instrumental response factors for amides and amide photo-oxidation products were derived from ion-molecule collision theory18 using calculated molecular properties.
3. Computational methods
3.1. Electronic structure calculations
Stationary points on the potential energy surfaces for reactions of the amides with OH were characterized using second-order Møller–Plesset perturbation theory (MP2) as well as BHandHLYP density functional theory. Pre- and post-reaction adducts were located by following the reaction path (IRC) from the saddle points. In addition, reaction enthalpies were calculated using G3 model chemistry,19 while dipole moments and isotropic polarizabilities (employed in calculations of ion-molecule reaction rate coefficients, see above) were obtained in B3LYP/aug-cc-pVTZ calculations. The G3 results are summarized in Table S3 (ESI†).
DFT and MP2 calculations are often capable of giving very good geometries for minima on reactive potential energy surfaces, but may be less suitable for transition states as these may have significant multi-reference character. We have therefore re-optimized the geometries of the transition states using CASPT2 with an active space consisting of the bonding and anti-bonding orbital of the bond being broken in the amide and the unpaired electron in the OH radical, i.e. 3 electrons in 3 orbitals.
Energies of the stationary points were improved using coupled cluster singles and doubles with explicitly correlated wave functions using the F12a approximation with perturbative triples scaled as recommended in the MOLPRO manual, denoted CCSD(T*)-F12a.20,21 Dunning's correlation consistent basis sets were used in all calculations.22,23
Unrestricted wave functions were used for the open-shell calculations. The G3, BHandHLYP, B3LYP and MP2 calculations were performed using Gaussian09,24 while the explicitly correlated coupled cluster calculations and CASPT2 calculations were performed using Molpro 2012.1.25
3.2. Statistical rate theory calculations
The reactions of amides with OH radicals proceed via pre-reaction complexes and the overall kinetics and may therefore be governed by forward reactions and back dissociation of the complex. Specific rate coefficients for the unimolecular forward reactions were calculated using RRKM theory based on energies and molecular data from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations. Rate coefficients for back dissociation of the complex were obtained via detailed balancing from capture rate coefficients of complex formation. The capture rate coefficients were calculated with long-range transition state theory (LRTST).26 Spin–orbit coupling in the OH radical (139.7 cm−1)27 was accounted for in the model by lowering the energy of the OH radical by half of the splitting and including the 2Π3/2 and 2Π1/2 spin–orbit states in the electronic partition function. It was assumed that spin–orbit coupling could be neglected in the pre-reaction adduct and in the transition states.
Lennard-Jones parameters of MF and DMF were approximated by the corresponding values for methyl acetate (ε/kB = 469.8 K, σ = 4.94 Å)28 and for collisional energy transfer an exponential-down model was assumed. The energy transfer parameter for collisions with He was set to 〈ΔEdown〉 = 200 cm−1. Variation of this value between 100 and 300 cm−1 lead only to minor changes (<2%) in the calculated rate coefficients.
The temperature and pressure dependence of the phenomenological rate coefficients for the bimolecular overall reactions was obtained from the eigenvalues of a corresponding master equation by employing the Bartis–Widom method29,30 as implemented in the MESMER code.31
4. Results
There are three possible product channels in the MF + OH reaction:| | | CH3NHCHO + OH → CH3NHĊO + H2O ΔH° = −100 kJ mol−1 | (1a) |
| | | → CH3ṄCHO + H2O ΔH° = −40 kJ mol−1 | (1b) |
| | | → ĊH2NHCHO + H2O ΔH° = −104 kJ mol−1 | (1c) |
and also three possible product channels in the DMF + OH reaction:| | | (CH3)2NCHO + OH → (CH3)2NĊO + H2O ΔH° = −98 kJ mol−1 | (2a) |
| | | → trans-ĊH2N(CH3)CHO + H2O ΔH° = −100 kJ mol−1 | (2b) |
| | | → cis-ĊH2N(CH3)CHO + H2O ΔH° = −100 kJ mol−1 | (2c) |
The reaction enthalpies listed refer to 298 K and to the lowest energy conformations of the species involved; they stem from G3 calculations.19 We note that the barrier for cis–trans isomerization of ĊH2N(CH3)CHO is 80 kJ mol−1.
4.1. Structures and energies of stationary points
Stationary points on the amide + OH potential energy surfaces (PES) were located in MP2 and BHandHLYP calculations employing different basis sets. The results of the calculations are summarized in Tables S4 and S5 (ESI†) while Fig. 3 and 4 show the energies of the stationary points calculated using CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ. Cartesian coordinates of the stationary points calculated using MP2/aug-cc-pVTZ are collected in Tables S6 and S7 (ESI†). The values from G3X-K32 model chemistry reported by Borduas et al.6 for MF are similar to the present results. The reactions of both amides with OH radicals are characterized by post- and pre-reaction van der Waals adducts and submerged barriers. To locate the pre- and post-reaction complexes, IRCs were followed in both BHandHLYP and MP2 calculations using the aug-cc-pVDZ basis set.
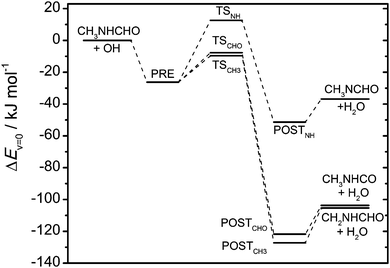 |
| | Fig. 3 Potential energy diagram (including zero-point energies) for the OH + MF reaction from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations; energies are relative to reactant energies. | |
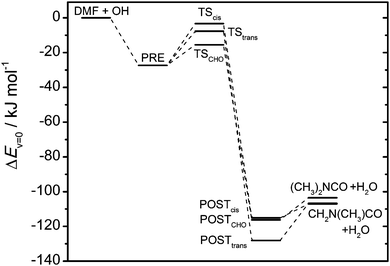 |
| | Fig. 4 Potential energy diagram (including zero-point energies) for the OH + DMF reaction from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations; energies are relative to reactant energies. | |
The transition states of H abstraction from the methyl (TSCH3) and the carbonyl (TSCHO) groups in MF are very similar in energy, and it is therefore difficult to calculate the branching ratio accurately as even small changes in the barrier heights have a large impact on the calculated branching ratio. It is intuitively clear that abstraction from the NH group (TSNH) will constitute only a minor route.
For DMF, the barrier of H abstraction from the carbonyl group is significantly lower than for the methyl groups (TScis and TStrans), and it is therefore expected that it will be the dominating reaction channel. However, it is not possible to rule out H abstraction from the methyl groups from the present calculations.
The results from the MP2 and BHandHLYP calculations (see ESI,† Tables S4 and S5) are generally in good agreement, although the methods predict different conformers of the DMF post reaction adducts as the most stable, but with very similar energies. A comparison of important transition state geometry parameters is shown in Fig. S1 (ESI†). The transition state geometries obtained with MP2/aug-cc-pVTZ and BHandHLYP/aug-cc-pVTZ are very similar; the largest differences in bond lengths for the bonds being formed and broken is 5 pm, and the largest difference in the improved (CCSD(T*)-F12a, see above) barrier heights is 4 kJ mol−1. BHandHLYP calculations predict slightly earlier transition states and lower barriers for all reactions. The CASPT2(3,3) calculations give geometries very similar to the MP2 results except for abstraction from the carbonyl group in DMF where CASPT2 gives a much later transition state, but a very similar improved barrier height (−14.8 kJ mol−1 for CASPT2 vs. −15.5 for MP2). The largest deviation between the barrier heights based on the CASPT2 and the MP2 and BHandHLYP geometries is 3.2 and 6.5 kJ mol−1, respectively.
4.2. Rate coefficients of the OH + MF and OH + DMF reactions
Fig. 5 shows experimental and calculated rate coefficients of the OH + MF reaction as a function of temperature. The experimental values exhibit a weak negative temperature dependence that can be parameterized as follows: kbim(OH + MF) = (1.3 ± 0.4) × 10−12 exp(3.7 kJ mol−1/(RT)) cm3 s−1. The detailed experimental conditions for the individual data points along with the measured rate coefficients are collected in Table S1 (ESI†). We also note that our experiments in the pressure range 30–600 mbar seem to indicate a very weak positive pressure dependence (increase of ∼20% in going from 30 to 600 mbar), which is illustrated in Fig. S2 (ESI†).
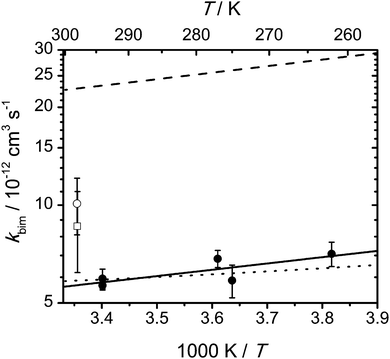 |
| | Fig. 5 Arrhenius plot of the rate coefficients for MF + OH; experimental values from this work at 600 mbar in helium (full circles), from ref. 6 at 1000 mbar in air (empty circle, with 20% uncertainty, see footnotea to Table 3), and from ref. 33 at 1000 mbar in air (empty square); the solid line corresponds to the Arrhenius equation kbim = 1.3 × 10−12 exp(3.7 kJ mol−1/(RT)) cm3 s−1, the dashed line represents the predicted rate coefficient from our master equation calculations based on the CCSD(T)-F12a//MP2 calculations, and the dotted line represents the calculated rate coefficient with the barrier heights adjusted to best reproduce the experimental values (see Table 1); error bars on the individual data points from our experiments denote two standard deviations and do not include possible systematic errors. | |
The rate coefficients calculated using the CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ results are about a factor of 5 higher than the experimental values. The calculated capture rate coefficient is rather large, kcap = 7.59 × 10−10 (T/298 K)−1/6 cm3 s−1 (ESI,† Table S8), but reducing it by a factor of 2 lowers the phenomenological rate coefficient only by less than 8% in the 250–300 K range. The relative branching fractions for H abstraction at the CHO and CH3 group hardly varies with a change in the capture rate coefficient and are calculated to be 91.5% and 8.5%, respectively, at T = 300 K and 89.8% and 10.2%, respectively, at T = 250 K. The contribution of H abstraction from the NH group is negligible.
Lowering the threshold energy for abstraction from the NH group by 4 kJ mol−1 and simultaneously increasing the threshold energies for abstraction from the CHO and the CH3 group by 4 kJ mol−1 changes the relative branching fractions merely to 89.7%, 10.2% and 0.1% for H abstraction from CHO, CH3, and NH, respectively, but results in a 75% reduction of the overall rate coefficient (at T = 300 K). Therefore, we conclude that the overall reaction is governed by the inner transition states and that abstraction from the NH group is not important at atmospheric conditions.
Fig. 5, in connection with Table 1, illustrates that a very good agreement between calculated and experimentally determined rate coefficients can be achieved with only moderate adjustment of the barriers. We note that with the adjusted values also the branching fractions observed in our photo-oxidation experiments are very well reproduced (see below).
Table 1 Calculated and adjusted transition state energies for OH + MF, relative to the energy of the reactants (unit: kJ mol−1)
| |
TSCH3 |
TSCHO |
TSNH |
|
CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ results.
Adjusted barrier heights to reproduce experimental rate coefficients and relative product branching fractions from our photo-oxidation study (see below) with our master equation calculations.
Excluded from analysis.
|
| Calculateda |
−9.6 |
−7.7 |
12.6 |
| Adjustedb |
−4.0 |
−4.1 |
12.6c |
Fig. 6 shows experimental and calculated rate coefficients of the OH + DMF reaction as a function of temperature. Also for this reaction system the experimental values exhibit a weak negative temperature dependence that can be parameterized as follows: kbim(OH + DMF) = (5.5 ± 1.7) × 10−13 exp(6.6 kJ mol−1/(RT)) cm3 s−1. The detailed experimental conditions for the individual data points along with the measured rate coefficients are collected in Table S2 (ESI†). We note that no discernible pressure dependence of the rate coefficient could be observed within our experimental uncertainty (see ESI,† Fig. S3). The agreement between the calculated and experimental rate coefficients is not as good as for the MF + OH reaction. This can be seen from a comparison of the dashed and solid lines in Fig. 5 and 6. Whereas for OH + MF this difference is about a factor of 5, the difference for OH + DMF is a factor of ∼10. Accordingly, rather large adjustments of the barriers are necessary to reproduce the experimental rates as can be seen from Table 2.
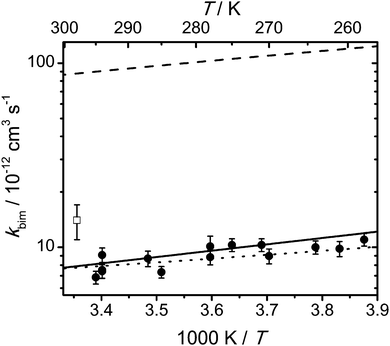 |
| | Fig. 6 Arrhenius plot of the rate coefficients for DMF + OH; experimental values from this work at 100 mbar in helium (full circles) and from ref. 33 at 1000 mbar in air (empty square); the solid line corresponds to an Arrhenius equation kbim = 5.5 × 10−13 exp(6.6 kJ mol−1/(RT)) cm3 s−1, the dashed line represents the predicted rate coefficient from our master equation calculations based on the CCSD(T)-F12a//MP2 calculations, and the dotted line represents the calculated rate coefficient with the barrier heights adjusted to best reproduce the experimental values (see Table 2); error bars on the individual data points from our experiments denote two standard deviations and do not include possible systematic errors. | |
Table 2 Calculated and adjusted transition state energies for OH + DMF, relative to the energy of the reactants (unit: kJ mol−1)
| |
TStrans |
TScis |
TSCHO |
|
CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ results.
Adjusted barrier heights to reproduce experimental rate coefficients and relative product branching fractions from our photo-oxidation study (see below) with our master equation calculations.
|
| Calculateda |
−7.7 |
−3.3 |
−15.5 |
| Adjustedb |
−7.9 |
+5.0 |
−6.7 |
The calculated capture rate coefficient for the DMF + OH reaction, kcap = 7.55 × 10−10 (T/298 K)−1/6 cm3 s−1, is very similar to that for the MF + OH reaction (Table S8, ESI†). But since the energy barrier for the lowest H abstraction step relative to the bimolecular reactants is considerably lower than in the case of MF + OH, a reduction of the capture rate coefficient by a factor of two also leads to a stronger decrease of the phenomenological rate coefficient by up to 27% (compared to up to 8% for MF + OH) in the temperature range 250–300 K.
The differences in the temperature dependence between the two reaction systems can be rationalized in terms of the potential energy surface. The pre-reaction complexes are nearly equally stable with respect to the bimolecular reactants, but the lowest energy barrier for hydrogen abstraction is somewhat larger for the OH + MF reaction than for the OH + DMF reaction (cf.Tables 1 and 2). This may lead to a somewhat stronger (negative) temperature dependence in the latter case. The differences in the pressure dependence are too small compared to our experimental uncertainty for a reasonable further analysis (cf. Fig. S2 and S3, ESI†).
Table 3 summarizes the currently available kinetic data for reactions of OH radicals with different amides. The rate coefficients reported by Solignac et al.33 are somewhat higher than those from both Koch et al.34 and the present work but agree within the combined error bars. An interesting feature of the data is that the values from relative measurements are consistently higher than those from absolute measurements. This is probably related to the nature of the experiments: in the relative rate measurements,5,6,33,35,36 the rate of amide removal is measured relatively to the removal rate of a reference compound, and any additional process that removes the amide will increase the observed rate. In the absolute measurements (this work and Koch et al.34), the measured quantity is the removal rate of OH radicals in an excess of amide, and any process removing the amide will give a smaller observed rate. Despite all our efforts to detect and prevent any wall loss, we cannot completely rule out that some of the amide has been lost by other processes before passing the detection volume in our photolysis cell. The negative activation energies from this work nicely agree with the other available values for N-methylacetamide, N,N-dimethylacetamide, N-methylpropanamide, and N,N-dimethylpropanamide.
Table 3 Rate coefficients and Arrhenius activation energies for the reactions of OH with different amides
| Amide |
k/10−12 cm3 s−1 |
E
a/kJ mol−1 |
Ref. |
|
T = 298 K, P = 1 bar (air), the errors given represent the standard deviation of multiple experiments, the authors estimate a total uncertainty closer to 20%.
T = 298 K. P = 1 bar (air).
Measurements at T = 294 K, P = 600 mbar (helium).
Measurements at T = 294 K, P = 30–600 mbar (helium).
T = 298 K, P = 1 bar (air).
T = 300 K, P = 130 mbar (helium).
T = 296 K, P = 1 bar (air).
T = 300 K and P = 1 bar (air).
|
| Formamide |
4.44 ± 0.46a |
|
6
|
|
N-Methylformamide |
10.1 ± 0.6a |
— |
6
|
| 8.6 ± 2.4b |
— |
33
|
| 5.7 ± 1.7c |
−3.7 |
This work |
|
N,N-Dimethylformamide |
14 ± 3b |
— |
33
|
| 8.5 ± 2.6d |
−6.6 |
This work |
| Acetamide |
3.5 ± 1e |
— |
5
|
| 0.77 ± 0.35a |
— |
6
|
|
N-Methylacetamide |
11 ± 3e |
— |
5
|
| 5.2e |
−4.1 |
34
|
| 5.42 ± 0.19a |
— |
6
|
|
N,N-Dimethylacetamide |
19 ± 3b |
— |
33
|
| 13.6f |
−5.2 |
34
|
| Propanamide |
1.78 ± 0.43a |
— |
6
|
|
N-Methylpropanamide |
7.6f |
−3.3 |
34
|
|
N,N-Dimethylpropanamide |
20.7f |
−4.4 |
34
|
| 1-Methyl-2-pyrrolidinone |
21.5 ± 3.6g |
|
35
|
| 22 ± 4h |
|
36
|
| 1-Methyl-2,2-pyrrolidinedione |
14 ± 3h |
|
36
|
| 2-Oxo-1-pyrrolidinecarboxaldehyde |
6 ± 1h |
|
36
|
4.3. CH3NHCHO photo-oxidation studies
Barnes et al.5 carried out OH and Cl atom initiated photo-oxidation experiments with MF employing long-path FTIR detection and reported (CHO)2NH and CH3NCO as the main products with yields of roughly 50% and 30%, respectively, and with variable amounts of the peroxynitrate-type compound, CH3NHC(O)OONO2, depending on the NOx-level in the experiment. Unfortunately, the lack of calibrated reference spectra prevented the authors from making a proper quantification of the product yields. Borduas et al.6 recently carried out NOx-free OH-initiated photo-oxidation experiments with MF employing PTR-MS detection and reported a CH3NCO yield around 40%; they further reported formamide (∼15% yield) and HNCO as oxidation products. Diformamide, (CHO)2NH, was only detected as a trace product and was not quantified.
Fig. 7 shows the time evolution of the main ion signals observed by PTR-ToF-MS during a photo-oxidation experiment. Fig. S4 and S5 (ESI†), respectively, illustrate the difference in PTR-ToF-MS mass spectra taken after and just before the photo-oxidation experiment, and the NO and NO2 mixing ratios during the experiment. The NO level is nearly constant around 25 ppbV during the photo-oxidation, whereas the NO2 level increases from a single-digit value to nearly 400 ppbV in the same period. Reacting compounds are MF (detected in its protonated form at m/z 60.042 and its 13C isotopologues at m/z 61.043) and isopropyl nitrite, which is detected at m/z 29.996 (NO+), m/z 43.055 (C3H7+), m/z 61.066 (C3H9O+) and m/z 79.074 (C3H11O2+). Signals that increased during the photo-oxidation experiment are m/z 58.029 (C2H4NO+, protonated methylisocyanate), m/z 74.024 (C2H2NO+, protonated N-formylformamide), m/z 76.041 (C2H2NO2+, protonated methylisocyanate water cluster) and m/z 77.032 (CH5N2O2+, protonated methylnitramine). We also observed an increase in the ion signals at m/z 43.018 (C2H3O+), m/z 45.034 (C3H9O+), m/z 45.992 (NO2+), m/z 59.049 (C3H7O+) and m/z 77.056 (C3H9O2+). These signals stem from compounds (acetaldehyde, acetone and nitric acid) typically formed in any smog chamber photo-oxidation experiment in the presence of isopropyl nitrite and NOx, i.e. even in the absence of MF. In our product analysis, we only included signals with a larger than 1% increase relative to decrease in the protonated MF signal. In spite of every effort made no trace of CH3NHC(O)OONO2 was ever detected in any of the present experiments.
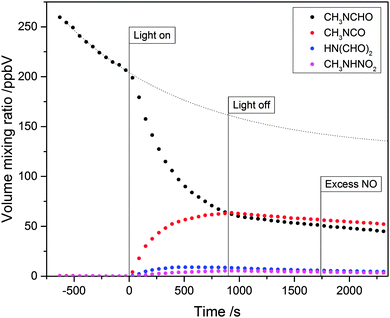 |
| | Fig. 7 Time evolution of N-methylformamide (MF) and the major degradation products during a photo-oxidation experiment. The thin dotted curve represents the extrapolated dilution and wall loss of MF. | |
Wall loss of MF was significant in the experiments. Additional loss was inflicted by synthetic air that was constantly added to compensate for instrumental sampling flows (kdilution = 1.13 × 10−4 s−1); analysis of the MF loss curve before and after the 15 min photo-oxidation period suggested an MF wall loss rate coefficient kwall![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) loss ≈ 2.6 × 10−4 s−1.
loss ≈ 2.6 × 10−4 s−1.
There is apparently a mismatch in Fig. 7 between the amount of N-methylformamide reacted and the amounts of products formed in the experiment. However, the apparent mismatch is deceptive: N-nitro methylamine, CH3NHNO2, formed with a ∼5% yield in the experiment shown, can only be formed in the reaction between CH3ṄH radicals and NO2, and even under conditions with high NO2 levels, as in the present case, the major loss of CH3ṄH radicals is due to reaction with O2 resulting in methanimine, CH2NH, which is extremely difficult to detect and to quantify due to polymerization and surface-induced reactions.
It is highly significant that there is no discernable change in the N-nitro methylamine volume mixing ratio upon addition of excess NO to titrate peroxy radicals present in the chamber. This indicates that either very little CH3NHC(O)OONO2 was ever formed during the experiment or that the lifetime of the compound is very short, or a combination of both of the above. FTIR-spectroscopic evidence for formation of CH3NHC(O)OONO2 in N-methylformamide photo-oxidation experiments5 is rather limited, and the weak bands reported as characteristic of CH3NHC(O)OONO2 actually have their counterparts in the IR spectrum of CH3NHNO2.37
Given the scarcity of well-documented mechanistic information on amide photo-oxidation chemistry, a first principles mapping of the reaction thermochemistry was undertaken and additional mechanism elucidating calculations carried out. The present quantum chemistry results have many similarities with those reported by Borduas et al.;6 there are, however, significant differences in approach, results and, consequently, in the mechanistic conclusions.
The CH3NHCHO reaction with OH radicals may, as already mentioned, proceed via hydrogen abstraction from either the CHO group, the NH group or the CH3 group. The N–H abstraction route is calculated to have a medium-sized barrier of around 12 kJ mol−1, while the C–H abstraction routes have barriers well below the entrance energy of the reactants. The quantum chemistry calculations therefore unambiguously show that the N–H abstraction route is of no importance in the CH3NHCHO + OH reaction at atmospheric conditions, and it will not be addressed any further here.
Route (1a), hydrogen abstraction from the CHO group, is expected to be followed by either radical dissociation leading to isocyanates or to the CH3ṄH radical, or by addition of O2:
| | | CH3NHCHO + OH → CH3NHĊO + H2O ΔH° = −100 kJ mol−1 | (1a) |
| | | CH3NHĊO → CH3NCO + H ΔH° = +116 kJ mol−1 | (3a) |
| | | → HNCO + ĊH3 ΔH° = +29 kJ mol−1 | (3b) |
| | | → CH3ṄH + CO ΔH° = +75 kJ mol−1 | (3c) |
| | | CH3NHĊO + O2 → CH3NHC(O)OȮ ΔH° = −157 kJ mol−1 | (4) |
The barriers to the N–H and N–C bond scission
reactions (3a) and (3b) are around 168 and 117 kJ mol
−1, respectively, which place both substantially above the entrance energy of the reactants in
(1a); the barrier to
reaction (3c) leading to CH
3ṄH + CO is barely 1 kJ mol
−1 above the entrance energy of the reactants. Considering the amount of internal energy available in the CH
3NHĊO radical upon its formation and the energy partitioning between CH
3NHĊO and H
2O, it can be concluded that these routes will not be important at atmospheric conditions. CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ results for
reactions (3a)–(3c) are illustrated in Fig. S6 and documented in Tables S9 and S10 (ESI
†).
The initially hot, chemically activated CH3NHC(O)OȮ* radical (* denotes rovibrational activation) may initiate two H transfer reactions followed by radical dissociations; the corresponding net reactions are:
| | | CH3NHĊO + O2 → CH3NCO + HO2 ΔH° = −93 kJ mol−1 | (5) |
| | | → CH2NH + CO2 + OH ΔH° = −271 kJ mol−1 | (6) |
The barrier to H transfer from the NH group is found to be far below the entrance energy of the reactants, and an IRC calculation links a post-reaction adduct between CH
3NCO and HO
2 on the exit side. The barrier to H transfer from the CH
3 group is also below the initial entrance energy of the reactants; subsequent dissociation of the resulting ĊH
2NHC(O)OOH radical to CH
2NH + CO
2 + OH proceeds
via another barrier below the entrance energy of the reactants.
Fig. 8 illustrates the energetics of the CH
3NHĊO + O
2 reaction (quantum chemistry results collected in Tables S11 and S12, ESI
†).
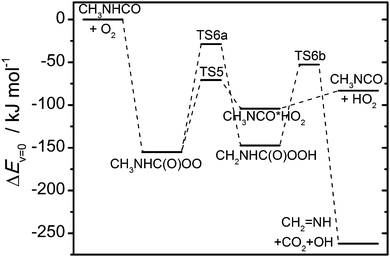 |
| | Fig. 8 Relative energies of stationary points on the potential energy surface of the CH3NHĊO + O2 reaction. Results from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations (including zero-point energies). | |
The branching between reactions (4)–(6) at atmospheric pressure was examined using a master equation model based on the potential energy surface shown in Fig. 8 and including the CH3NHC(O)OȮ + NO reaction as a bimolecular sink with a rate coefficient of 2 × 10−11 cm3 s−1, which is a typical value for RC(O)OȮ + NO reactions.38 With a rate coefficient for the CH3NHĊO + O2 reaction of around 5 × 10−12 cm3 s−1 (typical value for RĊ(O) + O2 reactions),38 the CH3NHĊO radicals will be thermalized before reaction with O2 at atmospheric conditions. The calculations show that reaction (6) is of no importance and that ca. 90% of the CH3NHĊO + O2 reaction results in CH3NCO. Increasing the energy of the saddle point TS5 by 4 kJ mol−1, decreases the CH3NCO yield to around 80%.
The CH3NHC(O)OȮ* radicals formed in reaction (4) and subsequently thermalized by collisions are expected to react with NO2 resulting in the formation of a meta-stable peroxynitrate, and with NO leading to CH3ṄH radicals:
| | | CH3NHC(O)OȮ + NO2 ⇆ CH3NHC(O)OONO2 ΔH° = −118 kJ mol−1 | (7) |
| | | CH3NHC(O)OȮ + NO → CH3NHC(O)Ȯ + NO2 ΔH° = −35 kJ mol−1 | (8) |
| | | CH3NHC(O)Ȯ → CH3ṄH + CO2 ΔH° = −81 kJ mol−1 | (9) |
The enthalpy of
reaction (7) is calculated to be −118 kJ mol
−1, which compares to −130 kJ mol
−1 for the corresponding (CH
3)
2NC(O)OONO
2 reaction (see later). The lifetime of (CH
3)
2NC(O)OONO
2 is around 14 minutes at 298 K and 1 bar.
39 Even allowing for a considerable uncertainty in the G3 results, the 12 kJ mol
−1 difference in reaction enthalpies for the two peroxynitrates in question suggests a much shorter lifetime for CH
3NHC(O)OONO
2 than for (CH
3)
2NC(O)OONO
2. The G3 results imply a lifetime of CH
3NHC(O)OONO
2 of around only 1 s at 298 K and 1 bar. In conclusion, the theoretical results suggest that not only will a minor amount of CH
3NHC(O)OȮ radicals be formed during
N-methylformamide photo-oxidation, but also that the lifetime of the corresponding peroxynitrate, CH
3NHC(O)OONO
2, will be very short.
The atmospheric fate of the CH3ṄH radical is well established from experimental and theoretical studies of the CH3NH2 + OH reaction:40–42
| | | CH3ṄH + O2 → CH2NH + HO2 ΔH° = −81 kJ mol−1 | (10) |
| | | CH3ṄH + NO2 → CH2NH + HONO ΔH° = −199 kJ mol−1 | (11a) |
| | | → CH3NHNO2 ΔH° = −213 kJ mol−1 | (11b) |
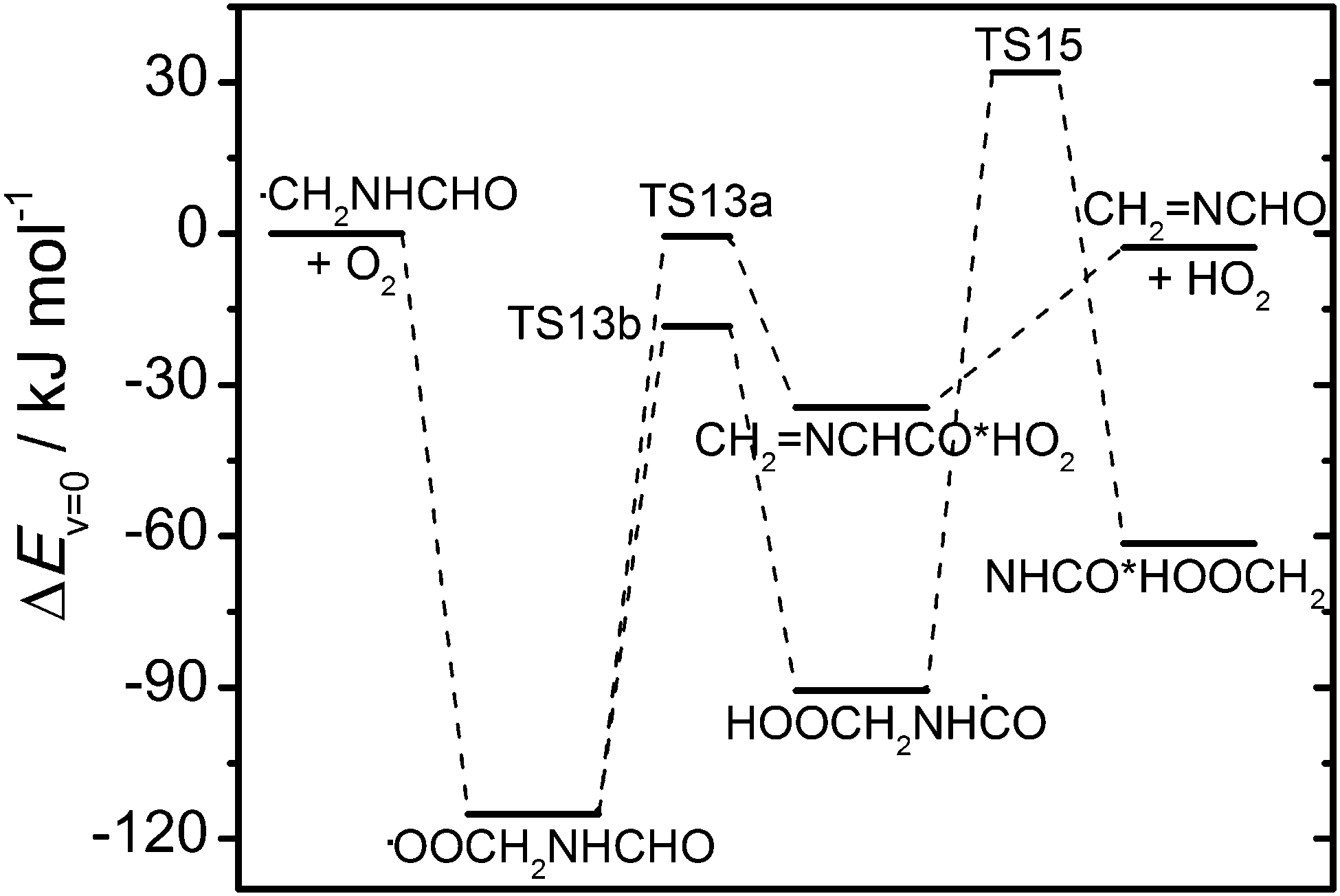
The other important abstraction route (1c) leading to the ĊH2NHCHO radical is expected to be followed by O2 addition forming a vibrationally excited peroxy radical that may initiate internal H abstraction reactions before reacting with NO (RO2 + HO2 reactions can be neglected with around 25 ppbV NO present in the chamber experiments):
| | | CH3NHCHO + OH → ĊH2NHCHO + H2O ΔH° = −104 kJ mol−1 | (1c) |
| | | ĊH2NHCHO + O2 → ȮOCH2NHCHO* ΔH° = −126 kJ mol−1 | (12) |
| | | ȮOCH2NHCHO* → CH2NCHO + HO2 ΔH° = +119 kJ mol−1 | (13a) |
| | | → HOOCH2NHĊO* ΔH° = +32 kJ mol−1 | (13b) |
| | | ȮOCH2NHCHO + NO → ȮCH2NHCHO + NO2 ΔH° = −53 kJ mol−1 | (14) |
Reaction (13a) proceeds
via a saddle point 0.6 kJ mol
−1 below the entrance energy of the reactants and a post-reaction complex on the exit side. The competing H transfer reaction from the CHO group has an around 18 kJ mol
−1 lower barrier towards formation of the intermediate HOOCH
2NHĊO radical, which in turn has a barrier around 32 kJ mol
−1 above the entrance energy of the reactants towards dissociation to HNCO
via another intermediate radical that can be described as a HNCO·CH
2OOH complex:
| | | HOOCH2NHĊO* → HNCO + OH + CH2O ΔH° = −96 kJ mol−1 | (15) |
CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ results for
reactions (13) and (15) are illustrated in
Fig. 9 and documented in Tables S13 and S14 (ESI
†).
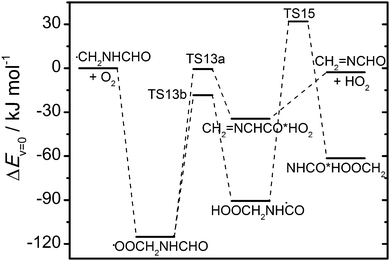 |
| | Fig. 9 Relative energies of stationary points on the potential energy surface of the ĊH2NHCHO + O2 reaction. Results from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations (including zero-point energies). | |
The branching between reactions (13a) and (14) at atmospheric pressure was examined using a master equation model based on the potential energy surface shown in Fig. 9 and including a bimolecular reaction between ȮOCH2NHCHO and NO with a rate coefficient of 9 × 10−12 cm3 s−1 (typical value for ROȮ + NO reactions).38 The results show that less than 1% CH2NCHO is formed in the reaction of O2 with thermalized ĊH2NHCHO radicals at 298 K (NO level assumed >1 ppbV). The ĊH2NHCHO radicals may, however, not be thermalized at the time of reaction with O2; reaction (1c) is exothermic by 104 kJ mol−1 and part of the reaction enthalpy could be available to ĊH2NHCHO in subsequent reactions. The master equation calculations show that with an additional 10, 20 or 30 kJ mol−1 available to reaction, the yield of CH2NCHO increases to respectively 6.6%, 26% and 56%. The actual amount of additional energy available to ĊH2NHCHO in reaction with O2 will implicitly depend on the rate coefficient of the reaction and explicitly on the energy partitioning in the preceding CH3NHCHO + OH reaction. Assuming equipartitioning of the reaction enthalpy, the ĊH2NHCHO radical will be formed with an additional internal energy of around 60 kJ mol−1 available for reaction. Assuming that the water molecule formed in reaction (1c) has one quantum of OH stretching vibration (∼3000 cm−1) and that the remaining reaction enthalpy is divided according to the equipartitioning principle then the ĊH2NHCHO radical will be formed with an additional internal energy of around 43 kJ mol−1. A typical rate coefficient for the reaction of a medium-sized alkyl radical with O2 is around 10−11 cm3 s−1,38 corresponding to a ĊH2NHCHO radical lifetime ∼20 ns at atmospheric conditions. Assuming an average energy transfer in collisions with N2 of 〈ΔEdown〉 = 250 cm−1, the initially 60 kJ mol−1 hot ĊH2NHCHO radicals will be thermalized in less than 3 ns at atmospheric conditions. Decreasing the barrier height of reaction (13a) by 4 kJ mol−1 increases the CH2NCHO yield to 2.6%. A conservative estimate is therefore that less than 5% of the ĊH2NHCHO + O2 reaction leads to CH2NCHO.
The ȮCH2NHCHO radicals may, in principle, either dissociate or undergo H abstraction by O2; the dissociation reaction is, however, highly endothermic ruling out this route at atmospheric conditions. Consequently, at atmospheric conditions the by far dominating primary product resulting from H abstraction from the CH3 group will be N-formylformamide:
| | | ȮCH2NHCHO → ṄHCHO + CH2O ΔH° = +90 kJ mol−1 | (16) |
| | | ȮCH2NHCHO + O2 → CHONHCHO + HO2 ΔH° = −179 kJ mol−1 | (17) |
In summary: the theoretical study shows that OH radicals initiate MF photo-oxidation by H abstraction from the CHO and CH
3 groups, and that H abstraction from the NH group is not important at atmospheric conditions. Abstraction from the CH
3 group leads, almost exclusively, to
N-formylformamide although minor amounts of CH
2NCHO cannot be ruled out. Abstraction from the CHO group is followed by a branching in the O
2 reaction with the CH
3NHCȮ radical leading to either CH
3NCO or
via the CH
3NHC(O)OO radical to CH
2NH and CH
3NHNO
2. Each route results in different products, and the branching ratios can therefore be quantified from the product distribution shown in
Fig. 7. The major routes in the OH-initiated photo-oxidation of MF are outlined in
Scheme 1 together with the branching ratios derived from the reactant and product time profiles shown in
Fig. 7. The possible formation of CH
2NCHO, which is an isomer of the major photo-oxidation product, CH
3NCO, could not be confirmed in the present experiments. It should be noted that the conservative upper estimate of its yield is 0.85% (0.05 × 0.17).
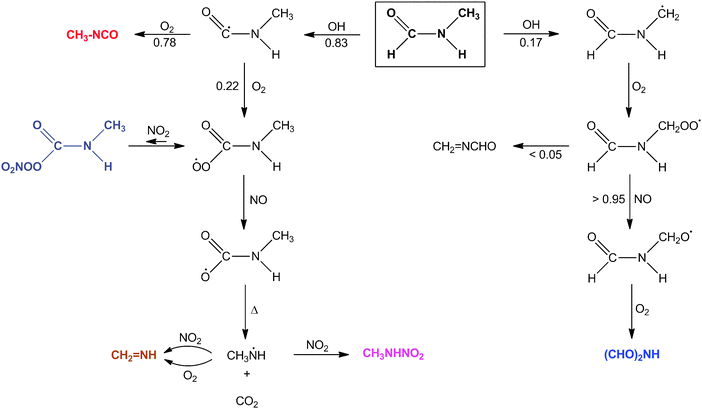 |
| | Scheme 1 Reaction scheme for OH initiated photo-oxidation of N-methylformamide (MF). Compounds in boldface colored type have been detected in photo-oxidation experiments. The grey colored (CH3)2NC(O)OONO2 is predicted to have a short lifetime and was not detected in the experiments. The branching ratios given have estimated uncertainties ±0.05. | |
CH2NH was observed in traces. CH2O and HCN were also observed in traces, but both compounds are difficult to quantify by PTR-ToF-MS. Our recent theoretical study on the atmospheric chemistry of CH2NH concludes unambiguously that HCN is the main product of methanimine photo-oxidation.43 However, no firm conclusion concerning CH2NH formation and subsequent reaction to HCN can be made from the available experimental data. Assuming branching in the CH3ṄH radical reactions with O2 and NO2 to be the same as in the corresponding (CH3)2Ṅ radical reactions,40,44 the observed amounts of CH3NCO, (CHO)2NH, CH3NHNO2 and the modeled amount of CH2NH accounts for >90% of MF reacted.
4.4. (CH3)2NCHO photo-oxidation studies
Barnes et al.5 carried out both OH and Cl atom initiated photo-oxidation studies of DMF and reported CH3N(CHO)2 as the major product in the degradation (∼70% yield) and unquantified amounts of (CH3)2NC(O)OONO2, depending on the NOx level in the experiment. The lack of calibrated reference spectra prevented the authors from making a proper quantification of the product yields.
Fig. 10 shows the time evolution of the main compounds detected by PTR-ToF-MS during a DMF photo-oxidation experiment, while Fig. S7 and S8 (ESI†), respectively, show the difference in PTR-ToF-MS mass spectra taken after and before the photo-oxidation experiment, and the NO and NO2 mixing ratios during the experiment. The NO level is nearly constant around 25 ppbV during the photo-oxidation, whereas the NO2 level increases from single-digit ppbV values to nearly 500 ppbV in the same period. Reacting compounds are DMF (detected in its protonated form at m/z 74.059 and its 13C isotope at m/z 75.068) and isopropyl nitrite which is detected at m/z 29.996 (NO+), m/z 43.054 (C3H7+), m/z 61.064 (C3H9O+) and m/z 79.074 (C3H11O2+). Signals that increased during the photo-oxidation experiment are m/z 44.049 (C2H6N+, 8% of total signal from protonated N-methylmethanimine), m/z 75.055 (C2H7N2O+, protonated N-nitroso dimethylamine), m/z 88.037 (C3H6NO2+, protonated N-formyl,N-methylformamide), m/z 91.050 (C2H7N2O2+, protonated N-nitro dimethylamine) and m/z 151.035 (C3H7N2O5+, protonated (CH3)2NC(O)OONO2). The signals at m/z 44.049 (C2H6N+, ∼92% of total signal), m/z 72.044 (C3H6NO+), m/z 89.043 (C3H7NO2+), and m/z 105.044 (C3H7NO2+) are produced from (CH3)2NC(O)OONO2. Signals from N-formyl,N-methylformamide oxidation are m/z 105.029 (C2H5O3N2+, protonated HC(O)N(CH3)NO2) and m/z 165.015 (C3H5O6N2+, protonated HC(O)N(CH3)C(O)OONO2). We also observed an increase in the ion signals at m/z 31.017 (CH3O+), m/z 43.017 (C2H3O+), m/z 45.033 (C3H9O+), m/z 45.991 (NO2+), m/z 59.049 (C3H7O+), m/z 60.049 (13C12C2H7O+), m/z 77.022 (C2H5O3+) and m/z 77.056 (C3H9O2+). As mentioned above, these signals stem from compounds (formaldehyde, acetaldehyde, acetone, nitric acid and peroxyacetylnitrate) typically formed in any smog chamber photo-oxidation experiment in the presence of isopropyl nitrite and NOx, i.e. even in the absence of (CH3)2NCHO. In our product analysis, we only included signals with a >1% increase relative to decrease in the protonated N,N-dimethylformamide signal.
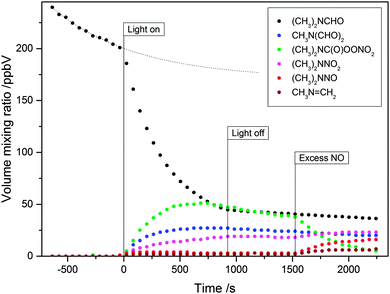 |
| | Fig. 10 Time evolution of the main ion signals observed during photo-oxidation of N,N-dimethylformamide (DMF). The thin dotted curve represents the extrapolated dilution and wall loss of DMF. | |
The wall loss of DMF was significant in the experiments. Additional loss was caused by instrumental sampling flows that were compensated for by constantly adding synthetic air (kdilution = 1.13 × 10−4 s−1); analysis of the MF loss curve before and after the photo-oxidation period gave a wall loss rate coefficient kwallloss ≈ 2.8 × 10−4 s−1. Also in the DMF experiments there is apparently a mismatch between the amount of DMF reacted and the amount of products formed, and, again, the apparent mismatch is deceptive. N-Nitro dimethylamine, (CH3)2NNO2, formed with a ∼5% yield in the experiment shown, can only be formed in the reaction between (CH3)2Ṅ radicals and NO2, and even under conditions with high NO2-levels, as in the present case, the major reaction of (CH3)2Ṅ radicals is with O2 resulting in N-methyl methanimine, CH3NCH2, which is difficult to quantify due to polymerization and surface induced reactions.
Fig. 10 shows a clear change in the (CH3)2NC(O)OONO2, CH3NCH2, (CH3)2NNO2 and (CH3)2NNO volume mixing ratios upon addition of excess NO titrating peroxy radicals present in the chamber. This will be addressed later.
Hydrogen abstraction from the CHO group in (CH3)2NCHO is expected to be followed by either O2 addition or dissociation of the (CH3)2NĊO radical:
| | | (CH3)2NCHO + OH → (CH3)2NĊO + H2O ΔH° = −98 kJ mol−1 | (2a) |
| | | (CH3)2NĊO + O2 → (CH3)2NC(O)OȮ* ΔH° = −136 kJ mol−1 | (18) |
| | | (CH3)2NĊO → CH3NCO + ĊH3 ΔH° = +49 kJ mol−1 | (19) |
The barrier to C–C scission is high, around 23 kJ mol
−1 above the entrance energy of the reactants in
(2a), and this route will therefore not be of importance at atmospheric conditions. Results from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations of
reaction (19) are collected in Tables S15 and S16 (ESI
†) and the energetics are illustrated in Fig. S9 (ESI
†).
The hot (CH3)2NC(O)OȮ* radical may initiate H transfer reactions from the CH3 groups followed by dissociation and OH regeneration:
| | | (CH3)2NC(O)OȮ → ĊH2(CH3)NC(O)OOH ΔH° = −11 kJ mol−1 | (20) |
| | | ĊH2(CH3)NC(O)OOH → CH2NCH3 + CO2 + OH ΔH° = −125 kJ mol−1 | (21) |
The barrier to H-transfer
(20) is found well below the entrance energy of the reactants. However, the path towards dissociation of the intermediate ĊH
2(CH
3)NC(O)OOH radical to CH
2NCH
3 + CO
2 + OH proceeds
via a barrier well above the entrance energy of the reactants. The internal H transfer route is consequently of no importance at atmospheric conditions.
Fig. 11 illustrates the energetics of the (CH
3)
2NĊO + O
2 reaction (quantum chemistry results collected in Tables S17 and S18, ESI
†).
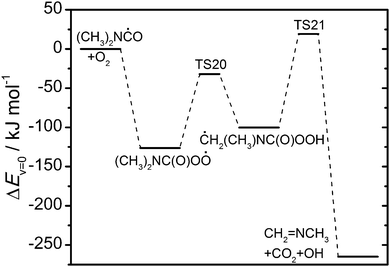 |
| | Fig. 11 Relative energies of stationary points on the potential energy surface of the (CH3)2NHC(O) + O2 reaction. Results from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations. | |
The thermalized peroxy radical may react with NO2 and NO resulting in the formation of a meta-stable peroxynitrate and eventually in the (CH3)2Ṅ radical:
| | | (CH3)2NC(O)OȮ + NO2 ⇆ (CH3)2NC(O)OONO2 ΔH° = −130 kJ mol−1 | (22) |
| | | (CH3)2NC(O)OȮ + NO → (CH3)2NC(O)Ȯ + NO2 ΔH° = −90 kJ mol−1 | (23) |
| | | (CH3)2NC(O)Ȯ → (CH3)2Ṅ + CO2 ΔH° = −58 kJ mol−1 | (24) |
Kirchner
et al.39 investigated the thermal stabilities of peroxynitrates and reported
k−21 = 1.2 × 10
−3 s
−1 at 298 K and 1 bar, and
Ea = (111.5 ± 5.8) kJ mol
−1. An analysis of the (CH
3)
2NC(O)OONO
2 decay upon the addition of excess NO, see
Fig. 10, gave a loss rate coefficient
k−21 = 2.86 × 10
−3 s
−1 at 304 K and 1 bar, which is in excellent agreement with the results of Kirchner
et al.39 for the same temperature.
The atmospheric reactions of the (CH3)2Ṅ radical are well established from previous studies of dimethylamine photo-oxidation:40–42
| | | (CH3)2Ṅ + O2 → CH3NCH2 + HO2 ΔH° = −71 kJ mol−1 | (25) |
| | | (CH3)2Ṅ + NO2 → CH3NCH2 + HONO ΔH° = −190 kJ mol−1 | (26a) |
| | | → (CH3)2NNO2 ΔH° = −203 kJ mol−1 | (26b) |
| | | (CH3)2Ṅ + NO → (CH3)2NNO ΔH° = −192 kJ mol−1 | (27a) |
| | 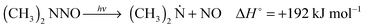 | (27b) |
Hydrogen abstraction from one of the CH
3 groups in (CH
3)
2NCHO is expected to be followed by O
2 addition. The
cis–
trans rotational barrier in ĊH
2NCH
3CHO is quite high (around 100 kJ mol
−1 from BHandHLYP/aug-cc-pVDZ calculations), which may prevent thermal equilibrium to be established before further reaction takes place; the energy difference between the two rotamers is, however, small. We will not make a distinction between the radical
cis- and
trans-rotamers here as they eventually end up as the same product.
| | | (CH3)2NCHO + OH → trans-ĊH2N(CH3)CHO + H2O ΔH° = −100 kJ mol−1 | (2b) |
| | | → cis-ĊH2N(CH3)CHO + H2O ΔH° = −100 kJ mol−1 | (2c) |
| | | ĊH2N(CH3)CHO + O2 → ȮOCH2N(CH3)CHO* ΔH° = −129 kJ mol−1 | (28) |
The hot peroxy radical, ȮOCH
2N(CH
3)CHO*, may initiate H transfer reactions; two routes are conceivable:
| | | ȮOCH2NCH3CHO* → HOOCH2NCH3Ċ(O)* ΔH° = +34 kJ mol−1 | (29a) |
| | | → HOOCH2N(ĊH2)CHO* ΔH° = +36 kJ mol−1 | (29b) |
The barriers to the H transfer
reactions (29) are found well below the entrance energy of the reactants in the O
2 addition
reaction (28), but the paths towards dissociation of the intermediates proceeds
via barriers well above,
Fig. 12 (quantum chemistry results are documented in Tables S19 and S20, ESI
†). These H transfer initiated routes are therefore of no importance at atmospheric conditions.
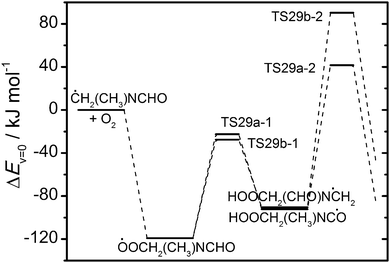 |
| | Fig. 12 Relative energies of stationary points on the potential energy surface of the ĊH2(CH3)NCHO + O2 reaction. Results from CCSD(T*)-F12a/aug-cc-pVTZ//MP2/aug-cc-pVTZ calculations. | |
With around 25 ppbV NO present in the chamber peroxy radical reactions with other peroxy radicals can be neglected: the oxyradical may either undergo H abstraction by O2 or dissociate:
| | | ȮOCH2NCH3CHO + NO → ȮCH2N(CH3)CHO + NO2 ΔH° = −48 kJ mol−1 | (30) |
| | | ȮCH2N(CH3)CHO + O2 → CHON(CH3)CHO + HO2 ΔH° = −183 kJ mol−1 | (31) |
| | | ȮCH2N(CH3)CHO → CH3ṄCHO + CH2O ΔH° = +76 kJ mol−1 | (32) |
The rate of ȮCH
2N(CH
3)CHO dissociation was estimated to be 1 × 10
−3 s
−1 employing the Méreau
et al.45 structure–activity relationship for alkoxy radical decomposition reaction. Assuming a typical value for
k30 = 1 × 10
−14 cm
3 s
−1, the O
2 abstraction reaction will therefore completely dominate the atmospheric fate of the ȮCH
2N(CH
3)CHO radical.
The major routes in the OH initiated photo-oxidation of N,N-dimethylformamide is outlined in Scheme 2 together with relative branching fractions derived from analysis of the time profiles presented in Fig. 10. The observed amounts of CH3NCO, (CHO)2NCH3, (CH3)2NNO, (CH3)2NNO2, and the modelled amount of CH2NCH3 accounts for >90% of DMF reacted.
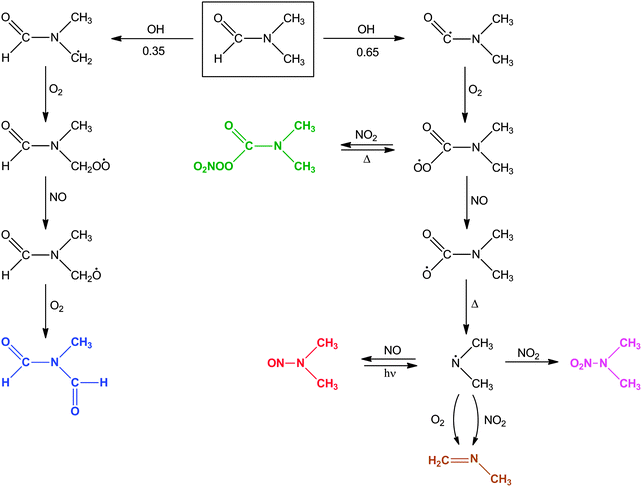 |
| | Scheme 2 Reaction scheme for OH initiated photo-oxidation of N,N-dimethylformamide. The branching ratios given have estimated uncertainties ±0.05. | |
5. Summary and conclusions
The rate coefficients of the reactions of OH radicals with N-methylformamide and N,N-dimethylformamide have been experimentally determined at T = 294 K to be (5.7 ± 1.7) × 10−12 cm3 s−1 and (8.5 ± 2.6) × 10−12 cm3 s−1, respectively. Taking [OH]24h = 106 cm−3, one obtains atmospheric lifetimes with respect OH reaction of 1.5 to 2 days. The major MF photo-oxidation product is methyl isocyanate, CH3NCO, which is not a benign compound in the environment. Methylnitramine, CH3NHNO2, is only a minor product. However, the potential to nitramine formation (18% yield of CH3ṄH radicals in photo-oxidation of MF) is almost as high as for methylamine (25% yield of CH3ṄH radicals).40,42 For DMF the situation is perhaps even more worrisome; we find the potential to formation of dimethylnitrosamine, (CH3)2NNO, and dimethylnitramine, (CH3)2NNO2, in DMF photo-oxidation actually being larger than for dimethylamine, 65% yield of (CH3)2Ṅ radicals in DMF photo-oxidation versus ∼40% in dimethylamine.40,42,44 Considering the recent recommendation by the Norwegian Institute of Public Health that the total amount of nitrosamines and nitramines should not exceed 0.3 ng m−3 in air and 4 ng L−1 in drinking water to ensure minimal or negligible risk of cancer for the public from exposure to these substances,46 immediately raises the question if emission regulations of MF and DMF should be implemented.
Acknowledgements
This work is part of the Atmospheric Degradation of Amines (ADA) project supported by Masdar, Statoil, Vattenfall, Shell and the CLIMIT program under contracts 193438, 201604 and 208122, and by the Research Council of Norway through a Centre of Excellence Grant (Grant No. 179568/V30). A. J. C. B. is grateful for an Erasmus Student Mobility Grant.
References
- U.S. Environmental Protection Agency, 2014.
- C. J. Nielsen, H. Herrmann and C. Weller, Chem. Soc. Rev., 2012, 41, 6684–6704 RSC.
- G. L. Kennedy, Crit. Rev. Toxicol., 2012, 42, 793–826 CrossRef CAS PubMed.
- OECD SIDS Dimethylformamide, UNEP 2001.
- I. Barnes, G. Solignac, A. Mellouki and K. Becker, ChemPhysChem, 2010, 11, 3844–3857 CrossRef CAS PubMed.
- N. Borduas, G. da Silva, J. G. Murphy and J. P. D. Abbatt, J. Phys. Chem. A DOI:10.1021/jp503759f.
- A. Chakir, G. Solignac, A. Mellouki and D. Daumont, Chem. Phys. Lett., 2005, 404, 74–78 CrossRef CAS PubMed.
- O. Welz and M. Olzmann, J. Phys. Chem. A, 2012, 116, 7293–7301 CrossRef CAS PubMed.
- A. L. Holloway, J. Treacy, H. Sidebottom, A. Mellouki, V. Daele, G. Le Bras and I. Barnes, J. Photochem. Photobiol., A, 2005, 176, 183–190 CrossRef CAS PubMed.
- M. Baasandorj, D. K. Papanastasiou, R. K. Talukdar, A. S. Hasson and J. B. Burkholder, Phys. Chem. Chem. Phys., 2010, 12, 12101–12111 RSC.
- C. Antoine, Comptes Rendus, 1888, 107, 681–684 Search PubMed.
-
NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg MD, 20899, retrieved December 2, 2014, http://webbook.nist.gov Search PubMed.
- R. Gopal and S. A. Rizvi, J. Indian Chem. Soc., 1968, 45, 13 CAS.
- J. Heinrich, J. Ilavski and J. Surovy, Chem. Zvesti, 1961, 15, 414–418 CAS.
- N. R. Jensen, J. Hjorth, C. Lohse, H. Skov and G. Restelli, Atmos. Environ., Part A, 1991, 25, 1897–1904 CrossRef.
- A. Jordan, S. Haidacher, G. Hanel, E. Hartungen, L. Mark, H. Seehauser, R. Schottkowsky, P. Sulzer and T. D. Mark, Int. J. Mass Spectrom., 2009, 286, 122–128 CrossRef CAS PubMed.
- M. Müller, T. Mikoviny, W. Jud, B. D'Anna and A. Wisthaler, Chemom. Intell. Lab. Syst., 2013, 127, 158–165 CrossRef PubMed.
- T. Su, J. Chem. Phys., 1994, 100, 4703 CrossRef CAS PubMed.
- L. A. Curtiss, K. Raghavachari, P. C. Redfern, V. Rassolov and J. A. Pople, J. Chem. Phys., 1998, 109, 7764–7776 CrossRef CAS PubMed.
- G. Knizia, T. B. Adler and H.-J. Werner, J. Chem. Phys., 2009, 130, 054104 CrossRef PubMed.
- T. B. Adler, G. Knizia and H.-J. Werner, J. Chem. Phys., 2007, 127, 221106 CrossRef PubMed.
- R. A. Kendall, T. H. Dunning, Jr. and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS PubMed.
- T. H. Dunning, Jr., J. Chem. Phys., 1989, 90, 1007–1023 CrossRef PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- H.-J. Werner, P. J. Knowles, G. Knizia, F. R. Manby and M. Schuetz, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 242–253 CrossRef CAS.
- Y. Georgievskii and S. J. Klippenstein, J. Chem. Phys., 2005, 122, 194103 CrossRef PubMed.
- J. A. Coxon and S. C. Foster, J. Mol. Spectrosc., 1982, 91, 243–254 CrossRef CAS.
-
B. E. Poling, J. M. Prausnitz and J. P. O'Connell, The Properties of Gases and Liquids, McGraw-Hill, Boston, 5th edn, 2001 Search PubMed.
- J. T. Bartis and B. Widom, J. Chem. Phys., 1974, 60, 3474–3482 CrossRef CAS PubMed.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2006, 110, 10528–10544 CrossRef CAS PubMed.
- D. R. Glowacki, C.-H. Liang, C. Morley, M. J. Pilling and S. H. Robertson, J. Phys. Chem. A, 2012, 116, 9545–9560 CrossRef CAS PubMed.
- G. da Silva, Chem. Phys. Lett., 2013, 558, 109–113 CrossRef CAS PubMed.
- G. Solignac, A. Mellouki, G. Le Bras, I. Barnes and T. Benter, J. Photochem. Photobiol., A, 2005, 176, 136–142 CrossRef CAS PubMed.
- R. Koch, W. U. Palm and C. Zetzsch, Int. J. Chem. Kinet., 1997, 29, 81–87 CrossRef CAS.
- S. M. Aschmann and R. Atkinson, Atmos. Environ., 1999, 33, 591–599 CrossRef CAS.
- G. Solignac, I. Magneron, A. Mellouki, A. Muñoz, M. M. Reviejo and K. Wirtz, J. Atmos. Chem., 2006, 54, 89–102 CrossRef CAS.
- M. M. Maguta, M. Aursnes, A. J. C. Bunkan, K. Edelen, T. Mikoviny, C. J. Nielsen, Y. Stenstrøm, Y. Tang and A. Wisthaler, J. Phys. Chem. A, 2014, 118, 3450–3462 CrossRef CAS PubMed.
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi and J. Troe, Atmos. Chem. Phys., 2006, 6, 3625–4055 CrossRef CAS.
- F. Kirchner, A. Mayer-Figge, F. Zabel and K. H. Becker, Int. J. Chem. Kinet., 1999, 31, 127–144 CrossRef CAS.
-
C. J. Nielsen, B. D'Anna, M. Karl, M. Aursnes, A. Boreave, R. Bossi, A. J. C. Bunkan, M. Glasius, A.-M. K. Hansen, M. Hallquist, K. Kristensen, T. Mikoviny, M. M. Maguta, M. Müller, Q. Nguyen, J. Westerlund, K. Salo, H. Skov, Y. Stenstrøm and A. Wisthaler, Summary Report: Photo-oxidation of Methylamine, Dimethylamine and Trimetahylamine, Climit project no. 201604 NILU OR 2/2011, ISBN 978-82-425-2357-0, NILU, 2011.
- L. Onel, L. Thonger, M. A. Blitz, P. W. Seakins, A. J. C. Bunkan, M. Solimannejad and C. J. Nielsen, J. Phys. Chem. A, 2013, 117, 10736–10745 CrossRef CAS PubMed.
- L. Onel, M. A. Blitz, M. Dryden, L. Thonger and P. W. Seakins, Environ. Sci. Technol., 2014, 48, 9935–9942 CrossRef CAS PubMed.
- A. J. C. Bunkan, Y. Tang, S. R. Sellevåg and C. J. Nielsen, J. Phys. Chem. A, 2014, 118, 5279–5288 CrossRef CAS PubMed.
- C. R. C. Lindley, J. G. Calvert and J. H. Shaw, Chem. Phys. Lett., 1979, 67, 57–62 CrossRef CAS.
- R. Méreau, M.-T. Rayez, F. Caralp and J.-C. Rayez, Phys. Chem. Chem. Phys., 2003, 5, 4828–4833 RSC.
-
M. Låg, B. Lindeman, C. Instanes, G. Brunborg and P. Schwarze, Health effects of amines and derivatives associated with CO2 capture, The Norwegian Institute of Public Health, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cp05805d |
|
| This journal is © the Owner Societies 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a