
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron capture dissociation and drift tube ion mobility-mass spectrometry coupled with site directed mutations provide insights into the conformational diversity of a metamorphic protein†
Sophie R.
Harvey
a,
Massimiliano
Porrini
b,
Robert C.
Tyler
c,
Cait E.
MacPhee
d,
Brian F.
Volkman
c and
Perdita E.
Barran
*e
aSchool of Chemistry, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK
bInstitut Européen de Chimie et Biologie (IECB), U869 ARNA – Inserm Chimie et Biologie des Membranes et des Nano-objets (CBMN), 33607 Pessac Cedex, France
cDepartment of Biochemistry, Medical College of Wisconsin, Milwaukee, WI 53226, USA
dSchool of Physics and Astronomy, University of Edinburgh, West Mains Road, Edinburgh, EH9 3JJ, UK
eSchool of Chemistry, Manchester Institute of Biotechnology, University of Manchester, Manchester, M1 7DN, UK. E-mail: perdita.barran@manchester.ac.uk
First published on 12th March 2015
Abstract
Ion mobility mass spectrometry can be combined with data from top-down sequencing to discern adopted conformations of proteins in the absence of solvent. This multi-technique approach has particular applicability for conformationally dynamic systems. Previously, we demonstrated the use of drift tube ion mobility-mass spectrometry (DT IM-MS) and electron capture dissociation (ECD) to study the metamorphic protein lymphotactin (Ltn). Ltn exists in equilibrium between distinct monomeric (Ltn10) and dimeric (Ltn40) folds, both of which can be preserved and probed in the gas-phase. Here, we further test this mass spectrometric framework, by examining two site directed mutants of Ltn, designed to stabilise either distinct fold in solution, in addition to a truncated form consisting of a minimum model of structure for Ltn10. The truncated mutant has similar collision cross sections to the wild type (WT), for low charge states, and is resistant to ECD fragmentation. The monomer mutant (CC3) presents in similar conformational families as observed previously for the WT Ltn monomer. As with the WT, the CC3 mutant is resistant to ECD fragmentation at low charge states. The dimer mutant W55D is found here to exist as both a monomer and dimer. As a monomer W55D exhibits similar behaviour to the WT, but as a dimer presents a much larger charge state and collision cross section range than the WT dimer, suggesting a smaller interaction interface. In addition, ECD on the W55D mutant yields greater fragmentation than for the WT, suggesting a less stable β-sheet core. The results highlight the power of MS to provide insight into dynamic proteins, providing further information on each distinct fold of Ltn. In addition we observe differences in the fold stability following single or double point mutations. This approach, therefore, has potential to be a useful tool to screen for the structural effects of mutagenesis, even when sample is limited.
Introduction
The development of ‘soft’ ionisation techniques such as electrospray (ESI)1,2 and nano-electrospray ionisation (n-ESI)3,4 revolutionised biological mass spectrometry (MS). It is now accepted as a powerful tool in the structural analysis of proteins and protein complexes that can probe solution-phase topologies and even in vivo active structures.5–10 In the early 1960s the first reports of combining the technique of ion mobility spectroscopy (IMS) to mass spectrometry were published.11–13 This hybrid technique known as ion mobility-mass spectrometry (IM-MS) provides an extra dimension of information over mass spectrometry alone, separating ions based not only on their mass-to-charge ratio but also on their size and shape, known as their rotationally averaged collision cross section (CCS). IM-MS has been successfully applied to study the conformations adopted by peptides and proteins in the gas-phase and, hence, is gaining importance as a biophysical tool.14–17 Furthermore, the CCS determined from experiment can be compared to theoretical CCS, obtained from coordinates of solved structures (either from NMR or X-ray crystallography) or from molecular modelling, providing confirmatory information on the conformations adopted by the gas-phase protein ions.18–21Recently studies which compare the results from IM-MS experiments, on the global fold of a protein, to amino acid level detail obtained from electron capture dissociation (ECD) experiments have been presented.22–24 ECD is a fast non-ergodic fragmentation process in which bond dissociation following activation occurs much faster than typical bond vibration and hence ECD is thought not to perturb the higher order structure of proteins, enabling protein structural studies to be performed.25,26 ECD has been shown previously to preferentially cleave the backbone of proteins without disruption of weaker non-covalent interactions, such as hydrogen bonding networks present in a protein:peptide complex,27 and has become an influential and sophisticated technique to study protein fold in the gas-phase at an amino acid level.28–31
Here we use the combination of DT IM-MS and ECD to investigate the two distinct folds of the chemokine Ltn. Chemokines are small secreted signalling proteins involved in immune response and are divided into four subclasses depending on the number and positioning of their conserved cysteine residues.32 Lymphotactin is a unique chemokine, containing only a single disulfide bond, as opposed to the two normally found in chemokines, and is the defining member of the C subclass of chemokines.33 Another unusual feature of Ltn is that it is a metamorphic protein,34 existing in two distinct conformations in equilibrium (Fig. 1) in a ligand-free state, each of which performs separate functions in vivo and are, therefore, essential for protein function.35
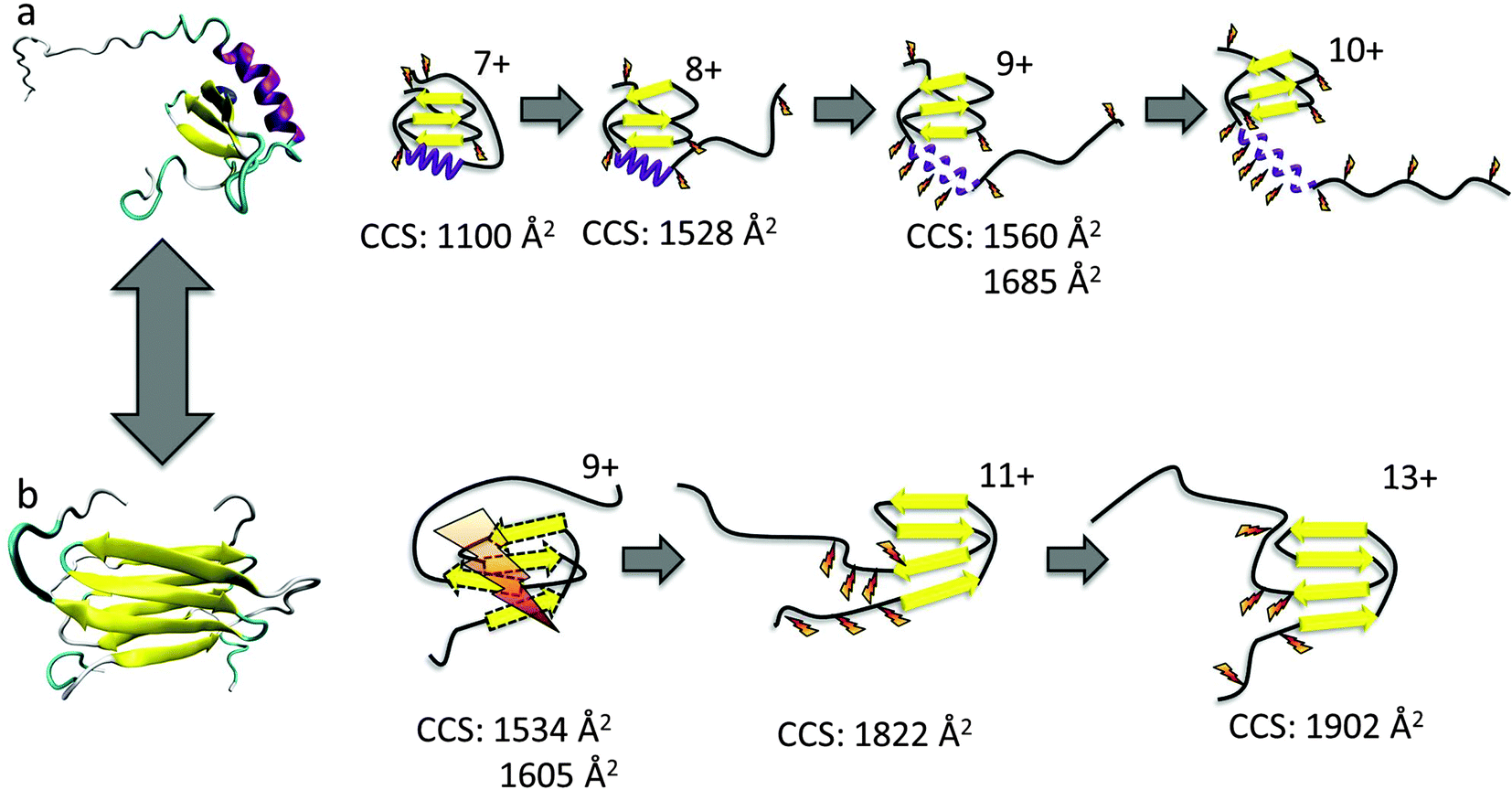 | ||
| Fig. 1 The conformational equilibrium of lymphotactin; (a) monomeric, Ltn10 and (b) dimeric, Ltn40. Cartoon representations of Ltn10 and Ltn40 unfolding pathways as determined through DT IM-MS and ECD, with yellow arrows representing β-sheet regions and purple spirals representing α-helical regions. For Ltn40 a single monomeric unit is shown for figure clarity. Favourable sites of ECD fragmentation are shown by orange ‘lightening’ bolts, for Ltn40 where fragments only map to a monomer unit with no secondary structure, the extensive fragmentation is depicted by a large lightning bolt and dashed lines represent a lack of defined structural elements. | ||
Ltn exists in equilibrium between a monomeric, conserved chemokine fold, known as Ltn10 and a distinctive dimeric fold, known as Ltn40.35,36 The interconversion between these two folds involves a complete restructuring of the core residues.37 We have studied both conformations of wild type (WT) Ltn using gas-phase techniques, which exclude conformational constriction due to buffers or additives and enable the intrinsic stability of each structural element in both Ltn10 and Ltn40 to be assessed. Our earlier work indicated that the Ltn10 fold could be transferred and retained in a solvent-free environment, with the intrinsically disordered (ID) tail being associated with the structural core at the low charge states of the protein and unfolding as the charge state increases.24 We hypothesized, and this was supported by ECD data, that the tail unfolding is then followed by unravelling of the α-helix, with little perturbation of the β-sheet region, as summarised in Fig. 1a. For Ltn40 a structural collapse is observed for the lowest charge state, leading to a small CCS and extensive backbone dissociation during ECD. Whilst the higher charge states of the dimer show greater stability suggesting the structural core remains stable and unperturbed in these conformations (Fig. 1b).
The studies presented here utilise DT IM-MS and ECD to examine the conformational equilibrium of this metamorphic protein further; through the study of specific mutants designed to mimic either the Ltn10 or Ltn40 fold, whilst limiting conformational interconversion. The results provide detailed insight into the unfolding of each distinct Ltn conformation, in conjunction with studying the effect of specific site mutations on both folds and allowing any subsequent increase or decrease in fold stability to be identified.
Results and discussion
WT 1–72
Initial studies focused on a truncated Ltn mutant (WT 1–72), which contains the structural core of the protein but does not contain the last 21 amino acids which form an intrinsically disordered tail in the WT protein. This construct enables us to study a minimum structural model for Ltn10, which contains the two main types of protein secondary structural elements; α-helix and β-sheets.We have established conditions which enable both WT monomer (Ltn10) and WT dimer (Ltn40) to be preserved, which we utilise for all mutants studied here.24 The mass spectrum of WT 1–72 shows that the protein presents mainly as a monomer (M1–72) and is capable of accepting between four and eight protons (Fig. 2a). The majority of the intensity in the WT 1–72 mass spectra is due to the [M1–72 + 5H]5+ and [M1–72 + 6H]6+ species, suggesting that these two charge states are particularly conformationally stable. A WT 1–72 dimer (D1–72) is also observed under these conditions but at much lower intensity than the monomer, presenting over the charge state range [D1–72 + 8H]8+ to [D1–72 + 12H]12+ (Fig. 2a) it is not discussed any further.
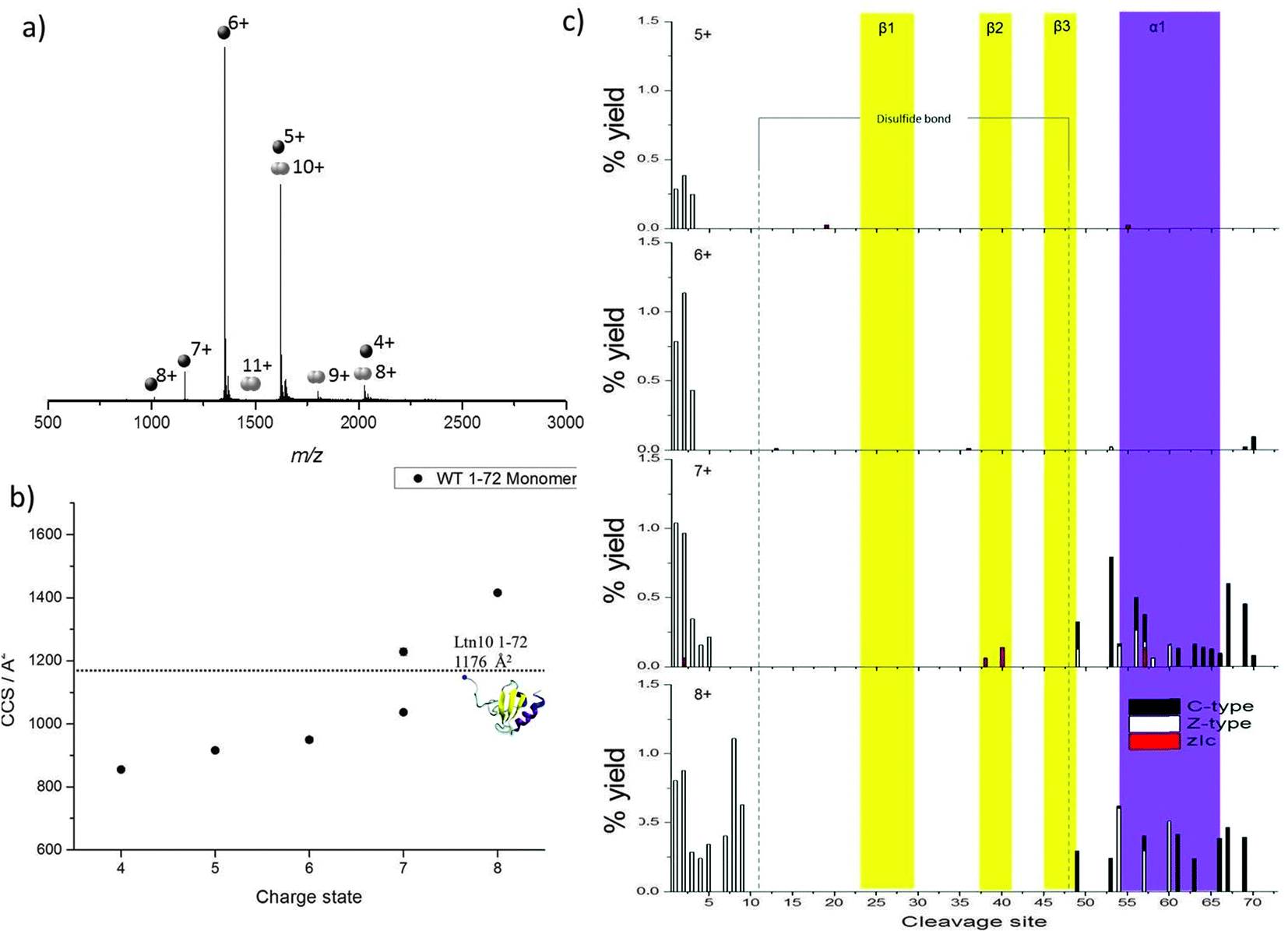 | ||
| Fig. 2 WT 1–72 Ltn (a) mass spectrum obtained on the DT IM-MS instrument for 100 μM WT 1–72 in 20 mM ammonium acetate (AmAc). (b) Average experimental CCS for M1–72 obtained from three different day repeats, error bars are calculated from the standard deviation and generally fall within symbol size. Dashed line represents theoretical CCS obtained from the clipped NMR structure (PDB 1J9O) removing residues 73–93. (c) Percentage yields calculated for zIc, c and z-type40 fragments as a function of cleavage site for M1–72 over the charge states [M1–72 + 5H]5+ to [M1–72 + 8H]8+. | ||
Previously, the CCS adopted by WT 1–72 were determined using DT IM-MS,24Fig. 2b and ESI,† Table S1. In summary, as the charge state increases so does the CCS, this is attributed to more extended solution-phase conformations being more solvent and charge accessible, hence accepting larger numbers of charges, and consequently having a larger CCS. Coulombic repulsion between charged groups can also have an effect in the gas-phase and can cause an increase in CCS with increasing charge. Over the charge state range [M1–72 + 4H]4+ to [M1–72 + 6H]6+ the CCS increases slightly (11%), which suggests that these species are present in a stable, compact, conformationally similar family in which the addition of charge does not significantly influence the conformations adopted. The experimental CCS observed are all smaller by ∼15% than the theoretical value (1176 Å2) obtained from NMR coordinates of the full length protein with residues 73–93 removed (PDB 1J9O). These smaller values indicate a compaction of structure further than that achieved by in vacuo minimising the NMR structure; additionally we anticipate a tethering of the loose N-terminus to the protein core.
Two distinct conformations are observed for the [M1–72 + 7H]7+ species, the more compact conformation appears similar in CCS to those observed for [M1–72 + 4H]4+ to [M1–72 + 6H]6+ suggesting this is probably of a similar, compact, possibly folded structure. The second larger conformation indicates WT 1–72 is more unfolded at this charge state. A further jump in CCS is observed moving to the [M1–72 + 8H]8+ species.
The charge states [M1–72 + 5H]5+ to [M1–72 + 8H]8+ were observed at high enough intensity for effective trapping and sufficient fragmentation efficiency for analysis by ECD fragmentation (ESI,† Fig. S1 and S2). Following ECD experiments, fragmentation maps were plotted in which the percentage yield of all fragment ions observed are plotted against cleavage site, considering c and z-type fragments as well as zIc fragments. Fragmentation yields were calculated considering remaining precursor, charge reduced species and fragment ions. zIc fragments are internal fragments, formed by multiple cleavages and which have a z-type N-terminus and c-type C-terminus,24 where “I” refers to the fact that they are internal. Internal fragments have been attributed previously to charge remote fragmentation38 and are thought to be particularly prevalent in sequences containing basic residues, and hence are likely to play an important role in the fragmentation of highly basic proteins such as chemokines. Furthermore, internal fragments were observed previously at significant intensity for WT lymphotactin.24
As charge state increases, increased fragmentation is observed attributed to a number of factors; mainly, ECD fragmentation is more efficient at higher charge states,39 furthermore, at higher charge states protein ions are likely to be more extended than their lower charged counter parts and hence there are a greater number of sites available for cleavage and dissociation. By mapping the fragmentation observed onto the secondary structural elements of the protein a detailed picture of how the protein restructures and, hence, the stability of each of the different structural elements, can be obtained as a function of charge. All identified c, z and zIc fragments for [M1–72 + 5H]5+ to [M1–72 + 8H]8+ are therefore shown in Fig. 2c, as a function of cleavage site onto which the regions involved in formation of each β-strand and the α-helix are mapped.
For [M1–72 + 5H]5+ and [M1–72 + 6H]6+, limited fragmentation and subsequent dissociation is observed, resulting in the identification of several c, z and zIc fragments. The low intensity is most likely due to the compact nature of these ions coupled with their low charge states and hence low ECD fragmentation efficiency. As charge state increases both the extent and the intensity of fragmentation observed increases in the N-terminal region suggesting this region is beginning to unfold from the structural core of the protein. In addition at the [M1–72 + 7H]7+ and [M1–72 + 8H]8+ charge states we observe an increase in the fragmentation in the α-helical region (highlighted in purple in Fig. 2c), for both c and z type fragments in addition to zIc fragments, suggesting this region is beginning to unravel from the structural core of the protein, losing any non-covalent stabilising interactions. This observation is consistent with our DT IM-MS observations in which we see a significant increase in CCS for the [M1–72 + 7H]7+ and [M1–72 + 8H]8+ species. For [M1–72 + 8H]8+ the extent of fragmentation occurring in the α-helix decreases in comparison to [M1–72 + 7H]7+, this is attributed to the lower signal intensity of the [M1–72 + 8H]8+ species, leading to fewer ions being trapped and subjected to ECD and highlights the difficulties of performing such ECD studies on multiple charge states. At all charge states very little fragmentation is observed in the β-sheet core (highlighted in yellow in Fig. 2c) suggesting this region retains its stabilising interactions even after desolvation and ionisation and is more stable than the α-helical region, in part attributed to the disulfide bond which would further stabilise this region.
These observations are similar to the trends observed for the WT protein in the Ltn10 conformation (Fig. 1); one notable difference is that in WT Ltn10 significant unravelling of the α-helix is not observed until the [MWT + 9H]9+ species whereas in WT 1–72 this unravelling is observed from the [M1–72 + 7H]7+ species. At lower charge states the intrinsically disordered tail is believed to be associated with the structural core of the WT protein and proposed to uncouple before the α-helix unravels. It is therefore plausible that the intrinsically disordered tail confers structural stability and protection to the WT Ltn10 fold and the absence of the tail in WT 1–72 causes this construct to unfold at a lower charge state. An additional explanation could be that in the WT 1–72, due to its smaller size, the charges would likely reside closer to each other and therefore Columbic repulsion of charges could induce unfolding at lower charge states than for the WT protein.
Probing the Ltn10 fold through specific site mutations
In order to further probe the Ltn10 fold, we next considered a mutant, known as CC3, designed to favour the monomeric form of Ltn, Ltn10.41 The CC3 mutant includes an extra disulfide bond based on the third disulfide in the CC chemokine, HCC-2. CC3 was engineered by introducing two extra cysteines in positions 21 and 59, with the aim that the addition of an extra disulfide bond, based on the conserved chemokine fold, would structurally constrain the protein and stop structural rearrangement to form the dimer fold.41 For this mutant very little dimer signal is observed, as expected, (Fig. 3a inset and ESI,† Fig. S3). The CC3 monomer presents over a range of charge states from [MCC3 + 5H]5+ to [MCC3 + 8H]8+, with the species which accept either six or seven protons being the most favourable and hence most intense. A small proportion of CC3 dimer is observed but at incredibly low intensity highlighting that for this mutant formation of the dimer is not as favourable as for the WT type protein, hence our analysis will focus on the monomeric CC3. | ||
| Fig. 3 CC3 Ltn. DT IM-MS results: average experimental CCS for MCC3 obtained from three different day repeats, error bars are calculated from the standard deviation and generally fall within symbol size. Dashed and dotted lines represents theoretical CCS obtained from NMR structures PDB 2HDM and 1J9O respectively. Insets: mass spectra obtained on DT IM-MS instrument of 100 μM protein (a) buffered CC3 in 20 mM Am. Ac and (c) buffered and supercharged CC3 in 20 mM Am. Ac +1% m-NBA ECD analysis: percentage yields calculated for c type, z type and zIc fragments as a function of cleavage site. (b) Buffered CC3 over the charge state range [MCC3 + 5H]5+ to [MCC3 + 8H]8+ (d) buffered and supercharged CC3 over the charge state range [MCC3 + 8H]8+ to [MCC3 + 11H]11+. For figure clarity a y-axis scale break is included for all CC3 fragmentation maps. | ||
Following DT IM-MS on the CC3 construct we find that it presents over a narrow range of CCS over the charge states observed, increasing by only 23% from smallest to largest species, (Fig. 3a and ESI,† Table S2). The experimental values obtained are in extremely good agreement with the theoretical value obtained minimising the NMR structure of this mutant (PDB 2HDM). The file used to determine the theoretical CCS, however, does not contain the final 18 amino acids in the intrinsically disordered tail. As experimental values match extremely well with this theoretical value without the full length tail we surmise, that as observed previously for the low charge states of WT Ltn10 (Fig. 1), the intrinsically disordered tail is wrapped around the structural core of the protein, but this occurs for all charge states of CC3 studied here under buffered conditions. If the full ID tail were to unfold from the structural core of the protein as a single unfolding event, as for WT Ltn10, a dramatic increase in CCS, between two charge states or two conformations of the same charge state, on the order of 40% would be expected (Fig. 1). The increase in CCS is not as significant for the CC3 mutant suggesting that this restructuring does not occur here. In fact in the CC3 mutant the helix is pinned to the core by the extra disulfide bridge and, therefore, the first unfolding pathway which couples tail and helix unfolding observed for the WT Ltn10 cannot occur for this mutant. Furthermore, this pinning of the α-helix to the structural core and corresponding conformational tightening may also further encourage the association of the tail to the structural core, limiting its unfolding from the core.
For ECD experiments the species [MCC3 + 5H]5+ to [MCC3 + 7H]7+ were observed at high enough intensity to be examined (ESI,† Fig. S4 and S5). Once again by considering c- and z-type fragments in combination with internal fragments a fragmentation map can be built-up to visualise the unfolding of the structurally constrained Ltn10 fold, shown in Fig. 3b in which the β-sheet regions and α-helix are again highlighted in yellow and purple, respectively. For all species studied very little fragmentation is observed, this is as expected due to the compact nature of these species and due to the fact that the species are relatively low charged in combination with the two disulfide bonds which can limit fragmentation and dissociation by ECD. Disulfide bonds have been shown to be preferentially cleaved in ECD,42 which can result in lower backbone fragmentation efficiency. Furthermore in compact proteins which contain numerous non-covalent interactions and those with multiple disulfide bonds, there is the possibility of fragmentation without subsequent dissociation, limiting the observed fragments. Despite this, information about how the CC3 mutant unfolds can still be obtained. At the lowest charge states, [MCC3 + 5H]5+ and [MCC3 + 6H]6+, minor fragmentation is observed at both termini, the higher charged species, [MCC3 + 7H]7+ and [MCC3 + 8H]8+, display slightly more fragmentation sites, consistent with these species being more extended or unfolded and hence capable of accepting a higher number of protons. The more extended nature of these species, would, therefore, provide more available sites for fragmentation and subsequent dissociation. Increased fragmentation is most evident along the N-termini and C-termini suggesting these regions are beginning to decouple from the core, coinciding with an increase in CCS observed for these two species. For both species internal fragments where the intrinsically disordered tail has been lost are observed, further supporting that this protein begins to unfold from both termini. Higher intensity fragmentation is observed as a result of unfolding of the C-terminus consistent with the findings for both the WT and WT 1–72 in which the protein was observed to unfold most extensively from the C-terminus.
For [MCC3 + 8H]8+ low intensity fragmentation in the α-helix is also observed suggesting this is the first secondary structural element to unfold from the structural core a finding which is consistent with both the WT Ltn10 and the truncated mutant WT 1–72. Less fragmentation in the central β-sheet core is observed than for the WT Ltn10 suggesting that there are still stabilising non covalent interactions here, which is consistent with our DT IM-MS experiments in which the [MCC3 + 8H]8+ species is only 23% larger than the most compact species. The lack of fragmentation in this region could also be due to the presence of the two disulfide bonds, which would also act to stabilise this region.
For CC3 minimal fragmentation occurs within the β-sheet core, however, there are a number of low intensity internal fragments which are observed within this region for the highest charge state probed under these conditions, namely the [MCC3 + 8H]8+ species. These fragments are of low abundance (19% of the total fragments for this species being due to fragmentation in this region and comprising 6% of the total intensity of fragments). When cleavage in this region occurs it results in zIc fragments generally consisting of more than one β-strand suggesting the fragments at least partially retain some of the stabilising interactions present in the solution fold. This observed stability is most likely due to the two disulfide bonds which would strengthen and stabilise this region, as well as stabilising non-covalent interactions present in this compact, constrained species. Considering the DT IM-MS and ECD data, and the fact that this species is mostly observed at low charge states, it is clear that this mutant is more compact and less prone to restructuring with respect to charge state than the WT protein. The extra disulfide bond in CC3 would confer this stability by essentially pinning the structural core together. To probe CC3 at higher charge states we added a ‘supercharging’ reagent, m-NBA (meta-nitrobenzyl alcohol).43
Further probing the unfolding landscape of Ltn10: supercharged CC3
Addition of 1% m-NBA shifted the highest charge state to [MCC3 + 13H]13+, (Fig. 3c inset and ESI,† Fig. S6). In order to probe the conformations of these ions, the CCS for the species [MCC3 + 5H]5+ to [MCC3 + 13H]13+ were then determined (Fig. 3c and ESI,† Table S3). An increase in CCS of up to 13% at like for like charge states is found, suggesting m-NBA promotes more extended structures at all charge states, not only the higher charge states promoted by this reagent. Previous studies have shown that addition of supercharging reagents can alter conformations of protein complexes.44 From Fig. 3c, it is evident that under these conditions we do not see any major increase in the CCS in a single step, for example the transition between z = 7 and z = 8 seen in WT Ltn in which CCS increases by ∼40%. Instead the CCS increases in smoother steps with respect to charge (from 1020 to 1828 Å2), clearly the higher charge states are promoting unfolding, however, the single stage tail-unfolding is not observed here and unfolding is a more continuous process with respect to charge.The experimental CCS obtained for the supercharged CC3 Ltn have been compared both to the theoretical CCS obtained from the minimised NMR coordinates for this mutant (PDB 2HDM) and for the WT Ltn10, the structure of which has the intrinsically disordered tail extended out from the structural core of the protein (PDB 1J9O). The theoretical CCS determined from the CC3 mutant compare well with the lowest charge states observed for CC3 under these conditions ([MCC3 + 5H]5+ and [MCC3 + 6H]6+), suggesting that despite the addition of m-NBA these species remain folded with the intrinsically disordered tail associated with the structural core. The theoretical CCS obtained from WT Ltn10 (1694 Å2) compares extremely well with the experimental CCS for CC3 Ltn [MCC3 + 12H]12+ and [MCC3 + 13H]13+ species (1777 and 1828 Å2), which suggests the increase in CCS can be attributed to unfolding of the intrinsically disordered tail, however, the continuous increase in CCS suggests this does not occur in a single step as observed for WT Ltn.
In order to determine the unfolding pathways accessed through the addition of m-NBA the [MCC3 + 8H]8+ to [MCC3 + 11H]11+ charge states were subjected to ECD fragmentation, Fig. 3d and ESI,† Fig. S7 and S8. It is interesting to first consider the [MCC3 + 8H]8+ species, which was probed under both buffered and supercharged conditions. Under supercharging conditions more extensive fragmentation is observed along the backbone of the α-helical region, suggesting that this region has become more unstructured and therefore more available for fragmentation under supercharged conditions. This finding is consistent with the observed increase of CCS for this species in the presence of m-NBA.
As we move to larger CCS, higher charged species, we observe more extensive fragmentation in both termini as well as along the intrinsically disordered tail region, emphasising that these regions are the first to unfold from the core of the protein. At all charge states studied under supercharged conditions zIc fragments are observed which correspond to the complete loss of the intrinsically disordered tail and regions of the N-termini, retaining the full structural core. This observation highlights that under these conditions, for all species of CC3 studied by ECD, the disordered regions are more weakly associated with the structural core of the protein and hence available for fragmentation than under buffered conditions.
Additionally, in the [MCC3 + 11H]11+ species we see greater fragmentation in the α-helix suggesting this region is becoming more unfolded and beginning to unravel from the structural core of the protein. We do observe fragmentation in the β-sheet region for this species, but at a lower level than that observed for the α-helix, consistent with our previous findings both for the WT Ltn and WT 1–72 constructs which showed that this region remains stable in the Ltn10 fold, due in part to the disulfide bonds. As the protein becomes more unfolded we see greater fragmentation including evidence for electron mediated reduction of disulfide bridges, which suggests they are more accessible. It is important to note that due to the conformational constriction implemented in this mutant by the addition of the second disulfide bridge, we do not see the unfolding pathway involving the complete loss of the α-helix and disordered tail in a single fragment, that is observed for the WT Ltn10, until we reach the most extended species, [MCC3 + 11H]11+. The extra disulfide bridge in CC3 successfully structurally constrains the Ltn10 fold and inhibits the unfolding, limiting the possibility of unstructuring the core residues needed for interconversion to Ltn40.41
Probing the Ltn40 fold through specific site mutation
The final mutant studied here, known as W55D Ltn, contains a single point mutation (tryptophan at position 55 is mutated to an asparagine) shown by NMR to shift the equilibrium towards the dimeric structure.35 The amino acid replacement is located in the beginning of the α-helix in the Ltn10 conformation and is designed to destabilise this conformation and push the equilibrium towards the dimeric fold (Ltn40), with loss of the tryptophan side chain disrupting the Ltn10 hydrophobic core and destabilising this fold. In our experiments, however, the most intense species is the monomer (MW55D) presenting over a range of charge states from [MW55D + 4H]4+ to [MW55D + 9H]9+, Fig. 4a. The dimeric species (DW55D) is also observed, at somewhat lower intensity than the monomer, over the charge state range [DW55D + 9H]9+ to [DW55D + 15H]15+ wider by three charge states than the WT dimer, Fig. 4b. The equilibrium of W55D monomer to dimer could not be significantly shifted towards the dimer through the alteration of pH over the range 2.8 to 9.8 or through increasing the buffer strength, ESI,† Fig. S9. The fact that the highest intensity peaks observed are due to the monomeric species is discussed in more detail later. | ||
| Fig. 4 W55D ltn. (a) Mass spectrum obtained on the DT IM-MS instrument for 100 μM W55D in 20 mM AmAc. (b) Average experimental CCS obtained from three different day repeats, error bars are calculated from the standard deviation and generally fall within symbol size. Dashed line represents theoretical CCS of Ltn40 obtained from the NMR structure (PDB 2JP1). Monomeric ECD analysis: percentage yields calculated for c type, z type and internal fragments as a function of cleavage site for MW55D over the charge states [MW55D+6H]6+ to [MW55D + 10H]10+. Fragments are mapped onto (c) Ltn10 fold and (d) Ltn40 fold. For figure clarity a y-axis break is included. | ||
From DT IM-MS it is clear that the change in monomeric CCS with respect to charge is different for W55D than for CC3, WT Ltn and WT 1–72 under buffered conditions. We no longer see a stable ensemble of species presenting over a narrow CCS range, which can then be followed by a major increase in CCS,24 instead the CCS is observed to increase almost uniformly with charge from 778 Å2 to 1407 Å2, Fig. 4b and ESI,† Table S4.
The dimeric form of the W55D Ltn mutant also appears notably different to the WT Ltn and presents over an extremely wide range of CCS, (Fig. 4b and ESI,† Table S4). In its dimeric form W55D Ltn presents over a CCS range of 1509 Å2 to 2519 Å2, with the most extended form observed ([DW55D + 16H]16+) being 32% larger than the most extended species observed for the WT Ltn40 ([DWT + 13H]13+). In this case, as no PDB file exists for the mutant dimer, theoretical comparisons were made with the WT dimer (PDB 2JP1), however, this file does not contain the intrinsically disordered tail and the theoretical value is therefore based on a structure which is missing 33 amino acids from each chain. From comparison of the experimental and theoretical CCSs we see that DW55D populates a wide range of conformations, both more compact and more extended than this theoretical value. Most conformations are far more extended than the theoretical CCS, which suggests that either the tail is extended out from the structural core and/or the whole structure is more flexible and, therefore, capable of existing in a larger range of conformations, which in turn suggests a smaller interface region stabilises the dimer. It is indeed likely that the wide range of CCS observed is due to a combination of both of these factors. It also appears that less dimer signal is observed for this mutant in comparison to the WT, perhaps suggesting that it is not as stable during ionisation and desolvation and is dissociating into monomer during this process. In order to further probe the flexibility and stability of the W55D mutant ECD analysis was performed both on the monomeric ([MW55D + 6H]6+ to [MW55D + 10H]10+) and dimeric ([DW55D + 9H]9+, [DW55D + 11H]11+ and [DW55D + 13H]13+) species of W55D Ltn, ESI,† Fig. S10 to S12.
Discussion will first focus on the monomeric form of W55D. The monomeric species presents us with an interesting challenge in identifying the protein fold; can we use this combination of techniques to determine if the W55D Ltn monomer possess an ECD/IM-MS signature of the conserved chemokine monomer fold, Ltn10, as with monomeric WT, WT 1–72, and CC3 Ltn, or are the monomeric species we observe due to dissociation of the dimer and hence exist as a single unit in the dimeric Ltn40 fold?
In order to determine which, if either, fold the monomeric species of W55D Ltn exists in we have mapped the fragmentation observed for the monomeric W55D over the charge states [MW55D + 6H]6+ to [MW55D + 10H]10+ onto both the monomeric fold and a single chain of the dimeric fold, Fig. 4c and d, respectively. As expected the monomer fragmentation increases as a function of charge, however, the trends observed can enable us to identify the conformation present. As we move to more extended and hence higher charged species extensive fragmentation is observed in the N-terminus suggesting this region is beginning to unfold, as observed for both Ltn10 and Ltn40.
The most extended species and, therefore, highest charged species ([MW55D + 10H]10+) displays extensive fragmentation into the core residues of both folds suggesting that this species is significantly unstructured. Therefore, we focus on the intermediate charge states where less Colombically driven unfolding is likely. Beginning with the [MW55D + 8H]8+ species we observe fragmentation in the region of the residues 52–65. This fragmentation is not consistent with the data from the Ltn10 (see for example Fig. 2c), where we see fragmentation consistently throughout the region that is helical in solution. Instead fragmentation appears to branch out from the β-sheet core in the Ltn40 fold, (comparing Fig. 4c with d). This is consistent with the observations made for the dimeric WT Ltn40 fold in which we see fragmentation branching out from the β-sheet core with higher intensity fragments in the first 9 amino acids after the β3 strand, and supported by NMR data which shows that these regions that flank the β-sheet are associated with the protein core allowing them to be solved. Additionally, if MW55D were to exist as Ltn10 we would expect to see more evidence for the first unfolding pathway of this fold, which involves unfolding of the α-helix and intrinsically disordered tail, with loss of both regions being observed in a single fragment ion. The fragmentation maps suggest that MW55D is likely to be existing as a single unit of the dimeric Ltn40 fold. This could be a result of dissociation of the dimer upon ionisation and transfer of the protein, suggesting that the dimer interface is not as stable as that of the WT type protein. If this is indeed the case we can surmise that over this time scale, in the gas phase, monomeric W55D also does not restructure to the Ltn10 conformation.
To probe dimeric W55D Ltn conformations, the charge states of the dimer where the net charge is an odd number and, therefore, are not coincident with monomer species were considered, namely [DW55D + 9H]9+, [DW55D + 11H]11+ and [DW55D + 13H]13+. As for the monomer, c-type, z-type and zIc fragment yields were considered, however, in this case fragmentation can occur either from the dimer remaining intact and fragmenting or alternatively it may dissociate to monomer and then fragment, both cases were considered here. As both monomeric units comprising the dimer are identical in sequence these experiments do not distinguish between the two chains and therefore the total fragment percentage yields are mapped onto a single monomeric unit, Fig. 5. At the lowest charge state studied here, [DW55D + 9H]9+, ECD leads to extensive dissociation of the dimer to monomer followed by extensive fragmentation along the backbone, represented by below axis histograms Fig. 5. The tendency of the lowest dimer charge state to dissociate to monomer was also observed for the WT protein and attributed to either an encounter complex that lacks a stable β-sheet core or a less stable, perhaps collapsed form of the dimer. This tendency of the dimer to dissociate to monomer upon ECD fragmentation is, however, observed for all charge states of the W55D Ltn dimer studied here which also suggests that this dimer interface is not as strong in this mutant as compared to the WT Ltn.
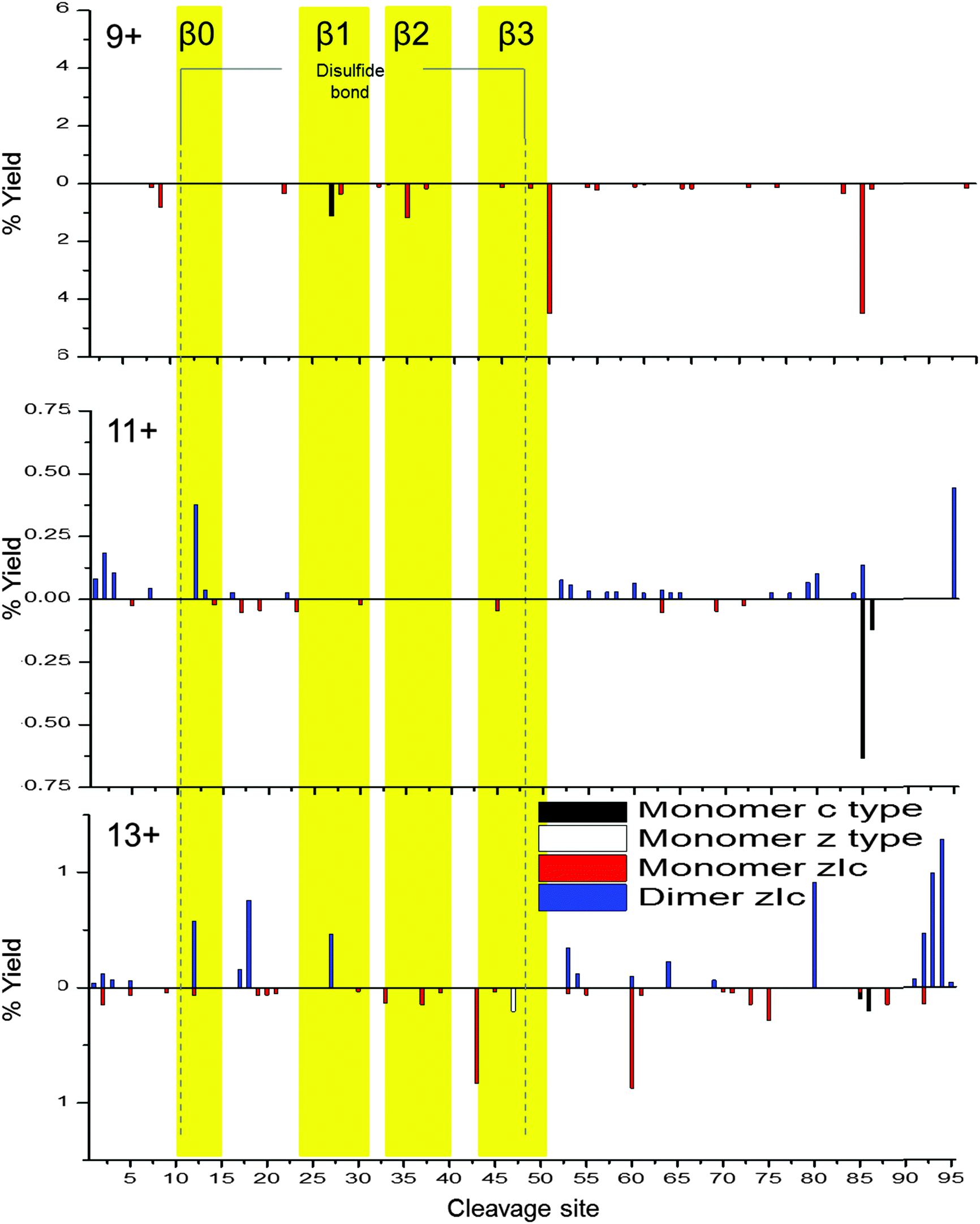 | ||
| Fig. 5 Percentage yields calculated for c type, z type and zIc fragments as a function of cleavage site for dimeric W55D over the charge states [D + 9H]9+, [D + 11H]11+ and [D + 13H]13+. Above axis histograms represent fragments formed while retaining the dimer interface. Below axis histograms represent fragments formed following dimer dissociation to monomer. For figure clarity, the y-axis scale varies with charge state. | ||
For the [DW55D + 11H]11+ the observed dimer fragmentation maps extremely well with the Ltn40 fold, with the majority of the fragmentation occurring at the termini and in the flexible regions just before the β-sheet core. This suggests that some dimeric species which survive the desolvation process are likely to be present in the Ltn40 fold. For [DW55D + 11H]11+ we begin to see fragmentation within the β0 strand, which increases in intensity as we go up in charge state to the [DW55D + 13H]13+ species. The increase in fragmentation along this region suggests it is the least stable of the β-strands and is the first to unfold from the structural core of the protein. This indicates that the Ltn40 conformation unfolds from the N-terminus, which is the converse of the Ltn10 conformation which appears to unfold first from the C-terminus. The ECD fragmentation maps allow us to rank the stability of each of the β-strands from least to most stable (β0 < β1 < β3 < β2), which could help in the design of future mutants aimed to mimic this fold but with increased stability and therefore decreased unfolding.
Visualising protein unfolding
By combining the DT IM-MS results with the ECD fragmentation maps for all the mutants studied here we can build up a detailed picture of the unfolding of both Ltn10 and Ltn40, giving insight into fold stability as a function of increased charge, Fig. 6. | ||
| Fig. 6 A cartoon representation of the unfolding of the Ltn10 and Ltn40 folds as shown by DT IM-MS and ECD, probed through specific mutants, (a) WT 1–72 (b) buffered CC3 (c) buffered and supercharged CC3 (d) monomeric W55D and (e) dimeric W55D, where the dimer is represented here by a single monomeric unit. Yellow arrows represent β-sheet regions and purple spirals represent α-helical regions. Orange ‘lightning bolts’ indicate sites where ECD fragmentation is most significant, extensive fragmentation along the backbone is depicted by a large lightning bolt and dashed lines represent a lack of defined structural elements. | ||
Using the methodology presented here we are able to compare the three mutants of this metamorphic protein. In summary, we find that in the absence of the intrinsically disordered tail unfolding of the Ltn10 fold still proceeds via the C-terminus in a similar manner as that observed for full length WT Ltn. For WT 1–72, however, we find unravelling of the α-helix occurs at lower charge states than for the full length WT Ltn10, suggesting the intrinsically disordered tail confers some stability or protection to the structural core and in particular the α-helix. We observe that the CC3 Ltn mutant is more structurally constrained than the WT Ltn, as expected for a protein with an additional disulfide bond, and exists in the Ltn10 fold. The positioning of this additional disulfide bond essentially pins the α-helix to the structural core of the protein and blocks the first unfolding pathway of the Ltn10 conformation. For the W55D Ltn our results suggest that the dimer interface of the Ltn40 fold is less stable in this mutant than in the WT protein, leading to dissociation of dimer to monomer both upon ionisation and transfer and as a result of ECD fragmentation. The fragmentation map of this mutant also suggests that for this conformation the first unfolding pathway involves the unfolding of the β0 strand and N-terminus. The combination of these two techniques and their application to mutagenesis studies can further in the intelligent design of mutants to stabilise protein fold and unfolding by highlighting susceptible regions.
Conclusions
The ability of this combination of techniques to distinguish significant effects of single and double point mutations on protein fold and unfolding highlights the wealth of information that can be obtained in this way. The power of using such techniques becomes evident when you consider the small sample quantities required for such studies, with as little as 25 μg of protein providing insight. Information can be obtained on the relative stabilities of the mutants in comparison to the WT protein and therefore has the potential to be highly influential in design of site directed mutants.Experimental procedures
Protein expression and purification
All recombinant human Ltn protein used in ECD and DT IM-MS investigations were expressed and purified as previously described.41,45 The lymphotactin mutants CC3 and W55D were prepared by site-directed mutagenesis using complementary primer pairs and the Stratagene QuickChange kit follow the manufacturer's instructions. All expression vectors were verified by DNA sequencing. Purified proteins were frozen, lyophilized and stored at −20 °C for subsequent study.DT IM-MS and theoretical calculations
For all ion mobility mass spectrometry experiments samples were ionised and introduced into the instrument using nano-electrospray ionisation. Nano-electrospray tips were made in-house using thin-walled glass capillaries (i.d. 0.5 mm) using a Flaming/Brown micropipette puller (Sutter Instrument Company, Novato, CA, USA). Sample solutions were ionised through a potential applied to a thin platinum wire (0.125 mm Goodfellow) inserted into the glass capillary.MS and DT IM-MS experiments were performed on an in-house modified Q-TOF (Micromass UK Ltd), adapted in order to carry out separations based on an ion's mobility, and to enable the temperature dependent CCS to be determined. The DT IM-MS instrument was previously modified to include a 5.1 cm long copper drift cell and supplementary ion optics situated post source but before the quadrupole analyser. The instrument and its operation have been described in detail elsewhere46 and further details are found in ESI.†
Theoretical CCSs were calculated from NMR structures (PDB identifiers 1J9O, 2JP1 and 2HDM for Ltn10, Ltn40 and monomeric CC3 mutant, respectively) using the trajectory method of MOBCAL code.47 Further details on the procedure are found in ESI.†
ECD
All high resolution mass spectrometry and ECD analysis were carried out on a 12 T SolariX FTICR (Bruker Daltonics). All samples were ionised using nano-electrospray ionisation, n-ESI was performed using a NanoMate (Advion biosciences) running in infusion mode and equipped with a HDA0 ESI chip (Advion biosciences). Analysis of WT 1–72 was carried out on 5 μM WT 1–72 in 100 mM ammonium acetate. Ion accumulation was between 3 and 10 seconds for each charge state studied, depending on the intensity of the species, and in all cases ECD fragmentation was summed over 300 scans. For W55D, analysis was performed on 30 μM W55D in 100 mM ammonium acetate. Ion accumulation was between 0.5 and 8 seconds for each charge state studied, depending on the intensity of the species, and in all cases ECD fragmentation was summed over 200 scans. For CC3, analysis was performed on 30 μM CC3 in 100 mM ammonium acetate for ‘buffered’ analysis and 30 μM CC3 in 100 mM ammonium acetate +1% m-NBA for ‘buffered and supercharged’ studies. In both cases ion accumulation was between 0.2 and 10 seconds, depending on the intensity of the species, and ECD fragmentation was summed over 200 scans. For this work it was not possible to collisionally activate the ions post ECD, to reveal cleaved sites on the protein backbone which perhaps have not dissociated due to non-covalent interactions. Future work could employ IRMPD to investigate this.Author contributions
SRH did all of the mass spectrometry experiments and analysis, and prepared the first draft of the manuscript. MP performed the calculations. RT expressed the proteins and with BFV designed the site specific mutants. PEB and CEM helped to analyse the data and PEB wrote the submitted form of this manuscript.Acknowledgements
The Schools of Chemistry and Physics at the University of Edinburgh, are thanked for an award of an EPSRC DTA studentship to SRH. Dr Logan Mackay and Dr David Clarke are thanked for assistance with FT-ICR MS. Dr Michel Laguerre (Institut Européen de Chimie et Biologie (IECB), Pessac, France) is thanked for providing access to computational resources. The authors would like to acknowledge NIH grant R01 AI013225 awarded to BFV.References
- M. Dole, L. L. Mack, R. L. Hines, R. C. Mobley, L. D. Ferguson and M. B. Alice, Molecular Beams of Macroions, J. Chem. Phys., 1968, 49, 2240–2249 CrossRef CAS PubMed.
- M. Yamashita and J. B. Fenn, Electrospray ion source. Another variation on the free-jet theme, J. Phys. Chem., 1984, 88, 4451–4459 CrossRef CAS.
- M. S. Wilm and M. Mann, Electrospray and Taylor-Cone theory, Dole's beam of macromolecules at last?, Int. J. Mass Spectrom. Ion Processes, 1994, 136, 167–180 CrossRef CAS.
- M. Wilm and M. Mann, Analytical properties of the nanoelectrospray ion source, Anal. Chem., 1996, 68, 1–8 CrossRef CAS.
- J. Fenn, M. Mann, C. Meng, S. Wong and C. Whitehouse, Electrospray ionization for mass spectrometry of large biomolecules, Science, 1989, 246, 64–71 CAS.
- C. S. Hoaglund-Hyzer, A. E. Counterman and D. E. Clemmer, Anhydrous protein ions, Chem. Rev., 1999, 99, 3037–3080 CrossRef CAS PubMed.
- A. J. R. Heck, Native mass spectrometry: a bridge between interactomics and structural biology, Nat. Methods, 2008, 5, 927–933 CrossRef CAS PubMed.
- J. L. P. Benesch, B. Ruotolo, D. A. Simmons and C. V. Robinson, Protein Complexes in the Gas Phase technology for structural genomics and proteomics, Chem. Rev., 2007, 107, 3544–3567 CrossRef CAS PubMed.
- V. L. Morton, P. G. Stockley, N. J. Stonehouse and A. E. Ashcroft, Insights into virus capsid assembly from non-covalent mass spectrometry, Mass Spectrom. Rev., 2008, 27, 575–595 CrossRef PubMed.
- Z. Ouyang, Z. Takáts, T. A. Blake, B. Gologan, A. J. Guymon, J. M. Wiseman, J. C. Oliver, V. J. Davisson and R. G. Cooks, Preparing Protein Microarrays by Soft-Landing of Mass-Selected Ions, Science, 2003, 301, 1351–1354 CrossRef CAS PubMed.
- K. B. J. McAfee and D. Edelson, Identification and Mobility of Ions in a Townsend Discharge by Time-resolved Mass Spectrometry, Proc. Phys. Soc., London, 1963, 81, 382 CrossRef CAS.
- P. Kebarle and A. M. Hogg, Mass Spectrometric study of ions at near atmospheric pressure I The ionic polymeriztion of ethylene.pdf, J. Chem. Phys., 1965, 42, 668–674 CrossRef CAS PubMed.
- A. M. Hogg and P. Kebarle, Mass Spectrometric study of ions at near atmospheric pressure II Ammonium Ions produced by the alpha radiolysis of ammonia and their solvation in the gas phase by ammonia and water molecules.pdf, J. Chem. Phys., 1965, 43, 449–456 CrossRef CAS PubMed.
- K. B. Shelimov, D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, Protein Structure in Vacuo: Gas-Phase Conformations of BPTI and Cytochrome c, J. Am. Chem. Soc., 1997, 119, 2240–2248 CrossRef CAS.
- D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, Naked Protein Conformations: Cytochrome c in the Gas Phase, J. Am. Chem. Soc., 1995, 117, 10141–10142 CrossRef CAS.
- T. Wyttenbach, G. Von Helden and M. T. Bowers, Gas-Phase Conformation of Biological Molecules Bradykinin, J. Am. Chem. Soc., 1996, 118, 8355–8364 CrossRef CAS.
- R. R. Hudgins, J. Woenckhaus and M. F. Jarrold, High resolution ion mobility measurements for gas phase proteins: correlation between solution phase and gas phase conformations, Int. J. Mass Spectrom. Ion Processes, 1997, 165–166, 497–507 CrossRef.
- S. R. Harvey, M. Porrini, C. Stachl, D. MacMillan, G. Zinzalla and P. E. Barran, Small-Molecule Inhibition of c-MYC:MAX Leucine Zipper Formation Is Revealed by Ion Mobility Mass Spectrometry, J. Am. Chem. Soc., 2012, 134, 19384–19392 CrossRef CAS PubMed.
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J.-E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Amyloid-β protein oligomerization and the importance of tetramers and dodecamers in the aetiology of Alzheimer's disease, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed.
- C. Bleiholder, N. F. Dupuis, T. Wyttenbach and M. T. Bowers, Ion mobility–mass spectrometry reveals a conformational conversion from random assembly to β-sheet in amyloid fibril formation, Nat. Chem., 2011, 3, 172–177 CrossRef CAS PubMed.
- N. A. Pierson, S. J. Valentine and D. E. Clemmer, Evidence for a Quasi-Equilibrium Distribution of States for Bradykinin Ions in the Gas Phase.pdf, J. Phys. Chem. B, 2010, 114, 7777–7783 CrossRef CAS PubMed.
- O. Skinner, F. McLafferty and K. Breuker, How Ubiquitin Unfolds after Transfer into the Gas Phase, J. Am. Soc. Mass Spectrom., 2012, 23, 1011–1014 CrossRef CAS PubMed.
- J. M. D. Kalapothakis, Y. Berezovskaya, C. G. Zampronio, P. A. Faull, P. E. Barran and H. J. Cooper, Unusual ECD fragmentation attributed to gas-phase helix formation in a conformationally dynamic peptide, Chem. Commun., 2014, 50, 198–200 RSC.
- S. R. Harvey, M. Porrini, A. Konijnenberg, D. J. Clarke, R. C. Tyler, P. R. Langridge-Smith, C. E. MacPhee, B. F. Volkman and P. E. Barran, Dissecting the Dynamic Conformations of the Metamorphic Protein Lymphotactin, J. Phys. Chem. B, 2014, 118, 12348–12359 CrossRef CAS PubMed.
- R. A. Zubarev, N. L. Kelleher and F. W. McLafferty, Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process, J. Am. Chem. Soc., 1998, 120, 3265–3266 CrossRef CAS.
- K. Breuker, H. B. Oh, C. Lin, B. K. Carpenter and F. W. McLafferty, Nonergodic and conformational control of the electron capture dissociation of protein cations, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14011–14016 CrossRef CAS PubMed.
- K. F. Haselmann, T. J. Jorgensen, B. A. Budnik, F. Jensen and R. A. Zubarev, Electron capture dissociation of weakly bound polypeptide polycationic complexes, Rapid Commun. Mass Spectrom., 2002, 16, 2260–2265 CrossRef CAS PubMed.
- D. M. Horn, K. Breuker, A. J. Frank and F. W. McLafferty, Kinetic Intermediates in the Folding of Gaseous Protein Ions Characterized by Electron Capture Dissociation Mass Spectrometry, J. Am. Chem. Soc., 2001, 123, 9792–9799 CrossRef CAS PubMed.
- K. Breuker, H. B. Oh, D. M. Horn, B. A. Cerda and F. W. McLafferty, Detailed unfolding and folding of gaseous ubiquitin ions characterized by electron capture dissociation, J. Am. Chem. Soc., 2002, 124, 6407–6420 CrossRef CAS PubMed.
- H. Oh, K. Breuker, S. K. Sze, Y. Ge, B. K. Carpenter and F. W. McLafferty, Secondary and tertiary structures of gaseous protein ions characterized by electron capture dissociation mass spectrometry and photofragment spectroscopy, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15863–15868 CrossRef CAS PubMed.
- K. Breuker, S. Brüschweiler and M. Tollinger, Electrostatic Stabilization of a Native Protein Structure in the Gas Phase, Angew. Chem., Int. Ed., 2011, 50, 873–877 CrossRef CAS PubMed.
- A. Zlotnik and O. Yoshie, Chemokines: a new classification system and their role in immunity, Immunity, 2000, 12, 121–127 CrossRef CAS.
- G. Kelner, J. Kennedy, K. Bacon, S. Kleyensteuber, D. Largaespada, N. Jenkins, N. Copeland, J. Bazan, K. Moore and T. Schall, et al. Lymphotactin: a cytokine that represents a new class of chemokine, Science, 1994, 266, 1395–1399 CAS.
- A. G. Murzin, Metamorphic Proteins, Science, 2008, 320, 1725–1726 CrossRef CAS PubMed.
- R. L. Tuinstra, F. C. Peterson, S. Kutlesa, E. S. Elgin, M. A. Kron and B. F. Volkman, Interconversion between two unrelated protein folds in the lymphotactin native state, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5057–5062 CrossRef CAS PubMed.
- E. S. Kuloglu, D. R. McCaslin, M. Kitabwalla, C. D. Pauza, J. L. Markley and B. F. Volkman, Monomeric solution structure of the prototypical ‘C’ chemokine lymphotactin, Biochemistry, 2001, 40, 12486–12496 CrossRef CAS PubMed.
- R. C. Tyler, N. J. Murray, F. C. Peterson and B. F. Volkman, Native-State Interconversion of a Metamorphic Protein Requires Global Unfolding, Biochemistry, 2011, 50, 7077–7079 CrossRef CAS PubMed.
- X. Li, C. Lin, L. Han, C. E. Costello and P. B. O'Connor, Charge remote fragmentation in electron capture and electron transfer dissociation, J. Am. Soc. Mass Spectrom., 2010, 21, 646–656 CrossRef CAS PubMed.
- A. T. Iavarone, K. Paech and E. R. Williams, Effects of charge state and cationizing agent on the electron capture dissociation of a peptide, Anal. Chem., 2004, 76, 2231–2238 CrossRef CAS PubMed.
- P. Roepstorff and J. Fohlman, Proposal for a common nomenclature for sequence ions in mass spectra of peptides, Biomed. Mass Spectrom., 1984, 11, 601 CrossRef CAS PubMed.
- R. L. Tuinstra, F. C. Peterson, E. S. Elgin, A. J. Pelzek and B. F. Volkman, An Engineered Second Disulfide Bond Restricts Lymphotactin/XCL1 to a Chemokine-like Conformation with XCR1 Agonist Activity†, Biochemistry, 2007, 46, 2564–2573 CrossRef CAS PubMed.
- R. A. Zubarev, N. A. Kruger, E. K. Fridriksson, M. A. Lewis, D. M. Horn, B. K. Carpenter and F. W. McLafferty, Electron capture dissociation of gaseous multiply-charged proteins is favored at disulfide bonds and other sites of high hydrogen atom affinity, J. Am. Chem. Soc., 1999, 121, 2857–2862 CrossRef CAS.
- A. T. Iavarone and E. R. Williams, Supercharging in electrospray ionization: effects on signal and charge, Int. J. Mass Spectrom., 2002, 219, 63–72 CrossRef CAS.
- H. Sterling, A. Kintzer, G. Feld, C. Cassou, B. Krantz and E. Williams, Supercharging Protein Complexes from Aqueous Solution Disrupts their Native Conformations, J. Am. Soc. Mass Spectrom., 2012, 23, 191–200 CrossRef CAS PubMed.
- F. C. Peterson, E. S. Elgin, T. J. Nelson, F. Zhang, T. J. Hoeger, R. J. Linhardt and B. F. Volkman, Identification and Characterization of a Glycosaminoglycan Recognition Element of the C Chemokine Lymphotactin, J. Biol. Chem., 2004, 279, 12598–12604 CrossRef CAS PubMed.
- B. J. McCullough, J. Kalapothakis, H. Eastwood, P. Kemper, D. MacMillan, K. Taylor, J. Dorin and P. E. Barran, Development of an Ion Mobility Quadrupole Time of Flight Mass Spectrometer, Anal. Chem., 2008, 80, 6336–6344 CrossRef CAS PubMed.
- M. F. Mesleh, J. M. Hunter, A. A. Shvartsburg, G. C. Schatz and M. F. Jarrold, Structural Information from Ion Mobility Measurements Effects of the Long-Range, J. Phys. Chem. A, 1996, 100, 16082–16086 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Further experimental details for the drift tube ion mobility mass spectrometry experiments, tables of experimental collision cross sections obtained from DT IM-MS experiments, spectra obtained and further information on the ECD fragmentation experiments can be found in ESI. In addition experimental details and theoretical CCS derivation can also be found in ESI. See DOI: 10.1039/c4cp05136j |
| This journal is © the Owner Societies 2015 |