
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The Lewis acidic and basic character of the internal HKUST-1 surface determined by inverse gas chromatography†
Alexander S.
Münch
and
Florian O. R. L.
Mertens
*
Institute of Physical Chemistry, Technical University Bergakademie Freiberg, Leipziger Straße 29, 09599 Freiberg, Germany. E-mail: florian.mertens@chemie.tu-freiberg.de
First published on 17th October 2014
Abstract
Metal–organic frameworks (MOFs) have demonstrated great utilizability in separation applications, as in the separation of small volatile compounds via gas chromatography (GC). In the present work, HKUST-1 (Hong Kong University of Science and Technology), one of the best investigated MOFs, is used as a stationary phase for the gas chromatographic separation of various analytes possessing different modes of interaction due to their differences in polarity and the presence of free electron pairs. The system was investigated by inverse gas chromatography (IGC) to demonstrate in general how MOF materials can be quantitatively and qualitatively characterized in respect to their Lewis basic and acidic properties. Applying IGC theory, the investigation of the separation problem of benzene and its completely hydrogenated analogue cyclohexane was used to the determine the donor properties of the MOF linker benzene-1,3,5-tricarboxylic acid and the separation of diethyl ether, diisopropyl ether, tetrahydrofuran, and di-n-propyl ether to determine the acceptor properties of the coordinatively unsaturated sites of the copper(II)-secondary building unit (SBU), i.e. the nodal points of the MOF lattice.
Introduction
Until now, the synthesis of a very large number of metal–organic frameworks with different and partially exotic topologies, that display, if the inapplicability of the standard BET analysis for microporous systems is ignored, BET values of up to 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 m2 g−1, is still the main objective of many research groups.1–7 Beside synthesis, many potential applications of this class of materials connected to the structural variability in combination with crystallinity and permanent porosity as well as the possibility to tune pore size and geometry for specific applications are conceivable and were intensively discussed in recent years.8–17 A specific field of MOF application that allows to utilize these features are the various types of chromatography, such as liquid chromatography16,17 and gas chromatography.18–20
000 m2 g−1, is still the main objective of many research groups.1–7 Beside synthesis, many potential applications of this class of materials connected to the structural variability in combination with crystallinity and permanent porosity as well as the possibility to tune pore size and geometry for specific applications are conceivable and were intensively discussed in recent years.8–17 A specific field of MOF application that allows to utilize these features are the various types of chromatography, such as liquid chromatography16,17 and gas chromatography.18–20
Recently, the separation of different analyte mixtures, such as those of xylene isomers,19,21n-alkanes,20,22,23 polychlorinated biphenyls,24 polycyclic aromatic hydrocarbons,24 and branched alkanes23,25 by MOF based capillary GC was demonstrated. In contrast to the interest in the separations, there have been almost no studies using the so called inverse gas chromatography with MOFs, a term that is used for GC investigations which do not primarily focus on separation problems but on the evaluation of physico-chemical properties of the material which the stationary phase is made of.20,21,26–30 HKUST-1 is one of the best investigated metal–organic frameworks in respect to crystal structure, properties, and possible applications.21,31–34 This MOF, originally reported by Chui et al.,35 consists of copper(II)-dimer units, where each copper ion is coordinated to four oxygen atoms of the organic linker benzene-1,3,5-tricarboxylate, forming a three dimensional crystalline and porous framework with three cavities possessing the diameters 4.7 Å, 10.3 Å, and 12.1 Å.36 One coordination site of the metal ions is unsaturated and therefore free to accommodate Lewis basic molecules, like water.35 The coordinated water molecules can easily be removed at mild conditions (by heating in vacuum or inert gas) from the coordination shell of the Cu2+ ions without any loss of porosity and crystallinity creating an unsaturated coordination site, a procedure that is called activation.37 Beside these Lewis acidic centers of the copper(II)-SBUs, HKUST-1 possesses also Lewis basic areas like the π-electron rich benzene rings of the linker molecules benzene-1,3,5-tricarboxylate.
Molecules, like aromatic compounds, can be coordinated by weak interactions such as π–π stacking and C–H–π interactions as largely explored in the field of supramolecular chemistry.38,39 The interaction between analytes and the crystalline lattice can be classified in several ways depending on the properties of the analyte. Analytes possessing a permanent dipole moment are characterized in addition to the nonspecific part of the van der Waals interaction (interactions with a 1/r6 dependence), which is the London- or dispersion interaction,40 by the so called specific part which should be in the case of HKUST-1 mainly the Debye-interaction.41 This type of interaction is defined as the angle averaged interaction between a permanent and an induced dipole. The other contribution to the specific part of the van der Waals interaction is the so called Keesom interaction,42 which results from averaged interaction between two permanent dipoles.
Since the linker molecules and metal centers in many MOFs can be exchanged and new functionalities can be introduced to some degree in a predictable manner (crystal engineering), it appears to be important that the effect of such modifications can be quantitatively evaluated. In this contribution we therefore report a procedure how specific and nonspecific interaction between the Lewis acidic (open metal sites of the copper-SBUs) and the Lewis basic (π-electron rich aromatic rings) parts of HKUST-1 and different analytes can be analyzed by means of inverse gas chromatography.
Theoretical background – inverse gas chromatography
Inverse gas chromatography43,44 has been widely applied in the determination of physico-chemical parameters in very different systems. From the measurement of the temperature and carrier gas flow dependent retention times, quantities characterizing the stationary phase – analyte interaction such as the thermodynamic adsorption enthalpy, ΔHads, and adsorption entropy, ΔSads, as well as kinetic parameters, such as mass transport and diffusion coefficients can be obtained. Once a routinely applicable procedure to prepare chromatographic columns and capillaries is established, IGC is an efficient method to investigate adsorption phenomena in porous media and thus in metal–organic frameworks as well.Determination of thermodynamic parameter values of the adsorption process
By plotting the logarithm of the retention volume gained from isothermal gas chromatographic measurements versus the inverse temperature, the enthalpy and entropy of adsorption are easily obtained from | (1) |
The retention volume VN was obtained from the adjusted retention times tr′ = (tr − tm), the gas flow Fa, and the application of the two correction terms reflecting the adjustment of the gas to the column temperature and the pressure drop experienced during the passage through the column:47,48
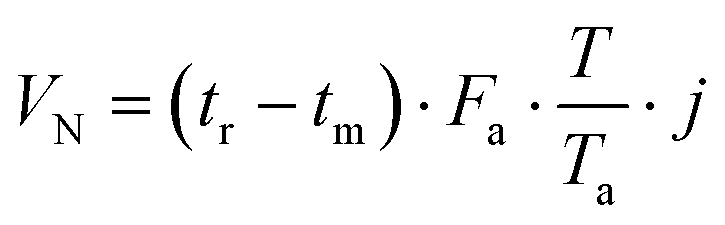 | (2) |
T a stands for ambient temperature and j is the James–Martin gas compressibility correction factor.
Starting from the desire to obtain heats of adsorption that can be compared to calorimetric measurements, which are usually performed under isochoric conditions, the heat of adsorption derived from eqn (1), which represents an enthalpy change, needs to be converted to a heat of adsorption that represents an internal energy change by |ΔHiso| = |ΔUdiff| + RTav (for details see ESI†).
IGC for the determination of surface interaction Gibbs free energies
Following the terminology used by Fowkes49 the interaction of an adsorbate can be split in the so called nonspecific interaction Gibbs free energy ΔGnonspec caused by the London-dispersion interaction and a term denoted as specific interaction Gibbs free energy ΔGspec caused by the Keesom- and Debye-interactions as well as all Gibbs free energy contributions caused by acid–basic interactions, hydrogen bonding, π–π-interactions, and steric effects:| |−ΔGinteract| = |−ΔGspec| + |−ΔGnonspec| | (3) |
Measuring the retention times as a function of the temperature, the interaction Gibbs free energy can be determined by calculating the net retention volume according to
| −ΔGinteract = RT · lnVN + C | (4) |
Calculation of the nonspecific ΔGnonspec and specific component ΔGspec of the interaction Gibbs free energy of adsorption ΔGinteract
In case that a polar analyte interacts with the surface of solids, dispersive and specific interactions occur and the problem arises how to split the nonspecific part of the interaction Gibbs free energy from the specific part. Both types of interactions, the specific and the nonspecific one, cannot be investigated separately. In the literature different approaches to solve this problem can be found50,52–56 that are all based on the same evaluation model. At first, the (linear) relationship between the Gibbs free energy of interaction, ΔGinteract, of n-alkanes possessing a specific property (f.e. the polarizability) that can be expected to scale with the nonspecific interaction strength is determined as a reference function. n-Alkanes are chosen because they display only nonspecific interactions. The choice of the specific property is the quantity that distinguishes the different reported approaches. In the next step, the reference function value at the value of the specific property of the polar analyte to be investigated is taken and the difference to ΔGinteract of the analyte to be investigated is calculated. Under the assumption that the dependence of the nonspecific interaction does strictly correlate with the specific property of the analytes, no matter what sort of molecule it is, the calculated difference is fully ascribed to the specific interactions. Fig. 1 illustrates the described procedure. | ||
| Fig. 1 Top: schematic representation of the procedure to determine the specific Gibbs free energy of adsorption, ΔGspec, of a polar analyte that can be characterized by a specific property that is assumed to generally scale with the nonspecific interaction strength, i.e. the London dispersion interaction. Bottom: Gibbs free energy of adsorption of diisopropyl ether (●), diethyl ether (×), tetrahydrofuran (*), and di-n-propyl ether (■) on HKUST-1. The reference line was established by linear regression from the dependence of the nonspecific interactions of the n-alkanes pentane, hexane, heptane on the scaled electronic polarizability (hν)0.5αO,L. The regression line is given by: −ΔGvdW = 3.96 × 1052 V0.5 J C−2/3 m−2 mol−1·(hν)0.5αO,L − 35737.10 J mol−1, R2 = 0.9974. | ||
Using the polarizability α as the specific property bears the advantage that this quantity practically does not vary with temperature,52 which is why it was chosen in this work for the analysis. The relationship between polarizability, the London dispersion interaction, and the corresponding −ΔGL can be described by the expression (see Donnet et al.52):
 | (5) |
From eqn (3) and (4) then follows:
 | (6) |
With K = 3NA/((2·4πε0)2·rS,L6) where the subscripts S and L refer to the solid surface and the analyte. Thus, with  being the specific property, −ΔGspec can be determined by first determining the prefactor
being the specific property, −ΔGspec can be determined by first determining the prefactor  , from the reference line of the n-alkanes by plotting their RT lnVN values versus
, from the reference line of the n-alkanes by plotting their RT lnVN values versus and then calculating the difference between −ΔGinteract and the value of the reference line at the value of the specific property of the analyte to be investigated. The slope of the linear function (6),
and then calculating the difference between −ΔGinteract and the value of the reference line at the value of the specific property of the analyte to be investigated. The slope of the linear function (6),  , is characteristic for a given solid surface and is related to the London dispersive interaction component of this surface.52 The required electronic deformation polarizabilities can be taken from suitable references,57 calculated either by using the Debye equation or by the increment method according to Miller et al.58
, is characteristic for a given solid surface and is related to the London dispersive interaction component of this surface.52 The required electronic deformation polarizabilities can be taken from suitable references,57 calculated either by using the Debye equation or by the increment method according to Miller et al.58
Characterization of the surface donor–acceptor properties by IGC
Plotting the interaction Gibbs free energy ΔGspec as a function of temperature, the specific enthalpy ΔHspec and the specific entropy ΔSspec of adsorption can be obtained from the intercept and the slope of the Gibbs Helmholtz equation:| ΔGspec = ΔHspec − T·ΔSspec | (7) |
The specific enthalpy of adsorption of an analyte on a specific surface of a solid can be correlated to the acid–base properties of this solid, here the stationary phase of the chromatographic capillaries used.50 The quantitative description of the donor and acceptor properties of the surface requires the determination of the acid and base constant KA and KB. The relation to the specific interaction is given according to Gutmann59 by
| −ΔHspec = KA·DN + KB·AN | (8) |
 | (9) |
Materials and methods
Analytes
The chromatographic separation experiments were performed using a mixture of benzene (Petrochemisches Kombinat Schwedt, >99.5%) and cylcohexane (VWR, ≥99.5%) at a ratio 1:1 and a 1:1:1:1 mixture of diethyl ether (VWR, 99.7%), diisopropyl ether (Merck, >99%), tetrahydrofuran (Acros Organics, 98%), and di-n-propyl ether (ABCR, 99%). The thermodynamic values were determined by collecting the retention times of the pure compounds. All analytes and analyte mixtures were mixed prior to the experiment with some methane for the simultaneous determination of the dead time.
HKUST-1 preparation
For the infrared spectroscopy and powder X-ray crystallography investigations of the analyte – HKUST-1 interaction, the use of a completely desolvated HKUST-1 material possessing high surface area is required. In a typical synthesis a solution of 0.210 g (1 mmol) benzene-1,3,5-tricarboxylic acid (ABCR, 98%) dissolved in 25 mL ethanol (VWR, >99.8%) was dropwise added at room temperature to a solution of 0.299 g (1.5 mmol) copper(II)-acetate monohydrate (Acros Organics, >98%) in 25 mL deionized water. Afterwards the suspension was stirred for 1 hour and then the blue precipitate was filtered off and washed with ethanol and diethyl ether. For activation, the HKUST-1 material was heated to 373 K under vacuum conditions. From the procedure described, a dark blue powder was obtained. Powder X-ray diffraction, IR-spectroscopy, and the modified BET surface area measurements for microporous systems were conducted. The results can be found in the ESI.†Capillary coating
All experiments were conducted with a gas chromatographic silica capillary of 30 m length and an inner diameter of 0.25 mm. In order to apply the coating procedure used, the silica surface of the column has to be terminated with hydroxyl groups, which was achieved by first pumping a 1.0 M solution of sodium hydroxide through the capillary for a period of 60 min followed by an intensive rinsing with water until the pH-value of the solution at the outlet indicated neutrality. The same procedure was repeated with 0.1 M solution of hydrochloric acid. Finally the column was dried in a stream of argon (excess pressure at the inlet 0.2 bar) at a temperature of 423 K for 24 hour.21After this surface activation procedure was conducted, HKUST-1 was deposited by a cyclic layer deposition procedure based on the controlled SBU-approach. For this procedure,21 a solution, called the precursor solution, of 299 mg (1.5 mol) copper(II)-acetate monohydrate in 150 mL ethanol and a solution, called the linker solution, of 210 mg (1 mmol) benzene-1,3,5-tricarboxylic acid dissolved in the same amount of ethanol were prepared. These solutions were pumped with an excess pressure at the inlet of 5 bar alternately through the capillary using a self-built coating apparatus (ESI†). Between every cycling step the liquid film was conditioned for 2 min followed by a rinsing or purging procedure with ethanol executed by the coating apparatus in order to avoid the formation of small HKUST-1 particles inside the steel pipes. The whole coating procedure progressed by the following sequence, which was 15 times repeated: 1 mL precursor solution (5 bar) → 2 min conditioning → cleaning of the coating apparatus → 1 mL precursor solution (5 bar) → 2 min conditioning → cleaning of the coating apparatus.
After this procedure the film was homogenized and strongly fixed onto the capillary walls by the following conditioning sequence: 2 hours argon (0.2 bar excess pressure) at 333 K → 2 hours argon (0.2 bar excess pressure) at 353 K → 2 hours argon (0.2 bar excess pressure) at 373 K.
Chromatographic setup
All GC experiments were performed using an HP 5890 series II gas chromatograph equipped with a flame ionization detector (FID). Helium (Praxair, 99.999%) was the carrier gas in all cases. The data evaluation was conducted with the EZChrome software from Agilent Technologies.The chromatograms for the determination of the adsorption enthalpies and entropies of cyclohexane and benzene were recorded using pure substances only and taken at a constant inlet gas flow of 5.31 mL min−1 at different temperature that varied from 443.15 K to 473.15 K. The chromatograms for the determination of the adsorption enthalpies and entropies of the ethers were also recorded using pure substances but at a constant inlet gas flow of 4.15 mL min−1 and temperatures that varied from 493.15 K to 523.15 K. During the gas chromatographic measurements the ambient temperature and pressure was 298 K and 0.969 bar, respectively. The dead time was determined by the retention time of methane.
Inductively coupled plasma optical emission spectroscopy (ICP-OES)
For the determination of a film thickness profile on the basis of the deposited amount of HKUST-1, a capillary with an inner diameter dc of 0.25 mm and a length L of 30 m was coated according to the procedure described above. After purging of the coated layer with deionized water, a piece of approximately 10 mm was cut off from each side of the column for collecting scanning electron microscope (SEM) images and energy dispersive X-ray spectroscopic (EDX) data. Afterwards the capillary was cut into pieces of 2 m each. The deposited HKUST-1 was removed from every section by very pure concentrated hydrochloric acid (suprapore, Merck) and the concentration of the copper(II)-ions in the obtained solutions was determined using a Varian Vista-Pro-CCD Simultaneous ICP-OES apparatus. The amount of HKUST-1 in each of the original capillary sections was then calculated from the obtained concentrations.Results and discussion
Characterization of the HKUST-1 coatings inside a capillary
In contrast to the published procedures to deposit a thin film of a metal–organic framework in capillaries made from fused silica by other authors, we chose the approach to coat GC capillaries with HKUST-1 based on the controlled SBU approach (CSA). Previous processes were based either on the solvothermal synthesis of little MOF-crystallites and the following deposition of the particles from a suspension onto the capillary wall via the interaction with the surface hydroxyl groups19,22 or on a cyclic layer deposition technique in which the MOF structure is formed directly during the coating procedure inside the capillary via a precipitations reaction anchoring the MOF layer via a bifunctional self-assembled monolayer (SAM) onto the fused silica.20,21,60 Because the pipes of the coating apparatus become easily congested with the precipitate applying this method, we switched to the simpler anchoring technique using the hydroxyl groups of the fused silica surface in combination with the CSA method as described in the section Materials and methods.After the coating of the capillary, SEM-images from the inner capillary surface near the inlet (Fig. 2a) and near the outlet (Fig. 2b) were taken from which respective film thicknesses of 200–300 nm and 150–200 nm were determined, indicating that more HKUST-1 is deposited at the beginning of the capillary than at the end. In order to track the decrease of the film thickness, the amount of HKUST-1 was determined as described earlier by acid treatment and ICP-OES measurements. The MOF coating thickness profile obtained is displayed in Fig. 2c and shows the presence of an approximately constant film thickness in the capillary sections from 2 m to 26 m measured from the capillary entry. The increased amount at the end of the capillary in such systems is in the literature often attributed to the solution acceleration at the end of the capillary.19
 | ||
| Fig. 2 Characterization of the HKUST-1 capillary coatings. A – SEM-image of material near the capillary inlet. B – SEM-image of material near the capillary outlet. C – Profile of the mass of deposited HKUST-1 material along the capillary as indicator of the corresponding film thickness variations of the stationary phase. | ||
Because of the comparably small amount of HKUST-1 on the fused silica surface, the small dimension, and the curved surface of the capillary, a direct structural verification of HKUST-1 on the substrate wall by PXRD was not possible. We therefore, as an indirect verification, separately mixed test samples of the reaction solutions to be used for the coating experiment and investigated the precipitate by PXRD. In addition the reaction solutions leaving the capillary outlet were collected in one flask and the precipitate formed there was investigated by PXRD as well. In both cases the diffractograms of HKUST-1 were obtained (see ESI†).
Gas chromatographic separation
With the coated capillary (L = 30 m, dc = 0.25 mm), a mixture of benzene (bp = 80.1 °C) and cyclohexane (bp = 80.7 °C) was separated at 473 K with a helium flow of 4.37 mL min−1. In order to avoid overcharging effects, only the methane enriched gas phase over a mixture of these liquid analytes was injected into the gas chromatograph. As can be seen in Fig. 3, the identification of the two detected peaks is easily carried out by the comparison of their retention times with those of the pure substances. The chromatogram shows that benzene displays a higher adjusted retention time (tr′ = 0.88 min) than its completely hydrogenated analog (tr′ = 0.67 min). If the separation mechanism is mainly dominated by differing London dispersive interactions, the order of the analyte retention times follows the order of their boiling points. From the fact that this order is reversed, one can assume that a type of specific interaction, such as π–π stacking interactions between the Lewis basic π-electron rings of the HKUST-1 linker, benzene-1,3,5-tricarboxylate, and the π-electron density of the analyte benzene may be present what would not be the case with the nonaromatic cyclohexane. | ||
| Fig. 3 I – Chromatographic separation of a mixture of methane (for the determination of the dead time in all chromatograms), cyclohexane, and benzene (a) pure benzene (b), and of pure cyclohexane (c). Experimental setup: column: HKUST-1@SiO2 (30 m × 0.25 mm × 0.15 μm); carrier: He, flow: 4.37 mL min−1, 76.2 cm s−1; oven: 473.15 K, isotherm; injector: split, 11.7:1 at 250 °C; detector: FID at 250 °C. II – Chromatographic separation of a mixture of methane, diisopropyl ether, diethyl ether, tetrahydrofuran, and di-n-propyl ether (black line) A. Chromatograms of pure substances except methane. B – Di-n-propyl ether. C – Tetrahydrofuran. D – Diethyl ether. E – Diisopropyl ether. Experimental setup: column: HKUST-1@SiO2 (30 m × 0.25 mm × 0.15 μm); carrier: He, flow: 4.14 mL min−1, 72.5 cm s−1; oven: 523.15 K, isotherm; injector: split, 12:1 at 250 °C; detector: FID at 250 °C. (* – impurity). | ||
As an example demonstrating the influence of the Lewis acidity of the open metal sites of HKUST-1 on chromatographic separations, we chose ethers with different alkyl chains as Lewis basic analyte portfolio. The ether oxygen atoms with their high electron densities should be capable of interacting with the free unsaturated coordination sites of the copper ions of the SBU of the investigated stationary phase. The corresponding chromatogram of a mixture of diisopropyl ether, diethyl ether, tetrahydrofuran, and di-n-propyl ether is also shown in Fig. 3. If the ether molecules were to interact mainly via dispersive interactions, they should more or less separate according to their boiling points, which would be in the order diethyl ether → tetrahydrofuran → diisopropyl ether → di-n-propyl ether in contrast to the measured sequence of diisoproyl ether → diethyl ether → tetrahydrofuran → di-n-propyl ether. Of particular interest is the difference between tetrahydrofuran and diisopropyl ether, which have similar boiling points, i.e. 65.8 °C and 69 °C, respectively. These significant differences point to the relevance of specific interactions. Considering the geometry of the analytes and the accessibility of the free electron pair of the oxygen atoms of the different analytes for the interaction with the open metal site of the MOF-SBU, it is conceivable that the electron density of the oxygen atom of the diisopropyl group is shielded more intensively by the branched alkyl groups than in the cases of diethyl ether and tetrahydrofuran. Similarly, one can assume that the electron density of the tetrahydrofuran oxygen is also less shielded than the one of diethyl ether because of the ring structure of the THF molecule.
Thermodynamic investigation of the adsorption process on HKUST-1
The thermodynamic parameters ΔGads, ΔUdiff, ΔHiso, and ΔSads obtained from the van't Hoff plots in Fig. 4 are summarized in Table 1. Benzene displays a higher absolute value of the enthalpy of adsorption |ΔHads| than cyclohexane indicating a stronger interaction presumably because of the π–π stacking interactions between the aromatic rings of the HKUST-1 linker and the benzene molecule. Given that π–π stacking interactions are comparable weak, they are app. 8–12 kJ mol−1, the difference of 4 kJ mole appears to be in accordance with this mode of interaction.61 The absolute value of ΔSads for the adsorption of benzene on HKUST-1 is also slightly higher than for cyclohexane, which is somewhat counterintuitive given the more flexible nature of the cyclohexane molecule compared to the benzene one and the small difference in the adsorption enthalpies. If one adopts the assumption of the presence of π–π stacking interactions, the possibly very good alignment of the benzene ring of the MOF linker and the analyte may explain the observed ΔSads values. In the case of the ether separations, it is noteworthy that, although diisopropyl ether eluates earlier than diethyl ether, its enthalpy of adsorption is higher. Since the retention time is determined by the adsorption Gibbs free energy ΔGads, this effect must be caused by entropic contributions. It is comprehensible that the alkyl chains of diisopropyl ether, which are longer than the ones of diethyl ether and also branched, possess more degrees of freedom that are affected/restricted by the adsorption process. In the case of the two subsequently eluated compounds tetrahydrofuran and di-n-propyl ether, both the enthalpy and entropy differences support the observed order. | ||
| Fig. 4 Van't Hoff plots for the investigated analytes. I – Benzene: lnVN = 6155.0 K T−1 − 12.3, R2 = 0.9998. Cyclohexane: lnVN = 5527.0 K T−1 − 11.0, R2 = 0.9999. II – Diisopropyl ether: lnVN = 9260.6 K T−1 − 17.0, R2 = 0.9971. Diethyl ether: lnVN = 9064.9 K T−1 − 16.1, R2 = 0.9921. Tetrahydrofuran: lnVN = 9331.1 K T−1 − 15.3, R2 = 0.9886. Di-n-propyl ether: lnVN = 10387.0 K T−1 − 16.8, R2 = 0.9899. | ||
| −ΔHiso in kJ mol−1 | −ΔUdiff in kJ mol−1 | −ΔSads in J (K mol)−1 | −ΔGads in kJ mol−1 | |
|---|---|---|---|---|
| Cyclohexane | 47.2 | 43.2 | 95.8 | 0.7 |
| Benzene | 51.2 | 47.2 | 102.1 | 1.7 |
| Diisopropyl ether | 77.0 | 72.8 | 141.0 | 5.4 |
| Diethyl ether | 75.4 | 71.2 | 134.0 | 7.3 |
| Tetrahydrofuran | 77.5 | 73.6 | 127.3 | 12.8 |
| Di-n-propyl ether | 85.5 | 81.3 | 139.6 | 14.6 |
Determination of the specific und nonspecific interaction Gibbs free energies of ether-adsorption on HKUST-1
Conducting analyses as described in the theoretical background section for the adsorption of the above listed ethers on the HKUST-1 surface, a division of the interaction Gibbs free energy in a part which describes the specific interaction, like donor–acceptor contacts, and a part caused by nonspecific, dispersive London-interactions can be obtained. For these analyses the method by Donnet et al.,52i.e. choosing the scaled electronic polarizability (hν)0,5αO,L as the specific property, was applied. A graphic summary for the four ethers investigated is given in Fig. 1 bottom.For the estimate of the contribution of the nonspecific interaction of the ether analytes, the general relationship between the scaled electronic polarizability of molecules and nonspecific interactions with HKUST-1 was established using the unbranched alkanes n-pentane, n-hexane, and n-heptane. It is noteworthy that ΔGinteract changes to negative values for longer chains. Since both the adsorption enthalpy ΔHads and the adsorption entropy ΔSads can be expected to be negative, the gain in enthalpy (absolute value) must outweigh the contribution to ΔGinteract by the change of ΔSads caused by every additional –CH2– unit at the given temperature.
The values for |−ΔGinteract|, |−ΔGspecific|, and |−ΔGnonspecific| of the four investigated ethers at 493.15 K are compiled in Table 2. The specific Gibbs free energies of adsorption of diethyl ether and tetrahydrofuran are higher than the one of diisopropyl ether.
| −ΔGinteract in kJ mol−1 | −ΔGspec in kJ mol−1 | −ΔGnonspec in kJ mol−1 | |
|---|---|---|---|
| Diisopropyl ether | 7.3 | 5.2 | 2.4 |
| Diethyl ether | 9.1 | 12.3 | −3.2 |
| Tetrahydrofuran | 14.6 | 23.5 | −8.9 |
| Di-n-propyl ether | 16.6 | 13.6 | 2.8 |
According to Table 2, the eluation order of the first three compounds, which is given by |−ΔGinteract|, is mainly determined by the specific interaction whereas the nonspecific part by itself would cause a reversed order. If one considers the geometric factors of the analysts, the result can be interpreted in the way that THF has a more exposed, i.e. less shielded by side chains, oxygen atom that can therefore more strongly interact with the open metal sites. In addition, this adsorption process is also entropically supported since the ring is already strongly restricted in its motion compared to the freely moving side chains of diethyl ether and diisopropyl ether. Hence, the adsorption process is accompanied with a rather low entropy change. In respect to the sequence of the first three eluated compounds, di-n-propyl breaks the trend of increasing specific interaction and decreasing nonspecific interaction. The specific enthalpy of adsorption for all ethers is app. between 50–100% of the nonspecific one (see Table 3). Due to the lack of data on the interaction of HKUST-1 with other oxygen containing organic molecules, one can only compare the interaction strength with small gas molecules such as water (51 kJ mol−1), CO2 (20 kJ mol−1), and CO (24 kJ mol−1),61 which is in the same range.
| −ΔHspec in kJ mol−1 | −ΔSspec in kJ mol−1 | |
|---|---|---|
| Diisopropyl ether | 21.1 | −32.1 |
| Diethyl ether | 30.6 | −37.0 |
| Tetrahydrofuran | 39.7 | −32.6 |
| Di-n-propyl ether | 28.6 | −30.1 |
Determination of the donor–acceptor character of the HKUST-1 surface
A quantitative measure for the donor and acceptor properties, i.e. the acid base characteristics, of the surface of the stationary phase, here HKUST-1, can be gained by determining the acid constant KA and base constant KB. In order to calculate these constants, the specific enthalpy of adsorption and the donor and acceptor numbers of Gutmann must be known.50,59 Only for benzene, diethyl ether, and tetrahydrofuran we found corresponding values in the literature and thus the analysis had to be limited to these compounds. Using the Gibbs–Helmholtz equation directly or a van't Hoff analysis, the specific enthalpy and entropy of adsorption can be determined by a linear fitting procedure with respect to temperature of −ΔGspec. The values of −ΔGspec, −ΔHspec, and −ΔSspec for benzene calculated from the interaction Gibbs free energy of adsorption in the temperature range from 493.15 K to 523.15 K are given in the ESI.† It is important to note that the split of −ΔGspec into the two different parts, −ΔHspec and −ΔSspec, via temperature dependent measurements cures the problem of potential entropy contamination between the reference compounds and the investigated analytes. The term entropy contamination is supposed to indicate that although it can be assumed that the nonspecific interaction enthalpy scales generically with the specific property, here the polarizability, this cannot necessarily be assumed for the entropy. Therefore the particular adsorption entropies of the reference molecules influence the split of −ΔGinteract into −ΔGspec and −ΔGnonspec. By subsequently splitting −ΔGspec into −ΔHspec and −ΔSspec the adsorption entropy of the reference molecules will only affect the determined value of −ΔSspec, and will leave −ΔHspec unaffected. As a consequence, the determination of KA and KB will be free of entropy contamination effects. With the values −ΔHspec and −ΔSspec of benzene, diethyl ether, and tetrahydrofuran and the corresponding acceptor and donor numbers (see ESI†), the constants KA and KB are available, as discussed in the theoretical section, from the corresponding linear fit, which is graphically depicted in Fig. 5. A value for KA of 1.6 (in arbitrary units) describing the acceptor properties of the HKUST-1 surface and a value for KB of 0.4 (in corresponding units) for the donor properties were obtained. From these numbers, one has to conclude that HKUST-1 has to be overall classified as a predominantly Lewis acidic MOF due to the presence of the coordinatively unsaturated open metal sites. Even if the result may perhaps intuitively be predictable, the investigation points to a means that also the quantitative comparison between different MOF materials becomes possible. Although these comparative studies are still rare, the importance of open metal sites for possible applications in catalysis and separations have been demonstrated and thus the importance of the tuning of the Lewis acidity of open metal sites is evident. An example for such a comparative study is given by the investigation of 8 open metal site MOFs in respect nitrogen and sulphur based fuel contaminant uptake by the group of De Vos.34b The study indicates that MIl-100 (Fe, Cr, Al) binds nitrogen based contaminants better than HKUST-1 while in the case sulphur ones the opposite appears to be true. It is therefore difficult to rank the MOFs in respect to their Lewis acidity. In addition, the results also proved to be very solvent dependent. In this respect, the presented IGC method displays itself, given the use of noble gases as analyte carriers, as a more general means for the determination of the Lewis acidity and basicity of MOFs and will allow to quantify subtle differences of MOF materials properties. | ||
Fig. 5 a – Determination of the specific adsorption enthalpy and the corresponding entropy from the Gibbs–Helmholtz equation according to the linear regression of −ΔGspecvs. T for the analytes benzene ( ), diethyl ether ( ), diethyl ether ( ), and tetrahydrofuran ( ), and tetrahydrofuran ( ). For the analytical functions see ESI.† b – Determination of the acidity and basicity in terms of KA and KB. DN(benzene) = 0.1; DN(tetrahydrofuran) = 20.0; DN(diethyl ether) = 19.2; AN(benzene) = 8.2; AN(tetrahydrofuran) = 8.0; AN(diethyl ether) = 3.9. Linear regression: −ΔHspec/AN = 1.6 DN/AN + 0.4 (in arbitrary units according the approach of Schultz et al.). ). For the analytical functions see ESI.† b – Determination of the acidity and basicity in terms of KA and KB. DN(benzene) = 0.1; DN(tetrahydrofuran) = 20.0; DN(diethyl ether) = 19.2; AN(benzene) = 8.2; AN(tetrahydrofuran) = 8.0; AN(diethyl ether) = 3.9. Linear regression: −ΔHspec/AN = 1.6 DN/AN + 0.4 (in arbitrary units according the approach of Schultz et al.). | ||
Conclusion
Beside the continued demonstration that HKUST-1 is a suitable material for gas chromatographic applications, the influence of donor and acceptor areas of the MOF-lattice on the adsorption of different analytes was studied using inverse gas chromatography. The determination of a London force governed reference system and the determination of the so called interaction Gibbs free energies of a set of analytes allowed the separation of their interactions into nonspecific and specific contributions. An analysis based on the use of the Gutmann donor and acceptor numbers of a specific analyte portfolio allowed a quantitative judgement of the Lewis acidity and basicity of the MOF material HKUST-1. The analysis applied to HKUST-1 can be seen as a procedure that can be used for the design of MOF materials with intentionally modified properties.Parameter definitions
(See ESI.†)Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft is gratefully acknowledged (Priority Program 1362 “Porous Metal–Organic Frameworks”). A. M. thanks Stephan Glante for his experimental help with the chromatographic measurements of the ethers.Notes and references
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191 RSC.
- A. Carné-Sanchez, I. Imaz, K. C. Stylianou and D. Maspoch, Chem. – Eur. J., 2014, 20, 5192 CrossRef PubMed.
- M. Zhang, M. Bosch, T. Gentle III and H.-C. Zhou, CrystEngComm, 2014, 16, 4069 RSC.
- L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248 RSC.
- M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294 RSC.
- R. B. Getman, Y.-S. Bae, C. E. Wilmer and R. Q. Snurr, Chem. Rev., 2011, 112, 703 CrossRef PubMed.
- M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2011, 112, 782 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2011, 112, 724 CrossRef PubMed.
- M. Shah, M. C. McCarthy, S. Sachdeva, A. K. Lee and H.-K. Jeong, Ind. Eng. Chem. Res., 2012, 51(5), 2179 CrossRef CAS.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- L. Alaerts, C. E. A. Kirschhock, M. Maes, M. A. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. E. M. Denayer and D. E. De Vos, Angew. Chem., 2007, 119, 4371 ( Angew. Chem., Int. Ed. , 2007 , 46 , 4293 ) CrossRef.
- L. Alaerts, M. Maes, P. A. Jacobs, J. E. M. Denayer and D. E. De Vos, Phys. Chem. Chem. Phys., 2008, 10, 2979 RSC.
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. F. Clancy, L. B. Lobovsky, O. M. Yaghi and S. Dai Poole, Angew. Chem., 2006, 118, 1418 ( Angew. Chem., Int. Ed. , 2006 , 45 , 1390 ) CrossRef.
- Z.-Y. Gu and X.-P. Yan, Angew. Chem., Int. Ed., 2010, 49, 1477 CrossRef CAS PubMed.
- A. S. Münch, J. Seidel, A. Obst, E. Weber and F. O. R. L. Mertens, Chem. – Eur. J., 2011, 17, 10958 CrossRef PubMed.
- A. S. Münch and F. O. R. L. Mertens, J. Mater. Chem., 2012, 22, 10228 RSC.
- N. Chang and X.-P. Yan, J. Chromatogr. A, 2012, 1257, 116 CrossRef CAS PubMed.
- N. Chang, Z.-Y. Gu and X.-P. Yan, J. Am. Chem. Soc., 2010, 132, 13645 CrossRef CAS PubMed.
- Z.-Y. Gu, J.-Q. Jiang and X.-P. Yan, Anal. Chem., 2011, 83, 5093 CrossRef CAS PubMed.
- L. Fan and X.-P. Yan, Talanta, 2012, 99, 944 CrossRef CAS PubMed.
- I. Gutiérrez, E. Díaz, A. Vega and S. Ordóñez, J. Chromatogr. A, 2013, 1274, 173 CrossRef PubMed.
- P. Mukhopadhyay and H. P. Schreiber, Colloids Surf., A, 1995, 100, 47 CrossRef CAS.
- F. Thielmann, J. Chromatogr. A, 2004, 1037, 115 CrossRef CAS PubMed.
- A. Voelkel, B. Strzemiecka, K. Adamska and K. H. Milczewska, J. Chromatogr. A, 2009, 1216, 1551 CrossRef CAS PubMed.
- C. F. Poole, in Gas Chromatography, Elsevier, Oxford, U. K., 1st edn, 2012, pp. 477–494 Search PubMed.
- K. Schlichte, T. Kratzke and S. Kaskel, Microporous Mesoporous Mater., 2004, 73, 81 CrossRef CAS PubMed.
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520 CrossRef CAS.
- R. Ahmad, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2009, 25, 11977 CrossRef CAS PubMed.
- (a) L. Alaerts, E. Séguin, H. Poelman, F. Thibault-Starzyk, P. A. Jacobs and D. E. De Vos, Chem. – Eur. J., 2006, 12, 7353 CrossRef CAS PubMed; (b) M. Meas, M. Trekels, M. Boulhout, S. Schouteden, F. Vermoortele, L. Alaerts, D. Heurtaux, Y.-K. Seo, Y. K. Hwang, J.-S. Chang, I. Beurroies, R. Denoyel, K. Temst, A. Vantomme, P. Horcajada, C. Serre and D. E. De Vos, Angew. Chem., Int. Ed., 2011, 50, 4210 CrossRef PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148 CrossRef CAS.
- D. Peralta, G. Chaplais, A. Simon-Masseron, K. Barthelet, C. Chizallet, A.-A. Quoineaud and G. D. Pirngruber, J. Am. Chem. Soc., 2012, 134, 8115 CrossRef CAS PubMed.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337 CrossRef CAS.
- M. Nishio, Y. Umezawa, K. Honda, S. Tsuboyama and H. Suezawa, CrystEngComm, 2009, 11, 1757 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885 RSC.
- F. London, Z. Phys., 1930, 63, 245 CrossRef CAS.
- P. J. W. Debye, Phys. Z., 1920, 21, 178 CAS.
- W. H. Keesom, Phys. Z., 1921, 22, 129 CAS.
- J. R. Conder and C. L. Young, in Physicochemical Measurements by Gas Chromatography, John Wiley & Sons, Chichester, 1979 Search PubMed.
- D. R. Lloyd, T. C. Ward and H. P. Schreiber, in Inverse Gas Chromatography-Characterization of Polymers and Other Materials (ACS Symposium Series No. 391), American Chemical Society, Washington, DC, 1989 Search PubMed.
- N. A. Katsanos, A. Lycourghiotis and A. Tsiatsios, J. Chem. Soc., Faraday Trans. 1, 1978, 74, 575 RSC.
- N. A. Katsanos, R. Thede and F. Roubami-Kalantzopoulou, J. Chromatogr. A, 1998, 795, 133 CrossRef CAS.
- S. A. Greene and H. Pust, J. Phys. Chem., 1958, 62, 55 CrossRef CAS.
- A. T. James and A. J. P. Martin, Biochem. J., 1952, 50, 679 CAS.
- F. M. Fowkes, J. Phys. Chem., 1962, 66, 382 CrossRef CAS.
- J. Schultz, L. Lavielle and C. Martin, J. Adhes., 1987, 23, 45 CrossRef CAS.
- C. M. Dorris and D. G. Gray, J. Colloid Interface Sci., 1980, 77, 353 CrossRef.
- J. B. Donnet, S. J. Park and H. Balard, Chromatographia, 1991, 31, 434 CAS.
- C. Saint Fluor and E. Papirer, J. Colloid Interface Sci., 1983, 91, 69 CrossRef.
- D. T. Sawyer and D. J. Brookman, Anal. Chem., 1968, 40, 1847 CrossRef CAS.
- S. Dong, M. Brendlé and J. B. Donnet, Chromatographia, 1989, 28, 469 CAS.
- E. Brendlé and E. Papirer, J. Colloid Interface Sci., 1997, 194, 207 CrossRef.
- T. M. Miller, in CRC Handbook of Chemistry and Physics, ed. W. M. Haynes, CRC Press, Boca Raton, 93rd edn, 2012, ch. 10, pp. 187–196 Search PubMed.
- K. J. Miller and J. A. Savchik, J. Am. Chem. Soc., 1979, 101, 7206 CrossRef CAS.
- V. Gutmann, in The Donor-Acceptor Approach to Molecular Interactions, Plenum Press, New York, 1987 Search PubMed.
- A. S. Münch, M. S. Lohse, S. Hausdorf, G. Schreiber, D. Zacher, R. A. Fischer and F. O. R. L. Mertens, Microporous Mesoporous Mater., 2012, 159, 132 CrossRef PubMed.
- M. O. Sinnokrot and C. D. Sherrill, J. Phys. Chem. A, 2006, 110, 10656–10668 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental details, retention times, linear regression functions, determined enthalpies, entropies, and other quantities are listed in the ESI. The characterization of HKUST-1 used for the adsorption experiments by PXRD, BET is also described in the ESI. See DOI: 10.1039/c4ce01327a |
| This journal is © The Royal Society of Chemistry 2015 |