
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBase catalysed decomposition of anthracene endoperoxide†
M.
Klaper
,
P.
Wessig
and
T.
Linker
*
Department of Chemistry, University of Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: linker@uni-potsdam.de
First published on 20th November 2015
Abstract
Catalytic amounts of a weak base are sufficient to induce the decomposition of anthracene endoperoxides to anthraquinone. The mechanism has been elucidated by isolation of intermediates in combination with DFT calculations. The whole process is suitable for the convenient generation of hydrogen peroxide under very mild conditions.
Endoperoxides 2 (EPOs)1 play an important role in organic chemistry,2 mechanistic studies,3 material sciences,4 and drug synthesis5 as well as in medicine6 and under physiological conditions.7 While nature has developed particular enzymes like prostaglandin-endoperoxide synthase,8 they can easily be synthesised via photooxygenation of the corresponding diene-system 1 with singlet oxygen (1O2) in the laboratory.9 EPOs are of current interest since they are under discussion for the use in photodynamic therapy (PDT) of cancer.10 Although their synthesis is well understood, the decomposition mechanism is still a matter of debate and various pathways have been proposed (Scheme 1).
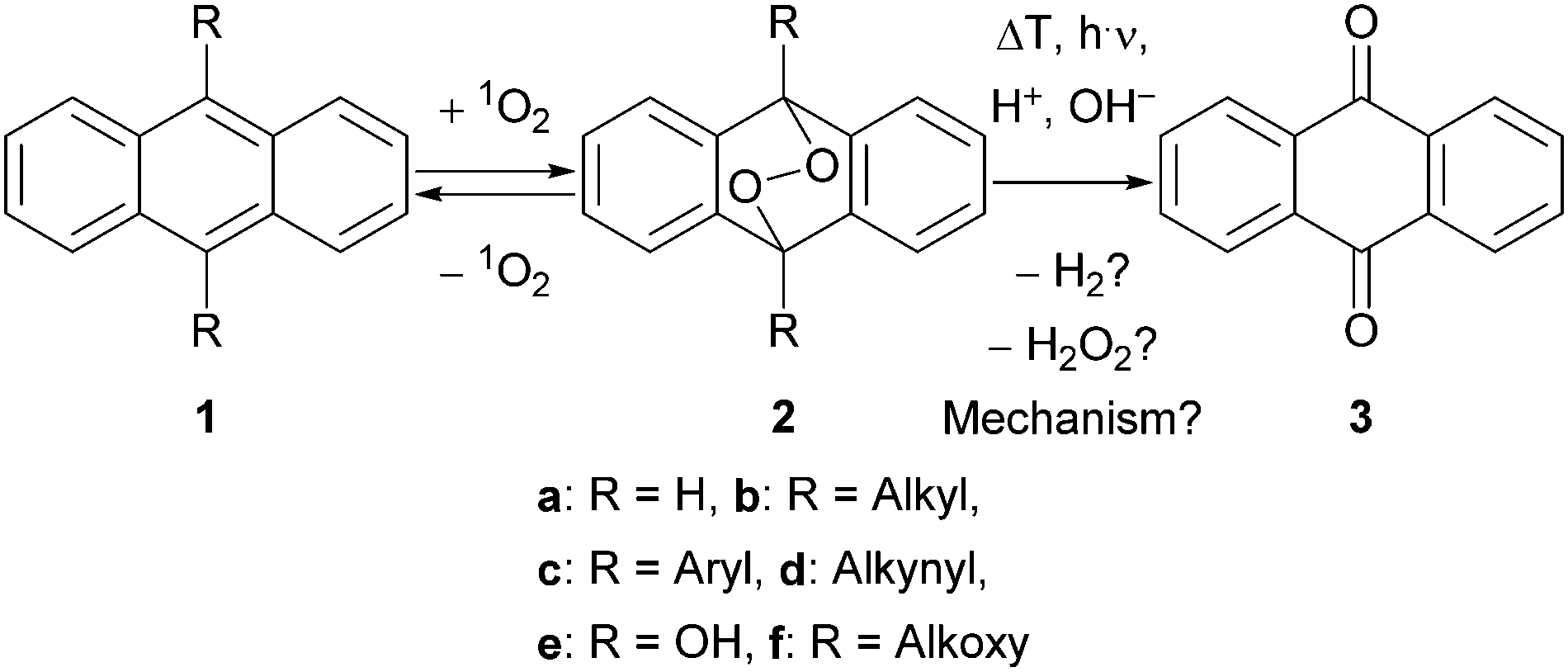 | ||
| Scheme 1 Photooxygenation of acenes 1 and the formation of anthraquinone (3). | ||
Thus, for R = Aryl (2c) the addition of 1O2 is completely reversible,11 which we applied in material sciences4 and for molecular rotors,12 before. If R = Alkynyl (2d), this cleavage pathway is even faster.13 On the other hand, for R = OH (2e) the formation of anthraquinone (AQ, 3) and hydrogen peroxide (H2O2) is obvious due to the hemi acetal structure. Intermediates like EPO 2e are important in the industrial production of H2O2 by the anthraquinone process as well.14 Furthermore, alkoxy substituted EPOs 2f are known to be unstable and hence applied to release caged (bio)molecules and as 1O2-sensitive linkers.15
However, the mechanism of the cleavage of alkyl- and unsubstituted EPOs 2b and 2a is much more complicated. Hence, many rearrangement products have been found under thermal conditions.16 Even more difficult is the explanation of AQ (3) formation at room temperature, which we often observe during workup and column chromatography. To clarify this interesting mechanism, Bendikov as well as González and Corral independently performed theoretical calculations from ground or excited states, respectively;17 a homolytic cleavage of the O–O bond was proposed with subsequent elimination of hydrogen gas (H2). However, this O–O-cleavage should be only possible at higher temperatures or under irradiation, due to a barrier of approx. 23 kcal mol−1.17a Furthermore, we never observed the formation of any gas under our conditions.
In contrast, a very conclusive paper, which addresses the photooxygenation of anthracene as well as the consecutive formation of AQ (3), both steps are explained by a photo-induced electron transfer18 together with the formation of hydrogen peroxide (H2O2) as a side product. But since the formation of AQ (3) can also be observed in the dark and without any sensitizer, we presume that the degradation was caused by a change in the pH-value due to irradiation. However, the observation of hydrogen peroxide formation is very important and in accordance with the commercial anthraquinone process.14 Furthermore, Heyne et al. investigated the degradation of 2a under photochemical conditions, leading to (a) 1a and 1O2 or (b) rearrangement products after homolytic O–O cleavage.19
Our group is working on differently substituted anthracene EPOs for many years now3,4,20 and we observed their degradation frequently along with the formation of AQ (3). Since the theoretically proposed formation of hydrogen gas,17 the photo-induced electron transfer18 or photochemical degradations19 are strongly sensitive towards the applied conditions and we could strictly exclude electron transfer or irradiation below 400 nm (maximum absorption range of 2a),19 we started a detailed mechanistic study towards the undesirable decomposition of EPO 2a as model compound.
Herein, we present our results from various experiments under aerobic and anaerobic conditions in combination with DFT calculations. We provide a conclusive mechanistic picture, which explains the formation of AQ (3) from EPO 2a under release of hydrogen peroxide. Thus, our results should not only be of general mechanistic interest, but could offer opportunities for new sources of reactive oxygen species (ROS) under very mild conditions; even physiological ones.
The EPO 2a (R = H)21 is accessible from the corresponding anthracene 1a in quantitative yield at −78 °C and is stable for weeks under ambient light, atmosphere and temperature after careful isolation. Thus, an unintentional decomposition can be ruled out. At the beginning, we initiated the degradation by acids; here stoichiometric amounts of sulfuric acid were necessary to obtain full conversion to AQ (3) (Scheme 2) in Schlenck equipment (ESI†).
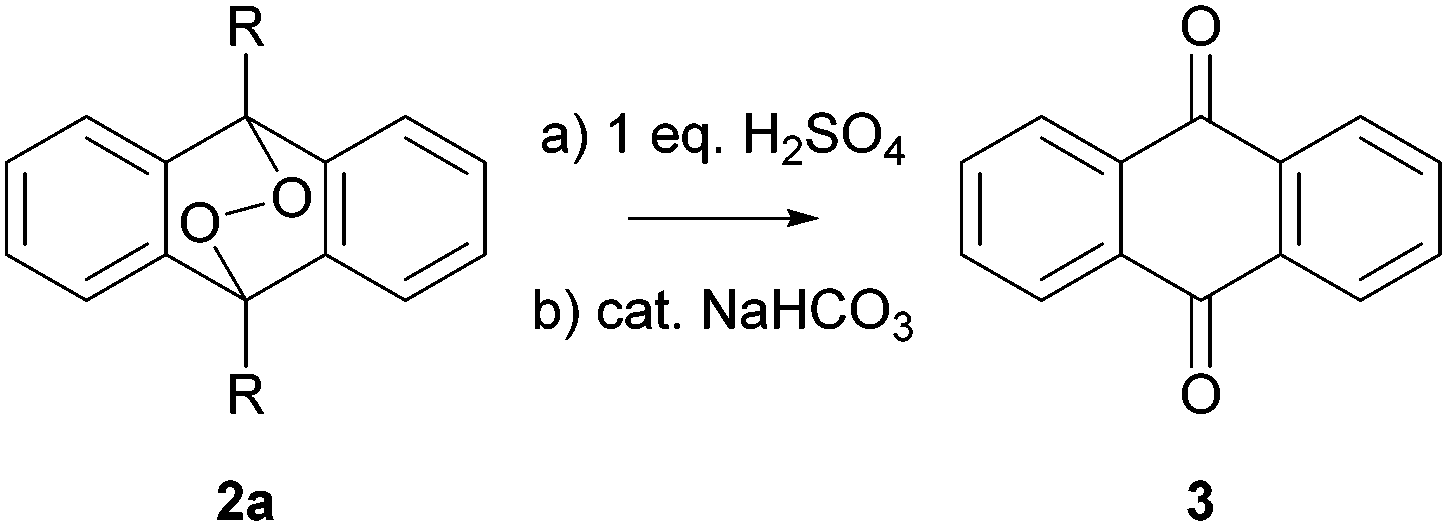 | ||
| Scheme 2 Acidic and basic decomposition of EPO 2a. | ||
On the other hand, bases like potassium hydroxide, sodium hydroxide, 1,8-diazabicycloundec-7-ene (DBU), triethyl amine (NEt3), potassium carbonate, sodium carbonate and even catalytic amounts of sodium bicarbonate afforded AQ (3) quantitatively. The reaction speed increases dramatically after a short induction period (∼30 s) and the whole transformation to AQ (3) is completed within 5 minutes. Obviously, Schlenck equipment was not sufficient to completely exclude oxygen in this process. Therefore, we transferred all reagents and equipment into a glove box, so that no oxygen could interfere. Here, we investigated the reactions with bases only since they gave clean reactions under catalytic conditions and are hence more prone to be responsible for the often observed formation of AQ (3) during workup. Additionally, we more focus on the inorganic bases since amine bases are known to act as weak reducing agents.22
In fact, when the reactions were repeated in the glove box in deuterated and carefully deoxygenated solvents, we observed a slow reaction of 2a with catalytic amounts of the above mentioned water free bases in dichloromethane-d2 (DCM-d2) or chloroform-d (CDCl3) within minutes, whereas the reaction in dimethyl sulfoxide (DMSO-d6) was very fast with a strong change in colour to deep red within seconds. On the other hand, we dissolved EPO 2a in pure DMSO-d6 for a blank test and observed no reaction taking place even after days; the EPO 2a did not decompose also after the addition of water.
In the NMR spectrum, both tautomers 4 and 5 of the first reaction cycle were detected in different compositions, depending on the solvent (Scheme 3). Both forms have been proposed in literature before to be cleavage products of 2a, but satisfying analytical data were never provided starting from the EPO 2a.18,23 Thus, we analysed this labile intermediate properly for the first time. We conclude this is due to the extreme oxidation-sensitivity of the tautomer 5.
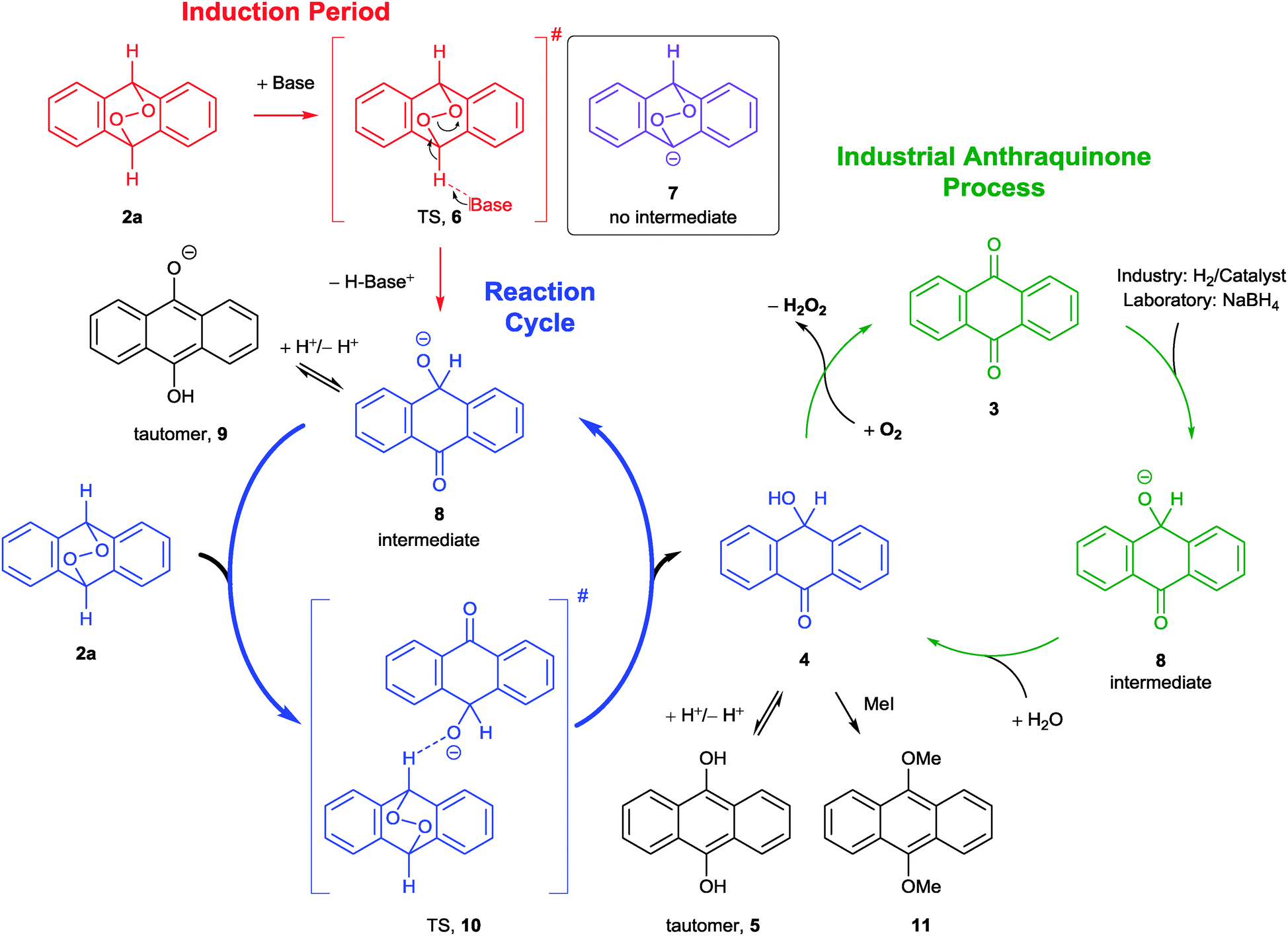 | ||
| Scheme 3 Mechanism for the base catalysed decomposition of endoperoxide 2a. | ||
We could prove this assumption by simply opening the NMR tubes; the red colour vanished very fast and AQ (3) was observed afterwards. Additionally, we could rule out 9,10-dihydro-9,10-dihydroxy anthracene as a decomposition product, proposed in an earlier photo cleavage study,24 by comparison with original material synthesised from 2a and thiourea (ESI†).25
Interestingly, p-hydroxy anthrone (4) was also postulated as an intermediate in the anthraquinone process, but has never been isolated.14 Consecutively, we repeated the reduction of AQ (3) with sodium borohydride (Scheme 3, green). While no reaction was observed in DCM-d2 or CDCl3, we found the same intermediates as in the case of the basolysis of the EPO 2a in DMSO-d6. Furthermore, in all cases an addition of trifluoroacetic acid-d1, hydrochloric acid (DCl in D2O or HCl in H2O),26 or methyl iodide (MeI) led quantitatively to green fluorescent 5 or 9,10-dimethoxy anthracene (11), respectively.
Since catalytic amounts of base are sufficient for full conversion of 2a, we propose an autocatalytic mechanism taking place (Scheme 3), which starts similar to a Kornblum–DeLaMare rearrangement.27 During the induction period (Scheme 3, red), a catalytic amount of base attacks at 2a abstracting one proton to form the anion 8 (Scheme 3, blue) over the transition state 6 (TS, red). Compound 7 (purple) is not the intermediate, since it is no stationary point on the potential energy surface (PES). Thus, the reaction runs in a SN2 manner directly through to the heterolytically opened anion 8, which is indeed an intermediate on the PES. The driving force of this reaction is the formation of a C–O double bond and a stable benzophenone structure at the expense of a weak O–O bond (for details see ESI†). Beyond this, the benzyl alcoholate 8 exists in equilibrium with the tautomeric form 9, which explains the deep red colour. During the intermolecular diffusion controlled reaction cycle, the intermediate 8 attacks another endoperoxide starting material 2a to form 10 which was found to be a second transition state on the PES in quantum chemical calculations. Then the TS 10 breaks down into p-hydroxy anthrone (4) and another anion 8. The p-hydroxy anthrone (4) is now in a solvent dependent equilibrium with 9,10-dihydroxy anthracene (5),23g,h comparable to the anthrone–anthranol–equilibrium.28
The energy difference of 4 and its constitution isomer 2a amounts to approximately −77 kcal mol−1 (ESI†), which should be the driving force of this very fast reaction. We were able to record the absorption, excitation and fluorescence spectra of the tautomer 5 (ESI†), which are consistent with literature data.29 Both of these molecules are extremely oxidation-sensitive and form AQ (3) and hydrogen peroxide in the presence of oxygen. After opening the sealed fluorescence cuvette, we observed a very fast disappearance of the fluorescence signal with a half-life time of 2.4 min (Fig. 1).
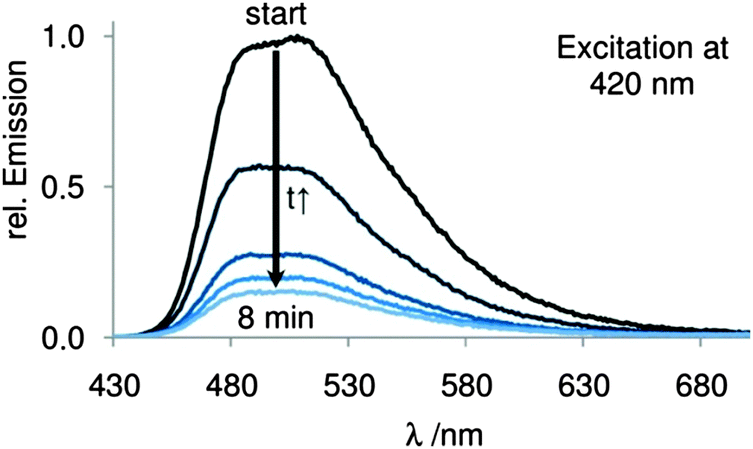 | ||
| Fig. 1 Fluorescence spectra of 9,10-dihydroxy anthracene (5) before (start) and after exposure to oxygen with chronological progress. | ||
By performing the reduction of AQ (3) with NaBH4 in the glove box, we could demonstrate that 4 has to be the intermediate in the industrial anthraquinone process as well (Scheme 3, green). The arising H2O2 from both reaction types (basolysis and reduction) was detected via several chemical and biomimetic methods (ESI†).30 We did not rely on the detection with potassium iodide, exclusively, since this reaction is not selective and EPOs show the same result.31
With this mechanism we can reasonably explain the formation of AQ (3) during the decomposition of EPO 2a. Even more, only a catalytic amount of the very weak base NaHCO3 is sufficient for this reaction, which is frequently used during workup and could illuminate the often observed unintentional formation of AQ (3). Furthermore, we verified the assumed intermediates 4 and 5, which are in accordance with comparable literature data.23,29 We were able to substantiate these results with the help of quantum chemical calculations. We found that the anion 7 lies too high on the PES to be the intermediate; instead we could show that the directly opened form 8, which exists in equilibrium with the tautomeric form 9, is the true intermediate for this step while 6 is the transition state for the first reaction cycle.
The overall reaction proceeds diffusion controlled and autocatalysed via the second transition state 10.
In conclusion, we could clarify the mechanism of the undesirable anthracene EPO decomposition under standard laboratory or work-up conditions, which proceeds even in the presence of catalytic amounts of sodium bicarbonate. Additionally, this type of EPO could be a very mild and convenient source of ROS, since it is long time storable at room and slightly elevated temperature. Furthermore, the decomposition and release of hydrogen peroxide can be triggered under extreme mild basic – possibly even physiological – conditions. In addition, this protocol can synergistically benefit from the formation of anthraquinone (3) as single byproduct, which is a structural motive in potent anti-cancer agents.32 Thus, EPOs of the type 2a should gain special interest in the field of cancer treatment by both, the release of reactive oxygen species and the formation of AQ (3).
We thank the University of Potsdam for generous financial support, Dr Heidenreich and Dr Starke for assistance in analyses and Dr Fudickar for helpful discussions.
Notes and references
-
(a)
W. Fudickar and T. Linker, PATAIS Chemistry of Functional Groups, John Wiley & Sons Ltd, Chichester, UK, 2014 Search PubMed
; (b) W. A. Pryor, Organic Free Radicals, American Chemical Society, Washington D.C., USA, 1978 Search PubMed
-
(a) B. K. Ohta and C. S. Foote, J. Am. Chem. Soc., 2002, 124, 12064–12065 CrossRef CAS PubMed
-
(a) M. Klaper and T. Linker, Chem. – Eur. J., 2015, 21, 8569–8577 CrossRef CAS PubMed
-
(a) W. Fudickar and T. Linker, Langmuir, 2010, 26, 4421–4428 CrossRef CAS PubMed
-
(a) H.-J. Chen and Y. Wu, Org. Lett., 2015, 17, 592–595 CrossRef CAS PubMed
-
(a) S. M. N. Efange, R. Brun, S. Wittlin, J. D. Connolly, T. R. Hoye, T. McAkam, F. L. Makolo, J. A. Mbah, D. P. Nelson, K. D. Nyongbela and C. K. Wirmum, J. Nat. Prod., 2009, 72, 280–283 CrossRef CAS PubMed
-
(a) C. L. Bos, D. J. Richel, T. Ritsema, M. P. Peppelenbosch and H. H. Versteeg, Int. J. Biochem. Cell Biol., 2004, 36, 1187–1205 CrossRef CAS PubMed
- R. G. Salomon, Acc. Chem. Res., 1985, 18, 294–301 CrossRef CAS
-
(a)
A. A. Frimer, Singlet Oxygen Vol. I. Physical-Chemical Aspects, CRC Press, Boca Raton, 1985 Search PubMed
-
(a) C. Pierlot, S. Hajjam, C. Barthélémy and J.-M. Aubry, J. Photochem. Photobiol., B, 1996, 36, 23–29 CrossRef
- J.-M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS PubMed
-
(a) D. Zehm, W. Fudickar and T. Linker, Angew. Chem., Int. Ed., 2007, 46, 7689–7692 CrossRef CAS PubMed
-
(a) W. Fudickar and T. Linker, J. Am. Chem. Soc., 2012, 134, 15071–15082 CrossRef CAS PubMed
-
(a) J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed
-
(a) A. Meyer and A. Mokhir, Angew. Chem., Int. Ed., 2014, 53, 12840–12843 CrossRef CAS PubMed
-
(a) J. Rigaudy, A. Defoin and J. Baranne-Lafont, Angew. Chem., Int. Ed., 1979, 18, 413–415 CrossRef
-
(a) A. R. Reddy and M. Bendikov, Chem. Commun., 2006, 1179–1181 RSC
- H. Kotani, K. Ohkubo and S. Fukuzumi, J. Am. Chem. Soc., 2004, 126, 15999–16006 CrossRef CAS PubMed
-
(a) A. Lauer, A. L. Dobryakov, S. A. Kovalenko, H. Fidder and K. Heyne, Phys. Chem. Chem. Phys., 2011, 13, 8723–8732 RSC
-
(a) W. Fudickar and T. Linker, Aust. J. Chem., 2013, 67, 320–327 CrossRef
- C. J. Brown and M. Ehrenberg, Acta Crystallogr., 1984, 40, 1059–1060 Search PubMed
-
(a) I. P. Beletskaya and A. V. Cheprakov, Chem. Rev., 2000, 100, 3009–3066 CrossRef CAS PubMed
-
(a) S. G. Semenov and M. V. Makarova, Russ. J. Gen. Chem., 2014, 84, 843–847 CrossRef CAS
- M. E. Sigman, S. Zingg, R. M. Pagni and J. H. Burns, Tetrahedron Lett., 1991, 32, 5737–5740 CrossRef CAS
- A. C. Spivey, C. G. Mañas and I. Mann, Chem. Commun., 2005, 4426–4428 RSC
- K. H. Meyer, Justus Liebigs Ann. Chem., 1911, 379, 37–78 CrossRef CAS
-
(a) N. Kornblum and H. E. DeLaMare, J. Am. Chem. Soc., 1951, 73, 880–881 CrossRef CAS
- Y. Ogata, Y. Kosugi and K. Nate, Tetrahedron, 1971, 27, 2705–2711 CrossRef CAS
-
(a) T. C. Werner, J. Chang and D. M. Hercules, J. Am. Chem. Soc., 1970, 92, 763–768 CrossRef CAS
-
(a) Y. Jiang, W. Wang, X. Li, X. Wang, J. Zhou and X. Mu, ACS Appl. Mater. Interfaces, 2013, 5, 1913–1916 CrossRef CAS PubMed
- A. Cimato, G. Facorro, F. Aquirre, A. Hager, T. De Paoli, J. Ihlo, H. A. Farach and C. P. Poole Jr., Spectrochim. Acta, Part A, 1998, 54, 2001–2008 CrossRef
- Q. Huang, G. Lu, H.-M. Shen, M. C. Chung and C. N. Ong, Med. Res. Rev., 2007, 27, 609–630 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterization data, NMR spectra, absorption spectra, fluorescence spectra and theoretical calculations. See DOI: 10.1039/c5cc08606j |
| This journal is © The Royal Society of Chemistry 2016 |