
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular iron complexes as catalysts for selective C–H bond oxygenation reactions
A. C.
Lindhorst
,
S.
Haslinger
and
Fritz E.
Kühn
*
Chair of Inorganic Chemistry/Molecular Catalysis, Technische Universität München (TUM), Department of Chemistry/Catalysis Research Center, Lichtenbergstr. 4, D-85747 Garching bei München, Germany. E-mail: fritz.kuehn@ch.tum.de; Fax: +49 89 289 13473; Tel: +49 89 289 13096
First published on 8th October 2015
Abstract
The selective oxygenation of C–H bonds is a promising and interesting task for both academia and chemical industry. Inspired by the efficiency and selectivity of naturally occurring enzymes, iron heme and nonheme complexes have proven to be valuable candidates for the development of environmentally friendly catalysts as alternative to traditional systems. This feature article summarises developments of the last decade regarding oxygenation reactions of aliphatic and aromatic substrates with various oxidants catalysed by molecular iron complexes. With a special focus on the catalytic performance of the systems, both the potential and the limitations as well as mechanistic aspects are discussed in some detail.
 A. C. Lindhorst | Anja C. Lindhorst received her BSc (2011) and MSc (2013) in chemistry from the Technische Universität München under the guidance of Peter Schieberle. In 2014 she joined the group of Fritz E. Kühn as a PhD student supported by the Fonds der Chemischen Industrie. Her research interests focus on the application of Fe NHC complexes in oxidation catalysis. |
 S. Haslinger | Stefan Haslinger graduated from Technische Universität München (TUM) as BSc (2010) and MSc (2012) in chemistry under the guidance of Bernhard Rieger. He worked on research projects in the field of homogeneous catalysis at Argonne National Laboratory, Illinois, as well as in Singapore and joined the group of Fritz E. Kühn at TUM as a PhD student in 2012. He co-authored several articles on the coordination chemistry and the electronic manipulation of Fe NHC complexes and their application in oxidation catalysis. |
 Fritz E. Kühn | Fritz E. Kühn studied chemistry at the Technische Universität München (TUM) and obtained his PhD in 1994. After a Humboldt postdoctoral fellowship at Texas A&M University, and a “Habilitation” in Munich, he became “Privatdozent” at the TUM in 2000. He was then appointed Principal Researcher at the Instituto Tecnologico e Nuclear (ITN) in Sacavem. Since 2006, he is Professor of Molecular Catalysis at TUM and since 2007 also Acting Chair of Inorganic Chemistry. His research areas are organometallic chemistry and catalysis. Fritz E. Kühn has published ca. 400 research articles, reviews and book contributions. His current h-index is 50. |
1. Introduction
The selective oxidative functionalization of hydrocarbons is a fundamental process in nature and holds high significance for the chemical industry.1–3 Raw materials obtained from crude oil and natural gas are converted to bulk chemicals and substrates for fine chemical synthesis. Technical processes often involve harsh reaction conditions and require reagents and oxidants in stoichiometric amounts.4 Moreover, difficulties posed by the inherent chemical inertness of the starting materials can result in the accumulation of mostly unwanted by-products. To render these processes more environmentally friendly and atom efficient, mild reaction conditions and the use of non-toxic oxidants such as dioxygen (preferably directly from air) are desirable.5In nature, the oxidation of alkanes and arenes is mediated by both heme and nonheme metalloenzymes.6–9 They exhibit excellent selectivity and operate under mild reaction conditions. Many of them are based on active iron sites with the most extensively studied examples belonging to the Cytochrome P450 (Cyt P450) superfamily, which enable the hydroxylation of a plethora of substrates employing dioxygen as the terminal oxidant.10,11 Their active site contains an FeIII porphyrin cofactor, which is coordinated axially by a cysteinate residue of the protein backbone, leaving a trans coordination site for the binding of O2 (Fig. 1).11 In nonheme enzymes the iron centre is coordinated by amino acid functionalities to form the active site. Prominent examples, capable of mediating the oxidation of C–H bonds are the methane monooxygenase (MMO), the Rieske dioxygenases, and the aromatic amino acid hydroxylases (Fig. 1). A common mechanistic feature of these enzymes is the formation of an iron(III)–peroxo intermediate, which directly reacts to form a high-valent iron–oxo species.6,10
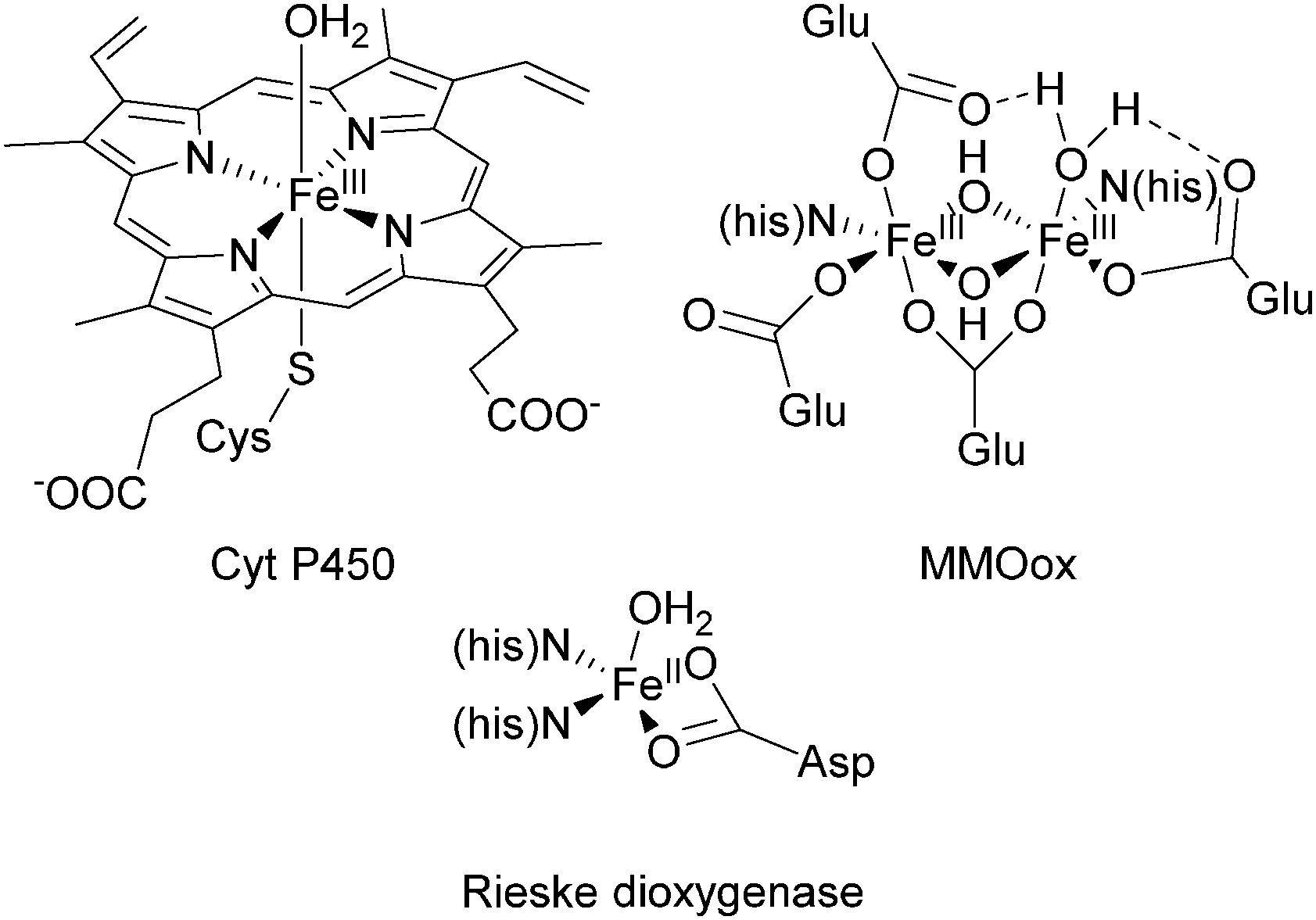 | ||
| Fig. 1 Structural environment of the active sites of Cyt P450, Rieske dioxygenase, and the oxidised form of methane monooxygenase.10,12,13 | ||
Inspired by the high catalytic efficiency of the enzymatic model systems, indicated by high conversions and selectivity, numerous biomimetic heme and nonheme complexes have been developed and applied in C–H bond oxygenation reactions.14,15 Due to its biological relevance, commonness in nature and unique properties iron is often the metal of choice. Forming a plethora of non-toxic and environmentally benign compounds, its use in catalytic processes helps to increase their sustainability.16,17 In this context the choice of oxidant also plays an important role. Depending on the application molecular oxygen, hydrogen peroxide (H2O2) or air are most desirable, with the latter two involving less safety risks.18,19 The use of tert-butyl hydroperoxide (tBHP) results in the formation of tert-butanol as by-product, which can be recycled and is therefore preferable to several other (e.g. heavy metal containing) oxidants.20
On the one hand, artificial catalyst systems aim at mimicking the structure and reactivity of natural enzymes, on the other hand they help to increase mechanistic understanding of the natural systems.15,21,22 For instance, active intermediates may be more stable in synthetic metal complexes and can be characterised spectroscopically or crystallographically. In terms of efficiency and selectivity biomimetic catalysts usually cannot compete with metalloenzymes as they tend to suffer from (oxidative) degradation outside of the protective protein surroundings. However, they allow higher flexibility regarding reaction conditions, substrates and tuning of selectivity.6 In the past decades many systems have been reported that catalyse the oxidation of hydrocarbons with varying oxidants. This article summarises key developments achieved in the field of homogeneous C–H bond oxygenation catalysed by molecular iron complexes in the last decade. The primary focus will be laid on the performance of the catalytic systems, however some mechanistic aspects will be discussed when necessary.
2. Oxygenation of aliphatic C–H bonds
The selective functionalization of saturated hydrocarbons plays an important role in the production of many commodity chemicals and in classical organic synthesis. For example, within the DuPont process cyclohexane is oxidised to a mixture of cyclohexanol and cyclohexanone, which constitute raw materials for the production of Nylon 6 and Nylon 6,6′ in a scale of mega tons per year.23–25 Dioxygen is used as the oxidant in combination with cobalt or manganese catalysts. Temperatures between 150 and 175 °C are required and the alkane conversion is kept at 3–8% to compensate for the low selectivity of the radical reaction. This demonstrates the need for catalytic systems with increased activity and selectivity, which is and has been a main goal of academic and industrial research through the last decades.Molecular heme and nonheme iron complexes have often been used to achieve such improvements.13,15,26,27 Main challenges regarding selectivity are caused by the inertness of the substrates. Nature and topology of the ligand supporting the iron centre are important features, which can be altered to tune catalytic activity and selectivity. The oxygenation of a hydrocarbon generally requires two individual steps, namely C–H bond cleavage and C–O bond formation.12,23 The first step is generally assumed to proceed via homolytic bond fission. The lifetime of the intermediate alkyl radical is decisive for the product distribution. A long lifetime is usually associated with a radical-based mechanism, where free radicals are formed directly from the oxidant, e.g. H2O2, by Fenton-type reactions. The selectivity of this reaction type is generally low as long-lived alkyl radicals are prone to unselective interaction with O2 for instance.12,23 A short lifetime relates to a more selective metal-centred oxidation mechanism. In many cases this includes the interaction of the iron catalyst with the oxidant to form an iron–peroxo species. This can either interact directly with the substrate or decompose to form the active oxidant (Scheme 1).12,23 High-valent iron–oxo species have often been proposed to be the active oxidant and their spectroscopic, structural, and electronic properties were investigated.13,28,29 However, often they cannot be tracked by experimental techniques under catalytic conditions and therefore model reactions have been established, which help to elucidate the prevalent oxidation mechanism.
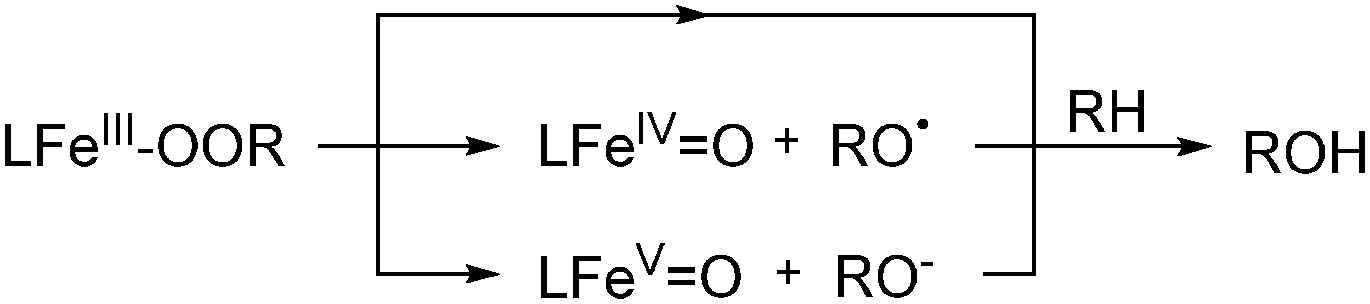 | ||
| Scheme 1 Oxidation of alkanes by a reactive iron–peroxo intermediate. | ||
The product distribution resulting from catalytic cyclohexane oxidation (Scheme 2), the so-called alcohol-to-ketone or A/K ratio, provides important information about alkyl radical lifetime. Long-lived cyclohexyl radicals can be trapped by O2 to form cyclohexyl peroxide, which may decompose in a Russell-type termination step to yield equal amounts of alcohol and ketone.23 Therefore, an A/K ratio close to one indicates a radical mechanism, whereas higher values support the involvement of a metal-centred oxidation mechanism. The regioselectivity can be evaluated by the oxidation of adamantane (Scheme 2) and is commonly expressed as the ratio of oxidised tertiary and secondary C–H bonds (3°/2°). Furthermore, the degree by which stereoinformation is retained during the oxidation of substituted cyclohexanes (cis-1,2-dimethylcyclohexane, cis-DMCH, Scheme 2), reflects the balance between epimerisation and C–O bond formation.12 A high amount of configuration retention (% RC) implies a short alkyl radical lifetime. The same accounts for the observation of kinetic isotope effects (KIE). In the competitive oxidation of cyclohexane and d12-cyclohexane with H2O2 a low KIE of 1–2 (4–5 for tBHP) points towards a radical-based mechanism, the presence of a metal-centred oxidation mechanism is indicated by a higher KIE of ∼7.12,23 Labelling experiments with 18O enriched reactants additionally provide information about the origin of the oxygen atoms incorporated in the product.
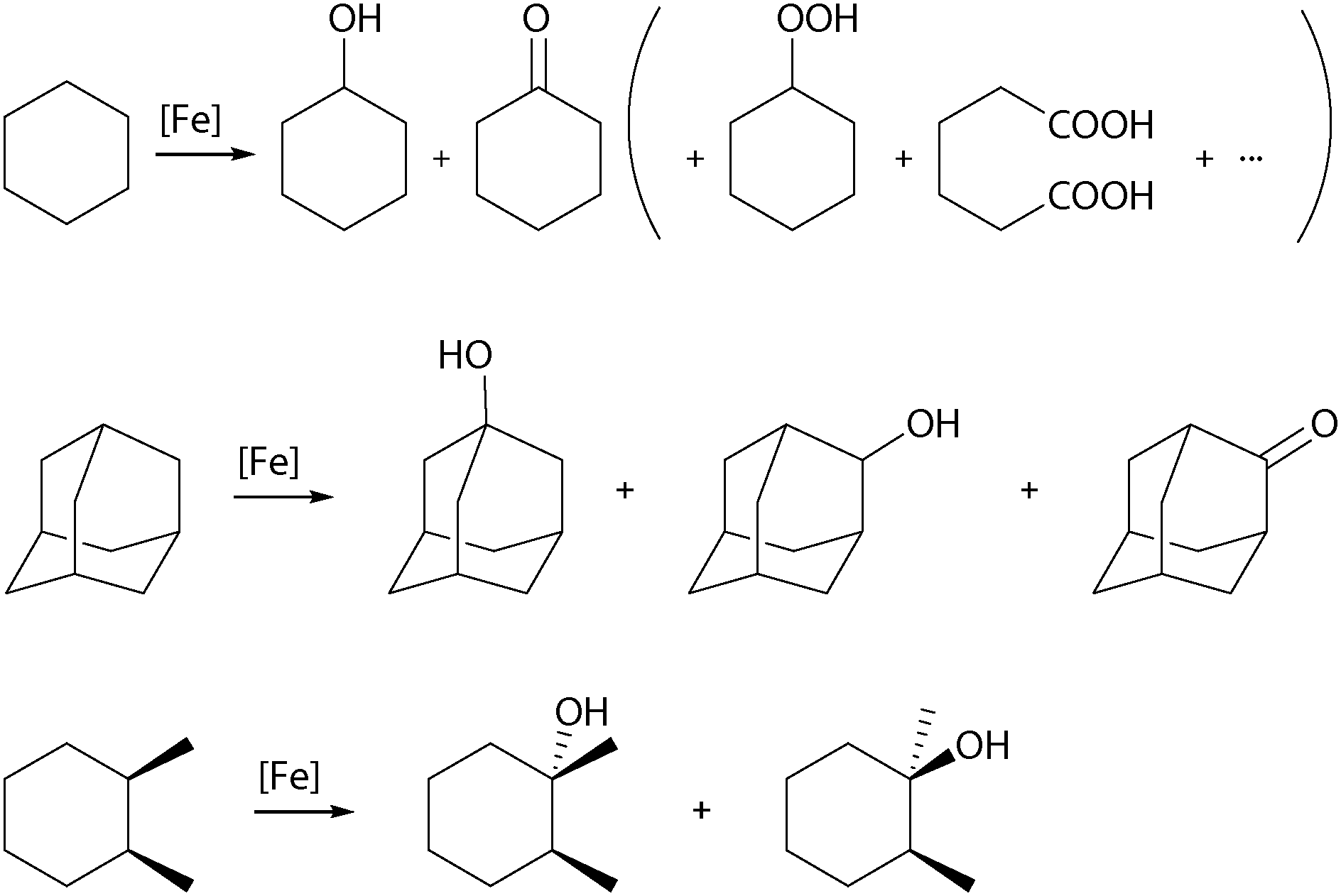 | ||
| Scheme 2 Benchmark substrates for iron-catalysed alkane oxidation reactions. | ||
2.1 Iron heme complexes and related derivatives
Iron complexes bearing porphyrin or closely related ligands such as phthalocyanines or corroles have been applied frequently in the oxidation of alkanes since the late 1970s.23,27,30 They resemble the active sites of naturally occurring hemoproteins such as Cyt P450, which are able to catalyse the oxidation of hydrocarbons under ambient conditions and therefore provide attractive inspiration for the design of biomimetic oxidation catalysts.Groves and co-workers reported the first example of the catalytic hydroxylation of non-activated alkanes by a mononuclear FeIII complex of 5,10,15,20-tetrakis(phenyl)porphyrin (1a, Fig. 2) with iodosylbenzene (PhIO) in 1979.31 Cyclohexane and adamantane were selectively oxidised to the corresponding alcohols in yields of 8 and 13% respectively (Table 1). These early investigations showed that both the nature of the ligand and the oxidant significantly influence catalytic performance. The introduction of bulky, substituted arene units in the meso positions and electron withdrawing substituents to the pyrrole β-position increased catalyst stability under oxidising conditions.30 Additional optimisation of the reaction conditions led to the development of catalyst systems exhibiting high reactivity towards hydroxylation of alkanes. For instance Wang et al.32 established the use of chloramine-T (N-chloro-para-toluenesulfonylamide) in combination with dioxygen as oxidant. Remarkably, cyclohexanol is formed exclusively from cyclohexane in 13% yield with [FeIII(1a)]Cl as the catalyst (2.6 turnovers). Similarly Bandyopadhyay et al.33 reported on the selective conversion of cyclohexane to cyclohexanol with tBHP in 47% yield based on the oxidant (19 catalytic turnovers) using a pentafluorophenyl substituted porphyrin (1b, Fig. 2) as ligand for an iron(III) complex. Investigations of the solvent influence revealed that the addition of water to the system significantly improves yields. Nakagaki and co-workers applied an acetalphenyl-substituted iron porphyrin (1c, Fig. 2) complex to the oxidation of cyclohexane with PhIO, which also could be successfully immobilized on silica while remaining catalytically active.34
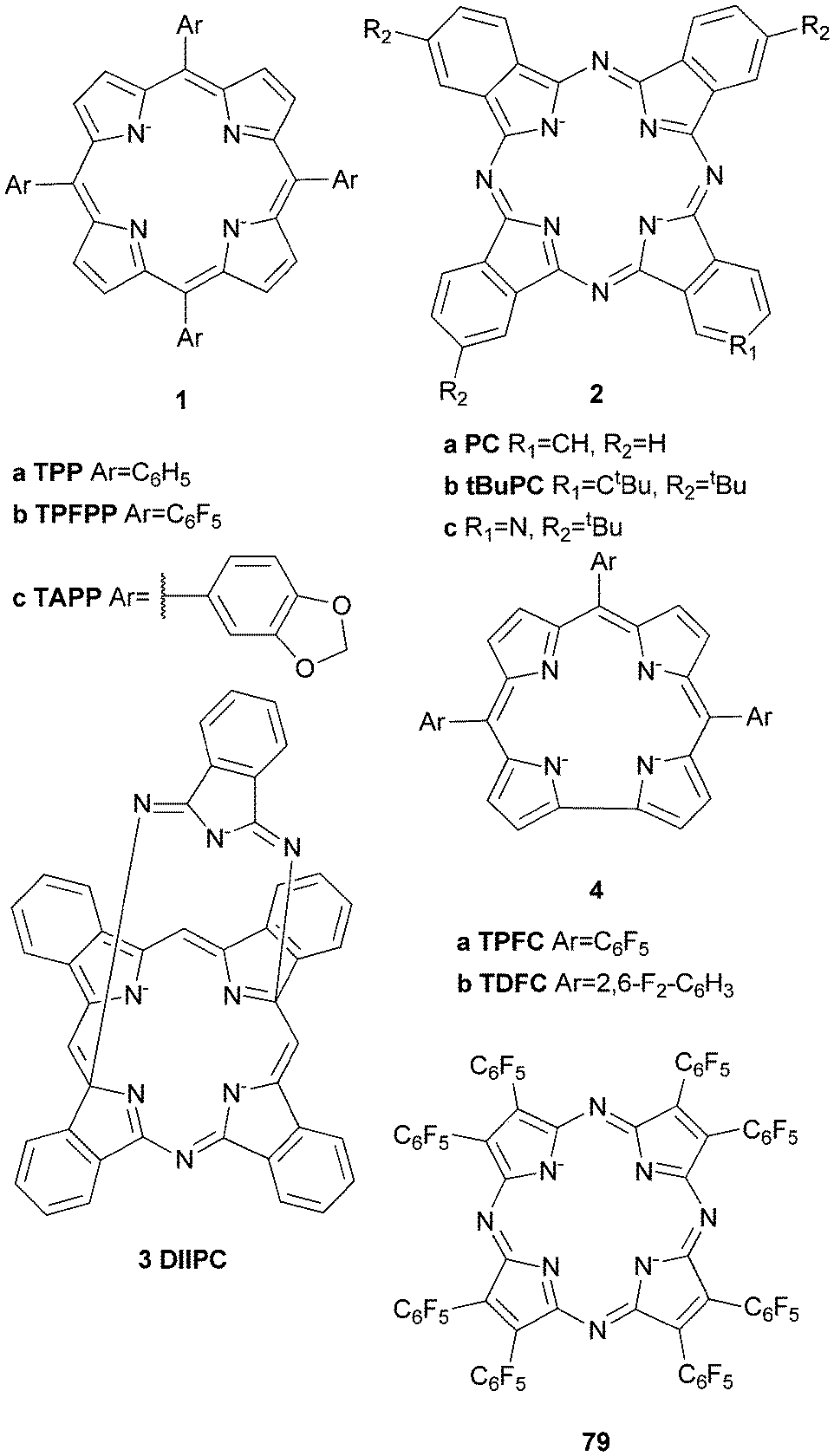 | ||
| Fig. 2 Examples of porphyrin, phthalocyanine, corrole and porphyrazine ligands used in iron-based oxidation catalysis. | ||
| Catalyst | Oxidant | % yielda | A/Kb | TOc | % RCd | 3°/2°![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) e e |
Ref. |
|---|---|---|---|---|---|---|---|
| a % yield of cyclohexanol and cyclohexanone from cyclohexane oxidation based on the minor component. b Ratio of cyclohexanol to cyclohexanone in cyclohexane oxidation. c Catalytic turnovers, moles of products from cyclohexane oxidation per mole of catalyst. d Retention of configuration of the tertiary bonds of cis-1,2-dimethylcyclohexane. e 3°/2° = 3 × (1-adamantol)/((2-adamantol) + (2-adamantone)). f Only alcohol detected. g S = solvent (MeOH or H2O). h Reaction carried out in acetone as solvent. i Ionic liquid used as solvent. | |||||||
| [FeIII(1a)Cl] | PhIO | 8 | —f | 0.5 | 48 | 31 | |
| [FeIII(3)S]g | H2O2 | 6.7 | 116 | 37 | |||
| [FeIV(4a)Cl] | tBHP | 27 | 0.58 | 23 | 10.2 | 40 | |
| [FeII(5a)(CH3CN)2](ClO4)2 | H2O2 | 32 | 5 | 3.2 | 100 | 17 | 48 and 49 |
| [FeII(5a)(OTf)2]h | H2O2 | 18 | 3.5 | 1.8 | 93 | 55 | |
| [FeII(10)(OTf)2] | H2O2 | 65 | 9.5 | 6.5 | 13 | 60 | |
| [FeII(14c)(CH3CN)2](ClO4)2 | H2O2 | 7.2 | 1.8 | 2.2 | 52 | ||
| [FeII(15c)(OTf)2] | H2O2 | 0.0078 | 2.25 | 3.9 | 53 | ||
| [FeII(16a)(CH3CN)2](ClO4)2 | H2O2 | 19.2 | 4.6 | 9.6 | 85 | 10.3 | 56 |
| [FeII(16a)(CH3CN)2](ClO4)2h | H2O2 | 21.6 | 4.4 | 10.8 | 82 | 24.9 | 56 |
| [FeII(17)(CH3CN)2](ClO4)2 | H2O2 | 25.5 | 9.2 | 2.5 | 74 | 59 | |
| [FeII(19h)(CH3CN)2](SbF6)2 | H2O2 | 40 | 7.3 | 4.0 | 40 | 75 | |
| [FeII(20c)(CH3CN)2](SbF6)2 | H2O2 | 24.4 | 5.0 | 122 | 156 | ||
| [FeII(22b)(OTf)2] | H2O2 | 42 | 0.15 | 42 | 99 | 72 | |
| [FeII(23)(CH3CN)](ClO4)2 | H2O2 | 12 | 7 | 50 | 96 | 6 | 78 |
| [FeII(24c)(OTf)2] | H2O2 | 65 | 12 | 6.5 | 93 | 30 | 82 |
| [FeII(24d)(OTf)2]i | H2O2 | 20 | 8 | 20 | 88 | ||
| [FeII(26)3](OTf)2 | tBHP | 27.3 | 0.6 | 27.3 | 27 | 19.9 | 90 |
| [FeII(28b)2][FeII(28b)Cl(H2O)2]2Cl4 | H2O2 | 1.6 | 6.6 | 317.3 | 93 | ||
| [FeII(29a)(CH3CN)2] | H2O2 | 34 | 1.5 | 34 | 17.0 | 95 | |
| [FeIII(31a)(H2O)](ClO4)2 | H2O2 | 78 | 11.3 | 15.6 | 18.8 | 99 | |
| [FeII(32)(CH3CN)](ClO4)2 | H2O2 | 23 | 1.2 | 2.3 | 101 | ||
| [FeII(33)(CH3CN)](ClO4)2 | H2O2 | 25 | 1.3 | 25 | 102 | ||
| [FeII(35a)](PF6)2 | H2O2 | 37 | 1.2 | 7.4 | 105 | ||
| [FeII(37a)(OTf)2] | H2O2 | 5.2 | 1.0 | 0.52 | 107 | ||
| {[FeII(37d)(CH3CN)]2O}(BF4)4 | H2O2 | 41 | 7.2 | 4.1 | 9.4 | 109 | |
| [FeII(39)Cl2] | H2O2 | 17.5 | 74.5 | 16.7 | 111 | ||
| [FeIII(41)Cl] | tBHP | 10.2 | 0.7 | 112.2 | 114 | ||
| [FeIII(42h)Cl] | H2O2 | 45.8 | 3.4 | 45.1 | 111 | ||
| [(FeIII)2(46)(O)(OBz)]ClO4 | mCPBA | 37 | 6.5 | 270 | 17.1 | 122 | |
| [FeIII(50b)3] | H2O2 | 16 | 1.1 | 32 | 125 | ||
| [(FeIII)2(51)(O)(H2O)2]2(ClO4)6 | H2O2 | 52 | 3.7 | 4.3 | 94 | 126 | |
| [FeII(FeIII)2(54)6Cl2](C5H12) | O2 | 14 | 7 | 129 | |||
| [(FeII)4(55a)(O)2](PF6)4 | H2O2 | 9.3 | 6 | 43 | 61.5 | 130 | |
| [FeII(64)(Ph2C(OH)COO)] | O2 | 55 | 10 | 100 | 11.7 | 141 | |
| {[FeII(65)Cl]2O}(ClO4)2 | H2O2 | 53 | 3.5 | 5.3 | 142 | ||
| FeII(H2O)6(BF4)2/67 | H2O2 | 98.7 | 1 | 98.7 | 144 | ||
| [FeII(71a)(CH3CN)](PF6)2 | H2O2 | 8 | 19 | 32 | 154 | ||
| [FeIII(75)](PBu3) | H2O2 | 15 | 6.1 | 75 | 155 | ||
Similarly to the hydroxylation reactions mediated by Cyt P450 enzymes, a high-valent iron(IV)–oxo porphyrin π-radical cation is understood as the active species in biomimetic iron heme model systems, although its exact nature strongly depends both on the reaction conditions and oxidant used.10
In 2011 a μ-nitrido mixed-valent iron phthalocyanine complex of ligand 2b was reported to be an active catalyst in the oxidation of methane to formic acid with H2O2 as oxidant.35 Phthalocyanines are structurally closely related to porphyrin ligands (Fig. 2). A detailed review of their use in oxidation catalysis was given by Sorokin.36 It is remarkable that although porphyrin iron μ-oxo and μ-nitrido dimers are considered as inactive, their phthalocyanine analogues have proven to be highly active catalysts in C–H bond activation. Similar to the porphyrin ligands, functionalization of the macrocycle enhances their stability against oxidative degradation.36 McGaff et al.37 for example described a “helmet” phthalocyaninato (3, Fig. 2) iron(III) complex, which was used as a cyclohexane oxidation catalyst with up to 116 catalytic turnovers using H2O2 as the oxygen source (Table 1).
Seldomly corrole-based iron complexes (4, Fig. 2) find application in the catalytic oxygenation of C–H bonds. The first example of an iron(IV) corrole compound, which was used to oxygenate benzylic C–H bonds was published in 1999; however, the respective activation of cyclohexane has been reported only in 2010.38 Since then various oxidants (e.g. mCPBA and tBHP) have been examined and it was found that the use of mCPBA as the terminal oxidant in the presence of 1.25 mol% meso–tris(pentafluorophenyl)corrolatoiron(IV)chloride leads to the exclusive formation of cyclohexanol from cyclohexane in yields of 50%.39–41
2.2 Iron nonheme complexes bearing pure N-donor ligands
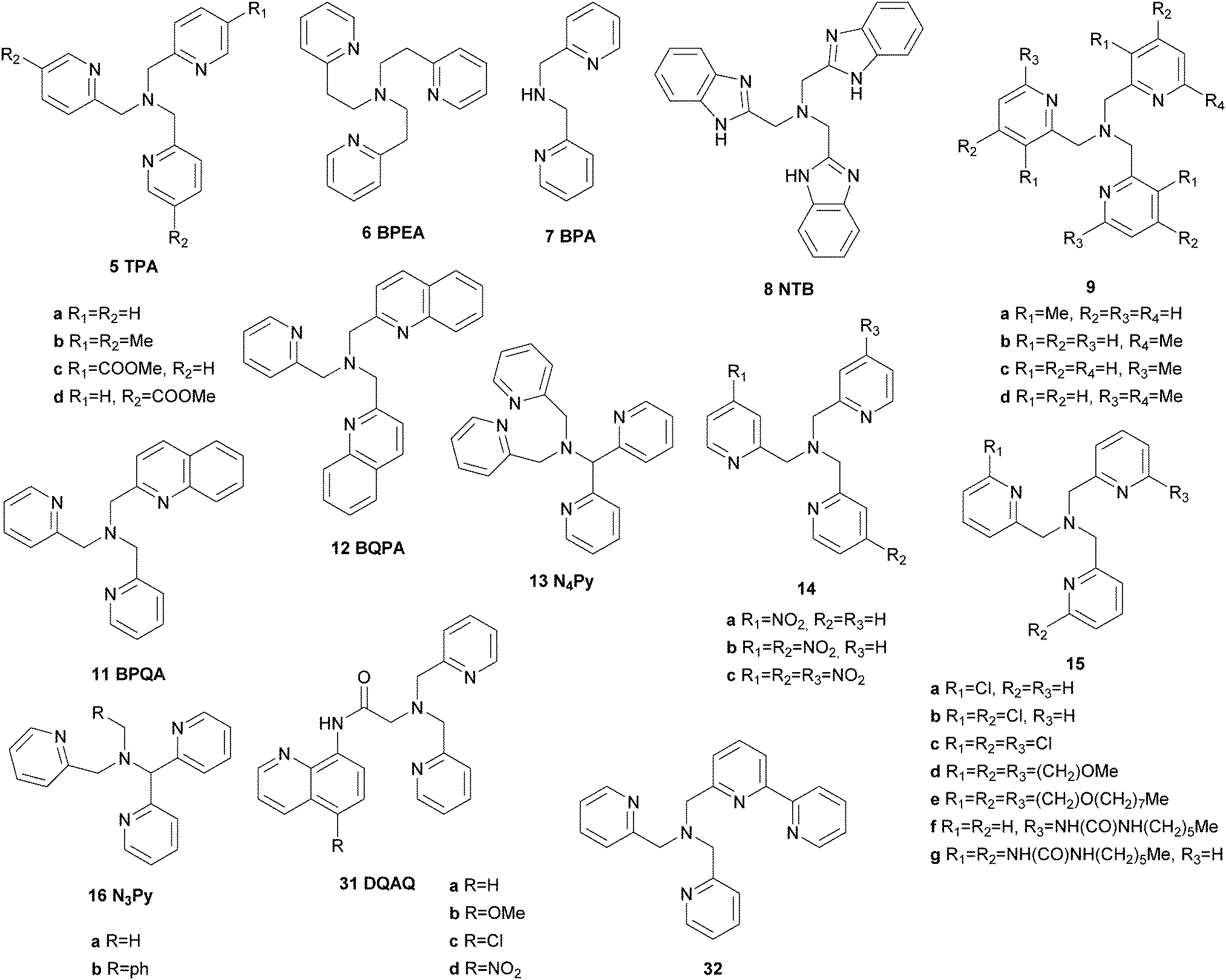
Iron complexes bearing multidentate N-donor ligands have been studied extensively as catalysts for the oxidation of alkane C–H bonds in the last 25 years and have proven great potential (Fig. 3). A broad variety of substrates can be covered nowadays ranging from model substrates such as cyclohexane or adamantane to complex biorelevant molecules and polymers. This allows their use in organic synthesis and environmental applications such as waste water treatment or degradation of pollutants.42,43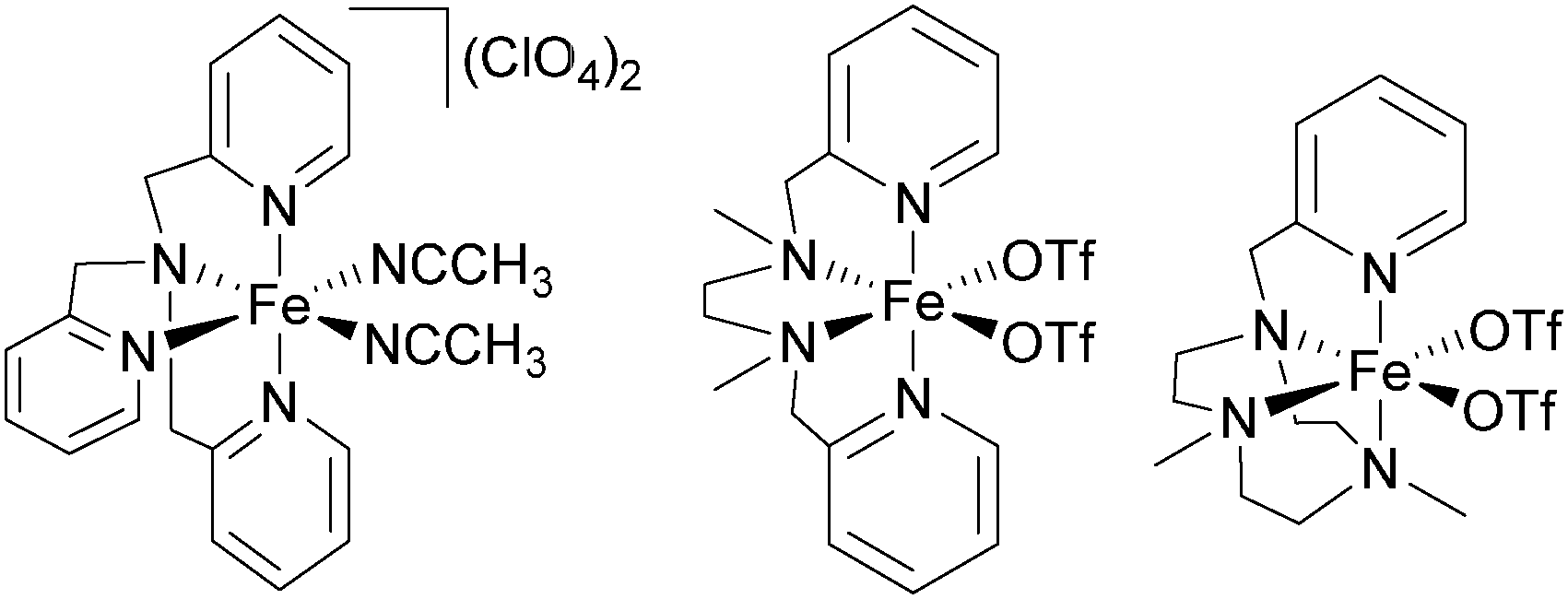 | ||
| Fig. 3 Prominent examples of iron-based alkane oxidation catalysts bearing tetradentate N-donor ligands.44,57,82 | ||
In the early 1990s Que and co-workers reported iron complexes [FeIII(5a)Cl2](ClO4) and [FeIII(5a)Br2](ClO4) to act as catalysts for the oxidation of cyclohexane with tBHP (Fig. 3).44,45 A series of ligands derived from the tris(2-pyridylmethyl)amine (TPA) scaffold (6–8, Fig. 4) was examined in the following years and their μ-oxo bridged diiron complexes exhibit more than 20 turnovers in cyclohexane oxidation with tBHP.46,47
 | ||
| Fig. 4 Pure N-donor ligands derived from the TPA structural motif used in iron-based oxygenation catalysis. | ||
An important breakthrough was achieved by the same group in 1997 when they reported on the stereospecific oxidation of alkanes using a ferrous TPA complex and H2O2 as the terminal oxidant.48 High A/K ratios and nearly 100% retention of configuration in the oxidation of cis-1,2-dimethylcyclohexane revealed the presence of a metal-centred mechanism rather than involvement of long lived alkyl radicals (Table 1). In this context the authors stressed the importance of cis labile coordination sites and a cis-α coordination of the ligand regarding catalyst stability and catalytic efficiency. These findings were supported by a range of iron(II) complexes carrying various TPA derived tetradentate and a pentadentate pyridine ligands (5b, 5c, 9–13, Fig. 4), which were tested in the oxidation of cyclohexane.49,50 The use of diluted H2O2 allowed minimisation of side reactions initiated by hydroxyl radical formation. The authors proposed the high-valent iron–oxo species FeV(O)(OH) as catalytically active species, which was formed via a ferric hydroperoxo intermediate. While at this stage evidence was found through mechanistic probe reactions and spectroscopic methods, it was confirmed later by density functional theory (DFT) calculations.51 The proposed mechanism for water-assisted oxidations catalysed by nonheme iron complexes bearing tetradentate N-donor ligands is depicted in Scheme 3. Inspired by the promising results obtained with this ligand system many studies dealing with derivatives thereof were published. For example, the introduction of up to three nitro groups to the pyridyl units of the TPA ligand (14, Fig. 4) was reported by Hitomi et al.52 However, with increasing number of nitro groups they observed a decrease in yield and selectivity for catalytic cyclohexane oxidation, presumably due to enhanced formation of hydroxyl radicals (Table 1). Similar results were obtained for chloro-substituted ligands 15a–c.53 TPA derived ligands 15d–g were designed to mimic enzymatic binding pockets by introduction of hydrophobic moieties (15d–f) and allow hydrogen bonding (15f and 15g).54 Unfortunately this did not lead to higher selectivity compared to the parent TPA complexes.
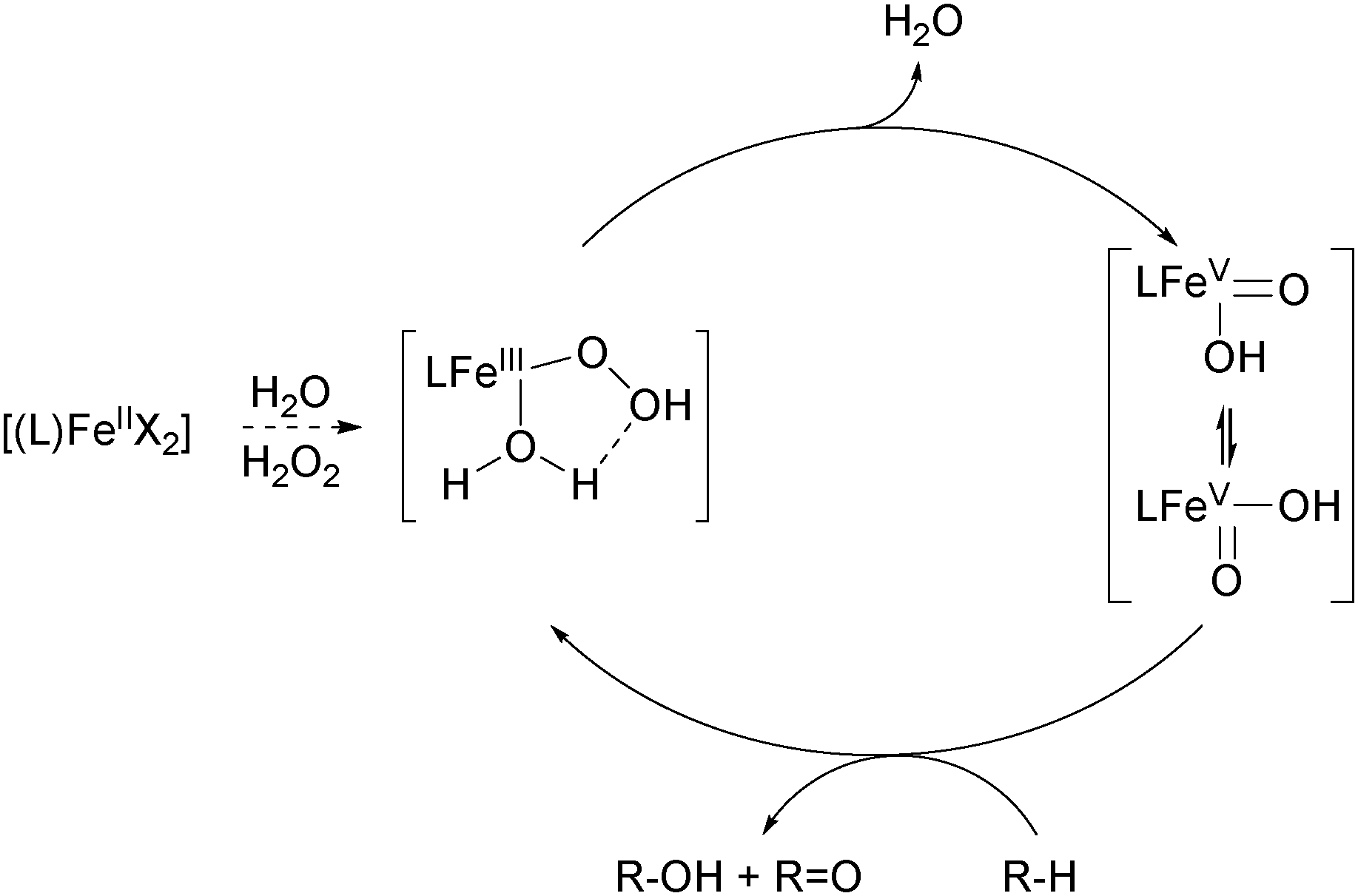 | ||
| Scheme 3 Mechanism of water-assisted oxidation of alkanes with H2O2 catalysed by iron(II) complexes bearing tetradentate N-donor ligands.6,13 | ||
Moreover, not only the ligands themselves but also the solvent has a decisive impact on the reaction mechanism and outcome as emphasised independently by Que et al. and by Feringa et al.55,56 For both TPA and N3Py (16) based complexes, the observed selectivity in the oxidation of cyclohexane and cis-1,2-dimethylcyclohexane is lower in acetone than in acetonitrile (Table 1). These results suggest that in acetone a competing reaction pathway becomes available, reducing the selectivity.56
Also in 1997 a μ-oxo bridged diiron(III) complex bearing a N,N′-dimethyl-N,N′-bis(2-pyridylmethyl)ethane-1,2-diamine (BPMEN) ligand 10 (Fig. 3) was reported to be more catalytically active than the TPA analogue in the oxidation of cyclohexane with H2O2.57 This structural motif was further employed by Ménage and co-workers who synthesised iron complexes incorporating a tetradentate N-(trimethoxybenzyl)ethane substituted BPMEN ligand (17).58,59 It became clear that beside the structure of the tetradentate N-donor ligand the nature of the additional monodentate ligands filling the coordination sphere of the metal cation strongly influences the balance between metal- and radical-based reaction pathways. Thus, with [FeII(17)(CH3CN)2](ClO4)2 as catalyst cyclohexane is oxidised yielding 25.5% cyclohexanol and cyclohexanone with an A/K ratio of 9.2 (Table 1). A series of ferrous iron complexes containing BPMEN derived ligands (18 and 19a–d, Fig. 5) was investigated by Britovsek et al.60,61 with respect to stability under oxidising conditions and catalytic efficiency. Complexes with a cis-α opposed to a cis-β or trans coordination geometry exhibit a significantly higher catalytic efficiency (Fig. 6).61 Oxidative ligand degradation was identified to be the main deactivation pathway and the authors underlined the importance of the presence of pyridine donors, which induce a strong ligand field and thereby stabilise reactive intermediates.62
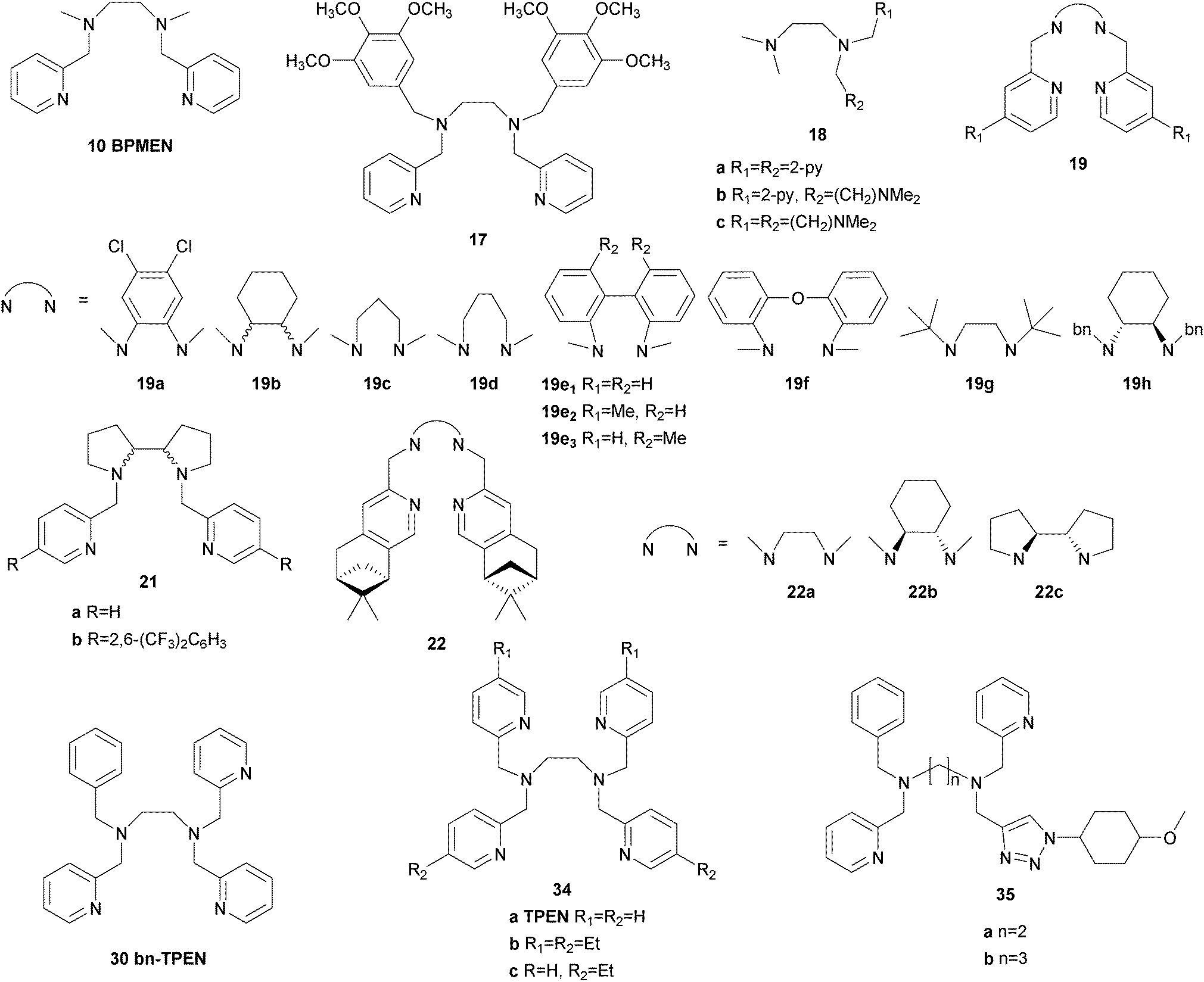 | ||
| Fig. 5 Pure N-donor ligands derived from the BPMEN structural motif used in iron-based oxygenation catalysis. | ||
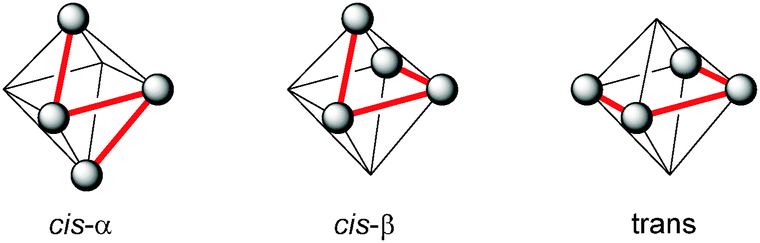 | ||
| Fig. 6 Coordination modes of linear tetradentate N-donor ligands.63 | ||
These findings were supported by further topological variations of the BPMEN ligand system.64–66 It was found that catalyst stability under oxidising conditions and can be altered by ligand tuning. Despite these efforts the best catalytic results in cyclohexane oxidation remained those achieved with the parent BPMEN ligand. Using [FeII(10)(OTf)2] as the catalyst 65% yield and an A/K ratio of 9.5 are reached in the oxidation of cyclohexane with H2O2 (Table 1).60 When the ethyl bridge connecting the two amine units of the BPMEN ligand is replaced by a biphenyl (19e) or biphenylether bridge (19f), the trans coordination of the remaining triflate ligands leads to a significant decrease in catalytic performance with A/K ratios of ∼1.63 Substitution of the bridging nitrogen donors by oxygen or sulphur atoms (20a–d, Fig. 11) results in a complete lack of catalytic activity.65 In this context Britovsek et al.60,65 proposed that a strong ligand field inducing primarily low spin iron complexes positively influences catalytic activity and selectivity.
A milestone was reached by Chen and White in 2007.67,68 For the first time an iron-based molecular catalyst was used to selectively oxidise functionalised organic molecules with H2O2 under synthetically applicable conditions. A large substrate excess is no longer required and a high tolerance towards functional groups is given with the chiral BPMEN derived complex [FeII((S,S)-21a)](SbF6)2 as catalyst. The presence of acetic acid alters both yields and stereoselectivity of the reactions. The selectivity is mainly guided by the electronic properties of the substrate C–H bonds rather than their steric demand. Thus electronically rich, tertiary bonds turned out to be more reactive than secondary or primary bonds despite their better sterical accessibility. However, suitable stereoelectronic modification of tertiary and secondary bonds within the substrate allowed the selective oxidation of the latter.68 By slow addition of the oxidant even natural products such as (−)-ambroxide and (+)-sclareolide are oxidised with higher yields and orthogonal site selectivity compared to the analogous enzyme-catalysed reactions.69 In Scheme 4 examples of the diastereoselective oxidation of complex substrates are shown to underline the synthetic utility of this system. By adding sterically demanding substituents to the ligand pyridyl units (21b, Fig. 5), control of the site-selectivity could be shifted from the substrate to the ligand as shown by Gormisky and White in 2013.70 Through the development of a structure-based reactivity model the observed selectivities could be reliably predicted for many substrates. In 2014 Gebbink and co-workers further demonstrated that the presence of the R,S-isomer of the PDP iron(II) catalyst does not have a negative impact on the catalytic efficiency of the system, but merely acts as a spectator.71 This implies a considerable simplification in catalyst synthesis and allows its preparation in multi-gram scale.
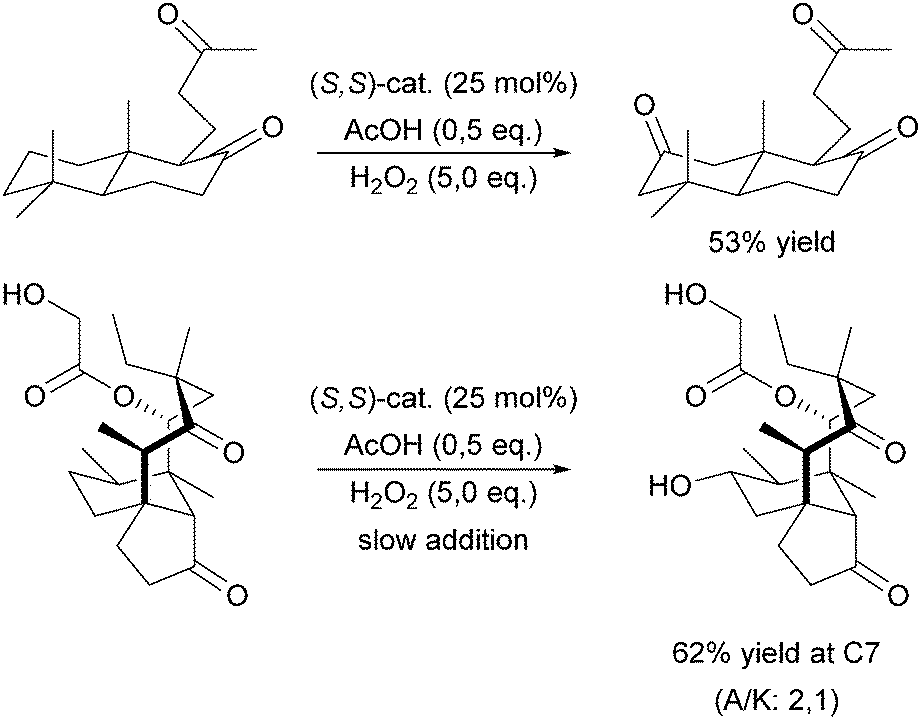 | ||
| Scheme 4 Selective oxidation of secondary C–H bonds in complex substrates using [FeII(21a)](SbF6)2 and H2O2.68 | ||
In 2009 Ribas, Costas and co-workers reported the efficient use of another series of BPMEN derived complexes bearing bulky pinene groups at remote positions of the ligand (22, Fig. 5).72 The isolation of the iron centre limits bimolecular degradation pathways and oxidation of various alkane substrates with H2O2 in acidic media was achieved with excellent stereoretention and high catalytic efficiency (Table 1). Furthermore, the oxidation of complex organic molecules under mild conditions was achieved with good yields using chiral iron complexes of ligands 19b, 22b and 22c.73,74 (−)-Ambroxide for example was oxidised with H2O2 under similar conditions applied by Chen and White68 but with lower catalyst loadings (Scheme 5). The selectivity of the reactions turned out to be guided by the special structure of the iron active site as well as the structural and electronic properties of the substrates.
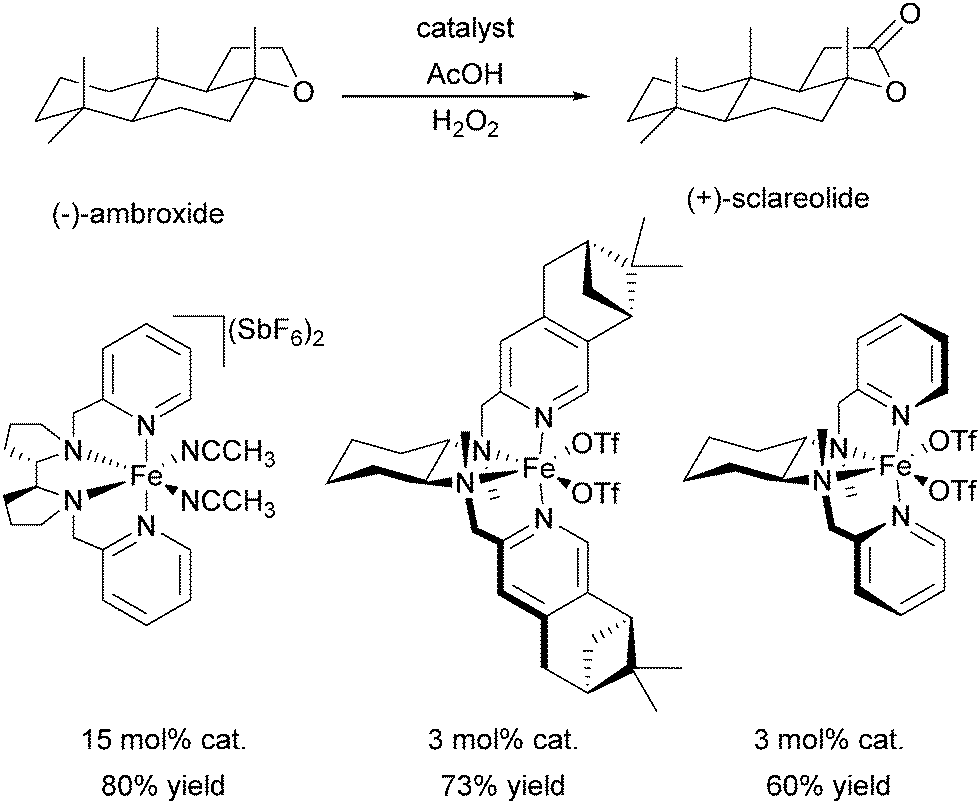 | ||
| Scheme 5 Oxidation of (−)-ambroxide with H2O2 by chiral iron(II) complexes bearing tetradentate BPMEN derived ligands.68,73,74 | ||
A similar methodology was applied by Goldsmith et al.75,76 who employed sterically demanding BPMEN derived ligands (19g and 19h, Fig. 5) to direct the selectivity of the corresponding iron complexes towards secondary instead of tertiary C–H bonds. This approach proved to be viable as the bulky ligand structure appeared to inhibit the oxidation of sterically congested C–H bonds, although in case of ligand 19g comparatively low catalytic turnovers were observed. Both iron complexes are also capable of promoting the oxidation of benzylic and allylic C–H bonds by using molecular oxygen as the terminal oxidant.77 In comparison to the H2O2-driven reaction, oxidation of alkanes with these systems proceeds much slower and substrates containing exclusively aliphatic primary and secondary C–H bonds cannot be activated.
Shteinman et al.78–81 introduced the tetradentate ligand TPCAH bearing an amide functionality (23, Fig. 7), which was also successfully applied in iron-based catalytic oxygenation of alkane substrates with H2O2. The mononuclear complex [FeII(23)(CH3CN)2](ClO4)2 exhibits remarkably high stereospecificity (Table 1).
 | ||
| Fig. 7 Pure N-donor ligands used in iron-based oxygenation catalysis. | ||
Another ligand family, which affords cis-α coordinated iron(II) complexes, has been described by Que, Costas and co-workers.82,83 The ligands they used are based on a methylpyridine derivatised triazacyclononane (PyTACN) scaffold (24, Fig. 7) and the corresponding iron complexes (Fig. 3) produced unprecedented catalytic efficiency in biomimetic oxidation reactions including cyclohexane oxidation (Table 1). Their reactivity can be tuned by modification of the ligand structure resulting in TONs of up to 64 in cyclohexane oxidation and a high degree of stereospecificity in the oxidation of cis-DMCH. These results implicate a metal-centred reaction pathway, which was proposed to occur via an alkyl radical rebound mechanism (Scheme 6).82 Support for the existence of the proposed reactive FeV(O)(OH) species could be provided by variable-temperature mass spectrometry.84 The oxidation of more complex organic molecules, such as biologically relevant terpenoids, has also been investigated. It could be shown, that the catalyst [FeII(24d)(OTf)2] displays enhanced selectivity towards the oxidation of methylene sites in acidic media.85 Further studies on the mechanism of iron complexes bearing PyTACN family ligands with different substitution patterns of the pyridyl unit have been published in 2013.86,87 The formerly proposed rebound mechanism could be ruled out by DFT calculations and it was shown that hydrogen abstraction and C–O bond formation occur in a concerted process (Scheme 6). Recently, ionic liquids were considered as valuable alternatives to traditionally used solvents such as acetonitrile.88 The oxidation of cyclohexane with H2O2 and [FeII(24d)(OTf)2] as the catalyst yields 20% oxygenated products with an A/K ratio of 8, indicating that the use of ionic liquids promotes a metal-centred reaction pathway (Table 1).
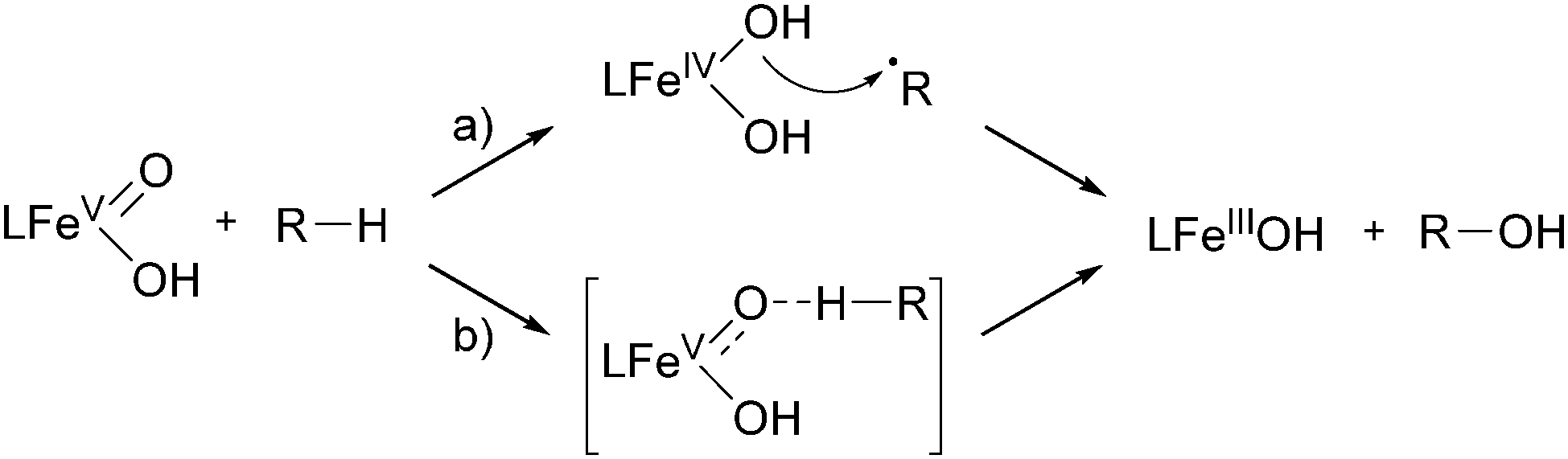 | ||
| Scheme 6 Oxidation of an alkane C–H bond by a FeV(O)(OH) species via a rebound (a) or concerted (b) mechanism.86 | ||
In addition to tetradentate N-donor ligands a few examples for ligands with lower and higher denticity have been reported in the context of iron-based catalytic C–H bond oxygenation. Shul'pin et al.89 described the in situ formation of an FeIII complex bearing bidentate 2,2′-bipyridine ligands (25, Fig. 7), which catalyses C–H bond oxidation. However, the selectivity of the catalytic system with H2O2 as the oxidant was rather low and free hydroxyl radicals were found to dominate alkane oxidation reactions. This also applies to catalytic alkane oxidation reactions with tBHP employing [FeII(26)3](OTf)2 as the catalyst, where 26 is a bidentate bis(imidazolyl)ketone (Table 1).90 Reedijk et al.91 observed the in situ formation of an iron complex bearing a tridentate ligand with a pyrazole unit (27). However, the catalytic efficiency of the complex in the oxidation of cyclohexane with H2O2 is comparatively low. The scorpionato type ligands 28a and 28b were used by Pombeiro and co-workers to afford half-sandwich and sandwich iron(II) complexes, which were tested as catalysts in cyclohexane oxidation.92,93 Despite coordinative saturation of the sandwich complexes they display high reactivity, exhibiting 385 turnovers in acidic media (Table 1). The closely related half-sandwich complexes reached even more turnovers but a radical mechanism is assumed to dominate the reaction as suggested by low A/K ratios. These results indicate that tridentate N-donor ligands are not as suitable as tetradentate N-donor ligands to support iron-based alkane oxidation catalysts, as they cover either 3 or 6 coordination sites of the iron centre. For pure N-donor ligands the presence of two cis-oriented labile coordination sites has proven to be an important feature, enabling the selective oxidation of aliphatic C–H bonds via a metal-centred, water assisted mechanism (Scheme 3).
A comparison of the catalytic efficiency of iron(II) complexes with tetra- and pentadentate N-donor ligands was drawn by Comba et al.94,95 Experimental and theoretical investigations indicate that the oxidation of alkanes with H2O2 catalysed by iron bispidine complexes bearing ligands 29a–29c (Fig. 7) proceeds along different pathways depending on the denticity of the ligand. The complex with tetradentate ligand 29a is more efficient than those with pentadentate ligands 29b or 29c (Table 1).95 However, both types of bispidine complexes are capable of promoting the oxidation of cis-1,2-dimethylcyclohexane with O2 as the terminal oxidant.96 Moreover, complexes of pentadentate ligands 13 and 30 as well as a number of tetradentate ligands (24c, 24d and 22c) were described to catalyse the oxidation of aliphatic C–H bonds with H2O as oxygen atom donor and Ce(IV) salts as single-electron oxidants.97,98 Despite a low A/K value of only 0.2 tertiary C–H bonds can be oxidised with a high degree of stereoretention using [FeII(24d)(OTf)2] as the catalyst, which rules out the formation of long-lived alkyl radicals.98
The first example of a mononuclear nonheme iron(III) complex, which catalyses selective C–H bond hydroxylation, was reported by Hitomi et al.99 in 2012. Containing a pentadentate amide ligand (31a, Fig. 4) the iron(III) complex efficiently catalyses the oxidation of cyclohexane (78% yield with respect to oxidant) with an A/K ratio of 11.3 (Table 1). The proposed reaction pathway includes the formation of an iron(V)–oxo active species generated from a ferric hydroperoxo intermediate. No prior oxidation of the metal atom, as it is often assumed for iron(II) complexes with cis-labile coordination sites, is required. Derivatisation of the quinoline unit of the ligand led to a series of complexes with tuneable electronic properties.100 With adamantane as a test substrate an increase in catalytic turnovers and selectivity for the oxidation of tertiary bonds was observed with rising electron deficiency of the catalyst.
Another pentadentate TPA derived ligand (32, Fig. 4) was described by Britovsek et al.101 in 2013. The corresponding ferrous complex exhibits moderate catalytic activity towards the oxidation of cyclohexane with H2O2 (Table 1). This was attributed to a short lifetime of the tentative iron(IV)–oxo species, which dimerises to form a μ-oxo bridged iron(III) complex. Xiang and co-workers reported the synthesis of an iron complex bearing the rigid pentadentate N-donor ligand 33.102 Cyclohexane is oxidised with 25% yield with respect to H2O2 and an A/K ratio of 1.3, pointing to the presence of free radicals during the reaction (Table 1).
Unusual iron complexes bearing hexadentate ligands (34a and 34b, Fig. 5) have been prepared by Banse and co-workers and reacted readily with peracids to form high-valent iron(IV)–oxo species that could be isolated and characterized spectroscopically.103,104 The selectivity of the complexes in alkane oxidation, however, is rather low and it was concluded that ligand oxidation proceeds through a bimolecular autoxidation mechanism. Lifetime of the catalysts under oxidising conditions could be improved by adding ethyl groups to the pyridyl moieties (34b), which rendered them more sterically demanding.104 When one of the pyridine units was replaced by a methoxyphenyltriazole moiety (35, Fig. 5), hexa-coordinated complexes were formed, with one donor coordinating only weakly.105 The triazole unit can be replaced by electrophilic anions and does not bind to the iron centre in the reaction intermediates, which is of possible interest for surface grafting.
Mono- and dinuclear iron(II) complexes of penta- to heptadentate oligopyridine ligands (36, n = 3–5) were prepared by Che et al.106 and applied in a large variety of oxidation reactions including alkene epoxidation as well as alkane, arene, and amine oxidation with oxone as the terminal oxidant. Unfortunately, the observed selectivities in alkane hydroxylation were low (A/K: 0.3).
There are few examples reported in the last decade dealing with the application of iron complexes with pure N-donor imine ligands in alkane oxidation catalysis. Imine ligands are good sigma donors and allow a high degree of π-backbonding. In 2005 Britovsek et al.107 reported a series of tridentate imine ligands bearing bulky substituents at the wingtips (37a and 37b, Fig. 8). They were used for the synthesis of coordination compounds of various transition metals, amongst them iron. The ferrous complexes exhibited an unusual trigonal bipyramidal geometry and moderate reactivity in the oxidation of cyclohexane (Table 1). The A/K ratios close to 1 indicate that the reaction mechanism is mainly guided by Fenton type chemistry.107 Similar observations were made by Reedijk and co-workers who used ligand 37c for the in situ formation of catalysts with different iron(II) and iron(III) salts.108 Surprisingly, in the absence of the ligand higher conversions of cyclohexane with H2O2 were observed while showing only slightly lower selectivity.
 | ||
| Fig. 8 Imine ligands used in iron-based oxygenation catalysis. | ||
On the contrary a metal-centred mechanism was assumed in alkane oxidation reactions using complexes of ligand 37d as catalysts.109 The monomeric complex [FeII(37d)(CH3CN)](BF4)2 is highly air sensitive and reacts readily with O2 to form a μ-oxo dimer, which yields 41% cyclohexanol and cyclohexanone with respect to H2O2 with an A/K ratio of 7.2 (Table 1).
Bauer et al.110 described the syntheses and application of iron complexes of the general formula [FeII(38)2(OTf)2] bearing bidentate iminopyridine ligands. Although selectivity in cyclohexane oxidation with H2O2 is low, activated methylene groups can be oxidised with tBHP and 3 mol% catalyst in yields of up to 91% (fluorene to fluorenone). Silva et al.111 reported an extraordinarily high selectivity towards the formation of cyclohexanol using [FeII(39)Cl2] and H2O2 in the presence of nitric acid (Table 1). 17.5% of cyclohexane is converted to the respective oxidation products with an A/K ratio of 74.5. An iron(II) complex containing two tridentate ligands of type 40 was described by Di Stefano and co-workers.112 The authors highlighted its easy preparation and applicability in alkane oxidation reactions with H2O2 in the presence of acetic acid as additive. However, the nature of the active species derived from the coordinatively saturated complex could not be revealed so far.
2.3 Iron nonheme complexes bearing C-, O- and S-donor ligands
There are many examples of iron-based oxidation catalysts bearing ligands with carbene, oxygen, or sulphur donors instead of or in addition to nitrogen donor atoms. Prominent examples are salen or carboxylate complexes.In contrast to many pure N-donor ligands, salen ligands mainly form pentacoordinate iron(III) complexes with one additional anionic ligand in the axial position.113 Complexes of this ligand class thereby closely resemble the active site of Cyt P450 enzymes. In 2006 Antunes and co-workers reported the capability of an iron(III) salen (41, Fig. 9) complex to catalyse the oxidation of cyclohexane with tBHP in the presence of acetic acid (Table 1).114 Yields of 10.2% cyclohexanol and cyclohexanone are reached after 24 hours reaction time. Higher yields of up to 23% can be achieved using PhIO as the oxidant and catalysts bearing chiral salen ligands of type 42a–42d (Fig. 9).115 Similarly to iron porphyrin complexes this increase in reactivity was attributed to the introduction of electron withdrawing substituents. However, both examples exhibit a lack of selectivity regarding the formation of alcohols and ketones as indicated by low A/K ratios. The exclusive formation of alcohols from adamantane with mCPBA as the oxidant and a high A/K ratio of 12.2 in cyclohexane oxidation was observed by Palaniandavar et al.116 who used a μ-oxo diiron(III) complex of ligand 42e as the catalyst. Assuming a reactivity distinct from Fenton chemistry, the authors proposed a high-valent iron salen π–cation radical as the active species. The use of H2O2 as the oxidant in the catalytic oxidation of alkanes with an iron complex of salen-type ligand 43 was described by Louloudi and co-workers.117,118 In homogeneous phase the reaction with cyclohexane affords oxygenated products in 12.2% yield (2.4 turnovers) with A/K of 1.8. Furthermore, Silva et al.111 reported iron(III)salen complexes of ligands (42f–42h, Fig. 9) to be capable of C–H bond oxidation with H2O2 in acidic media. The catalysts feature high catalytic efficiency and good selectivity towards the formation of alcohols (Table 1). In contrast to that alkane oxidation reactions catalysed by a Schiff base iron(III) complex bearing salicylidene derived ligand 44 are dominated by a hydroxyl radical-based mechanism.119
 | ||
| Fig. 9 Salen and phenolate O-donor ligands used in iron complexes for catalytic C–H bond oxidation. | ||
Caradonna et al.120,121 reported the application of a diiron(II) complex bearing tetracoordinate phenolate ligand 45 (Fig. 9) in cyclohexane oxidation catalysis using 2-methyl-1-phenylprop-2-yl hydroperoxide (MPPH) as oxygen atom donor. The complex shows high catalytic efficiency (230 turnovers) and selectivity for alcohol formation. An observed decrease of the product formation rate prior to full consumption of oxidant and/or substrate was attributed to product inhibition rather than decomposition of the catalyst.121 Rational ligand design was exerted in the case of bisphenolate ligand 46, which forms a doubly bridged diiron(III) complex, thus resembling the active site of MMO type enzymes.122 The oxidation of aliphatic substrates with this catalyst was achieved using mCPBA with high turnovers and good selectivity (Table 1). However, when H2O2 or tBHP are used as oxidants the catalytic efficiency is significantly reduced, which implies that differing reaction pathways must be present depending on the oxidant. On the other hand Reedijk et al.123 described the iron(III) complex bearing phenolate ligand 47 to be capable of catalysing the oxidation of alkanes with H2O2 as the oxidant. This also holds for the water-soluble iron(III) complexes of hydrazone ligands 48 and 49, which provide a coordination environment inspired by nonheme metalloenzymes.124 An alteration of the catalytic performance of a series of symmetrically coordinated 8-quinolinolato (50, Fig. 9) complexes could be observed by visible light irradiation during catalytic oxidation reactions with H2O2 (Table 1).125 This effect was particularly pronounced when dihalogenated ligands 50c–50e were used and it applies not only to aliphatic but also aromatic substrates.
Ligand systems incorporating carboxylate groups often act as bridging agents between two iron centres. This renders them particularly useful when mimicking dinuclear active sites of enzymes such as MMO. Nordlander, Shteinman et al.126 reported the syntheses and characterisation of a series of tetranuclear iron(III) complexes bearing octadentate ligand 51, which contains a benzoic acid group (Fig. 10). Those complexes, where the carboxylate unit of the ligand bridges two iron centres, catalyse the oxidation of cis-DMCH with H2O2 displaying a consistently high degree of configuration retention (Table 1). Moreover, cyclohexane oxidation is achieved in high yields and selectivity (A/K: 3.7). The same group later described a similarly bridged diiron complex containing ligand 52, which exhibits slightly lower catalytic efficiency.127 Equal amounts of alcohol and ketone are formed in the catalytic oxidation of cyclohexane with H2O2 using a hexanuclear iron(III) complex carrying nitrobenzoate ligands (53, Fig. 10).128 A mixed-valent trinuclear iron complex bearing tetramethylreductic acid ligands (H2TMRA, 54) was used for the oxidation of cyclohexane with O2 as the terminal oxidant (Table 1).129 Under addition of excess H2TMRA the reaction yields 14% products with an A/K ratio of 7. Shul'pin et al.130 explored the application of di- and tetranuclear iron complexes containing ligand 55a, which is structurally closely related to the successful PyTACN motif. The tetrameric complex proves to be more robust than the dimeric and even methane can be oxidised under moderate reaction conditions (50 °C, 60 bar). On the other hand, the mononuclear iron complexes carrying two tridentate oxazoline ligands of type 56a or the structurally closely related, phosphine containing ligands 56b–56d, exhibit only modest reactivity in the oxidation of benzylic C–H bonds with H2O2 or tBHP respectively.131,132 Antunes and co-workers reported a series of tri- and tetradentate ligands of type 57a–57d (Fig. 10).133–135 The corresponding iron complexes catalyse the oxidation of cyclohexane with H2O2 and mainly produce cyclohexanol and cyclohexanone. For example with 0.1 mol% [FeIII(57a)Cl3] as the catalyst 11.4% alcohol and 8.1% ketone with respect to the substrate are obtained.133 Nevertheless, significant amounts of side products such as cyclohexyl hydroperoxide (12.1%) and adipic acid (2.3%) are formed, which cannot be prevented by neither optimisation of reaction conditions nor microwave irradiation.
 | ||
| Fig. 10 Carboxylate ligands used in iron complexes for catalytic C–H bond oxidation. | ||
High sensitivity of catalytic hydrocarbon oxidation reactions towards the catalyst structure as well as the experimental conditions was also emphasised by Reedijk et al.136 who investigated the catalytic efficiency of mono- and dimeric iron complexes bearing pyrazine-2-carboxylic acid ligands (PCA, 58, Fig. 10). The latter was used as additive for the in situ formation of highly active hydrocarbon oxidation catalysts from hexa- and undecanuclear iron(III)-2,4,5-trimethoxybenzoate (59) complexes.137 Cyclohexane is oxidised with H2O2 in the presence of the precatalysts and pyrazine-2-carboxylic acid with exceptionally high turnovers of up to 2210 but low selectivity for alcohol formation. A surprisingly high selectivity for the oxidation of secondary compared to tertiary C–H bonds of methylcyclohexanes with H2O2 was observed for iron compounds containing carboxylate ligands of type 60.138 Moreover, visible light irradiation significantly accelerates cyclohexane oxidation. L-proline derived ligands 61, 62a, and 62b were designed to mimic the coordination environment of nonheme iron enzymes, but instead of achieving a metal-centred oxidation mechanism, cyclohexene is oxidised via a radical mechanism in allylic position.139
A high-valent iron–oxo species could be observed spectroscopically and by mass spectrometry during hydrocarbon oxidation by iron complexes bearing the independent ligands 55b and Racac (63, Fig. 10) with oxone.140 The combination of a neutral, robust N-donor and the redox active acetylacetonate (acac) derivative allows even the oxidation of challenging substrates such as ethane and propane. Recently Chatterjee and Paine observed the formation of an electrophilic FeIV oxidant, which evolved from the reaction of a ferrous benzilate complex containing scorpionato ligand 64 with molecular oxygen in the presence of a Lewis acid.141 It catalytically oxidises cyclohexane and other alkanes in high yields and exhibited excellent selectivity for cyclohexanol formation (Table 1).
Furthermore, iron coordination compounds carrying neutral NxOy ligands and their potential application as catalysts in C–H bond oxidation have been described (Fig. 11). In the presence of H2O2 the μ-oxo bridged diiron complex [[FeIII(65)Cl]2O](ClO4)2 catalyses the oxidation of cyclohexane with a conversion of 53% with respect to the oxidant and an A/K ratio of 3.5 (Table 1).142 A similar iron complex reported by Sun and co-workers bearing only one additional nitrogen donor unit (N4O, 66) shows lower catalytic efficiency with H2O2 and requires the use of mCPBA as oxidant.143 Further exploiting the above mentioned imine ligand qualities, Reedijk et al.144 used an iron complex bearing ligand 67 and observed full conversion of cyclohexane with H2O2 (Table 1). Equal amounts of cyclohexanol and cyclohexanone are formed, which was explained by the presence of free radicals. Gebbink and co-workers came to the same conclusion when they described an iron(III) complex with N3O2 ligand 62c, which adopts an unusual heptacoordinate coordination geometry.145 The catalytic performance of iron(II) complexes bearing NxO ligands of varying denticity (x = 2–4, 68a–68c, 69, Fig. 11) was compared by Bauer and co-workers.146 At low catalyst loadings the complexes with tetra- and pentadentate ligands perform better than those with tridentate ligands. However, low A/K ratios indicating a radical-based mechanism were observed throughout all catalytic experiments. This also applies to reactions catalysed by the ferrous complex of ligand 70, which displays a rare heptacoordinate geometry.147
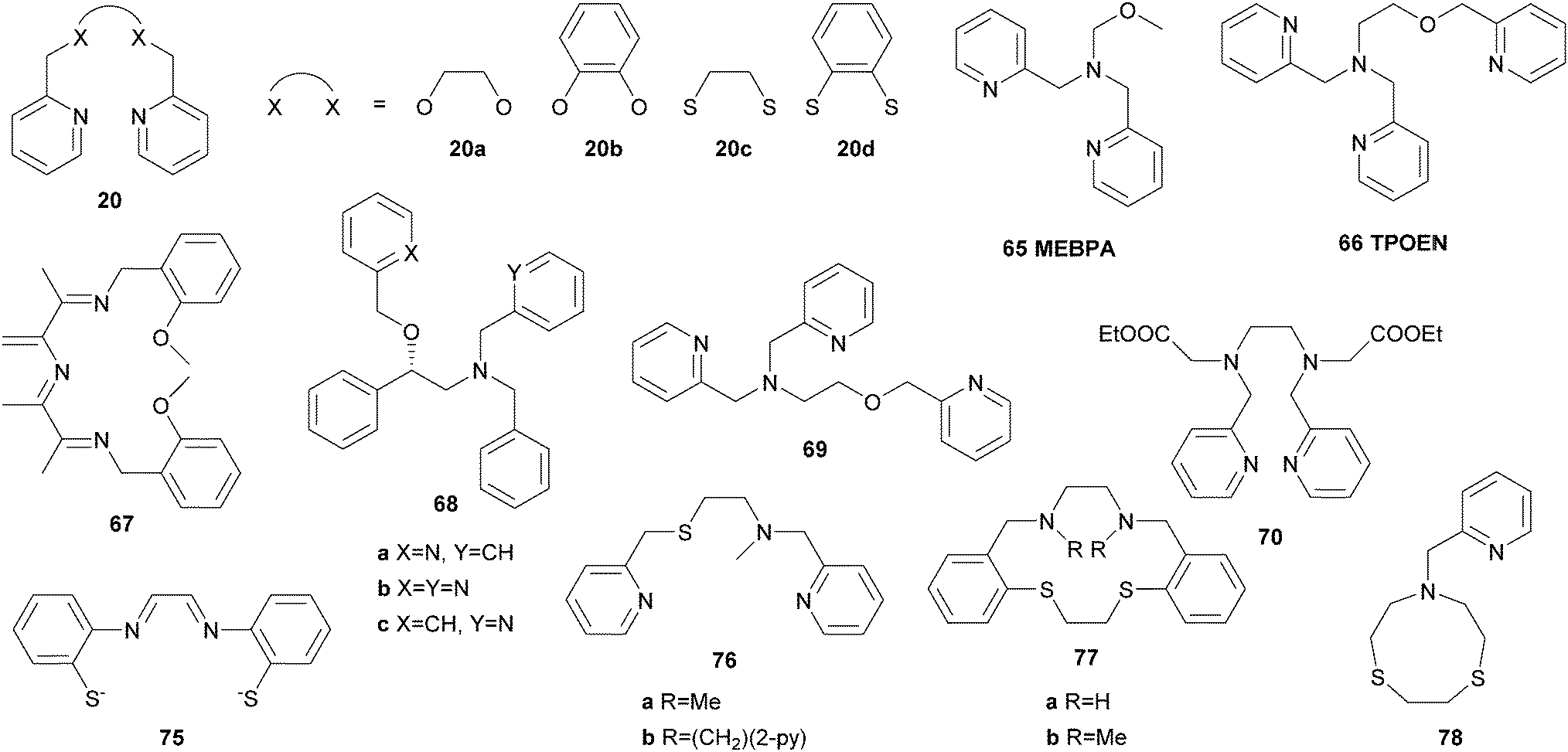 | ||
| Fig. 11 O- and S-donor ligands used in iron complexes for catalytic C–H bond oxidation. | ||
Our group has reported the synthesis and characterisation of a series of iron complexes supported by cyclic and acyclic tetradentate N-heterocyclic carbene ligands 71–74 (Fig. 12).148–151 The octahedral coordination geometry of iron is completed by coordination of labile acetonitrile solvent molecules. For ligands 71a, 71b, 73, and 74 the iron complexes exhibit trans-labile sites, for 71c and 72bcis-labile sites as a consequence of the coordination mode of the respective tetradentate ligand. In case of ligand 72a a fluctuation between equatorial and sawhorse-type coordination is observed. It has been shown that the electronic structure of the Fe(II) complexes can be influenced by target-oriented variation of different coordinating additives in the labile sites.152
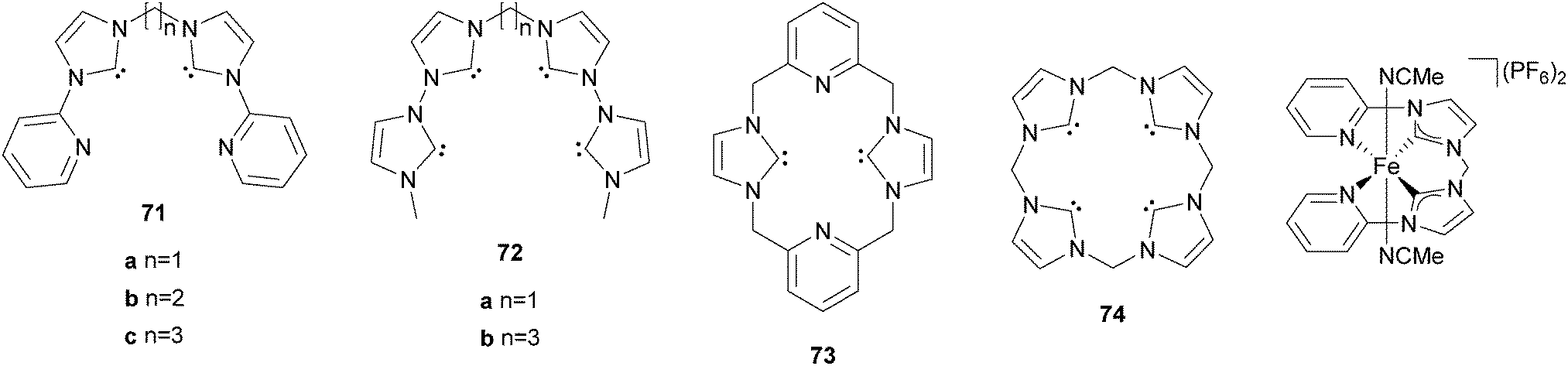 | ||
| Fig. 12 N-heterocyclic carbene ligands and a corresponding iron complex used for catalytic oxidation reactions. | ||
The ferrous complex [FeII(71a)(CH3CN)2](PF6)2 was the first organometallic iron compound to be used as olefin epoxidation catalyst and also exhibits high efficiency in the oxygenation of alkanes with H2O2 as the oxidant.153,154 Under ambient conditions and at low catalyst loadings cyclohexane is oxidised with up to 32 turnovers and high selectivity towards the formation of cyclohexanol and cyclohexyl hydroperoxide ((A + H)/K = 19). Substitution of one of the axial acetonitrile units by a phosphine or isonitrile ligand stabilises the catalyst under oxidising conditions without altering the selectivity, which translates to increased turnovers.154
As the active site of Cyt P450 enzymes exhibits a coordinating thiolate in addition to the porphyrin moiety, ligands bearing sulphur donor atoms have been applied to mimic catalytic and structural features of those enzymes. Pombeiro et al.155 reported the use of a ferric complex containing the tetradentate N2S2 ligand 75 as catalyst for the oxidation of cycloalkanes with H2O2 as the terminal oxidant. In the presence of PCA as co-catalyst cyclohexanol and cyclohexanone are produced from cyclohexane in yields up to 16.3% based on the substrate while the selectivity varies significantly depending on the reaction time and temperature (Table 1). Although Britovsek et al.65 reported [FeII(20c)(OTf)2] to be catalytically inactive, Pombeiro and co-workers demonstrated the opposite for the analogous complexes bearing chloride and acetonitrile instead of triflate ligands (Table 1).156 High selectivity towards alcohol formation and up to 1450 turnovers can be achieved by use of acid additives, in particular trifluoroacetic acid (TFA) and triflic acid (HOTf). Under similar reaction conditions iron(II) complexes containing BPMEN derived or macrocyclic ligands (76–78, Fig. 11), and also a dinuclear iron(I) thiophenolate carbonyl complex has been found to catalyse hydrocarbon oxidation reactions with H2O2.157–159 In general, these reports demonstrate the importance of suitable reaction conditions and an enhancement of catalytic efficiency upon acid addition. Radical pathways are dominant in these reactions.
3. Oxygenation of aromatic C–H bonds
Despite the extensive use of molecular iron complexes for the catalytic oxidation of aliphatic C–H bonds, fewer investigations have been made on the selective oxidation of aromatic substrates.160 On the one hand this may be due to the higher bond strength of aromatic compared to aliphatic C–H bonds (e.g., BDEC–H of 112.9 kcal mol−1 for benzene versus 99.5 kcal mol−1 for cyclohexane161). On the other hand the higher reactivity of the phenolic products compared to the non-activated substrates can lead to undesirable formation of catechols, hydroquinones, benzoquinones or even tars.162Many recent reports deal with mechanistic aspects of biomimetic arene oxygenation reactions catalysed by heme and nonheme iron complexes. The proposed oxidising species all derive from ferric hydroxo intermediates and identify as high-valent iron–oxo complexes in several cases. Taking this as a starting point different reaction pathways have been proposed, which involve either an initial hydrogen atom abstraction (path a, Scheme 7) or the formation of a metal-based intermediate (path b, c, Scheme 7).163,164 An inverse KIE observed for the competitive oxidation of benzene and d6-benzene and a negative Hammett ρ value indicate the formation of a cationic or radical σ-complex (path b), whereas a normal kinetic isotope effect points either towards an epoxide formation (KIE ∼ 1.2, path c) or an initial hydrogen atom abstraction (path a).165,166
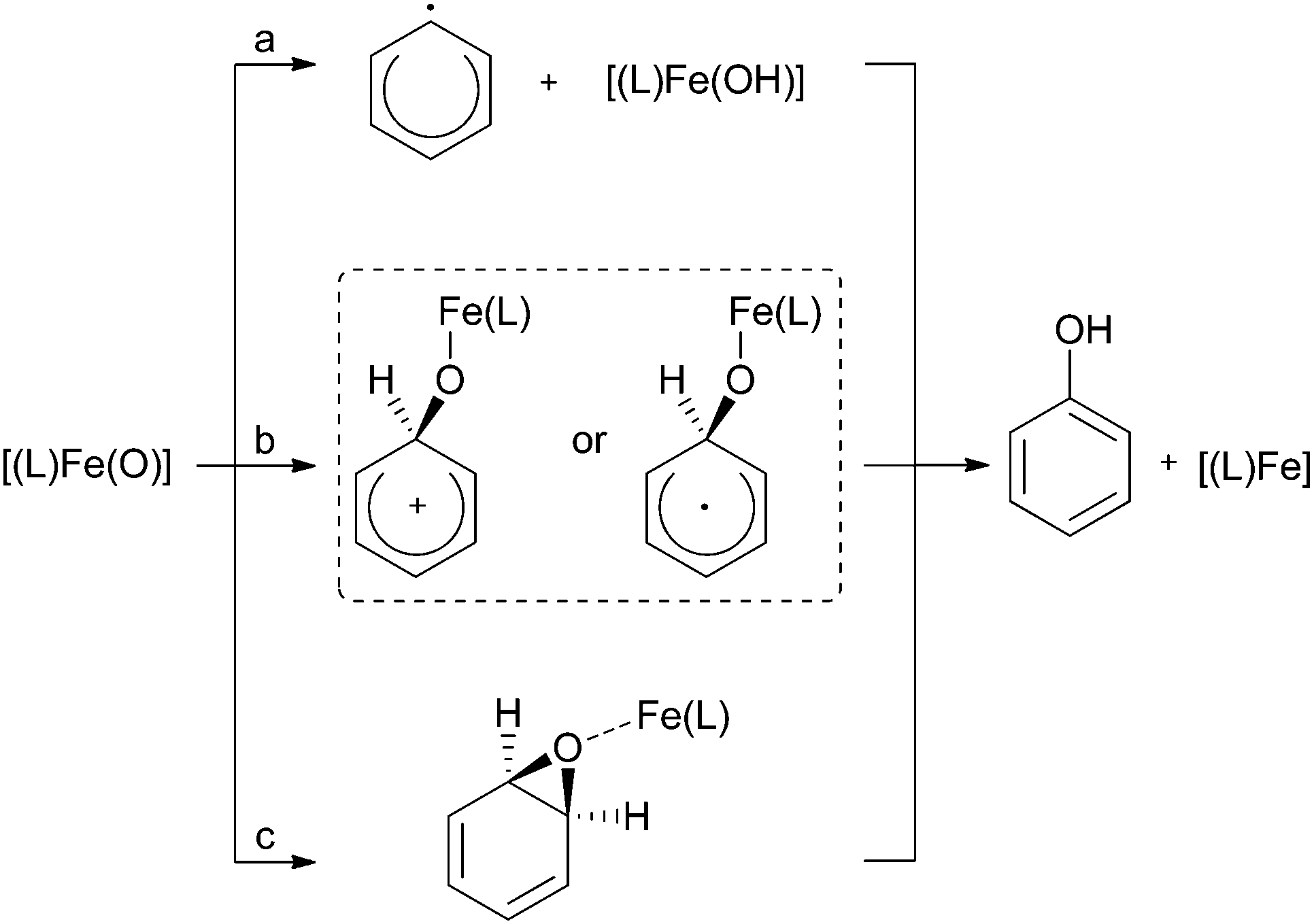 | ||
| Scheme 7 Proposed mechanisms for aromatic ring hydroxylation by a high-valent iron–oxo species.163,164 | ||
Moreover, the observation of NIH shift reactions – named after the National Institute of Health where they were first observed in enzymatic reactions167 – can be explained by a metal-centred oxidation pathway. d3-Benzene as a probe substrate to detect NIH shifts has been established by Kudrik and Sorokin (Scheme 8).168 The detection of mono- and trideuterated benzoquinone is used as an indicator for the replacement of a deuterium atom to an adjacent carbon atom.
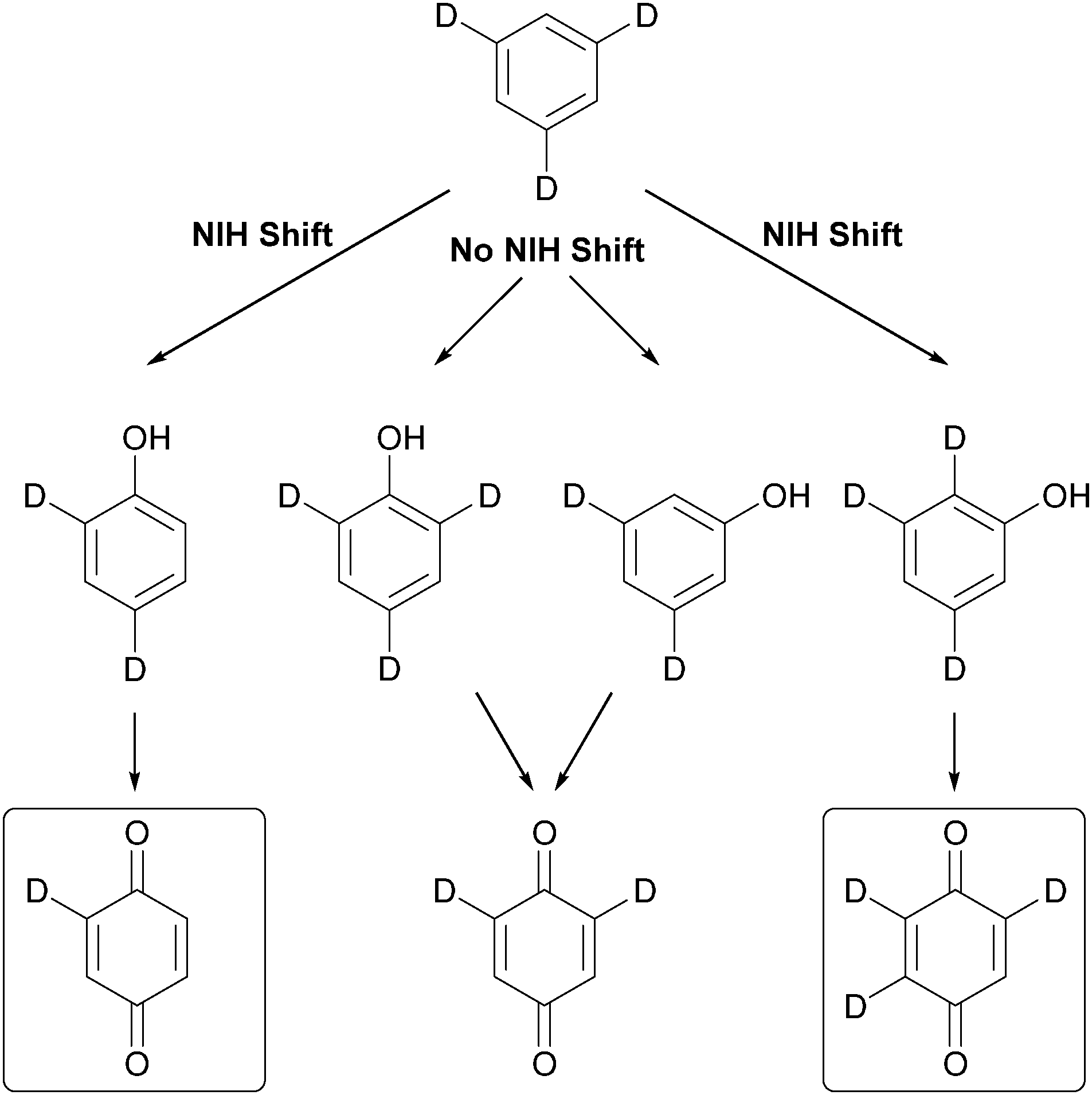 | ||
| Scheme 8 Detection of NIH shift with d3-benzene as a model substrate.168 | ||
3.1 Iron heme complexes and related derivatives
Iron heme complexes, bearing substituted porphyrin and phthalocyanine ligands, have been used since the early 1990s for the catalytic oxidation of aromatic substrates.30,36,169–171 However, the reaction mechanisms for aromatic substrates feature great complexity and in the last decade numerous experimental and theoretical investigations have been made to elucidate the nature of the active oxidising species. Mostly iron(IV)–oxo π-radical cations, analogous to those formed by Cyt P450 enzymes, are believed to be the catalytically active species. Amongst others, this was supported by Jang, Nam and co-workers who performed kinetic measurements of arene hydroxylation reactions under stoichiometric conditions.163 As catalyst they used an iron(IV)–oxo porphyrin π-radical cation complex, which was generated in situ by the reaction of PhIO and an iron(III) complex bearing ligand 1a (Fig. 2). The authors also observed an inverse KIE and a large negative Hammett ρ value, indicating that the reaction involves an electrophilic attack of the iron species and the formation of a σ-complex (path b, Scheme 7). Further evidence was gained by kinetic experiments under catalytic conditions and 18O labelling studies using mCPBA as the oxidant.172 By DFT methods Kumar et al.173 investigated the influence of axial ligands on the kinetics and mechanism of aromatic hydroxylation reactions mediated by iron(III) porphyrin complexes. They found that the nature of the axial ligand, being a cysteine residue in the naturally occurring enzyme, determines the basicity of the iron centre and thus influences the reaction rate.Practical use of iron porphyrin complexes can be made for environmental applications. Polycyclic aromatic hydrocarbons (PAHs), which constitute an important class of pollutants and possess carcinogenic properties, can be oxidised to the corresponding quinone derivatives using metalloporphyrin complexes and H2O2 as the oxygen source (Scheme 9).174,175 Giri and Chauhan observed that iron(III) porphyrin complexes bearing electron withdrawing substituents exhibit the highest catalytic efficiency. The mild reaction conditions in the absence of additives and the use of a commercially available and benign oxidant as well as a reusable catalyst renders this process very environmentally friendly.
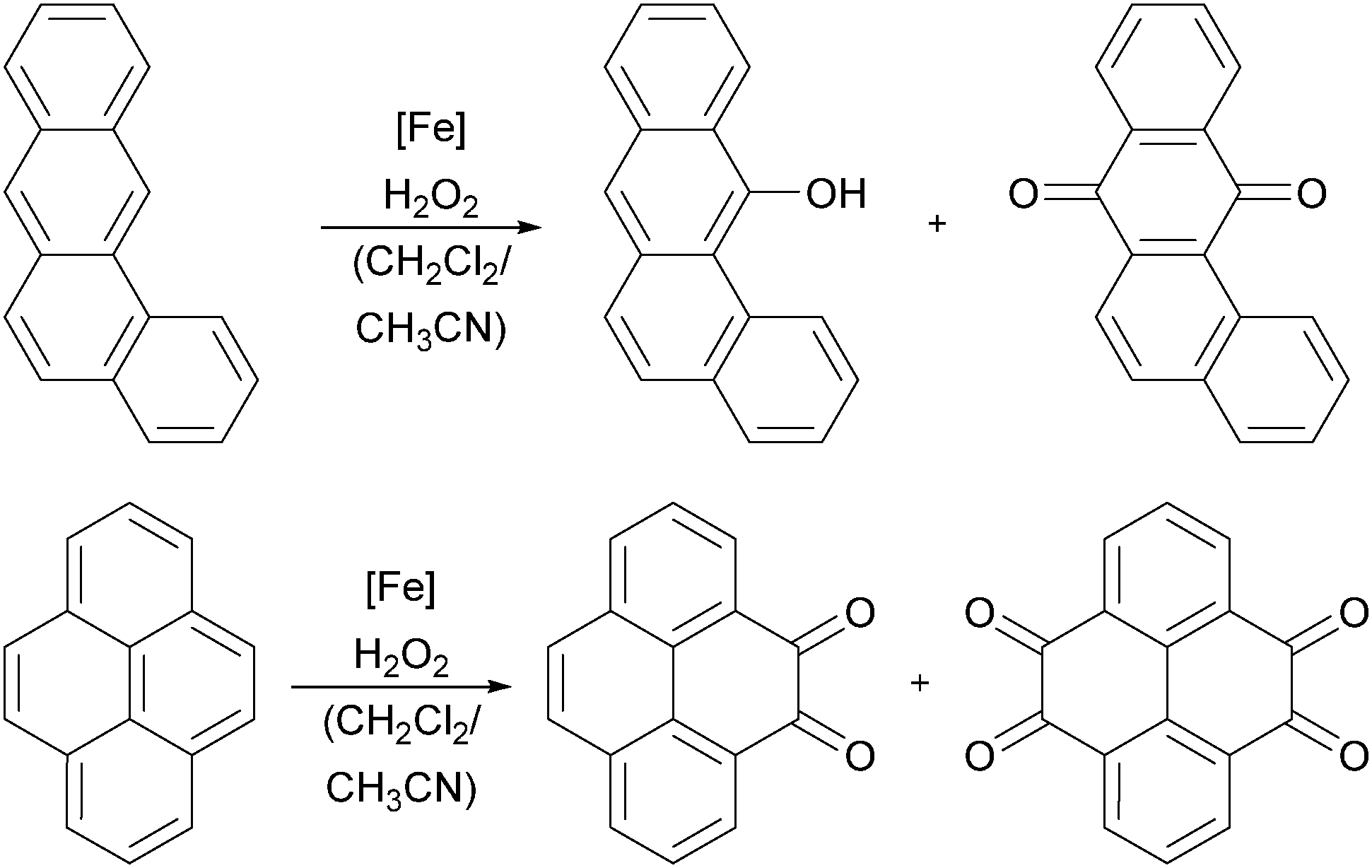 | ||
| Scheme 9 Oxidation of benzanthracene (top) and pyrene (bottom) by iron(III) porphyrin complexes and H2O2.174 | ||
In addition to metalloporphyrins, structurally related phthalocyanine and porphyrazine iron(III) complexes were found to catalyse the oxidation of aromatic C–H bonds. For example, Kudrik and Sorokin reported the catalytic oxidation of benzene with H2O2 by a μ-nitrido bridged iron tetra(tert-butylphthalocyanine) (2a, Fig. 2) complex.168 20% of benzene was converted with 12 turnovers and a NIH shift could be detected. The formation of benzene oxide during the reaction and a normal KIE indicate that the mechanism differs from that of iron porphyrin mediated reactions. The use of alternative oxidation reagents such as PhIO, iodosylbenzene sulfate or tetrabutylammonium oxone enables the oxidation of a variety of aromatic substrates catalysed by μ-oxo diiron complexes bearing phthalocyanine ligands 2b and 2c (Fig. 2).176,177 The systems prove to be very reactive and benzene can be oxidised to para-benzoquinone with 38% yield and 100% selectivity at a catalyst loading of 5 mol%.177 A common problem shared by these catalysts is the deactivation via oxidative degradation of the ligand scaffold. By elimination of all ligand C–H bonds, which was achieved using perfluorinated porphyrazine ligand 79 (Fig. 2), the stability under oxidative conditions could be increased.178
3.2 Iron nonheme complexes bearing pure N-donor ligands
In analogy to alkane oxidation reactions, nonheme iron complexes have been reported to act as catalysts for ring hydroxylation of aromatic substrates with various oxidants. Many of these contain pure N-donor ligands with varying denticity. Several mononuclear iron complexes with polyazadentate ligands (19c (Fig. 7), 80, 81 (Fig. 13)) catalyse the oxidation of benzene and substituted derivatives.179,180 Good selectivity for ring oxidation instead of benzylic oxidation in case of toluene or demethylation in case of anisole is observed. De Visser, Nam and co-workers used iron(II) complexes of the pentadentate ligands 13 and 30 to demonstrate that the corresponding iron(IV)–oxo species are active oxidants in arene oxygenation reactions.164 Similar to the above described iron porphyrin complexes the authors claimed that the reaction mechanism includes an electrophilic attack followed by the formation of a radical or cationic σ-complex (path b, Scheme 7), indicated by an inverse KIE and a large negative Hammett ρ value. An initial proton abstraction mechanism (path a, Scheme 7) could be ruled out on the basis of these findings. This also accounts for aromatic hydroxylation reactions mediated by BPMEN 10-supported iron catalysts as reported by Makhlynets and Rybak-Akimova.181 In this case, however, the iron(IV)–oxo complex was shown to be catalytically inactive and a different intermediate derived from a well-characterized ferric hydroperoxo species was assumed to be the active oxidant.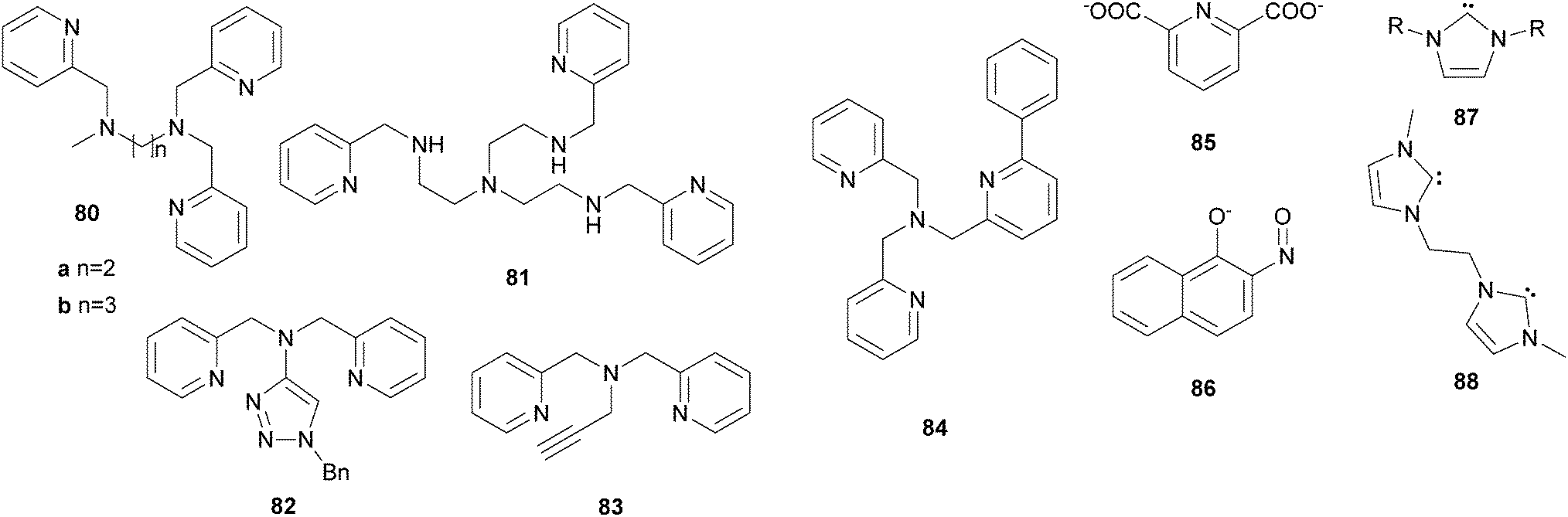 | ||
| Fig. 13 Examples of ligands used for iron-based arene oxidation reactions. | ||
Furthermore, iron(II) complexes bearing hexadentate ligands 34a or 34c (Fig. 5) were reported to be efficient catalysts for arene ring hydroxylation reactions with H2O2.182 Using 5 mol% [FeII(34a)](PF6)2 as the catalyst benzene is converted to phenol with 46% yield based on the oxidant, which can be enhanced by addition of reducing agents such as 1-naphtol. Complexes containing polyaza-ligands of lower denticity (19c, Fig. 5, 80a and 80b, Fig. 13) on the other hand exhibit reduced catalytic efficiency. In a later study a reaction mechanism was proposed involving the formation of a pair of caged oxidants including an iron(IV)–oxo species and a hydroxyl radical, which perform aromatic hydroxylation in a concerted manner.183 Che and co-workers demonstrated the versatility of their catalytic system, comprising mono- and diiron complexes with ligands of type 36 (Fig. 7) and oxone as the oxidant, by application to aromatic substrates.106 Several, mostly electron-rich, benzene derivatives can be oxidised to the corresponding quinones with good selectivity. Naphthoquinone for example is formed from naphthalene in 47% yield with 65% selectivity at a catalyst loading of 5 mol%. In addition to aliphatic C–H bond activation, hexacoordinate iron complexes bearing ligands 35a or 35b (Fig. 5) can also be used for the oxidation of anisole with H2O2.105 High affinity for ring hydroxylation is observed as well as a large ortho/para selectivity. Liu et al.184 used iron(II) complexes of ligands 7, 82, and 83 to relate electronic properties to catalytic performance. It could be shown that complexes with more negative reduction potentials exhibit a higher catalytic efficiency in benzene oxidation with H2O2 as oxygen source.
A close resemblance of the reactivity of nonheme aryl amino acid hydroxylases has been identified for iron complexes bearing phenyl derivatised TPA ligands of type 84 (Fig. 13).185 Intramolecular ortho-hydroxylation is observed after addition of tBHP and mechanistic investigations revealed that these systems are potent functional models, which support a better understanding of the enzymes' reactivity.185–187
3.4 Iron nonheme complexes bearing C-, O- and S-donor ligands
The replacement of nitrogen by other electron donating atoms such as oxygen, sulphur or carbenes allows the synthesis of manifold iron complexes, which can be used as catalysts in aromatic oxygenation reactions. Beller et al.188 observed the in situ formation of a catalyst from FeCl3, pyridine-2,6-dicarboxylic acid (85, Fig. 13), and a benzyl amine, which mediates the efficient and selective oxidation of non-activated aromatic substrates to the corresponding quinones. Among those were 2,3,5-trimethylbenzoquinone and 2-methylnaphtoquinone, which are industrially important intermediates for vitamin synthesis. An iron(II) complex bearing three bidentate 1-nitroso-2-naphtolate units (86, Fig. 13) was reported to be an efficient catalyst for benzene hydroxylation by Li et al.189 Starting with the observation of several by-products, such as hydroquinone and catechol, systematic optimisation of the reaction conditions led to exclusive formation of phenol in 10% yield. Structurally related catalysts containing bidentate quinolinolato ligands 50a–50e (Fig. 9) cannot reach this high degree of selectivity, although a similar, radical-based reaction mechanism has been proposed.125As already mentioned for alkane oxidation reactions, iron–NHC complex [FeII(71a)(CH3CN)2](PF6)2 was also applied for the oxygenation of aromatic substrates with H2O2 as the oxidant.153,190 In the presence of 1 mol% catalyst benzene and toluene are converted to phenols in yields of 11.2 and 14.7% respectively. In the case of toluene a high selectivity for aromatic instead of benzylic oxidation is observed. Based on reactivity studies and the observation of an inverse KIE the participation of a high-valent iron–oxo species in the catalytic reaction was proposed. Zhang et al.191 used mono- and bidentate carbene ligands 87 and 88 (Fig. 13) to form dinuclear iron complexes, which are further stabilised by a butane-2,3-dithiolate. Oxidation of benzene yields up to 26.7% phenol with H2O2 as the terminal oxidant. Again, a correlation between the reduction potential and the catalytic performance was observed, with the complex bearing two carbene donors and the most negative potential being most active.
Pombeiro and co-workers applied the iron(III) complex bearing N2S2 ligand 75 not only for alkane but also for arene oxidation.155 Under mild conditions a yield of 20% phenol and 110 turnovers were reported for the oxidation of benzene with H2O2 and PCA as additive. No formation of by-products has been observed.
4. Summary and outlook
The selective oxidation of organic C–H bonds is essential for many industrial and biological processes. Intense research efforts have been made in order to establish more efficient catalyst systems and increase sustainability in the synthesis of both commodity chemicals and organic molecules.A large number of iron complexes were applied as catalysts for the oxidation of aliphatic hydrocarbons with various oxidants. In particular nonheme iron catalysts bearing tetradentate N-donor ligands have shown potential for high efficiency. The first iron complex reported to catalyse the oxidation of alkanes with H2O2 by a metal-centred mechanism, is supported by the tetradentate TPA ligand 1a.48 Its reactivity and selectivity was soon surpassed by iron complexes containing ligands BPMEN (10) or Me,MePyTACN (24c); the latter yielding 65% oxygenated products from the oxidation of cyclohexane with H2O2 with an A/K ratio of 12 and a high degree of stereoselectivity.57,82,83 For these catalysts a coordination geometry exhibiting cis labile sites has been considered to be crucial to mediate oxidation with H2O2 by a water-assisted mechanism with high selectivity. They are still used as benchmark systems for newly developed catalysts. The successful application of some molecular iron complexes with predictable reactivity for the oxidation of complex organic molecules underlined the potential of iron-based catalysts in oxidation chemistry. It was reported that iron complexes bearing tetradentate N-donor ligands allowed the oxidation of natural products with high regio- and stereoselectivity under reaction conditions feasible for organic synthesis.42,67,68,73,74 However, catalyst degradation resulting in low turnover numbers (<100 for most systems) and the balance between metal-centred and radical-based reaction pathways pose major difficulties. Often generation of free radicals limits selectivity and leads to the formation of undesirable by-products. Therefore, in the past 10 years numerous investigations have been conducted to elucidate mechanistic aspects, including the nature of the active oxidising species. For selective, metal-centred oxidation pathways typically a high-valent iron–oxo complex has been proposed as the catalytically active species.
Depending on the application, problems regarding the oxidation of aromatic substrates are caused by the enhanced reactivity of the phenolic products. However, iron complexes have been found, which exhibit reactivity distinct from Fenton chemistry and catalyse the selective oxidation of arene C–H bonds. Among those are several catalysts containing N-donor ligands, which are well known for their high efficiency in alkane oxidation, but also other ligand classes have proven high potential.182 Heme-related phthalocyanine complexes for example also catalyse the selective oxidation of benzene to phenol with H2O2 as well as iron complexes bearing O-donor ligands or N-heterocyclic carbene ligands.168,189,190 Practical applications for environmental purposes, such as the oxidation of polycyclic aromatic hydrocarbons, were also reported.175 Furthermore, extensive mechanistic investigations contributed to a better understanding of synthetic as well as biological systems regarding reaction pathways and the identity of catalytically active intermediates and thus built a basis for a more rational catalyst design.
Despite those achievements most biomimetic and synthetic catalyst systems still lag behind enzymes with respect to reactivity and selectivity, and only very few practical applications have been realised. Therefore, further improvement is certainly necessary to develop efficient, environmentally friendly catalysts capable of oxidising C–H bonds with benign oxidants as alternatives to traditionally used systems.
Acknowledgements
The authors gratefully acknowledge financial support by the Fonds der Chemischen Industrie (FCI) for A.C.L. and of the TUM graduate school.References
- F. Jia and Z. Li, Org. Chem. Front., 2014, 1, 194–214 RSC.
- M. Bordeaux, A. Galarneau and J. Drone, Angew. Chem., Int. Ed., 2012, 51, 10712–10723 CrossRef CAS PubMed.
- A. Company, J. Lloret, L. Gómez and M. Costas, in Alkane C-H Activation by Single-Site Metal Catalysis, ed. P. J. Pérez, Springer, Heidelberg, 2012, ch. 5, vol. 38, pp. 143–228 Search PubMed.
- R. Sheldon, Metal-Catalyzed Oxidations of Organic Compounds: Mechanistic Principles and Synthetic Methodology Including Biochemical Processes, Elsevier Science, Amsterdam, 2012 Search PubMed.
- C. L. Hill, Nature, 1999, 401, 436–437 CrossRef CAS PubMed.
- L. Que Jr. and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef PubMed.
- P. R. Ortiz de Montellano, Chem. Rev., 2010, 110, 932–948 CrossRef CAS PubMed.
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que Jr., Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed.
- A. A. Shteinman, Russ. Chem. Rev., 2008, 77, 945–966 CrossRef CAS PubMed.
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed.
- B. Meunier, S. P. de Visser and S. Shaik, Chem. Rev., 2004, 104, 3947–3980 CrossRef CAS PubMed.
- S. Tanase and E. Bouwman, in Advances in Inorganic Chemistry, ed. R. van Eldik and J. Reedijk, Academic Press, Waltham, 2006, vol. 58, pp. 29–75 Search PubMed.
- K. P. Bryliakov and E. P. Talsi, Coord. Chem. Rev., 2014, 276, 73–96 CrossRef CAS PubMed.
- M. Christmann, Angew. Chem., Int. Ed., 2008, 47, 2740–2742 CrossRef CAS PubMed.
- W. N. Oloo and L. Que Jr, in Comprehensive Inorganic Chemistry II, ed. K. Poeppelmeier and J. Reedijk, Elsevier, Amsterdam, 2nd edn, 2013, ch. 6.26, pp. 763–778 Search PubMed.
- C.-L. Sun, B.-J. Li and Z.-J. Shi, Chem. Rev., 2011, 111, 1293–1314 CrossRef CAS PubMed.
- K. Riener, S. Haslinger, A. Raba, M. P. Högerl, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Rev., 2014, 114, 5215–5272 CrossRef CAS PubMed.
- H. Srour, P. Le Maux, S. Chevance and G. Simonneaux, Coord. Chem. Rev., 2013, 257, 3030–3050 CrossRef CAS PubMed.
- J. Piera and J.-E. Bäckvall, Angew. Chem., Int. Ed., 2008, 47, 3506–3523 CrossRef CAS PubMed.
- J.-E. Bäckvall, Modern Oxidation Methods, Wiley-VCH, Weinheim, 2004 Search PubMed.
- E. Y. Tshuva and S. J. Lippard, Chem. Rev., 2004, 104, 987–1012 CrossRef CAS PubMed.
- M. Fontecave, S. Ménage and C. Duboc-Toia, Coord. Chem. Rev., 1998, 178–180(part 2), 1555–1572 CrossRef CAS.
- M. Costas, K. Chen and L. Que Jr, Coord. Chem. Rev., 2000, 200–202, 517–544 CrossRef CAS.
- P. V. Saji, C. Ratnasamy and S. Gopinathan, US Pat., 6392093 (B1), 2002 Search PubMed.
- G. Duca, Homogeneous Catalysis with Metal Complexes: Fundamentals and Applications, Springer, Heidelberg, 2012 Search PubMed.
- E. P. Talsi and K. P. Bryliakov, Coord. Chem. Rev., 2012, 256, 1418–1434 CrossRef CAS PubMed.
- M. Costas, Coord. Chem. Rev., 2011, 255, 2912–2932 CrossRef CAS PubMed.
- A. R. McDonald and L. Que Jr, Coord. Chem. Rev., 2013, 257, 414–428 CrossRef CAS PubMed.
- S. V. Kryatov, E. V. Rybak-Akimova and S. Schindler, Chem. Rev., 2005, 105, 2175–2226 CrossRef CAS PubMed.
- B. Meunier, Chem. Rev., 1992, 92, 1411–1456 CrossRef CAS.
- J. T. Groves, T. E. Nemo and R. S. Myers, J. Am. Chem. Soc., 1979, 101, 1032–1033 CrossRef CAS.
- S.-J. Li and Y.-G. Wang, Tetrahedron Lett., 2005, 46, 8013–8015 CrossRef CAS PubMed.
- A. Singh, A. Agarwala, K. Kamaraj and D. Bandyopadhyay, Inorg. Chim. Acta, 2011, 372, 295–303 CrossRef CAS PubMed.
- G. M. Ucoski, K. A. D. F. DCastro, K. J. Ciuffi, G. P. Ricci, J. A. Marques, F. S. Nunes and S. Nakagaki, Appl. Catal., A, 2011, 404, 120–128 CrossRef CAS PubMed.
- A. B. Sorokin, E. V. Kudrik and D. Bouchu, Chem. Commun., 2008, 2562–2564 RSC.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152–8191 CrossRef CAS PubMed.
- E. S. Brown, J. R. Robinson, A. M. McCoy and R. W. McGaff, Dalton Trans., 2011, 40, 5921–5925 RSC.
- Z. Gross, L. Simkhovich and N. Galili, Chem. Commun., 1999, 599–600 RSC.
- A. N. Biswas, P. Das, A. Agarwala, D. Bandyopadhyay and P. Bandyopadhyay, J. Mol. Catal. A: Chem., 2010, 326, 94–98 CrossRef CAS PubMed.
- A. N. Biswas, A. Pariyar, S. Bose, P. Das and P. Bandyopadhyay, Catal. Commun., 2010, 11, 1008–1011 CrossRef CAS PubMed.
- A. Pariyar, S. Bose, A. N. Biswas, P. Das and P. Bandyopadhyay, Catal. Commun., 2013, 32, 23–27 CrossRef CAS PubMed.
- M. C. White, Science, 2012, 335, 807–809 CrossRef CAS PubMed.
- S. McArthur and M. C. Baird, Eur. Polym. J., 2014, 55, 170–178 CrossRef CAS PubMed.
- R. A. Leising, R. E. Norman and L. Que Jr, Inorg. Chem., 1990, 29, 2553–2555 CrossRef CAS.
- R. A. Leising, B. A. Brennan, L. Que Jr, B. G. Fox and E. Münck, J. Am. Chem. Soc., 1991, 113, 3988–3990 CrossRef CAS.
- T. Kojima, R. A. Leising, S. Yan and L. Que Jr, J. Am. Chem. Soc., 1993, 115, 11328–11335 CrossRef CAS.
- R. A. Leising, J. Kim, M. A. Pérez and L. Que Jr, J. Am. Chem. Soc., 1993, 115, 9524–9530 CrossRef CAS.
- C. Kim, K. Chen, J. Kim and L. Que Jr, J. Am. Chem. Soc., 1997, 119, 5964–5965 CrossRef CAS.
- K. Chen and L. Que Jr, J. Am. Chem. Soc., 2001, 123, 6327–6337 CrossRef CAS PubMed.
- K. Chen, M. Costas and L. Que Jr, J. Chem. Soc., Dalton Trans., 2002, 672–679 RSC.
- A. Bassan, M. R. A. Blomberg, P. E. M. Siegbahn and L. Que Jr, Chem. – Eur. J., 2005, 11, 692–705 CrossRef CAS PubMed.
- Y. Hitomi, S. Furukawa, M. Higuchi, T. Shishido and T. Tanaka, J. Mol. Catal. A: Chem., 2008, 288, 83–86 CrossRef CAS PubMed.
- H. Jaafar, B. Vileno, A. Thibon and D. Mandon, Dalton Trans., 2011, 40, 92–106 RSC.
- G. Guisado-Barrios, A. M. Z. Slawin and D. T. Richens, J. Coord. Chem., 2010, 63, 2642–2658 CrossRef CAS PubMed.
- A. Mairata i Payeras, R. Y. N. Ho, M. Fujita and L. Que Jr, Chem. – Eur. J., 2004, 10, 4944–4953 CrossRef CAS PubMed.
- M. Klopstra, G. Roelfes, R. Hage, R. M. Kellogg and B. L. Feringa, Eur. J. Inorg. Chem., 2004, 846–856 CrossRef CAS PubMed.
- T. Okuno, S. Ito, S. Ohba and Y. Nishida, J. Chem. Soc., Dalton Trans., 1997, 3547–3551 RSC.
- Y. Mekmouche, S. Ménage, C. Toia-Duboc, M. Fontecave, J.-B. Galey, C. Lebrun and J. Pécaut, Angew. Chem., Int. Ed., 2001, 40, 949–952 CrossRef CAS.
- Y. Mekmouche, S. Ménage, J. Pécaut, C. Lebrun, L. Reilly, V. Schuenemann, A. Trautwein and M. Fontecave, Eur. J. Inorg. Chem., 2004, 3163–3171 CrossRef CAS PubMed.
- G. J. P. Britovsek, J. England and A. J. P. White, Inorg. Chem., 2005, 44, 8125–8134 CrossRef CAS PubMed.
- J. England, C. R. Davies, M. Banaru, A. J. P. White and G. J. P. Britovsek, Adv. Synth. Catal., 2008, 350, 883–897 CrossRef CAS PubMed.
- M. Grau, A. Kyriacou, F. Cabedo Martinez, I. M. de Wispelaere, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2014, 43, 17108–17119 RSC.
- G. J. P. Britovsek, J. England and A. J. P. White, Dalton Trans., 2006, 1399–1408 RSC.
- J. England, G. J. P. Britovsek, N. Rabadia and A. J. P. White, Inorg. Chem., 2007, 46, 3752–3767 CrossRef CAS PubMed.
- J. England, R. Gondhia, L. Bigorra-Lopez, A. R. Petersen, A. J. P. White and G. J. P. Britovsek, Dalton Trans., 2009, 5319–5334 RSC.
- O. Y. Lyakin, K. P. Bryliakov, G. J. P. Britovsek and E. P. Talsi, J. Am. Chem. Soc., 2009, 131, 10798–10799 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2007, 318, 783–787 CrossRef CAS PubMed.
- M. S. Chen and M. C. White, Science, 2010, 327, 566–571 CrossRef CAS PubMed.
- N. A. Vermeulen, M. S. Chen and M. C. White, Tetrahedron, 2009, 65, 3078–3084 CrossRef CAS PubMed.
- P. E. Gormisky and M. C. White, J. Am. Chem. Soc., 2013, 135, 14052–14055 CrossRef CAS PubMed.
- V. A. Yazerski, P. Spannring, D. Gatineau, C. H. M. Woerde, S. M. Wieclawska, M. Lutz, H. Kleijn and R. J. M. Klein Gebbink, Org. Biomol. Chem., 2014, 12, 2062–2070 CAS.
- L. Gómez, I. Garcia-Bosch, A. Company, J. Benet-Buchholz, A. Polo, X. Sala, X. Ribas and M. Costas, Angew. Chem., Int. Ed., 2009, 48, 5720–5723 CrossRef PubMed.
- L. Gómez, M. Canta, D. Font, I. Prat, X. Ribas and M. Costas, J. Org. Chem., 2013, 78, 1421–1433 CrossRef PubMed.
- M. Canta, D. Font, L. Gómez, X. Ribas and M. Costas, Adv. Synth. Catal., 2014, 356, 818–830 CrossRef CAS PubMed.
- Y. He, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2011, 50, 12651–12660 CrossRef CAS PubMed.
- Q. Zhang, J. D. Gorden and C. R. Goldsmith, Inorg. Chem., 2013, 52, 13546–13554 CrossRef CAS PubMed.
- Y. He and C. R. Goldsmith, Chem. Commun., 2012, 48, 10532–10534 RSC.
- E. A. Gutkina, T. B. Rubtsova and A. A. Shteinman, Kinet. Catal., 2003, 44, 106–111 CrossRef CAS.
- E. A. Turitsyna, O. N. Gritsenko and A. A. Shteinman, Kinet. Catal., 2007, 48, 53–59 CrossRef CAS.
- E. A. Turitsyna and A. A. Shteinman, Russ. Chem. Bull., 2011, 60, 2094–2099 CrossRef CAS.
- A. A. Shteinman, Russ. J. Inorg. Chem., 2012, 57, 1328–1334 CrossRef CAS.
- A. Company, L. Gómez, M. Güell, X. Ribas, J. M. Luis, L. Que Jr. and M. Costas, J. Am. Chem. Soc., 2007, 129, 15766–15767 CrossRef CAS PubMed.
- A. Company, L. Gómez, X. Fontrodona, X. Ribas and M. Costas, Chem. – Eur. J., 2008, 14, 5727–5731 CrossRef CAS PubMed.
- I. Prat, J. S. Mathieson, M. Güell, X. Ribas, J. M. Luis, L. Cronin and M. Costas, Nat. Chem., 2011, 3, 788–793 CrossRef CAS PubMed.
- I. Prat, L. Gómez, M. Canta, X. Ribas and M. Costas, Chem. – Eur. J., 2013, 19, 1908–1913 CrossRef CAS PubMed.
- I. Prat, A. Company, V. Postils, X. Ribas, L. Que Jr, J. M. Luis and M. Costas, Chem. – Eur. J., 2013, 19, 6724–6738 CrossRef CAS PubMed.
- I. Prat, A. Company, T. Corona, T. Parella, X. Ribas and M. Costas, Inorg. Chem., 2013, 52, 9229–9244 CrossRef CAS PubMed.
- S. Lentini, P. Galloni, I. Garcia-Bosch, M. Costas and V. Conte, Inorg. Chim. Acta, 2014, 410, 60–64 CrossRef CAS PubMed.
- G. B. Shul'pin, C. C. Golfeto, G. Süss-Fink, L. S. Shul'pina and D. Mandelli, Tetrahedron Lett., 2005, 46, 4563–4567 CrossRef PubMed.
- P. C. A. Bruijnincx, I. L. C. Buurmans, Y. Huang, G. Juhász, M. Viciano-Chumillas, M. Quesada, J. Reedijk, M. Lutz, A. L. Spek, E. Münck, E. L. Bominaar and R. J. M. Klein Gebbink, Inorg. Chem., 2011, 50, 9243–9255 CrossRef CAS PubMed.
- G. de Ruiter, J. S. Costa, K. Lappalainen, O. Roubeau, P. Gamez and J. Reedijk, Inorg. Chem. Commun., 2008, 11, 787–790 CrossRef CAS PubMed.
- T. F. S. Silva, E. C. B. A. Alegria, L. M. D. R. S. Martins and A. J. L. Pombeiro, Adv. Synth. Catal., 2008, 350, 706–716 CrossRef CAS PubMed.
- T. F. S. Silva, M. F. Guedes da Silva, G. S. Mishra, L. M. D. R. S. Martins and A. J. L. Pombeiro, J. Organomet. Chem., 2011, 696, 1310–1318 CrossRef CAS PubMed.
- P. Comba, M. Maurer and P. Vadivelu, J. Phys. Chem. A, 2008, 112, 13028–13036 CrossRef CAS PubMed.
- P. Comba, M. Maurer and P. Vadivelu, Inorg. Chem., 2009, 48, 10389–10396 CrossRef CAS PubMed.
- P. Comba, Y.-M. Lee, W. Nam and A. Waleska, Chem. Commun., 2014, 50, 412–414 RSC.
- Y.-M. Lee, S. N. Dhuri, S. C. Sawant, J. Cho, M. Kubo, T. Ogura, S. Fukuzumi and W. Nam, Angew. Chem., 2009, 121, 1835–1838 CrossRef PubMed.
- I. Garcia-Bosch, Z. Codolà, I. Prat, X. Ribas, J. Lloret-Fillol and M. Costas, Chem. – Eur. J., 2012, 18, 13269–13273 CrossRef CAS PubMed.
- Y. Hitomi, K. Arakawa, T. Funabiki and M. Kodera, Angew. Chem., Int. Ed., 2012, 51, 3448–3452 CrossRef CAS PubMed.
- Y. Hitomi, K. Arakawa and M. Kodera, Chemistry, 2013, 19, 14697–14701 CrossRef PubMed.
- E. Wong, J. Jeck, M. Grau, A. J. P. White and G. J. P. Britovsek, Catal. Sci. Technol., 2013, 3, 1116–1122 CAS.
- J. Xiang, H. Li and J.-S. Wu, Z. Anorg. Allg. Chem., 2014, 640, 1670–1674 CrossRef CAS PubMed.
- M. Martinho, F. Banse, J.-F. Bartoli, T. A. Mattioli, P. Battioni, O. Horner, S. Bourcier and J.-J. Girerd, Inorg. Chem., 2005, 44, 9592–9596 CrossRef CAS PubMed.
- A. Thibon, J.-F. Bartoli, S. Bourcier and F. Banse, Dalton Trans., 2009, 9587–9594 RSC.
- N. Ségaud, J.-N. Rebilly, K. Sénéchal-David, R. Guillot, L. Billon, J.-P. Baltaze, J. Farjon, O. Reinaud and F. Banse, Inorg. Chem., 2013, 52, 691–700 CrossRef PubMed.
- P. Liu, Y. Liu, E. L.-M. Wong, S. Xiang and C.-M. Che, Chem. Sci., 2011, 2, 2187–2195 RSC.
- G. J. P. Britovsek, J. England, S. K. Spitzmesser, A. J. P. White and D. J. Williams, Dalton Trans., 2005, 945–955 RSC.
- B. Retcher, J. S. Costa, J. Tang, R. Hage, P. Gamez and J. Reedijk, J. Mol. Catal. A: Chem., 2008, 286, 1–5 CrossRef CAS PubMed.
- S. Tanase, J. Reedijk, R. Hage and G. Rothenberg, Top Catal., 2010, 53, 1039–1044 CrossRef CAS.
- P. Shejwalkar, N. P. Rath and E. B. Bauer, Dalton Trans., 2011, 40, 7617–7631 RSC.
- A. R. Silva, T. Mourao and J. Rocha, Catal. Today, 2013, 203, 81–86 CrossRef CAS PubMed.
- G. Olivo, G. Arancio, L. Mandolini, O. Lanzalunga and S. Di Stefano, Catal. Sci. Technol., 2014, 4, 2900–2903 CAS.
- N. S. Venkataramanan, G. Kuppuraj and S. Rajagopal, Coord. Chem. Rev., 2005, 249, 1249–1268 CrossRef CAS PubMed.
- G. C. Salomão, M. H. N. Olsen, V. Drago, C. Fernandes, L. Cardozo Filho and O. A. C. Antunes, Catal. Commun., 2006, 8, 69–72 CrossRef PubMed.
- A. N. Biswas, P. Das, S. K. Kandar, A. Agarwala, D. Bandyopadhyay and P. Bandyopadhyay, Catal. Commun., 2009, 10, 708–711 CrossRef CAS PubMed.
- R. Mayilmurugan, H. Stoeckli-Evans, E. Suresh and M. Palaniandavar, Dalton Trans., 2009, 5101–5114 RSC.
- G. Bilis, K. C. Christoforidis, Y. Deligiannakis and M. Louloudi, Catal. Today, 2010, 157, 101–106 CrossRef CAS PubMed.
- G. Bilis and M. Louloudi, Bioinorg. Chem. Appl., 2010, 2010 Search PubMed.
- D. S. Nesterov, O. V. Nesterova, M. F. C. Guedes da Silva and A. J. L. Pombeiro, Catal. Sci. Technol., 2015, 5, 1801–1812 CAS.
- T. L. Foster and J. P. Caradonna, J. Am. Chem. Soc., 2003, 125, 3678–3679 CrossRef CAS PubMed.
- L. C. Gregor, G. T. Rowe, E. Rybak-Akimova and J. P. Caradonna, Dalton Trans., 2012, 41, 777–782 RSC.
- T. Nagataki, Y. Tachi and S. Itoh, J. Mol. Catal. A: Chem., 2005, 225, 103–109 CrossRef CAS PubMed.
- S. Nayak, P. Gamez, B. Kozlevčar, A. Pevec, O. Roubeau, S. Dehnen and J. Reedijk, Polyhedron, 2010, 29, 2291–2296 CrossRef CAS PubMed.
- M. N. Kopylovich, T. C. O. MacLeod, M. Haukka, G. I. Amanullayeva, K. T. Mahmudov and A. J. L. Pombeiro, J. Inorg. Biochem., 2012, 115, 72–77 CrossRef CAS PubMed.
- Y. Wang, Z. Fu, X. Wen, C. Rong, W. Wu, C. Zhang, J. Deng, B. Dai, S. R. Kirk and D. Yin, J. Mol. Catal. A: Chem., 2014, 383–384, 46–52 CrossRef CAS PubMed.
- E. A. Gutkina, V. M. Trukhan, C. G. Pierpont, S. Mkoyan, V. V. Strelets, E. Nordlander and A. A. Shteinman, Dalton Trans., 2006, 492–501 RSC.
- M. Jarenmark, E. A. Turitsyna, M. Haukka, A. A. Shteinman and E. Nordlander, New J. Chem., 2010, 34, 2118 RSC.
- G. Trettenhahn, M. Nagl, N. Neuwirth, V. B. Arion, W. Jary, P. Pöchlauer and W. Schmid, Angew. Chem., Int. Ed., 2006, 45, 2794–2798 CrossRef CAS PubMed.
- Y. Kim, X. Feng and S. J. Lippard, Inorg. Chem., 2007, 46, 6099–6107 CrossRef CAS PubMed.
- V. B. Romakh, B. Therrien, G. Süss-Fink and G. B. Shul'pin, Inorg. Chem., 2007, 46, 3166–3175 CrossRef CAS PubMed.
- M. D. Godbole, M. P. Puig, S. Tanase, H. Kooijman, A. L. Spek and E. Bouwman, Inorg. Chim. Acta, 2007, 360, 1954–1960 CrossRef CAS PubMed.
- M. Lenze and E. B. Bauer, J. Mol. Catal. A: Chem., 2009, 309, 117–123 CrossRef CAS PubMed.
- N. M. F. Carvalho, A. Horn Jr. and O. A. C. Antunes, Appl. Catal., A, 2006, 305, 140–145 CrossRef CAS PubMed.
- N. M. F. Carvalho, H. M. Alvarez, A. Horn Jr. and O. A. C. Antunes, Catal. Today, 2008, 133–135, 689–694 CrossRef CAS PubMed.
- G. L. Parrilha, S. S. Ferreira, C. Fernandes, G. C. Silva, N. M. F. Carvalho, O. A. C. Antunes, V. Drago, A. J. Bortoluzzi and A. Horn Jr, J. Braz. Chem. Soc., 2010, 21, 603–613 CrossRef CAS.
- S. Tanase, P. Marques-Gallego, W. R. Browne, R. Hage, E. Bouwman, B. L. Feringa and J. Reedijk, Dalton Trans., 2008, 2026–2033 RSC.
- M. N. M. Milunovic, L. M. D. R. S. Martins, E. C. B. A. Alegria, A. J. L. Pombeiro, R. Krachler, G. Trettenhahn, C. Turta, S. Shova and V. B. Arion, Dalton Trans., 2013, 42, 14388–14401 RSC.
- S. Cheng, J. Li, X. Yu, C. Chen, H. Ji, W. Ma and J. Zhao, New J. Chem., 2013, 37, 3267 RSC.
- V. J. Dungan, B. M. L. Poon, E. S. Barrett and P. J. Rutledge, Tetrahedron Lett., 2013, 54, 1236–1238 CrossRef CAS PubMed.
- C.-W. Tse, T. W.-S. Chow, Z. Guo, H. K. Lee, J.-S. Huang and C.-M. Che, Angew. Chem., Int. Ed., 2014, 53, 798–803 CrossRef CAS PubMed.
- S. Chatterjee and T. K. Paine, Angew. Chem., Int. Ed., 2015, 54, 9338–9342 CrossRef CAS PubMed.
- S. Tanase, C. Foltz, R. de Gelder, R. Hage, E. Bouwman and J. Reedijk, J. Mol. Catal. A: Chem., 2005, 225, 161–167 CrossRef CAS PubMed.
- F. Li, M. Wang, C. Ma, A. Gao, H. Chen and L. Sun, Dalton Trans., 2006, 2427–2434 RSC.
- J. Tang, P. Gamez and J. Reedijk, Dalton Trans., 2007, 4644–4646 RSC.
- S. Gosiewska, H. P. Permentier, A. P. Bruins, G. van Koten and R. J. M. K. Gebbink, Dalton Trans., 2007, 3365–3368 RSC.
- M. Lenze, S. L. Sedinkin and E. B. Bauer, J. Mol. Catal. A: Chem., 2013, 373, 161–171 CrossRef CAS PubMed.
- Q. Zhang and C. R. Goldsmith, Inorg. Chim. Acta, 2013, 406, 301–306 CrossRef CAS PubMed.
- A. Raba, M. Cokoja, S. Ewald, K. Riener, E. Herdtweck, A. Pöthig, W. A. Herrmann and F. E. Kühn, Organometallics, 2012, 31, 2793–2800 CrossRef CAS.
- M. R. Anneser, S. Haslinger, A. Pöthig, M. Cokoja, J.-M. Basset and F. E. Kühn, Inorg. Chem., 2015, 54, 3797–3804 CrossRef CAS PubMed.
- D. T. Weiss, P. J. Altmann, S. Haslinger, C. Jandl, A. Pöthig, M. Cokoja and F. E. Kühn, Dalton Trans., 2015 10.1039/C5DT02386F.
- I. Klawitter, M. R. Anneser, S. Dechert, S. Meyer, S. Demeshko, S. Haslinger, A. Pöthig, F. E. Kühn and F. Meyer, Organometallics, 2015, 34, 2819–2825 CrossRef CAS.
- S. Haslinger, J. W. Kück, E. M. Hahn, M. Cokoja, A. Pöthig, J. M. Basset and F. E. Kühn, Inorg. Chem., 2014, 53, 11573–11583 CrossRef CAS PubMed.
- J. W. Kück, A. Raba, I. I. E. Markovits, M. Cokoja and F. E. Kühn, ChemCatChem, 2014, 6, 1882–1886 CrossRef PubMed.
- S. Haslinger, A. Raba, M. Cokoja, A. Pöthig and F. E. Kühn, J. Catal., 2015, 331, 147–153 CrossRef CAS PubMed.
- R. R. Fernandes, M. V. Kirillova, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2009, 353, 107–112 CrossRef CAS PubMed.
- R. R. Fernandes, J. Lasri, M. F. C. Guedes da Silva, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Appl. Catal., A, 2011, 402, 110–120 CrossRef CAS PubMed.
- R. R. Fernandes, J. Lasri, A. M. Kirillov, M. F. C. Guedes da Silva, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, Eur. J. Inorg. Chem., 2011, 3781–3790 CAS.
- R. R. Fernandes, J. Lasri, M. F. C. Guedes da Silva, J. A. L. da Silva, J. J. R. Frausto da Silva and A. J. L. Pombeiro, J. Mol. Catal. A: Chem., 2011, 351, 100–111 CrossRef CAS PubMed.
- E. E. Karslyan, L. S. Shul'pina, Y. N. Kozlov, A. J. L. Pombeiro and G. B. Shul'pin, Catal. Today, 2013, 218–219, 93–98 CrossRef CAS PubMed.
- Y. Kaneko, J.-I. Kadokawa, H. Tagaya and K. Chiba, Recent Res. Dev. Pure Appl. Chem., 1998, 2, 563–572 CAS.
- Y. R. Luo, Handbook of Bond Dissociation Energies in Organic Compounds, CRC Press, Boca Raton, 2002 Search PubMed.
- K. Schröder, K. Junge, B. Bitterlich and M. Beller, in Iron Catalysis, ed. B. Plietker, Springer, Heidelberg, 2011, ch. 3, vol. 33, pp. 83–109 Search PubMed.
- M.-J. Kang, W. J. Song, A.-R. Han, Y. S. Choi, H. G. Jang and W. Nam, J. Org. Chem., 2007, 72, 6301–6304 CrossRef CAS PubMed.
- S. P. de Visser, K. Oh, A.-R. Han and W. Nam, Inorg. Chem., 2007, 46, 4632–4641 CrossRef CAS PubMed.
- K. H. Mitchell, C. E. Rogge, T. Gierahn and B. G. Fox, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3784–3789 CrossRef CAS PubMed.
- K. R. Korzekwa, D. C. Swinney and W. F. Trager, Biochemistry, 1989, 28, 9019–9027 CrossRef CAS.
- G. Guroff, J. Renson, S. Udenfriend, J. W. Daly, D. M. Jerina and B. Witkop, Science, 1967, 157, 1524–1530 CAS.
- E. V. Kudrik and A. B. Sorokin, Chem. – Eur. J., 2008, 14, 7123–7126 CAS.
- I. Artaud, K. B. Aziza, C. Chopard and D. Mansuy, J. Chem. Soc., Chem. Commun., 1991, 31–33, 10.1039/C39910000031.
- M.-N. Carrier, C. Scheer, P. Gouvine, J.-F. Bartoli, P. Battioni and D. Mansuy, Tetrahedron Lett., 1990, 31, 6645–6648 CrossRef CAS.
- S. Tsuchiya and M. Seno, Chem. Lett., 1989, 263–266 CrossRef CAS.
- A.-R. Han, Y. J. Jeong, Y. Kang, J. Y. Lee, M. S. Seo and W. Nam, Chem. Commun., 2008, 1076–1078 RSC.
- D. Kumar, G. N. Sastry and S. P. de Visser, J. Phys. Chem. B, 2012, 116, 718–730 CrossRef CAS PubMed.
- N. G. Giri and S. M. S. Chauhan, Catal. Commun., 2009, 10, 383–387 CrossRef CAS PubMed.
- A. Sorokin and B. Meunier, Eur. J. Inorg. Chem., 1998, 1269–1281 CrossRef CAS.
- H. M. Neu, M. S. Yusubov, V. V. Zhdankin and V. N. Nemykin, Adv. Synth. Catal., 2009, 351, 3168–3174 CrossRef CAS PubMed.
- H. M. Neu, V. V. Zhdankin and V. N. Nemykin, Tetrahedron Lett., 2010, 51, 6545–6548 CrossRef CAS PubMed.
- M. S. Yusubov, C. Celik, M. R. Geraskina, A. Yoshimura, V. V. Zhdankin and V. N. Nemykin, Tetrahedron Lett., 2014, 55, 5687–5690 CrossRef CAS PubMed.
- J.-F. Bartoli, F. Lambert, I. Morgenstern-Badarau, P. Battioni and D. Mansuy, C. R. Chim., 2002, 5, 263–266 CrossRef CAS.
- V. Balland, D. Mathieu, N. Pons-Y-Moll, J. F. Bartoli, F. Banse, P. Battioni, J.-J. Girerd and D. Mansuy, J. Mol. Catal. A: Chem., 2004, 215, 81–87 CrossRef CAS PubMed.
- O. V. Makhlynets and E. V. Rybak-Akimova, Chem. – Eur. J., 2010, 16, 13995–14006 CrossRef CAS PubMed.
- A. Thibon, J.-F. Bartoli, R. Guillot, J. Sainton, M. Martinho, D. Mansuy and F. Banse, J. Mol. Catal. A: Chem., 2008, 287, 115–120 CrossRef CAS PubMed.
- A. Thibon, V. Jollet, C. Ribal, K. Sénéchal-David, L. Billon, A. B. Sorokin and F. Banse, Chem. – Eur. J., 2012, 18, 2715–2724 CrossRef CAS PubMed.
- B. Xu, W. Zhong, Z. Wei, H. Wang, J. Liu, L. Wu, Y. Feng and X. Liu, Dalton Trans., 2014, 43, 15337–15345 RSC.
- M. P. Jensen, S. J. Lange, M. P. Mehn, E. L. Que and L. Que Jr, J. Am. Chem. Soc., 2003, 125, 2113–2128 CrossRef CAS PubMed.
- S. Ménage, J.-B. Galey, G. Hussler, M. Seité and M. Fontecave, Angew. Chem., Int. Ed. Engl., 1996, 35, 2353–2355 CrossRef PubMed.
- A. Bassan, M. R. Blomberg and P. E. Siegbahn, Chemistry, 2003, 9, 4055–4067 CrossRef CAS PubMed.
- K. Möller, G. Wienhöfer, K. Schröder, B. Join, K. Junge and M. Beller, Chem. – Eur. J., 2010, 16, 10300–10303 CrossRef PubMed.
- X. Wang, T. Zhang, B. Li, Q. Yang and S. Jiang, Appl. Organomet. Chem., 2014, 28, 666–672 CrossRef CAS PubMed.
- A. Raba, M. Cokoja, W. A. Herrmann and F. E. Kühn, Chem. Commun., 2014, 50, 11454–11457 RSC.
- Y. Wang, T. Zhang, B. Li, S. Jiang and L. Sheng, RSC Adv., 2015, 5, 29022–29031 RSC.
| This journal is © The Royal Society of Chemistry 2015 |