
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomimetic self-assembling acylphthalocyanines†
Hong-Guang
Jin
,
Mihaela Carmen
Balaban
,
Sabine
Chevallier-Michaud
,
Michel
Righezza
and
Teodor Silviu
Balaban
*
Aix-Marseille Université, CNRS UMR 7313, Centrale Marseille, Chirosciences, Service 442, Avenue Escadrille Normandie-Niemen, F-13013 Marseille, France. E-mail: ts.balaban@univ-amu.fr
First published on 26th June 2015
Abstract
We synthesized a series of biomimetic self-assembling phthalocyanines equipped with carbonyl groups as recognition motifs, a central zinc atom and diverse solubilizing alkyl chains mimicking for the first time with these robust pigments the natural chlorosomal bacteriochlorophylls. Upon self-assembly a very broad and red-shifted Q-band absorption extending to over 900 nm is put into evidence.
Highly ordered self-assembling functional dyes with unique photoelectric properties as a consequence of excitonic interactions between neighboring dye monomers are desirable for materials science.1 A well-defined manner connecting these dye monomers are crucial for a device to transfer the excitation energy to a designated acceptor for efficient energy utilization.2 Compared to traditional covalent bonds, the supramolecular self-assembly based on noncovalent interactions is an useful approach to reach this goal, including (i) relative easy synthesis of monomers, (ii) the self-healing and thermodynamically stable self-assemblies. Phthalocyanines (Pc's),3 are the most stable and robust light-harvesting system with high absorption coefficients in the visible and near-infrared region. However, Pc's based devices have been rather limited due to their poor solubility in common organic solvents and their disordered aggregation on the corresponding surface,4 meanwhile, in contrast with other dyes,5 incorporating specific recognition sites into Pc's to induce self-assembly, is still synthetically challenging.
Natural light-harvesting systems function with high efficiencies, also under low-light illumination conditions. Green photosynthetic bacteria can scavenge photons even at one hundred meters under water surface6 thanks to their special organelles, called chlorosomes, which agglomerate homologues of bacteriochlorophylls (BChl's) c, d, and e as functional pigments (Fig. 1a). In polar solvents, the monomeric forms prevail having an intense and sharp Qy-band at ∼670 nm. Upon self-assembly, this band is red-shifted to ∼740 nm and increases by up five times its full-width-at-half-maximum mainly due to inhomogeneous broadening.7 Our laboratory has mimicked8 and extended the self-assembly algorithm9 (Fig. 1b) by employing different recognition motifs as well as various solubilizing groups grafted onto porphyrins or chlorins which self-assemble in the same way as the natural BChl's. We report here on equipping Pc's with acyl recognition groups which will then induce self-assembly under appropriate conditions in a similar manner as the natural BChl's and our previously reported 3,13-diacetyl-Zn-porphyrins for which by means of single crystal X-ray structures we could unravel their supramolecular architecture, essential for light-harvesting.10
 | ||
Fig. 1 (a) Natural self-assembling BChl's where R8 and R12 can be homologous groups such as methyl, ethyl, n-propyl or isobutyl. Farnesol is depicted as the long-chain alcohol esterifying the 17-propionic acid but also other fatty alcohols such as stearol, cetol, phytol, geranyl-geraniol, etc. can be encountered in various bacteria. Asterisks denote stereocenters. (b) Single crystal structure of a BChl mimic: 3,13-diacetylporphyrin having Zn replacing the central Mg atom self-assembled by Zn⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C < ligations.10 A tetramer is shown with the partially disordered 3,5-di-tert-butyl groups truncated at their ipso carbon for clarity. Broad and red-shifted absorption bands characterize this stair-case-like architecture, typical for J-aggregates.11 (c) Targeted constructs in this work whereby a diacylated ZnPc is statistically monoreduced to a ketol with alkyl chains of various lengths for tuning the solubility. C < ligations.10 A tetramer is shown with the partially disordered 3,5-di-tert-butyl groups truncated at their ipso carbon for clarity. Broad and red-shifted absorption bands characterize this stair-case-like architecture, typical for J-aggregates.11 (c) Targeted constructs in this work whereby a diacylated ZnPc is statistically monoreduced to a ketol with alkyl chains of various lengths for tuning the solubility. | ||
Herein we present a series of self-assembling tetra-acylated ZnPc's with long alkyl chains as solubilizing groups and carbonyl groups as the recognition groups mimicking the self-assembly algorithm of BChl's. When the self-assembly operates, a broad and red-shift near-infrared absorption onset at over 900 nm can be seen in the electronic absorption spectra. The temperature dependence of the equilibrium between the monomer and the aggregated species was also studied.
Prior to our studies,12 no acyl-Pc's had been described in the literature. This is probably due to the reactivity of the carbonyl groups under the strongly basic and high temperature conditions required for the templated cyclo-tetramerization by divalent metal salts of the Pc synthons. In a shot-gun approach we tried in vain to acylate under various Friedel–Crafts conditions the unsubstituted CuPc. Due to its insolubility in various solvents, unreacted material was always recovered. In concentrated sulfuric acid, apparently a good solvent for CuPc, neither anhydrides nor acyl chlorides reacted with or without preformed complexes of AlCl3.
We then turned our attention to a multistep synthetic sequence which was optimized as shown in Scheme 1. Friedel–Crafts acylation of ortho-dichlorobenzene, which is available in bulk quantities, with various acyl chlorides, also available on an industrial scale, provided the 4-acyl-1,2-dichlorobenzene derivatives 2a–d with R = CnH2n+1 (n = 1, 5, 11, 15). Direct conversion to the phthalodinitriles occurred unproblematically by Pd-catalysis but the cyclotetramerization reaction to the corresponding Pc's, although it occurred in good yields, was accompanied by a Meerwein–Pondorf–Verley type reaction. Thereby some of the carbonyl groups became reduced to the secondary alcohols, giving much too complex reaction mixtures, albeit their self-assembly could be undoubtedly proven.12
 | ||
| Scheme 1 Synthetic route of the desired tetraacylated ZnPc-6a–d. Yields for procedures (a)–(c) performed several times on a multigram scale are detailed in the ESI.† The overall yields for ZnPc-6a–d are based on the procedures (d) and (e) having 4a–d as starting material. | ||
Therefore we restored to protect at the early 4-acyl-1,2-dichlorobenzene stage the carbonyl groups with neopentyl glycol, which can eventually be easily removed. Cyanation involving palladium catalysis of these derivatives led to the precursor phthalodinitriles 4a–d in moderate to high yields. Their cyclotetramerization gave the tetracarbonyl-protected ZnPc-5a–d. Finally, deprotection of the carbonyl groups proved to be possible with TiCl4 furnishing the desired tetraacylated ZnPc-6a–d. The synthetic intermediates, excluding 2a, are also novel compounds which could have interesting applications as they can be easily obtained cheaply in large quantities.
After succeeding in equipping the series of ZnPc-6a–d with carbonyl groups and a central zinc atom as recognition groups, we then performed self-assembly experiments of these compounds in nonpolar solvents. With the increasing solubility brought about by the longer alkyl chains, the ZnPc-6b–d could self-assemble in solvents of low polarity such as n-heptane, while ZnPc-6a could also self-assemble in moderately polar solvents such as dichloromethane and chloroform (Fig. S3, ESI†). Below, we present ZnPc-6d as an example of the self-assembly process.
We performed UV-Vis absorption measurements in dry n-heptane at room temperature for ZnPc-6d (Fig. 2). The pure sample powder and the cuvettes were thoroughly vacuum dried and the prepared solution was also sonicated for 2 min before the measurement. From the absorption spectra, a very broad and red-shifted Q-band absorption with an onset at over 900 nm could be seen, which is rare among the phthalocyanines. When two drops of methanol were added to the 3 mL cuvette, only a partial disassembly occurred because methanol can ligate and compete to the coordination of the central zinc ion. The Pc assemblies are thus much more robust than our previous porphyrinic BChl mimics where stoichiometric amounts of Zn-coordinating alcohols or even adventitious water were sufficient to monomerize the aggregates.8,9 Further additions of five drops of methanol led to phase separation which could be prevented by further adding five drops of dichloromethane thus ensuring a homogeneous solution. Thereby, the aggregates disassemble completely, giving intense monomeric Q-bands, with no aggregates absorbing above 750 nm. Noteworthy is that during the disassembly process, the absorption bands and fluorescent emission attributed to the monomer were also intensified gradually (Fig. S6, ESI†). Meanwhile, in the absence of Zn-coordinating solvents, slow growth of aggregates from a few hundred nanometres to visible fluffs which flocculate in the n-heptane solution could also be seen by naked eye or microscopy (Fig. S7, ESI†). Gentle shaking or short sonication restores homogeneous solutions which were stable for various spectroscopic studies.
 | ||
| Fig. 2 Absorption spectra of ZnPc-6d in dry n-heptane (3.3 × 10−5 M, black line), after addition of two drops of methanol (red line) and followed by five more drops of methanol and five drops dichloromethane (the green line). A 3 mL cuvette with 1 cm pathlength is shown before (at left) and after addition of methanol and gentle shaking (at right). A microscopic image of the fluffs characteristic of these macro aggregates is provided in the ESI† (Fig. S7). Instead of methanol, pyridine or THF can be used to disassemble the aggregates (Fig. S9, ESI†). | ||
All of the above phenomena resembled both that of the natural BChl's and of our previous mimics. In order to further study the self-assembly and disassembly process, variable-temperature UV-Vis absorption measurements were performed from T = 20 to 95 °C. As shown in Fig. 3, an equilibrium could be put into evidence. During the temperature-rise period, the aggregates gradually disassemble to monomers while in the temperature-fall period, the monomers again self-assemble almost fully reversibly, to the aggregates. Upon heating the aggregates gradually disassemble in a fashion resembling the melting of DNA with a monotonous variation (red curves in the insets of Fig. 3). Evidence for a cooperative and autocatalytic aggregate growth process is provided by the sigmoidal shape of the absorbance upon cooling (blue traces in the insets of Fig. 3). These show an inflexion point at 65 °C (thin black lines) at both monitored wavelengths. Very similar trends were earlier encountered for the natural BChl c, where a detailed kinetic modeling provided the size of a critical nucleus of about 14 molecules.13 Assemblies larger than this critical nucleus grow to macroscopic sizes, while smaller oligomers disassemble.
 | ||
| Fig. 3 Temperature-variable UV-Vis absorption of ZnPc-6d (3.3 × 10−5 M) in dry n-heptane containing ∼2% dry dichloromethane. Above is the temperature-rise period and below is the temperature-fall period. The solid arrows reflect the electronic absorption's trend. Insets show the variable temperature evolution of the aggregate maxima monitored at 780 nm and that of the monomeric building blocks monitored at 620 nm. Black dotted traces show the almost perfect reversibility of the absorbance after a heating/cooling cycle. | ||
In order to verify that the biomimetic self-assembly is indeed operating, we used the same conditions for diluting into dry n-heptane the carbonyl protected compound ZnPc-5d. In this case only monomeric absorption spectra were observed with no red-shifts or band broadening (Fig. S8, ESI†). However, the ketals ZnPc-5, due to steric hindrance, prevent the formation of face-to-face H-aggregates11d,e which could translate into the blue-shifted shoulders and peaks of the Q-bands encountered in the spectra of the deprotected ZnPc-6 after addition of Zn-coordinating solvents such as methanol, THF or pyridine (Fig. S9, ESI†).
High-performance liquid chromatography (HPLC) optimizations have been performed on different columns in order to achieve baseline separation of the regioisomers of ZnPc-5a–d and ZnPc-6a–d. Since the pioneering studies of Michael Hannack14 and Clifford Leznoff,15 very few authors report efficient resolutions of such regioisomers and usually a statistical mixture is implicitly assumed when a nonsymmetrically substituted phthalodinitrile is cyclotetramerized. In our hands, we often encountered serious deviations from the 50%, 25%, 12.5% and 12.5% for our Cs, C2v, C4h, and D2h regioisomers, respectively (A4 in Scheme 2).
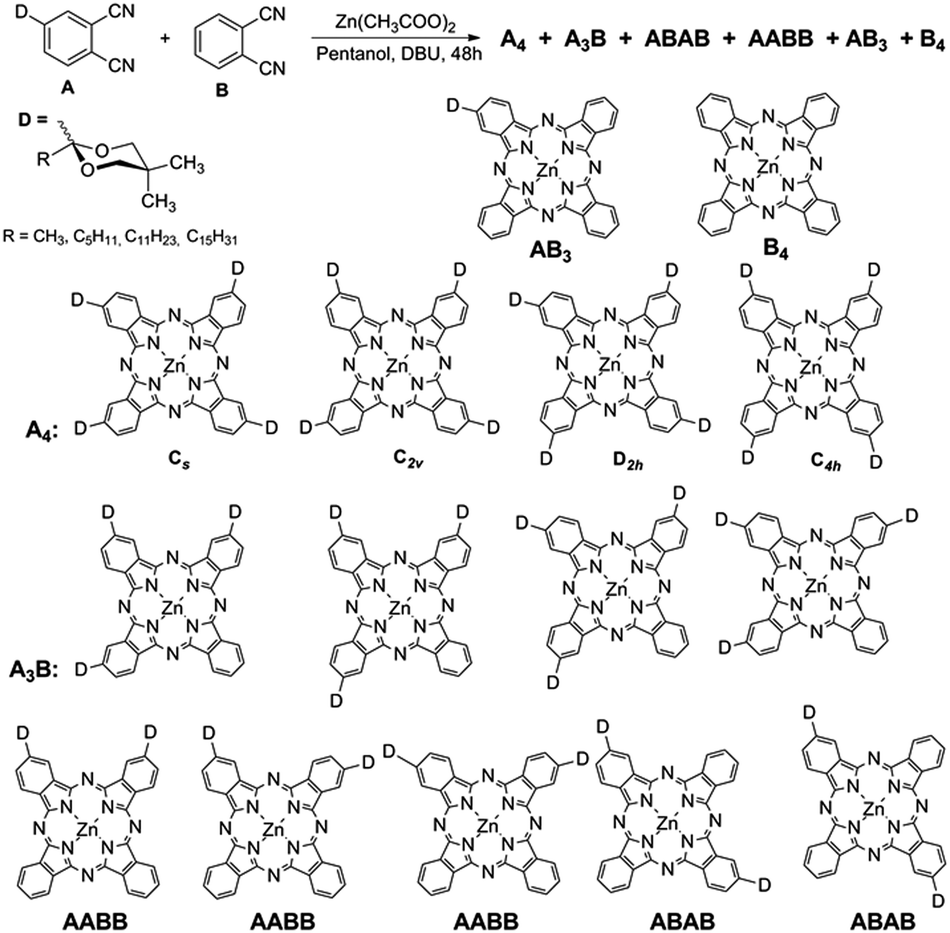 | ||
| Scheme 2 Mixed condensation of two different phthalodinitriles A and B. D stands for the 1,3-dioxolane protecting group which after removal gives the acylated phthalocyanines with RCO being either acetyl, hexanoyl, lauroyl or palmitoyl residues. | ||
We did had previous experience with reverse phase HPLC separations of similarly substituted water-soluble Pc's.16 An elegant study of water soluble Pc's, with a view on photodynamic therapy applications, stands out by the very careful experimentations and the efficient HPLC separations.17
Even more complex mixtures resulted in our hands when the protected phthalodinitriles 4a–d (A) were submitted to a mixed condensation with the unsubstituted phthalodinitrile (B in Scheme 2). In these cases, beside the four regioisomers A4 of type ZnPc-5, highly colored Pc's having one, two, or three dioxolane (D) protected acyl residues were formed, beside some unsubstituted ZnPc (B4 in Scheme 2) which could be easily separated due to its limited solubility. HPLC coupled with mass-spectrometric and diode-array UV-Vis detectors allow analytical detective-like investigations of such mixtures which upon careful assignments become “informed” libraries18 of Pc's (Fig. S12–S15, ESI†). We envisaged that the different regioisomers could have different self-assembly properties. Indeed, the UV-Vis spectra of the four A4 isomers show Q-bands with different intensities (Fig. S13, ESI†), and in future work, by combining with theoretical calculations, an assignment of the regioisomers will be attempted. Up-scaling of the HPLC-separations need to be performed in order to allow also NMR-based assignments.
In a truly biomimetic approach, we envisaged working with such mixtures because in the natural chlorosomes a similar heterogeneity was optimized by evolution. In the Cloroflexus aurantiacus antennas, the same BChl c core chromophore is esterified at the 173 position by various long chain aliphatic alcohols such as cetyl, stearyl, oleyl, geranyl–geranyl etc.19 in various proportions depending on the habitat. On the contrary, in Chlorobaculum tepidum chlorosomes, the heterogeneity is ensured by different R8 and R12 substituents while the sole esterifying alcohol is farnesol (Fig. 1a). While R8 changes from ethyl, propyl to isobutyl, R12 can be methyl or ethyl. Depending on the illumination conditions and, most intriguingly, on the stereochemistry of the 3-hydroxyethyl group involved in the self-assembly algorithm, the composition of these chlorosomal BChl homologues can vary significantly. We have also shown previously that the isolated pure enantiomers self-assemble less readily than their scalemic mixtures or racemates.20
With this heterogeneity in mind we performed a sodium borohydride reduction by using it in substoichiometric amounts on the mixture of Pc's so that only partial reduction of the acyl groups occurred to the corresponding racemic 2-hydroxyalkanols. Gratifyingly, we could put into evidence by HR-MS a compound whose mass corresponds to the monoreduced diacyl derivative. Although the exact desired geometry as shown in Fig. 1c, is not yet ascertained (as both AABB and the targeted ABAB types have the same mass), the fact that upon self-assembly, the typical chlorosomal red-shift and line broadening occurs, indicates that the self-assembly algorithm operates correctly and that regioisomers which do not have the ideal geometry, leading to possible defects, will be reversibly disassembled from the final structure. This self-healing is typical for supramolecular assemblies in a thermodynamically driven process. We can thus preferentially select the species which will self-assemble effectively, and after repeated cycles of centrifugations and re-suspensions, the Pc's that do not self-assemble are simply discarded with the supernatant.
In summary, we firstly optimized a way to synthesize for the first time highly soluble acylphthalocyanines by employing carbonyl-protected groups, and secondly, due to the recognition ability of the carbonyl groups, the central zinc atom and the solubilizing alkyl chains, these could self-assemble in the same way as the natural BChl's. The supramolecular self-assemblies of these acylphthalo-cyanines in n-heptane show a very broad near-infrared Q-band absorption with an onset at over 900 nm.
H.-G. Jin gratefully acknowledges his PhD fellowship from the China Scholarship Council. Dr Valèrie Monnier and Christophe Chendo are thanked for the high-resolution mass spectra recorded at the Spectropole in Marseille. Prof. Harry L. Anderson is thanked for helpful discussions on cooperative aggregation mechanisms.
Notes and references
- (a) J.-M. Lehn, Science, 2002, 295, 2400 CrossRef CAS PubMed; (b) A. C. Grimsdale and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS PubMed; (c) P. Kambhampati, Acc. Chem. Res., 2011, 44, 1 CrossRef CAS PubMed; (d) J. Rawson, A. C. Stuart, W. You and M. J. Therien, J. Am. Chem. Soc., 2014, 136, 17561 CrossRef CAS PubMed.
- (a) G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517 CrossRef CAS PubMed; (b) M. Z. Papiz, S. M. Prince, T. Howard, R. J. Cogdell and N. W. Isaacs, J. Mol. Biol., 2003, 326, 1523 CrossRef CAS; (c) A. W. Roszak, T. D. Howard, J. Southall, A. T. Gardiner, C. J. Law, N. W. Isaacs and R. J. Cogdell, Science, 2003, 302, 1969 CrossRef CAS PubMed.
- (a) Phthalocyanines – Properties and Applications, ed. C. C. Leznoff and A. P. B. Lever, VCH, New York, vol. 1, 1989, vol. 2, 1992; vol. 3,1993; vol. 4, 1996 Search PubMed; (b) Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, vol. 15–20, 2003 Search PubMed; (c) Handbook of Porphyrin Science with Applications to Chemistry, Physics, Materials Science, Engineering, Biology and Medicine, K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, vol. 1–35, 2010–2015 Search PubMed; (d) M. V. Martinez-Diaz, G. de la Torre and T. Torres, Chem. Commun., 2010, 46, 7090 RSC; (e) M. G. Walter, A. B. Rudine and C. C. Wamser, J. Porphyrins Phthalocyanines, 2010, 14, 759 CrossRef CAS; (f) J. Mack and N. Kobayashi, Chem. Rev., 2011, 111, 281 CrossRef CAS PubMed; (g) G. de la Torre, G. Bottari, M. Sekita, A. Hausmann, D. M. Guldi and T. Torres, Chem. Soc. Rev., 2013, 42, 8049 RSC.
- (a) M. E. Ragoussi, J. J. Cid, J. H. Yum, G. de la Torre, D. Di Censo, M. Grätzel, M. K. Nazeeruddin and T. Torres, Angew. Chem., Int. Ed., 2012, 51, 4375 CrossRef CAS PubMed; (b) T. Ikeuchi, H. Nomoto, N. Masaki, M. J. Griffith, S. Mori and M. Kimura, Chem. Commun., 2014, 50, 1941 RSC.
- (a) J. A. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251 CrossRef CAS PubMed; (b) K. Kameyama, M. Morisue, A. Satake and Y. Kobuke, Angew. Chem., Int. Ed., 2005, 44, 4763 CrossRef CAS PubMed.
- (a) J. Overmann, H. Cypionka and N. Pfennig, Limnol. Oceanogr., 1992, 37, 150 CrossRef CAS; (b) A. K. Manske, J. Glaeser, M. M. M. Kuypers and J. Overmann, Appl. Environ. Microbiol., 2005, 71, 8049 CrossRef CAS PubMed.
- (a) M. I. Bystrova, I. N. Mal'gosheva and A. A. Krasnovskii, Mol. Biol., 1979, 13, 440 Search PubMed; (b) K. M. Smith, L. A. Kehres and J. Fajer, J. Am. Chem. Soc., 1983, 99, 1387 CrossRef.
- (a) T. S. Balaban, A. D. Bhise, M. Fischer, M. Linke-Schaetzel, C. Roussel and N. Vanthuyne, Angew. Chem., Int. Ed., 2003, 42, 2140 CrossRef CAS PubMed; (b) T. S. Balaban, M. Linke-Schaetzel, A. D. Bhise, N. Vanthuyne and C. Roussel, Eur. J. Org. Chem., 2004, 3919 CrossRef CAS PubMed.
- (a) M. C. Balaban and T. S. Balaban, J. Porphyrins Phthalocyanines, 2007, 11, 277 CrossRef CAS; (b) J. Szmytkowski, J. Conradt, H. Kuhn, C. M. Reddy, M. C. Balaban, T. S. Balaban and H. Kalt, J. Phys. Chem. C, 2011, 115, 8832 CrossRef CAS.
- T. Jochum, C. M. Reddy, A. Eichhofer, G. Buth, J. Szmytkowski, H. Kalt, D. Moss and T. S. Balaban, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 12736 CrossRef CAS PubMed.
- (a) E. E. Jelley, Nature, 1936, 138, 1009–1010 CrossRef CAS PubMed; (b) G. Z. Scheibe, Angew. Chem., 1936, 49, 563 CAS; (c) G. Scheibe, A. Mareis and H. Ecker, Naturwissenschaften, 1937, 25, 474 CrossRef CAS; (d) A. Mishra, R. K. Behera, P. K. Behera, B. K. Mishra and G. B. Behera, Chem. Rev., 2000, 100, 1973 CrossRef CAS PubMed; (e) F. Würthner, T. E. Kaiser and C. R. Saha-Möller, Angew. Chem., Int. Ed., 2011, 50, 3376 CrossRef PubMed.
- (a) M. C. Balaban and T. S. Balaban, US0054166 A1, 2011; (b) M. C. Balaban and T. S. Balaban, EP2292623A1, 2011.
- T. S. Balaban, J. Leitich, A. R. Holzwarth and K. Schaffner, J. Phys. Chem. B, 2000, 104, 1362 CrossRef CAS.
- (a) M. Sommerauer, C. Rager and M. Hanack, J. Am. Chem. Soc., 1996, 118, 10085 CrossRef CAS; (b) S. Rodríguez-Morgade and M. Hanack, Chem. – Eur. J., 1997, 3, 1042 CrossRef PubMed; (c) B. Görlach, M. Dachtler, T. Glaser, K. Albert and M. Hanack, Chem. – Eur. J., 2001, 7, 2459 CrossRef.
- C. C. Leznoff, C. R. McArthur and Y. N. Qin, Can. J. Chem., 1993, 71, 1319 CrossRef CAS PubMed.
- K. Arnold, T. S. Balaban, M. N. Blom, O. T. Ehrler, S. Gilb, O. Hampe, J. E. van Lier, J. M. Weber and M. M. Kappes, J. Phys. Chem. A, 2003, 107, 794 CrossRef CAS.
- Z. Jiang, W. He, H. Yao, J. Wang, N. Chen and J. Huang, J. Porphyrins Phthalocyanines, 2011, 15, 140 CrossRef CAS.
- O. Ramström and J.-M. Lehn, Nat. Rev. Drug Discovery, 2002, 1, 26 CrossRef PubMed.
- F. Fages, N. Griebenow, K. Griebenow, A. R. Holzwarth and K. Shaffner, J. Chem. Soc., Perkin Trans. 1, 1990, 2791 RSC.
- T. S. Balaban, A. D. Bhise, G. Bringmann, J. Bürck, C. Chappaz-Gillot, A. Eichhöfer, D. Fenske, D. C. G. Götz, M. Knauer, T. Mizoguchi, D. Mössinger, H. Rösner, C. Roussel, M. Schraut, H. Tamiaki and N. Vanthuyne, J. Am. Chem. Soc., 2009, 131, 14480 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, product characterization, spectral data. See DOI: 10.1039/c5cc04602e |
| This journal is © The Royal Society of Chemistry 2015 |