
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electron-deficient fullerenes in triple-channel photosystems†
Javier
López-Andarias
a,
Altan
Bolag
b,
Christoph
Nançoz
b,
Eric
Vauthey
b,
Carmen
Atienza
a,
Naomi
Sakai
b,
Nazario
Martín
*a and
Stefan
Matile
*b
aDepartamento de Química Orgánica, Universidad Complutense, IMDEA-Nanoscience, Madrid, Spain. E-mail: nazmar@quim.ucm.es
bSchool of Chemistry and Biochemistry, University of Geneva, Geneva, Switzerland. E-mail: stefan.matile@unige.ch
First published on 7th April 2015
Abstract
Fullerenes of increasing electron deficiency are designed, synthesized and evaluated in multicomponent surface architectures to ultimately build gradients in LUMO levels with nine components over 350 meV down to −4.22 eV.
The development of synthetic methods to build large multicomponent architectures is important because biological function originates from structures of highest sophistication and our ability to build organic materials on the same level of precision is quite limited.1–3 To help to improve this situation, we became interested in learning how to grow multicomponent architectures directly on solid surfaces. This is of interest to control directionality. Over the years, synthetic methods such as zipper assembly,4 self-organizing surface-initiated polymerization (SOSIP),5 templated stack-exchange (TSE)6 and templated self-sorting (TSS)7 have been introduced. SOSIP-TSE emerged as the method of choice to build photosystems with co-axial molecular channels to transport holes and electrons, so-called supramolecular n/p-heterojunctions (SHJs).6 Transcribing a more complex lesson from nature, double-channel photosystems have been equipped with antiparallel redox gradients of up to three components to drive holes and electrons apart after their generation with light.8 Moreover, the synthesis of triple-channel architectures has been achieved recently with SOSIP-TSE.9
In triple-channel architectures with [60]fullerene stacks for electron transport1,2 along with oligothiophene stacks for hole transport1,3 and co-axial naphthalenediimide (NDI) stacks for electron transport, photocurrents increased with decreasing energy of the LUMO of [60]fullerenes 1–4 (Fig. 1).10 This finding called for the synthesis of more electron-deficient fullerenes. Moreover, the availability of π-acidic fullerenes with solubility in polar solvents and linkers for dynamic covalent11 interfacing in complex systems is of general interest to transport electrons1–3,8,10 and bind, transport and transform anions.12 Here, we report the synthesis of five π-acidic fullerenes to complete a nine-component redox gradient over 350 meV down to −4.22 eV, and demonstrate their compatibility with triple-channel SOSIP-TSE architectures.
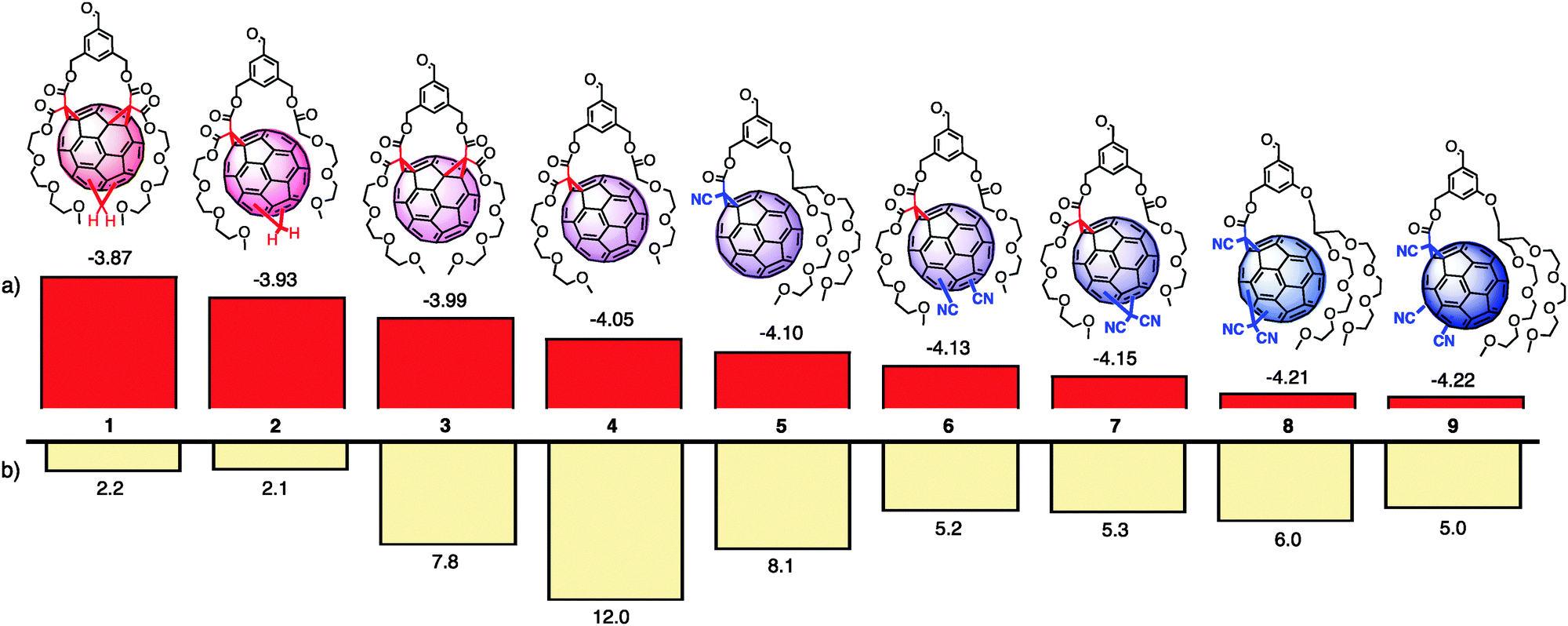 | ||
| Fig. 1 Structure of the new electron-deficient [60]fullerenes 5–9 compared to the previously reported electron-rich 1–4.10 (a) Energy of the LUMO in eV against vacuum, assuming −5.1 eV for Fc+/Fc. Determined by DPV (Fig. S1, ESI†). (b) Short-circuit photocurrent density JSC in μA cm−2 generated in triple-channel photosystems together with oligothiophene donors (HOMO: −5.9 eV, LUMO: −3.2 eV, see Fig. 2). | ||
All electron-deficient [60]fullerenes 5–9 contain two triethyleneglycol (TEG) tails to assure solubility in aprotic polar solvents and a peripheral benzaldehyde to connect to multicomponent surface architectures.10 To gradually increase their π-acidity, one, two or three electron-withdrawing cyano groups were attached either directly to the aromatic truncated icosahedron13 or as a substituent of the methano-14 or malonate-derived cyclopropanes (Fig. 1).15 Synthetic methods to introduce these cyano acceptors were available for all variations.13–15 The preparation of 6 and 7 requires the standard Bingel reaction with the respective malonate. Meanwhile, in 5, 8 and 9, the Bingel reaction is done with cyanoacetates instead of malonates. Compared to Bingel fullerenes 6 and 7, this modification caused the lack of one TEG tail, therefore, to restore solubility in aprotic apolar solvents, a new solubilizer carrying two TEG tails had to be introduced. Only fullerene 5 is a monodisperse compound. In fullerenes 6–9, the position of the additional withdrawing substituents is not defined with respect to the Bingel cyclopropane, i.e., they exist as mixtures of regioisomers. Synthetic and analytical details of all other new fullerenes can be found in the Schemes S1 and S2, ESI.†
The energy levels of the LUMO of the new electron-deficient fullerenes 5–9 were determined by differential pulse voltammetry (DPV) against the Fc+/Fc couple as an internal standard (Fig. S1, ESI†).10 Not surprisingly, highest LUMO energies were found at −4.10 eV for 5 with only one cyano acceptor without direct connection to the aromatic system. The two cyano acceptors attached directly to the fullerene core in 6 caused a decrease of 30 meV to −4.13 eV. Interestingly, two cyano groups in the cyclopropane in 7 further increased π-acidity to −4.15 eV. Fullerenes with three cyano acceptors were the most π-acidic, with two cyano acceptors in the cyclopropane of 8 being slightly weaker than two cyano acceptors directly attached to the fullerene surface of 9. Together with the previously reported fullerenes 1–4,10 these results yield an electron transfer cascade of nine components of high structural similarity, covering 350 meV down to −4.22 eV.
The new electron-deficient fullerenes were engineered into multicomponent surface architectures under the conditions developed for the electron-rich series (Fig. 2 and Scheme S3, ESI†). As exemplified for the most π-acidic 9, oxime formation with the reported quaterthiophene gave dyad 10 (Fig. 2). The acetal in 10 was hydrolysed under acidic conditions, and the liberated aldehyde was used for templated stack exchange (TSE)6,10 with dynamic covalent hydrazone chemistry.11 The obtained triple-channel architecture 11 is expected to contain hole-transporting oligothiophene stacks between electron-transporting channels composed of co-axial strings of fullerenes and NDIs. Triple-channel architectures with all other new fullerenes were prepared analogously. As expected for the bulky dyads, TSE yields were between 51 and 74% (Fig. S2 and S3, ESI†).
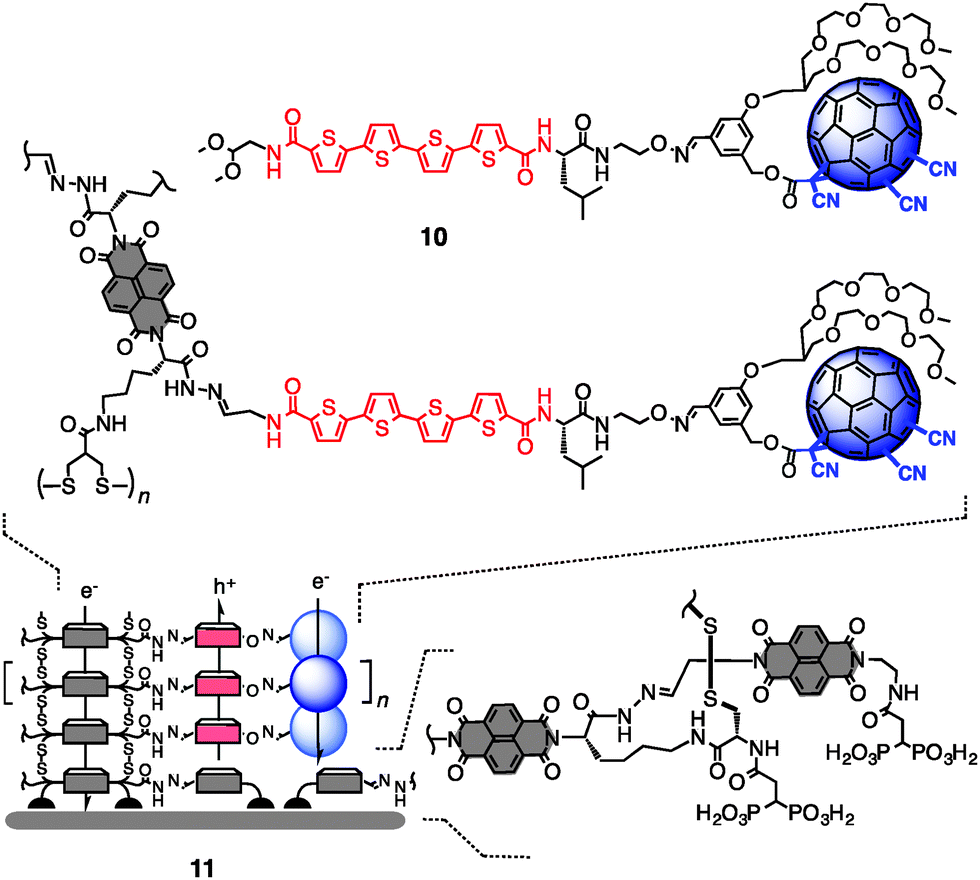 | ||
| Fig. 2 Structure of dyad 10 and schematic architecture of triple-channel photosystem 11 exemplified for the most π-acidic fullerene 9. | ||
Photocurrent generation by the new photosystems was measured under the conditions used before to assure the comparability of the results.10 Without going into details, photosystems were characterized as the working electrode with a platinum wire as the counter electrode and triethanolamine as the hole acceptor in solution. Results obtained with this set-up have served well to extract trends in proof-of-principle studies but they are not comparable with results obtained for optimized optoelectronic devices.9 The highest short-circuit photocurrent densities JSC = 8.1 μA cm−2 were obtained with the photosystem containing the least π-acidic fullerene 5 (Fig. 1b). All other photosystems with more electron-deficient fullerenes 6–9 had a similar activity around JSC = 5.5 ± 0.5 μA cm−2. These results suggested that the activity in triple-channel photosystem 11 does not directly increase with increasing π-acidity of the fullerene, although hole transfer to the oligothiophene after excitation of the fullerene should improve.10,16 Further analysis did not reveal distinct differences with regard to charge recombination efficiencies (35–64%), open-circuit voltages (270–320 mV) or activation energies (196–258 meV, Fig. S4, S5 and Table S2, ESI†). Characterization of dyads in solution by femtosecond fluorescence up-conversion did not provide helpful explanations for the observed trends. Compared to slow fluorescence decay of isolated quaterthiophenes (τ = 550 ps), fluorescence lifetimes of all dyads analogous to 10 were much shorter. The lifetimes of the dyad with fullerene 4 (τ1 = 1.4 ps, τ2 = 16.9 ps) and the more π-acidic fullerene 9 (τ1 = 1.4 ps, τ2 = 17.2 ps) were roughly identical (Table S3, ESI†). Only a slightly longer lifetime of dyads with the most π-basic fullerene 1 (τ1 = 2.2 ps, τ2 = 30.0 ps, Table S3, ESI†) could support slow charge separation as the origin of the exceptionally poor activity of the respective triple-channel photosystem (JSC = 2.2 μA cm−2, Fig. 1b). This conclusion would also agree with previous observations in the action spectra.10
Inspection of the structures of nine fullerenes under evaluation revealed that best activities were found for fullerenes with the best preserved π-system, i.e., fullerenes 4 (JSC = 12.0 μA cm−2) and 5 (JSC = 8.1 μA cm−2) with just one cyclopropane added. Photosystems with fullerene 3, characterized by a defined positioning of the two cyclopropanes, generated significant photocurrent as well (JSC = 7.8 μA cm−2). All mixtures of regioisomers gave clearly weaker activity (Fig. 1b). Only in the systems with directly comparable structures and structural disorder and with clearly different LUMO levels (230 meV), i.e. photosystems with the more π-basic fullerene 2 (JSC = 2.1 μA cm−2) and the more π-acidic fullerene 7 (JSC = 5.3 μA cm−2), photocurrents did increase significantly with increasing π-acidity. These observations suggested that the preserved integrity and structural homogeneity of the fullerenes are most important for their activity in the described triple-channel photosystems, whereas decreasing LUMO energies are clearly beneficial10,16 but overall less important.
Taken together, the synthesis of a series of electron-deficient fullerenes allowed us to build a redox gradient of nine components over 350 meV down to −4.22 eV. Incorporated into triple-channel photosystems, decreasing LUMO energies give higher activity for strictly comparable fullerenes, but the preserved integrity and structural homogeneity of the strained π-system are more important. Possible origins of these trends include poorer organization of the strings of fullerenes with regioisomers and thus reduced charge mobility, or weakened electron affinity with decreasing tension in the π-system. After all, the −4.22 eV reached with fullerene 9 is quite remarkable in the context of fullerenes but not further impressive in the general context. For example, two cyano acceptors placed in the core of naphthalenediimides afford LUMOs at −4.78 eV.12 The observation that structural integrity and homogeneity are essential for function is interesting and important. They support the general implication from nature that the construction of complex architectures with molecular level precision could provide access to organic materials with interesting activities, and thus encourage continuing the development of synthetic methods.4–10 However, the activities observed for all components are clearly sufficient for the construction of triple-channel architectures with gradients in the fullerene channel composed of up to nine components. This is a tantalizing number that goes far beyond lessons from nature and precedence from double-channel architectures.8 We also hope that the reported series will be of use in other functional systems and for other purposes, ranging from optoelectronic devices1–3 to binding, transport and catalysis with anion-π interactions on π-acidic fullerene surfaces.12
We thank Santiago Lascano and Duy-Hien Tran for contributions to synthesis, the NMR and Mass Spectrometry platforms for services, the University of Geneva, the European Research Council (ERC Advanced Investigator), the Swiss National Centre of Competence in Research (NCCR) Molecular Systems Engineering, the Swiss NCCR Chemical Biology and the Swiss NSF for financial support (S.M.). We also thank the European Research Council (ERC-320441-Chirallcarbon), Ministerio de Economía y Competitividad (MINECO) of Spain (project CTQ2011-24652; Ramón y Cajal granted to C.A. and FPU granted to J.L.-A.) and the CAM (FOTOCARBON S2013/MIT-2841). N.M. is indebted to the Alexander von Humboldt Foundation.
Notes and references
- (a) E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC; (b) F. Würthner and K. Meerholz, Chem. – Eur. J., 2010, 16, 9366–9373 CrossRef PubMed; (c) D. Bonifazi, O. Enger and F. Diederich, Chem. Soc. Rev., 2007, 36, 390–414 RSC; (d) P. M. Beaujuge and J. M. J. Fréchet, J. Am. Chem. Soc., 2011, 133, 20009–20029 CrossRef CAS PubMed; (e) J. L. Delgado, P.-A. Bouit, S. Filippone, M. A. Herranz and N. Martín, Chem. Commun., 2010, 46, 4853–4865 RSC.
- (a) H. Hayashi, I. V. Lightcap, M. Tsujimoto, M. Takano, T. Umeyama, P. V. Kamat and H. Imahori, J. Am. Chem. Soc., 2011, 133, 7684–7687 CrossRef CAS PubMed; (b) F. G. Brunetti, C. Romero-Nieto, J. López-Andarias, C. Atienza, J. L. López, D. M. Guldi and N. Martín, Angew. Chem., Int. Ed., 2013, 52, 2180–2184 CrossRef CAS PubMed; (c) C. Reiriz, R. J. Brea, R. Arranz, J. L. Carrascosa, A. Garibotti, B. Manning, J. M. Valpuesta, R. Eritja, L. Castedo and J. R. Granja, J. Am. Chem. Soc., 2009, 131, 11335–11337 CrossRef CAS PubMed; (d) O. Vostrowsky and A. Hirsch, Chem. Rev., 2006, 106, 5191–5207 CrossRef CAS PubMed; (e) F. B. Kooistra, J. Knol, F. Kastenberg, L. M. Popescu, W. J. H. Verhees, J. M. Kroon and J. C. Hummelen, Org. Lett., 2007, 9, 551–554 CrossRef CAS PubMed; (f) D. M. Guldi, B. M. Illescas, C. Atienza, M. Wielopolski and N. Martín, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC; (g) F. Würthner, Z. Chen, F. J. M. Hoeben, P. Osswald, C. You, P. Jonkheijm, J. von Herrikhuyzen, A. P. H. J. Schenning, P. van der Schoot, E. W. Meijer, E. Beckers, S. Meskers and R. A. J. Janssen, J. Am. Chem. Soc., 2004, 126, 10611–10618 CrossRef PubMed; (h) D. M. Guldi, I. Zilbermann, G. Anderson, A. Li, D. Balbinot, N. Jux, M. Hatzimarinaki, A. Hirsch and M. Prato, Chem. Commun., 2004, 726–727 RSC.
- A. Mishra and P. Bäuerle, Angew. Chem., Int. Ed., 2012, 51, 2020–2067 CrossRef CAS PubMed.
- N. Sakai, A. L. Sisson, T. Bürgi and S. Matile, J. Am. Chem. Soc., 2007, 129, 15758–15759 CrossRef CAS PubMed.
- N. Sakai, M. Lista, O. Kel, S. Sakurai, D. Emery, J. Mareda, E. Vauthey and S. Matile, J. Am. Chem. Soc., 2011, 133, 15224–15227 CrossRef CAS PubMed.
- (a) N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 18542–18545 CrossRef CAS PubMed; (b) N. Sakai, P. Charbonnaz, S. Ward and S. Matile, J. Am. Chem. Soc., 2014, 136, 5575–5578 CrossRef CAS PubMed.
- (a) M. Lista, J. Areephong, N. Sakai and S. Matile, J. Am. Chem. Soc., 2011, 133, 15228–15231 CrossRef CAS PubMed; (b) E. Orentas, M. Lista, N.-T. Lin, N. Sakai and S. Matile, Nat. Chem., 2012, 4, 746–750 CrossRef CAS PubMed.
- H. Hayashi, A. Sobczuk, A. Bolag, N. Sakai and S. Matile, Chem. Sci., 2014, 5, 4610–4614 RSC.
- G. Sforazzini, E. Orentas, A. Bolag, N. Sakai and S. Matile, J. Am. Chem. Soc., 2013, 135, 12082–12090 CrossRef CAS PubMed.
- A. Bolag, J. Lopez-Andarias, S. Lascano, S. Soleimanpour, C. Atienza, N. Sakai, N. Martín and S. Matile, Angew. Chem., Int. Ed., 2014, 53, 4890–4895 CrossRef CAS PubMed.
- A. Wilson, G. Gasparini and S. Matile, Chem. Soc. Rev., 2014, 43, 1948–1962 RSC.
- (a) A. Vargas Jentzsch, A. Hennig, J. Mareda and S. Matile, Acc. Chem. Res., 2013, 46, 2791–2800 CrossRef CAS PubMed; (b) Y. Zhao, N. Sakai and S. Matile, Nat. Commun., 2014, 5, 3911 CAS; (c) Y. Zhao, C. Beuchat, J. Mareda, Y. Domoto, J. Gajewy, A. Wilson, N. Sakai and S. Matile, J. Am. Chem. Soc., 2014, 136, 2101–2111 CrossRef CAS PubMed.
- M. Keshavarz-K, B. Knight, G. Srdanov and F. Wudl, J. Am. Chem. Soc., 1995, 117, 11371–11372 CrossRef CAS.
- (a) T.-H. Zhang, P. Lu, F. Wang and G.-W. Wang, Org. Biomol. Chem., 2003, 1, 4403–4407 RSC; (b) Y. Zhang, Y. Matsuo, C.-Z. Li, H. Tanaka and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 8086–8089 CrossRef CAS PubMed.
- M. Urbani, J. Iehl, I. Osinska, R. Louis, M. Holler and J.-F. Nierengarten, Eur. J. Org. Chem., 2009, 3715–3725 CrossRef CAS.
- T. E. Kang, H.-H. Cho, C.-H. Cho, K.-H. Kim, H. Kang, M. Lee, S. Lee, B. S. Kim, C. Im and B. J. Kim, ACS Appl. Mater. Interfaces, 2013, 5, 861–868 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc01551k |
| This journal is © The Royal Society of Chemistry 2015 |