
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA rationally designed amino-borane complex in a metal organic framework: a novel reusable hydrogen storage and size-selective reduction material†
Xinbo
Wang
,
Linhua
Xie
,
Kuo-Wei
Huang
and
Zhiping
Lai
*
Division of Physical Science and Engineering, King Abdullah University of Science and Technology (KAUST), Thuwal, 23955-6900, Saudi Arabia. E-mail: zhiping.lai@kaust.edu.sa
First published on 23rd February 2015
Abstract
A novel amino-borane complex inside a stable metal organic framework was synthesized for the first time. It releases hydrogen at a temperature of 78 °C with no volatile contaminants and can be well reused. Its application as a size-selective reduction material in organic synthesis was also demonstrated.
Over the past several decades, hydrogen has emerged as an ideal successor to gasoline because of its lightweight and clean combustion properties.1 The current hydrogen storage approaches such as high pressure and cryogenics, however, have suffered disadvantages including large bulky size as well as high safety risks. One of the milestones for hydrogen storage was the development of light-weight solids that could reversibly release H2. Especially ammonia-borane (AB) (NH3BH3), which releases hydrogen upon heating according to reaction (1), is considered to be a promising hydrogen storage material due to its stability and high gravimetric content of hydrogen (19.6 wt%).2 However, several issues still need to be addressed before it can be used, involving decreasing the dehydrogenation temperature below 85 °C, and preventing the formation of volatile by-products, such as borazine (N1BxHx), ammonia, and diborane. The current methods to overcome these issues can be summarized into three general approaches. One is to chemically modify AB in the form of RNHBH3,3–6 where R can be an alkyl group, heterocycles, ionic liquid, or a metal cation. A second approach is the catalytic dehydrogenation of AB with different mechanistic pathways,7–11 including activation by base, acid, ionic liquids, transition metal complexes, or metal nanoparticles. A third approach is to physically load AB into microporous materials such as mesoporous silicas12 and metal organic frameworks (MOFs).13–21 The nano-confinement as well as the unique interactions between AB and the framework were found to be beneficial to reduce the dehydrogenation temperature and the formation of by-products.
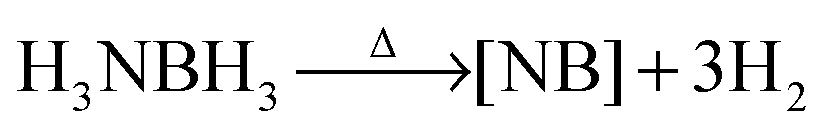 | (1) |
The recently developed MOFs are very attractive for adsorption not only because of their high surface area, high porosity, and tunable pore size, but also because of their easy functionality that allows to introduce functional groups to their frameworks by design.22,23 In fact, functional groups have been widely used to enhance the adsorption loading rate, and to fine-tune the adsorption/desorption kinetics. A number of amino substituted MOFs have been reported in the literature,24–26 such as IRMOF-3, DMOF-1-NH2, UMCM-1-NH2 and UiO-66-NH2. Therefore, our approach in principle can serve as a general method to utilize MOFs as a platform to store borane. However, UiO-66-NH2 is of particular interest because of its good stability. UiO-66-NH2 is constructed with hexameric Zr6O4(OH)4(–CO2)12 clusters and 2-aminoterephthalic acid containing tetrahedral (approximately 8 Å) and octahedral cavities (approximately 11 Å) accessible by microporous triangular windows (approximately 6 Å).27 The framework of UiO-66-NH2 is stable at temperatures up to 300 °C. It is also stable in most organic solvents, water, and acidic aqueous solutions due to strong bonding between oxophilic Zr4+ ions and carboxylate groups of the ligands as well as the inherent high connectivity of its framework.26 The good stability of UiO-66-NH2 has enabled its wide range of applications in such as post-synthetic modifications,26 metal nanoparticle dispersions,28 and direct usage in heterogeneous catalysis.29
The UiOAB complex can also find potential usage in organic synthesis. Borane–amine adducts are important stock compounds that have found broad range of applications in academic and industrial processes, such as hydroboration of olefins, reduction of carbonyl compounds, and as precursors for polyaminoboranes.30 Unlike other diborane compounds, such as borane–tetrahydrofuran (BH3–THF) and borane–dimethyl sulfide (BH3–Me2S), that often suffer low stability, high volatility, flammability and sometimes an unpleasant odor, borane–amine adducts are typically stable in air, easy to handle, and environmentally benign31 due to a tighter boron–nitrogen bond. Although enantioselective reduction of carbonyl compounds has been well established with borane reagents such as the CBS-reduction reaction,32 to the best of our knowledge, a general substrate-size-selective protocol has not been previously reported for either borane or borane–amine adducts. The UiOAB complex containing borane–amine substructures inside its uniform cavity may offer a good opportunity to achieve this goal with a molecular sieving effect that has been observed in other MOFs and zeolite systems.33–35 To demonstrate this idea, the selective reduction of aldehyde compounds with different molecular sizes was selected as a prototype system.
The MOF-amino-borane complex, namely UiOAB, was obtained by simply treating UiO-66-NH2 with borane in diethyl ether medium (see the Experimental details in the ESI†). Elemental analysis using ICP-OES (Table S1, ESI†) revealed that 4.55% of borane was loaded in UiOAB, which is equivalent to 1.2 borane per amine group inside the framework. The more than stoichiometric amount of borane loading indicates that there are at least two types of amine-borane complexes formed. The solid state 11B NMR of UiOAB (Fig. 1, right) contains two main peaks at −17.2 and −22.5 ppm, which indicated that there were two types of boron states corresponding to –NH2BH3 and –NH2B2H5, respectively. The FTIR spectrum (Fig. 1, left) of UiOAB exhibited a new broad peak near 2300 cm−1, which is due to the characteristic peak of B–H symmetric stretching vibrations and indicates that UiO-66-NH2 has reacted with borane to yield UiOAB. After thermolysis and washing with methanol, this band disappeared in UiO-66-NH2-Re. In fact, the FTIR spectrum of UiO-66-NH2-Re contained nearly the same peaks as UiO-66-NH2. In addition, the ICP-OES and 11B NMR spectrum of UiO-66-NH2-Re exhibit no additional signals for boron, indicating that the adsorbed borane was completely removed. All of these characterization results confirmed that the desired MOF-amino-borane complex UiOAB was formed.
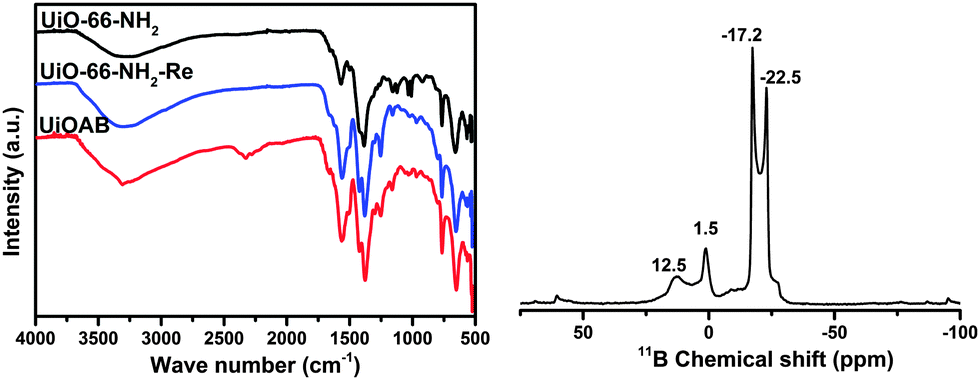 | ||
| Fig. 1 Left: FTIR spectra of UiO-66-NH2 (black), UiOAB (red) and UiOAB after thermolysis at 90 °C and washing with methanol, which is referred to as UiO-66-NH2-Re (blue). Right: solid state 11B NMR of UiOAB. | ||
Borane is a well-known reductant that may react with carboxylate groups, which is one of the main building blocks of UiO-66-NH2, and possibly leads to a structural collapse.36 Therefore, PXRD was recorded in each step to monitor the structural change. The PXRD pattern (Fig. 2, left) of UiO-66-NH2 matches well with simulated UiO-66. The XRD patterns of UiOAB and UiO-66-NH2-Re are nearly identical to that of UiO-66-NH2. All of the patterns have sharp diffraction peaks and flat baselines, which indicate that the synthesized UiO-66-NH2 powder exhibited good crystallinity and remained intact during borane loading and hydrogen release. However, the PXRD peaks of UiOAB after TGA analysis during which it was heated to 200 °C and maintained at this temperature for 6 hours, nearly disappeared, which indicates that the structure collapsed at this temperature. This temperature is much lower than that reported in the literature26 (i.e., approximately 300 °C, which was determined from TGA analysis). The lower decomposition temperature is most likely due to two reasons. First, the structural collapse temperature determined by PXRD is typically lower than that determined by TGA analysis. Second, boron can promote radical oxidation of the carboxylate group in the framework, which has been commonly observed in organic chemistry.37,38 Nevertheless, since the temperatures for borane loading and hydrogen release (which will be discussed in the next paragraph) are much below 200 °C, UiOAB is stable enough for borane storage and reuse.
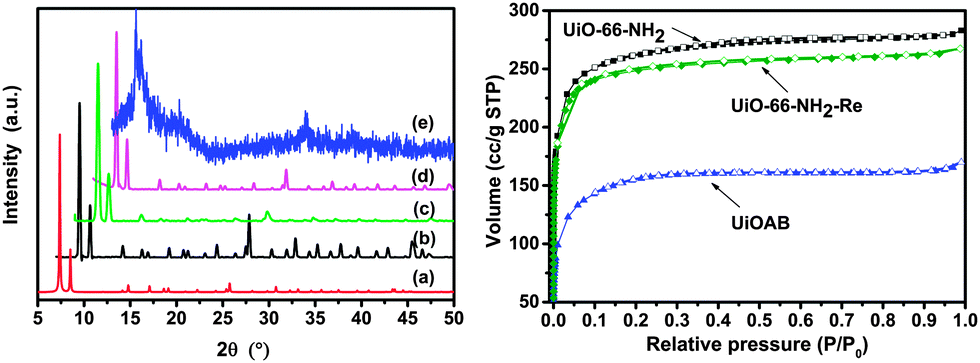 | ||
| Fig. 2 Left: PXRD patterns of UiO-66-NH2 at different stages. Each pattern was shifted 2 degrees from each other to the right for clarity. (a) Simulated UiO-66-NH2 pattern from cif-file CCDC 837796, (b) as-synthesized UiO-66-NH2, (c) as-synthesized UiOAB, (d) UiO-66-NH2-Re, and (e) UiOAB after TGA-QMS analysis during which UiOAB was heated from room temperature to 200 °C at a ramping rate of 1 °C min−1 under 20 ml min−1 argon flow. Right: N2 uptake isotherm at 77 K; adsorption (filled) and desorption (open). | ||
Fig. 3 shows the SEM images of the crystal morphologies at different stages. The as-synthesized UiO-66-NH2 particles have an octahedral morphology with an average particle size of 1 micrometer. The size and morphology were well preserved in UiOAB. However, some particles were broken into approximately half their original size after hydrogen release in UiO-66-NH2-Re, which is most likely due to the volume change that occurs during the adsorption–desorption process. This volume change is a common challenge for the adsorption process. The BET surface area and pore volume of the as-synthesized UiO-66-NH2 was determined to be 976 m2 g−1 and 0.4377 cm3 g−1, respectively, which is similar to the values reported in the literature.26 After loading with borane, these values sharply decreased to 298 m2 g−1 and 0.1618 cm3 g−1, respectively, in UiOAB, which demonstrated that the pores were largely occupied by borane. For UiO-66-NH2-Re, these two values recovered to 834 m2 g−1 and 0.4136 cm3 g−1, respectively. Therefore, most of the pores were re-opened after hydrogen release. Both the BET adsorption (Fig. 2, right) and PXRD data demonstrated the excellent stability and reusability of UiO-66-NH2 in our hydrogen production strategy.
 | ||
| Fig. 3 SEM images of (a) UiO-66-NH2, (b) UiOAB, and (c) UiO-66-NH2-Re. | ||
Thermogravimetric analysis coupled with quadrupole mass spectrometry (TGA-QMS) was used to investigate hydrogen release (Fig. 4) from both neat AB and UiOAB. The results indicated that neat AB releases hydrogen mainly at two temperatures. The first temperature is at 114 °C, and the second at 149 °C. Neat AB also produces a large quantity of volatile by-products, such as ammonia, diborane, and borazine. In contrast, the dehydrogenation of UiOAB started at a temperature below 50 °C, which is the lowest detectable limit of the equipment, and only one temperature peak was observed at 78 °C. Therefore, in comparison to neat AB, the dehydrogenation temperature was significantly reduced. In contrast to the approach where AB is physically absorbed inside the mesoporous silica or MOF channels, the amine-borane complex of UiOAB can be considered to be a single point of molecular AB. This may explain why there is only one hydrogen releasing peak in our system. The strong chemical bonding between borane and amino groups also explains why there are no volatile by-products generated during the decomposition process.
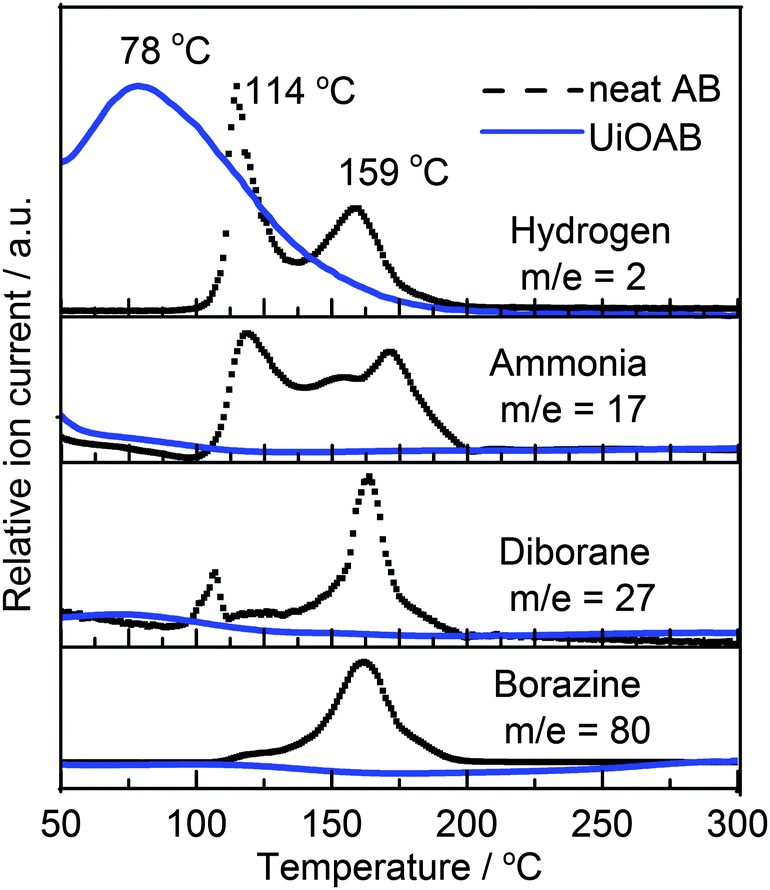 | ||
| Fig. 4 TGA-QMS spectra of neat AB (black dashed line) and UiOAB (blue solid line) during the dehydrogenation process. | ||
The area under the two hydrogen peaks of neat AB in Fig. 4 is equivalent to approximately 10 wt% of hydrogen, as reported in the literature.13 Using this as a reference, the total amount of hydrogen released from UiOAB is approximately 2.2 wt%. Because the borane loading in UiOAB is 4.55%, as determined by ICP-OES, therefore, almost all hydrogen in borane adsorbed in UiOAB are released. Strong reducing agents such as LiAlH4 and hydrazine and harsh conditions are always required to regenerate AB from the dehydrogenation products of poly-borazylene.39 While in our system, the AB complexes are spatially separated by each other so that polymerization is fundamentally avoided. After dehydrogenation, UiO-66-NH2 can be regenerated by simply soaking in methanol, as confirmed by IR, solid 11B NMR/MAS, N2 isothermal physisorption as well as by SEM as above mentioned. Table S2 summarizes the reported hydrogen release results from AB physically loaded in different MOF systems. Compared to most reported results, our work showed a higher hydrogen loading rate and a lower hydrogen release temperature. From all these results it can be clearly seen that no matter whether AB is physically loaded, as reported in the literature, or chemically bonded, as reported in our studies, the nano-confinement provided by the MOF structure played an important role in reducing the hydrogen release temperature and the generation of contaminants.
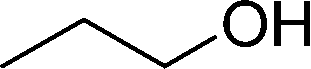
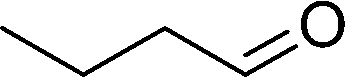
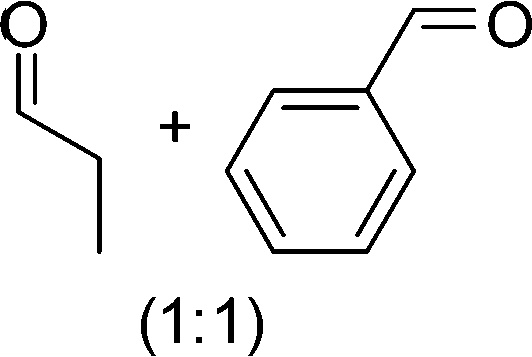
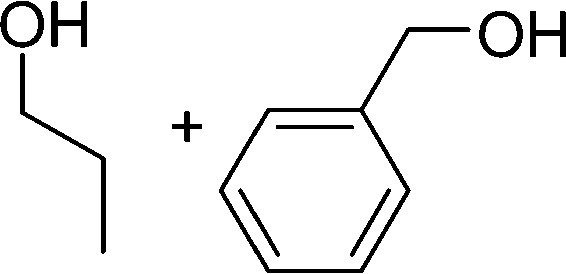
DFT calculations of the N2 physisorption isotherm data indicated a decrease in pore size from 7.3 Å in UiO-66-NH3 to 5.3 Å in UiOAB. This pore size is bigger than the kinetic diameter of linear alkyl aldehydes such as propanal (∼4.7 Å) and butyral (∼5.0 Å), but smaller than aromatic aldehydes such as benzaldehyde (∼5.9 Å) and phenanthrene-9-carbaldehyde (∼8.9 Å). So a substrate size selective reduction was expected and demonstrated. The aldehyde reduction reactions were conducted in methanol at 60 °C for 4 hours. Under these conditions our control experiments showed that when borane, sodium borane hydride and ammonia borane were used as reducing agents all these aldehydes were converted quantitatively to the related alcohol products without notable selectivity. However, when UiOAB was used as a reducing agent, the results in Table 1 show that for small sized propanal and butyral, the conversion is more than 95%, but for large benzaldehyde, the conversion decreased to 35%. The results indicate that propanal and butyral can freely enter the pores of UiOAB. Benzaldehyde can also enter the pores, presumably due to the structural flexibility that commonly observed in the MOF structure, but in much slow rate. For this reaction we further extended the reaction time to another 12 hours. Since as shown in Fig. 4, UiOAB starts to decompose at a temperature below 60 °C, to maintain enough amount of UiOAB in the reaction medium, we added another 1 eq. of UiOAB. At the end, the conversion of benzaldehyde was increased to 80%. When a more steric bulky molecule, phenanthrene-9-carbaldehyde, is used, no product was detected. To further demonstrate the size-selective effect, a mixture of propanal and benzaldehyde in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio was studied under the standard reaction conditions (Table 1, entry 5). A high product selectivity of 49 was obtained. All of these results clearly demonstrated the potential usage of UiOAB as a size-selective reduction agent for organic synthesis.
:1 mole ratio was studied under the standard reaction conditions (Table 1, entry 5). A high product selectivity of 49 was obtained. All of these results clearly demonstrated the potential usage of UiOAB as a size-selective reduction agent for organic synthesis.
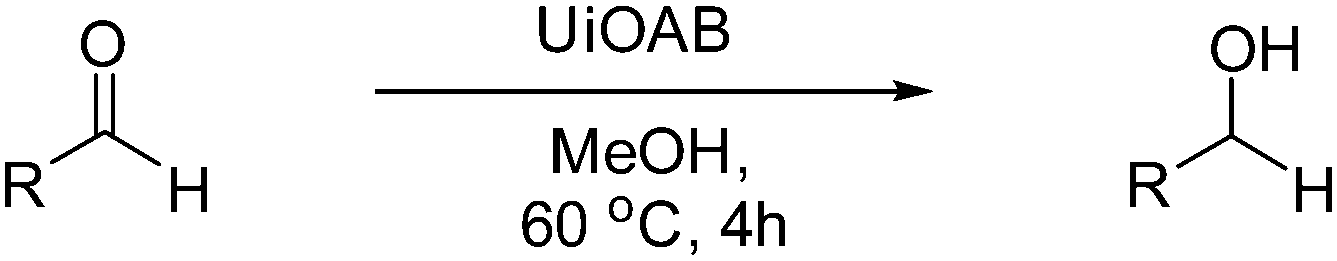

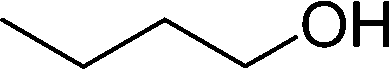
In summary, a microporous metal–organic framework UiOAB, which contains amino-borane complexes chemically bonded to its internal framework, was rationally designed and synthesized. This novel material can act as both a reusable hydrogen chemical storage material and a size-selective reduction agent. The dehydrogenation temperature was substantially decreased from 114 °C to 78 °C compared to neat AB, and also no volatile by-products were generated during the process. The enhancement in dehydrogenation kinetics and inhibition of volatile compounds confirmed the nano-confinement effect of the spatial isolation by chemical immobilization. In the reduction reactions, large substrates exhibited significantly reduced activities and conversions compared to small substrates, demonstrating that the reaction occurs primarily within the pores, which leads to a sharp molecular sieving effect.
Financial support from KAUST Competitive Grant URF/1/1378 and Baseline fund BAS/1/1375 is gratefully acknowledged.
References
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353 CrossRef CAS PubMed.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613 RSC.
- M. E. Bowden, I. W. Brown, G. J. Gainsford and H. Wong, Inorg. Chim. Acta, 2008, 361, 2147 CrossRef CAS PubMed.
- S. Li, Z. Tang, Q. Gong, X. Yu, P. R. Beaumont and C. M. Jensen, J. Mater. Chem., 2012, 22, 21017 RSC.
- G. Chen, L. N. Zakharov, M. E. Bowden, A. J. Karkamkar, S. M. Whittemore, E. B. Garner, T. C. Mikulas, D. A. Dixon, T. Autrey and S.-Y. Liu, J. Am. Chem. Soc., 2015, 1, 134 CrossRef PubMed.
- W. Luo, P. G. Campbell, L. N. Zakharov and S.-Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326 CrossRef CAS PubMed.
- F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746 CrossRef CAS PubMed.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287 CrossRef CAS PubMed.
- M. E. Bluhm, M. G. Bradley, R. Butterick, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS PubMed.
- D. W. Himmelberger, C. W. Yoon, M. E. Bluhm, P. J. Carroll and L. G. Sneddon, J. Am. Chem. Soc., 2009, 131, 14101 CrossRef CAS PubMed.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS PubMed.
- A. Gutowska, L. Y. Li, Y. S. Shin, C. M. M. Wang, X. H. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578 CrossRef CAS PubMed.
- L. Gao, C.-Y. V. Li, H. Yung and K.-Y. Chan, Chem. Commun., 2013, 49, 10629 RSC.
- S. Gadipelli, J. Ford, W. Zhou, H. Wu, T. J. Udovic and T. Yildirim, Chem. – Eur. J., 2011, 17, 6043 CrossRef CAS PubMed.
- R.-Q. Zhong, R.-Q. Zou, T. Nakagawa, M. Janicke, T. A. Semelsberger, A. K. Burrell and R. E. Del Sesto, Inorg. Chem., 2012, 51, 2728 CrossRef CAS PubMed.
- X.-l. Si, L.-x. Sun, F. Xu, C.-l. Jiao, F. Li, S.-s. Liu, J. Zhang, L.-f. Song, C.-h. Jiang and S. Wang, Int. J. Hydrogen Energy, 2011, 36, 6698 CrossRef CAS PubMed.
- G. Srinivas, J. Ford, W. Zhou and T. Yildirim, Int. J. Hydrogen Energy, 2012, 37, 3633 CrossRef CAS PubMed.
- G. Srinivas, W. Travis, J. Ford, H. Wu, Z.-X. Guo and T. Yildirim, J. Mater. Chem. A, 2013, 1, 4167 CAS.
- Z. Li, G. Zhu, G. Lu, S. Qiu and X. Yao, J. Am. Chem. Soc., 2010, 132, 1490 CrossRef CAS PubMed.
- H. M. Jeong, W. H. Shin, J. H. Park, J. H. Choi and J. K. Kang, Nanoscale, 2014, 6, 6526 RSC.
- Z. Li, W. Liu, H. Yang, T. Sun, K. Liu, Z. Wang and C. Niu, RSC Adv., 2015, 5, 10746 RSC.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O’Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef CAS PubMed.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. O. Yazaydin, R. Q. Snurr, M. O’Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424 CrossRef CAS PubMed.
- M. Savonnet, D. Bazer-Bachi, N. Bats, J. Perez-Pellitero, E. Jeanneau, V. Lecocq, C. Pinel and D. Farrusseng, J. Am. Chem. Soc., 2010, 132, 4518 CrossRef CAS PubMed.
- C. J. Doonan, W. Morris, H. Furukawa and O. M. Yaghi, J. Am. Chem. Soc., 2009, 131, 9492 CrossRef CAS PubMed.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700 RSC.
- P. A. P. Mendes, F. Ragon, A. E. Rodrigues, P. Horcajada, C. Serre and J. A. C. Silva, Microporous Mesoporous Mater., 2013, 170, 251 CrossRef CAS PubMed.
- L. Shen, W. Wu, R. Liang, R. Lin and L. Wu, Nanoscale, 2013, 5, 9374 RSC.
- F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. De Vos, Chem. Commun., 2011, 47, 1521 RSC.
- A. Staubitz, A. P. Robertson, M. E. Sloan and I. Manners, Chem. Rev., 2010, 110, 4023 CrossRef CAS PubMed.
- W. J. Atkins, E. R. Burkhardt and K. Matos, Org. Process Res. Dev., 2006, 10, 1292 CrossRef CAS.
- E. J. Corey, R. K. Bakshi and S. Shibata, J. Am. Chem. Soc., 1987, 109, 5551 CrossRef CAS.
- S. Horike, M. Dinca, K. Tamaki and J. R. Long, J. Am. Chem. Soc., 2008, 130, 5854 CrossRef CAS PubMed.
- J. M. Roberts, B. M. Fini, A. A. Sarjeant, O. K. Farha, J. T. Hupp and K. A. Scheidt, J. Am. Chem. Soc., 2012, 134, 3334 CrossRef CAS PubMed.
- A. V. A. Kumar and S. K. Bhatia, Phys. Rev. Lett., 2006, 96 Search PubMed.
- N. M. Yoon, C. S. Pak, H. C. Brown, S. Krishnam and T. P. Stocky, J. Org. Chem., 1973, 38, 2786 CrossRef CAS.
- C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415 CrossRef CAS PubMed.
- P. Renaud, A. Beauseigneur, A. Brecht-Forster, B. Becattini, V. Darmency, S. Kandhasamy, F. Montermini, C. Ollivier, P. Panchaud, D. Pozzi, E. M. Scanlan, A.-P. Schaffner and V. Weber, Pure Appl. Chem., 2007, 79, 223 CrossRef CAS.
- A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner, J. C. Gordon, T. Nakagawa, K. C. Ott, J. P. Robinson and M. Vasiliu, Science, 2011, 331, 1426 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional figures. See DOI: 10.1039/c5cc00193e |
| This journal is © The Royal Society of Chemistry 2015 |