
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conserved hydrogen bonding in tetrahydrocarbazolone derivatives: influence of solution-state assembly on crystal form nucleation†
Robert M.
Edkins
ab,
Elliott
Hayden
c,
Jonathan W.
Steed
a and
Katharina
Fucke
*c
aDepartment of Chemistry, Durham University, South Road, Durham, DH1 3LE, UK
bInstitut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany
cSchool of Medicine, Pharmacy and Health, Durham University, University Boulevard, Stockton-on-Tees, TS17 6BH, UK. E-mail: katharina.fucke@durham.ac.uk
First published on 17th February 2015
Abstract
Two tetrahydocarbazolone derivatives were found to show multiple unsolvated crystal forms. A persistent dimer motif was detected in solution by FTIR spectroscopy that is maintained in the kinetic crystal forms. Rationally introduced steric bulk induces the formation of a more stable catemeric form.
Different crystal forms (polymorphs) may vary substantially in their physico-chemical characteristics, including melting point, chemical and physical stability, solubility and dissolution rate, the latter of which represents both a challenge and an opportunity for the pharmaceutical industry.1 Thus, polymorphism studies are an ongoing focus during the development of new drug compounds, and studies of the solute–solute and solute–solvent interactions prior to crystallisation,2 as well as those during the nucleation process itself have become highly topical.3 Bioactive molecules and pharmaceuticals typically have multiple functional groups, enabling them to interact with receptors and thus show pharmacological action. Furthermore, as drug molecules become ever larger, they tend to show increased flexibility. These two factors make investigations and predictions of the crystallisation behaviour of most drug molecules inherently difficult.4 Therefore, to understand the factors leading to polymorphism, model compounds with reduced complexity have to be investigated, which allow the influence of, e.g., individual functional groups to be deconvoluted.5
In this study, we chose to investigate tetrahydrocarbazolone derivatives (Scheme 1), as they represent core fragments of many antibacterial and antiviral drugs and prodrugs,6 whilst having a rigid core with only one hydrogen-bond (HB) donor and one HB acceptor functionality. We were particularly interested in understanding whether these compounds show predictable supramolecular synthons7 that are robust enough to persist from a self-assembled pre-aggregated structure in solution to the solid state,8 and therefore influence the observed polymorphism, and whether it is possible to perturb these synthons by manipulation of the steric bulk of the molecule in order to affect the crystallisation outcome.
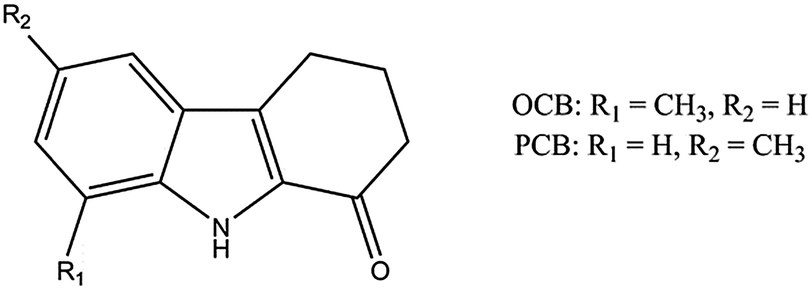 | ||
| Scheme 1 Molecular structure of the tetrahydrocarbazolone derivatives investigated in this study. | ||
A search for known crystal structures of the tetrahydrocarbazolone fragment in the Cambridge Structural Database9 returned a total of 22 crystal structures (search inputs are shown in the ESI†). A total of 19 of these exhibit hydrogen-bonded centrosymmetric dimer formation through two complementary HBs between the N–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups. In each case the interaction between donor and acceptor is not completely linear but exhibits a lateral offset of the acceptor in the direction of the aromatic ring (Fig. S1, ESI†). A search for the cyclopentenone analogue found seven structures, all of which contain the dimer motif; however, no hits were found for the cycloheptenone analogue. Simplifying the search to partially saturated indol-7-one derivatives found an additional 15 structures, with only four structures showing dimer formation, whilst a search for partially saturated quinolin-8-one derivatives gave eight structures, of which two contain the dimer synthon. However, the structures in the latter datasets have a high number of HB donor and acceptor groups, which make the realisation of other, competing synthons possible. Several hydrates were also retrieved that do not form the dimer motif, as a result of interactions with the water molecules. Overall, the high prevalence of the dimer motif for this group of related compounds indicates that this synthon is quite robust.
O groups. In each case the interaction between donor and acceptor is not completely linear but exhibits a lateral offset of the acceptor in the direction of the aromatic ring (Fig. S1, ESI†). A search for the cyclopentenone analogue found seven structures, all of which contain the dimer motif; however, no hits were found for the cycloheptenone analogue. Simplifying the search to partially saturated indol-7-one derivatives found an additional 15 structures, with only four structures showing dimer formation, whilst a search for partially saturated quinolin-8-one derivatives gave eight structures, of which two contain the dimer synthon. However, the structures in the latter datasets have a high number of HB donor and acceptor groups, which make the realisation of other, competing synthons possible. Several hydrates were also retrieved that do not form the dimer motif, as a result of interactions with the water molecules. Overall, the high prevalence of the dimer motif for this group of related compounds indicates that this synthon is quite robust.
With this in mind, we initially chose a tetrahydrocarbazolone derivative with a methyl group in the 7-position of the indole ring (‘ortho-methyl carbazolone’, OCB), i.e. in the direct vicinity of the HB-donating N–H group. This substitution pattern was anticipated to weaken the hydrogen-bonded dimers due to steric hindrance and reduce the possibility of realising the seemingly stabilising offset of the molecules against one another. We note that steric destabilisation of otherwise robust supramolecular synthons has been implicated previously in the formation of unusual helical structures of both carboxylic acids10 and benzyl alcohols11 and more generally in determining the structures of bulky monoalcohols.12
A polymorphism screen of OCB was carried out using slow and fast cooling experiments. For both experiment types, a saturated solution at boiling point was prepared in a total of 20 solvents of differing polarity and HB-accepting and -donating strengths. In addition, crystallisation from the melt and by sublimation using hot-stage microscopy was performed. Four different unsolvated crystal forms were identified and characterised by FTIR spectroscopy and powder X-ray diffraction (see ESI,† Fig. S2 and S3). All forms could be crystallised from solution, as well as from the melt. The different modifications can be differentiated by FTIR spectroscopy, even though the overall changes are small. The most pronounced difference is between the N–H vibrations of form I and the group of forms II, III and IV in the region 3350–3150 cm−1. Forms II, III and IV show peaks at 3286, 3291 and 3288 cm−1, respectively, while the peak of form I is shifted to considerably lower energy at 3227 cm−1. Thus, it was concluded that the HB-donor interactions of form I have to be significantly different from those of forms II, III and IV, which themselves should be closely related to one another.
The crystallisation experiments yielded crystals of sufficient size and quality to obtain X-ray single-crystal structures. All four crystal forms are presented in Fig. 1, of which form III has been described previously by Archana et al. (CSD ref. SUZGIE).13
 | ||
| Fig. 1 Crystal packing of the four polymorphs of OCB as determined by single-crystal X-ray diffraction. | ||
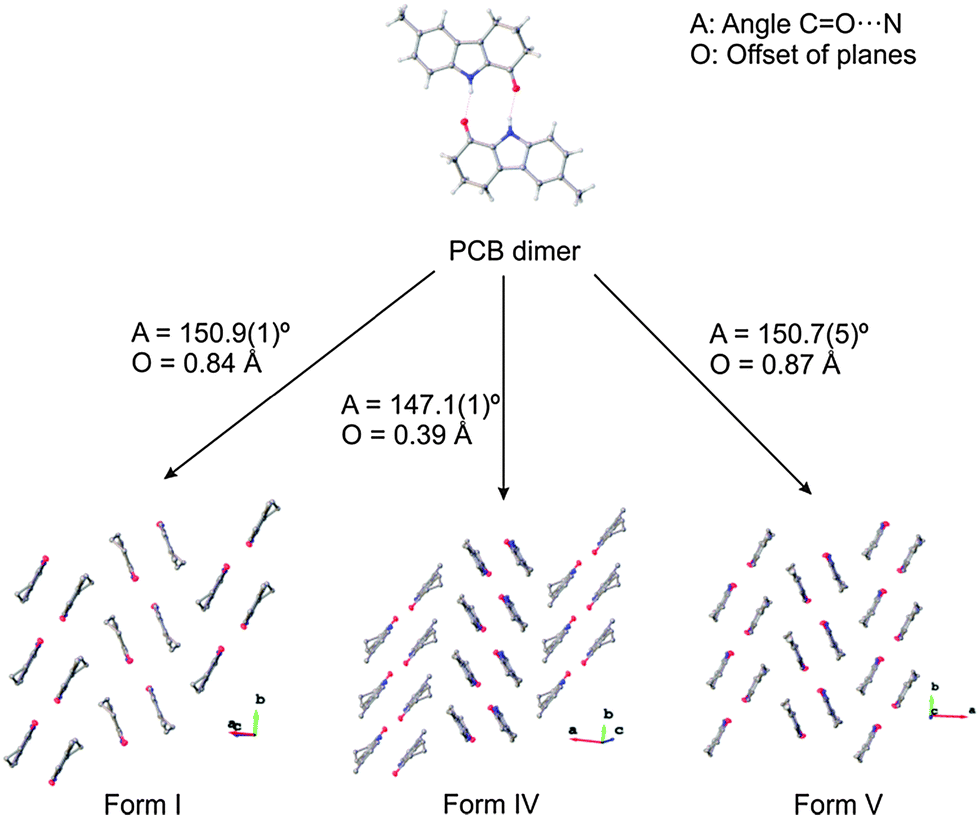
OCB crystal forms II, III and IV (Fig. 1) consist of the expected dimers generated by crystallographic inversion centres. The planes of the two molecules in all dimers are parallel, but show different plane-to-plane offsets. Due to staggering, the HB angle (D⋯A) changes from 156° in form II to 165° in forms III and IV. The N⋯O distance is consistently 2.84–2.86 Å. As a secondary motif, the dimers stack into a herringbone pattern generated by glide planes; however, the angles between the stacks vary widely. Form II shows the shallowest angle of 89.3° between neighbouring stacks; the stacks in form IV are oriented at 67.4°, while form III has the steepest packing with an angle of 46.5° (Fig. 1). The only short contacts between sets of dimers are weaker C–H⋯π interactions.
Form I, however, shows catemer formation, i.e. infinite chains of hydrogen-bonded molecules that are generated by a 21 screw operation along the crystallographic a-axis with an angle between the molecular planes of 88.4°. The hydrogen bonds are nearly linear on the donating side (N–H⋯O angle of 166°) but rather angled on the accepting side (CO⋯N angle of 113°), with a distance of 2.871(3) Å. The chains are packed in alternating stacks, which are related by a 21 screw operation along the crystallographic c-axis.
This behaviour of OCB mirrors that of carbamazepine, an anti-convulsant and mood-stabilising drug, for which stable dimer and catemer crystal forms were predicted in silico.14 However, the catemer form proved to be elusive for a long time and was only obtained recently by seeding the vapour phase with a catemer-forming structural analogue.15 While the catemer of OCB crystallised without cross-seeding, it requires considerable time to nucleate – up to several weeks in some solvents. It was also observed that samples of forms III and IV transformed into form I over time when stored as a suspension at room conditions (Table S1, ESI†). Form II and IV were additionally found to transform into form III under the same conditions. Thus, despite evident kinetic inhibition, it is concluded that form I is the thermodynamically stable polymorph at room temperature, followed by form III as the most stable dimer crystal form in this system. This fact highlights the importance of a thorough polymorphism screening, because the occurrence of a stable polymorph after a long nucleation time could easily be missed and could lead to incidents such as that of ritonavir.16
Non-empirical lattice energy calculations using the Partial Charges and Chemical Hardness Algorithm PACHA17 give insight into the relative strengths of the HBs in the crystal forms. Surprisingly, the stabilising energies of the dimers and the catemer vary only within the range 19.9–23.8 kJ mol−1, with the thermodynamically stable form I not having the strongest interactions. However, the ranking of the calculated densities of the crystal structures obeys the density rule of Burger and Ramberger,18i.e., the most stable form is the densest, and thermomicroscopic investigations confirmed that form I which melts at 174 °C has the highest melting point of all OCB crystal forms (form II: 168 °C, form III: 169–170 °C, form IV: 172 °C), thus suggesting monotropic relationship of all crystal forms with form I, with the latter being the most stable over the whole temperature range.
Having determined that form I has a longer induction time than the other forms, the hypothesis was postulated that OCB exists as dimers in solution, the melt and the vapour phase, which crystallise into the crystal forms II, III and IV upon rapid nucleation. If the crystallisation experiment is performed slowly however, so that the solution can age, the dimers convert into catemers, and subsequently the most stable polymorph, form I, crystallises. To prove initial solution dimerisation, an FTIR titration was performed by starting with a 0.542 M solution of OCB in chloroform (close to saturation) and conducting a serial dilution. At the highest concentration, two different N–H vibrations can clearly be observed, namely a sharp feature at 3460 cm−1 and a broader peak around 3300 cm−1 (Fig. 2). According to quantum calculations (vide infra), the higher energy peak corresponds to the OCB monomer, while the lower energy feature originates from the dimer. This assignment is corroborated by the titration: with decreasing concentration, the lower energy peak vanishes to the point that at a concentration of 0.018 M it is not detectable above the background. The monomer N–H vibration, however, is still observable at this low concentration (Fig. S4, ESI†).
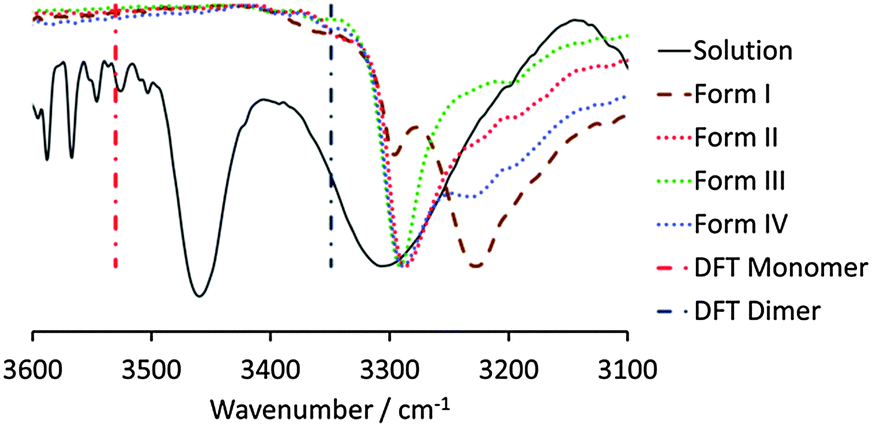 | ||
| Fig. 2 N–H region of the normalised transmission FTIR spectra of a 0.542 M solution of OCB in chloroform and the four crystal forms. Vertical lines depict the DFT-calculated N–H vibrations of the monomer and dimer. | ||
When the solution IR spectra are compared with the solid-state spectra of forms I–IV, it can be seen that the species formed in solution is indeed the dimer, as the N–H vibration is very similar in energy to that of the three dimer crystal forms, forms II, III, and IV. The N–H vibration of form I is shifted to significantly lower energy, and is clearly distinct from that observed in solution, excluding the possibility of catemer-like chains in chloroform solution.
A similar preliminary dimer formation is anticipated in the gas phase, as crystals formed by sublimation are form III, i.e. the most stable modification showing the dimer motif. This crystallisation method is the only one that did not result in the formation of form I under any conditions. Crystallisation from the melt typically resulted in one of the dimer polymorphs, but form I could be grown by annealing the melt at temperatures above the melting points of the dimer modifications (>170 °C). The overall crystallisation behaviour therefore supports the hypothesis that, before crystallisation occurs, the amorphous phase consists of OCB dimers, be it in solution, the vapour phase or the melt.
In order to investigate the influence of the methyl group in the vicinity of the HB donor on the resultant crystallisation behaviour, another tetrahydrocarbazolone molecule was investigated, in which the methyl group was changed from the 7- to the 5-position of the indole ring (‘para-methyl carbazolone’, PCB, Scheme 1). A crystallisation screen was carried out in a similar way to OCB (Table S3, ESI†) and the resulting crystal forms were characterised by FTIR spectroscopy and powder X-ray diffraction (Fig. S5 and S6, ESI†). At least five different polymorphs could be identified, with some of them occurring concomitantly and some indication of additional modifications that could not be obtained as pure phases. The crystal forms were named forms I to V according to their order of appearance in time. Forms I, IV and V could only be crystallised from solution, while forms II and III were only obtained from the melt. FTIR spectroscopic investigations indicated that all forms exhibit the dimer motif, as all N–H vibrations coincide within the narrow range of 3369 to 3366 cm−1, comparable to the dimer forms of OCB. Even though the existence of a catemer form cannot be completely excluded, we can be sure that it is not accessible in a reasonable experimental space. In addition, cross-seeding experiments of PCB melt or solutions with the catemer form of OCB, form I, resulted in the crystallisation of a random but pure dimer modification of PCB, and thus, unlike the analogous case of carbamazepine, a catemer form cannot be easily produced by this method.
Powder X-ray diffractograms showed that samples crystallised from solution and identified as form I are generally a mixture of several forms including predominantly form I as well as form V and additional phases, indicating that the energetic differences between the PCB polymorphs are small and that more modifications are likely to exist. Further investigations into this behaviour are underway.
Three modifications could be crystallised in sufficient quality to determine their X-ray crystal structures, namely form I, form IV and form V (Fig. 3 and Table S4, ESI†). These crystal structures are very closely related and show almost identical packing, corroborating the possibility that more energetically closely related crystal forms exist. In particular, forms I and V, which crystallise concomitantly, reveal the same geometry of the dimers and have the same packing with the only subtle difference being that both forms crystallise in layers, which in form I are oriented so that the methyl group of adjacent dimers are on opposite sides, while in form V these are on the same side. This results in different space groups for the two structures (form I in P21/n, form V in Pbca). Even though the chemical structures of OCB and PCB are similar, the packing schemes in the respective crystal structures are very different, and no pair of them is isostructural.
 | ||
| Fig. 3 Crystal packing of the three polymorphs of PCB as determined by single-crystal X-ray diffraction. | ||
The structures of OCB, PCB and their respective R22(10) hydrogen-bonded homodimers were optimised in the gas phase using DFT calculations (B3LYP/6-311+G(d), GD3BJ dispersion correction). The dimerisation energies in the gas phase are calculated to be −70.2 and −74.4 kJ mol−1 for OCB and PCB, respectively. The slightly less exothermic dimerisation energy of OCB is presumably due to the destabilising steric interaction of the ortho-methyl group. This introduces a lateral displacement of the dimer within the near coplanar structure of ca. 0.45 Å, due to a close contact between the carbonyl and methyl groups of the two monomers (Fig. S7, ESI†). The calculated hydrogen-bonding distances are respectively 1.859 (O–H)/2.871 (N–O) and 1.847 (O–H)/2.826 (N–O) Å for OCB and PCB, in good agreement with the experimental X-ray structures. Frequency calculations on the optimised monomer and dimer structures gave insight into our FTIR spectroscopic studies of OCB, namely that the N–H stretch of the monomer is higher in energy (3530 cm−1) than that of the hydrogen-bonded dimer (3349 cm−1).
We believe that the destabilising steric interaction present in the dimeric structure of OCB that is absent in PCB is the driving force towards the formation of the catemeric structure, i.e. due to a weakening of the hydrogen bonding in the dimer, the catemer modification of OCB, in which this steric interaction is less pronounced, becomes lower in energy relative to the dimer structures and, thus, experimentally accessible. Kinetically, however, it is slow to form due to the entropic cost of forming chains. Furthermore, it can be presumed that to form a catemer chain would first require the breaking of hydrogen bonds in the initially formed dimers that will have a large activation energy due to the generation of unsaturated or weakly solvated hydrogen bond acceptors and donors.
In addition, the dimer structure of FOCB, i.e. where the CH3 of OCB is replaced with the larger CF3 group, was optimised in the gas phase at the same level of theory. The binding in the dimer (−55.7 kJ mol−1) is weaker than that of the OCB dimer, from which we might predict that this compound would be expected to form a catemeric structure; this hypothesis will be tested in future work. The replacement of a methyl group with its bio-isostere CF3 is commonly performed in the development of pharmaceuticals, due to the greater metabolic stability of the latter; thus, while such a replacement may be desirable from a pharmacokinetic view point, its influence on solid-state behaviour, and, subsequently, on the formulation of the drug into a dosage form, is less often considered.
In conclusion, this study presents the polymorphism of two methyl-tetrahydrocarbazolone derivatives, and the influence of the position of methylation on the existence of supramolecular synthons. Ortho-methylated tetrahydrocarbazolone (OCB) can exist in four different crystal structures, three of which show the anticipated dimer formation. The thermodynamically stable polymorph, however, crystallises in a catemer motif but has a considerably longer nucleation time. This behaviour is reminiscent of carbamazepine, which was predicted to form a stable catemeric modification, but could only be crystallised as such by seeding with a structural analogue. The existence of a more stable polymorph with slower crystallisation kinetics can cause severe manufacturing problems in the pharmaceutical industry, as was the case with ritonavir. When moving the methyl group from the ortho- to the para-position (PCB), only dimer formation was observed, while the different polymorphs become very close in energy and concomitant crystallisation occurred. Thus, subtle changes in molecular structure can have profound influences on crystallisation behaviour. It is also predicted that a bio-isosteric replacement of the CH3 group of OCB with CF3 will further stabilise the catemer, highlighting a potential problem for the design of new drugs and their subsequent formulation into a dosage form.
We acknowledge the Royal Society of Chemistry for an Undergraduate Summer Research Bursary (E.H.), Durham University for a Durham Doctoral Fellowship (R.M.E.) and the Pro-vice Chancellor Seedcorn fund (K.F.), funding from the Engineering and Physical Sciences Research Council (EP/F063229/1, K.F. and J.W.S.) and the Alexander von Humboldt Foundation for a Postdoctoral Research Fellowship (R.M.E.). We also thank Diamond Light Source for synchrotron beam time allocated to the regional team of Newcastle and Durham Universities on I19 (MT 6749) and the instrument scientists for support.
Notes and references
- (a) D. J. W. Grant, in Polymorphism in Pharmaceutical Solids, ed. H. G. Brittain, Marcel Dekker Inc., New York, 1999, pp. 1–33 Search PubMed; (b) J. Bernstein, Cryst. Growth Des., 2011, 11, 632 CrossRef CAS.
- (a) R. C. Burton, E. S. Ferrari, R. J. Davey, J. L. Finney and D. T. Bowron, J. Phys. Chem. B, 2010, 114, 8807 CrossRef CAS PubMed; (b) A. Mattei, X. Mei, A.-F. Miller and T. Li, Cryst. Growth Des., 2013, 13, 3303 CrossRef CAS; (c) S. A. Kulkarni, E. S. McGarrity, H. Meekes and J. H. ter Horst, Chem. Commun., 2012, 48, 4983 RSC.
- (a) R. J. Davey, N. Blagden, S. Righini, H. Alison, M. J. Quayle and S. Fuller, Cryst. Growth Des., 2000, 1, 59 CrossRef; (b) R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Angew. Chem., Int. Ed., 2013, 52, 2166 CrossRef CAS PubMed; (c) D. Gebauer, M. Kellermeier, J. D. Gale, L. Bergstrom and H. Colfen, Chem. Soc. Rev., 2014, 43, 2348 RSC; (d) R. A. Sullivan, R. J. Davey, G. Sadiq, G. Dent, K. R. Back, J. H. ter Horst, D. Toroz and R. B. Hammond, Cryst. Growth Des., 2014, 14, 2689 CrossRef CAS.
- S. L. Price, Chem. Soc. Rev., 2014, 43, 2098 RSC.
- (a) K. Fucke, J. A. K. Howard and J. W. Steed, Chem. Commun., 2012, 48, 12065 RSC; (b) K. Fucke, G. J. McIntyre, C. Wilkinson, M. Henry, J. A. K. Howard and J. W. Steed, Cryst. Growth Des., 2012, 12, 1395 CrossRef CAS; (c) K. Fucke, G. J. McIntyre, M.-H. Lemée-Cailleau, C. Wilkinson, A. J. Edwards, J. A. K. Howard and J. W. Steed, Chem. – Eur. J., 2015, 21, 1036 CrossRef CAS PubMed.
- (a) X. Li and R. Vince, Bioorg. Med. Chem., 2006, 14, 2942 CrossRef CAS PubMed; (b) G. Periyasami, R. Raghunathan, G. Surendiran and N. Mathivanan, Bioorg. Med. Chem. Lett., 2008, 18, 2342 CrossRef CAS PubMed; (c) K. S. Gudmundsson, P. R. Sebahar, L. D. A. Richardson, J. G. Catalano, S. D. Boggs, A. Spaltenstein, P. B. Sethna, K. W. Brown, R. Harvey and K. R. Romines, Bioorg. Med. Chem. Lett., 2009, 19, 3489 CrossRef CAS PubMed.
- G. R. Desiraju, J. Am. Chem. Soc., 2013, 135, 9952 CrossRef CAS PubMed.
- (a) C. A. Hunter, J. F. McCabe and A. Spitaleri, CrystEngComm, 2012, 14, 7115 RSC; (b) S. F. Alshahateet, M. M. Bhadbhade, R. Bishop and M. L. Scudder, CrystEngComm, 2015, 17, 877 RSC.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, B58, 380 CrossRef CAS PubMed.
- J. N. Moorthy, R. Natarajan, P. Mal and P. Venugopalan, J. Am. Chem. Soc., 2002, 124, 6530 CrossRef CAS PubMed.
- J. N. Moorthy, S. Mandal and P. Venugopalan, Cryst. Growth Des., 2012, 12, 2942 CAS.
- C. P. Brock and L. L. Duncan, Chem. Mater., 1994, 6, 1307 CrossRef CAS.
- R. Archana, E. Yamuna, K. J. Rajendra Prasad, A. Thiruvalluvar and R. J. Butcher, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o2299 CAS.
- A. J. Florence, A. Johnston, S. L. Price, H. Nowell, A. R. Kennedy and N. Shankland, J. Pharm. Sci., 2006, 95, 1918 CrossRef CAS PubMed.
- J. B. Arlin, L. S. Price, S. L. Price and A. J. Florence, Chem. Commun., 2011, 47, 7074 RSC.
- S. R. Chemburkar, J. Bauer, K. Deming, H. Spiwek, K. Patel, J. Morris, R. Henry, S. Spanton, W. Dziki, W. Porter, J. Quick, P. Bauer, J. Donaubauer, B. A. Narayanan, M. Soldani, D. Riley and K. McFarland, Org. Process Res. Dev., 2000, 4, 413 CrossRef CAS.
- M. Henry, ChemPhysChem, 2002, 3, 561 CrossRef CAS.
- A. Burger and R. Ramberger, Mikrochim. Acta, 1979, 2, 259 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1041114–1041120. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc10453f |
| This journal is © The Royal Society of Chemistry 2015 |