
The controlled synthesis of plasmonic nanoparticle clusters as efficient surface-enhanced Raman scattering platforms†
Seunghoon
Lee
ab,
Jong Wook
Hong
ab,
Su-Un
Lee
ab,
Young Wook
Lee
ab and
Sang Woo
Han
*ab
aDepartment of Chemistry and KI for the NanoCentury, KAIST, Daejeon 305-701, Korea. E-mail: sangwoohan@kaist.ac.kr
bCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science (IBS), Daejeon 305-701, Korea
First published on 20th April 2015
Abstract
A facile aqueous synthesis method for the preparation of Au nanoparticle clusters by the controlled galvanic replacement of Ag nanoparticles with Au precursors is described. The prepared clusters showed both significantly enhanced surface-enhanced Raman scattering activity and stability.
Au nanoparticles (AuNPs) have attracted considerable research interest as optical materials due to their inherent localized surface plasmon resonance (LSPR) characteristics.1,2 In this regard, they have been extensively explored for numerous applications including bio/chemical/environmental sensors, surface-enhanced Raman scattering (SERS) spectroscopy, photothermal therapy, and polymer photovoltaics.3–6 Manipulation of the plasmonic characteristics of AuNPs is the most critical issue in such applications. Accordingly, tremendous efforts have been devoted to the control of the LSPR properties of AuNPs via controlling their size, shape, dielectric environment, and spatial arrangement, because these parameters explicitly determine their plasmonic characteristics such as the LSPR profile and SERS efficiency.1,3 In particular, the controlled assembly of AuNPs has recently proved to be a promising strategy to construct functional plasmonic platforms with specific optical properties, such as Fano resonance and promoted SERS activity.2,7–10 For instance, compared with individual AuNPs, AuNP clusters (AuNPCs) consisting of a number of AuNPs show highly efficient SERS properties because they are rich in interparticle gaps, which can act as “hot spots” for strong electromagnetic field enhancement.11,12 Furthermore, AuNPCs are thought to be very stable SERS platforms for biosensing applications. AuNPs have been commonly employed to obtain SERS signals from biomarkers or SERS-marker molecules in biological fluids or tissue matrices.13 However, the intensity of SERS signals changes irregularly during the measurement and decays significantly with time because the AuNPs aggregate with each other in an uncontrolled manner under high salt conditions.4,14 In stark contrast, AuNPCs would maintain sufficient SERS signals for a prolonged time under physiological conditions, as they are already composed of assembled AuNPs.
So far, charge interaction and surfactant- or DNA-mediated assembly of constituent AuNPs have been mostly adopted to produce AuNPCs in solutions or on solid substrates.7,15–17 Here, we report a novel synthesis method for the preparation of AuNPCs. Controlled synthesis of AuNPCs could be realized in the aqueous phase at room temperature under mild chemical conditions by fine control over the galvanic replacement of Ag nanoparticles (AgNPs) with Au precursors (Scheme 1). The structure of sacrificial AgNPs explicitly determines the final morphology of AuNPCs. The prepared AuNPCs exhibited both highly promoted SERS activity and stability for various analytes in water and phosphate-buffered saline (PBS) solution compared to AuNPs. Furthermore, single-molecule SERS events could be detected using the AuNPCs.
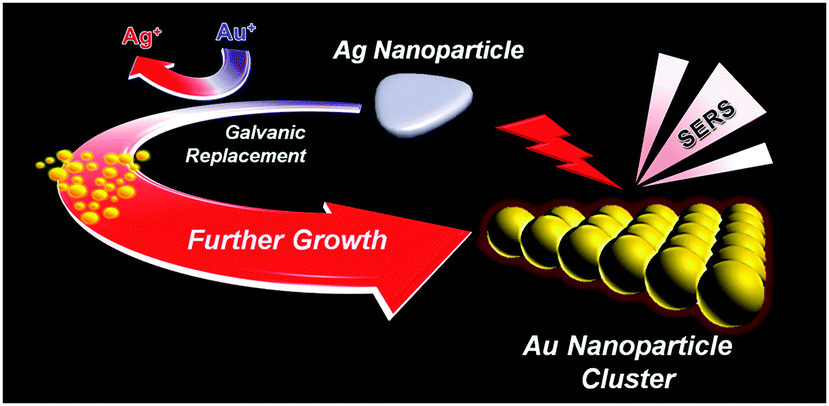 | ||
| Scheme 1 Formation of AuNPCs via the controlled galvanic replacement of AgNPs with Au precursors. | ||
To synthesize AuNPCs, an aqueous solution containing HAuCl4, KI, citrate, and poly(vinyl pyrrolidone) (PVP) was prepared first as a growth solution, and then ascorbic acid (AA) and H2O2 were added to this solution (see the ESI†). The pale-yellow color of the growth solution faded upon the addition of AA, implying the partial reduction of Au3+ to Au+.18 In a standard synthesis of AuNPCs, AgNPs having a prismatic shape (Ag nanoprisms) with an average edge length of 35 ± 8 nm (Fig. S1a†), which were prepared by following the reported procedure,19 were quickly injected into the growth solution and left undisturbed at room temperature. The color of the reaction mixture changed to purple in less than 10 s, indicating the formation of a certain Au nanostructure. Fig. 1a and b show, respectively, the representative SEM and TEM images of the product, demonstrating the successful formation of AuNPCs with a prism-like shape overall through galvanic replacement reaction between Ag nanoprisms and Au precursors [see also the low-magnification SEM image of the product (Fig. S2†)]. The inductively coupled plasma-atomic emission spectroscopy (ICP-AES)-determined Au composition of the AuNPCs was higher than 98%. The average cluster size of the prepared AuNPCs was 99 ± 24 nm. Each AuNPC contained AuNPs with an average particle size of 21 ± 3 nm (see the inset of Fig. 1b). The average number of AuNPs per cluster was 22 ± 6. A close inspection of the junction points between particles within the clusters showed that the gap sizes between particles were in the range of 0.5–2 nm with an average value of 1.1 nm (Fig. S3 and S4†). The dynamic light scattering-determined Z-average diameter of the AuNPCs was 106.7 nm (Fig. S5†), further indicating that the structures are assembled in solution and not during deposition. The prepared clusters were very stable. In fact, we found that the AuNPCs maintained their structure at least for ∼2 weeks in water and PBS solution (phosphate buffer: 10 mM, NaCl: 154 mM, pH = 7.4) (Fig. S6†). Furthermore, the assembly structure of AuNPCs cannot be destroyed by sonication for a prolonged time (Fig. S7†). The structural integrity of the AuNPCs is mainly due to strong van der Waals forces between particles, which can hold the particles together. The UV-vis extinction spectrum of the AuNPCs showed a broad SPR peak at 561 nm (Fig. S8†). On the other hand, the heat treatment of as-prepared AuNPCs demonstrated that the AuNPCs are indeed composed of isolated AuNPs (Fig. S9†).
 | ||
| Fig. 1 SEM (left column) and TEM (right column) images of AuNPCs prepared from (a, b) Ag nanoprisms and (c, d) oblate-shaped AgNPs. High-magnification TEM image of a single AuNPC is shown in the inset of (b). The scale bar indicates 20 nm. | ||
Notably, AuNPCs with different structures could be readily generated by using AgNPs with different morphologies, such as oblate-shaped AgNPs (Fig. S1b†) and large Ag nanoprisms (Fig. S1c†), as templates (Fig. 1c and d and Fig. S10†). This shows that the structure of sacrificial AgNPs determines the final morphology of AuNPCs. In general, the synthesized AuNPCs were larger than pristine AgNPs, revealing that AuNPCs were formed through galvanic replacement reaction followed by overgrowth of AuNPs via the reduction of remaining Au precursors in growth solution on the surface of growing NPs (vide infra).
Noticeably, the present synthesis route did not yield hollow or frame structures, which are commonly generated by the galvanic replacement of AgNPs with Au precursors.20–22 The formation of AuNPCs under our experimental conditions can be attributed to several factors. First, decreasing the reduction potential of Au precursors both through the complexation of Au ions with I−![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 23 and the pre-reduction of Au3+ to Au+ by AA is the key to the successful formation of AuNPCs. Analogous experiments in the absence of I− and/or without the pre-reduction process did not produce AuNPCs; instead ill-defined NPs were generated, where some of them had hollow features (Fig. S11†). This reveals that reducing the extent of galvanic replacement reaction by lowering the reduction potential of Au precursor accounts for the AuNPC formation. In previous studies, including ours, such control over the kinetics of galvanic replacement has been utilized to yield uncommon nanostructures.23–26 Second, employing suitable stabilizing agents, i.e., PVP and citrate, plays a crucial role in the formation of AuNPCs. In fact, the characteristic assembly structure was not formed when the reaction proceeded in the absence of PVP or citrate (Fig. S12†). Nanoparticle agglomerates or frame-like structures were obtained during the synthesis in the absence of PVP or citrate, respectively, which seem to originate from the extensive coalescence of constituent AuNPs. These observations imply that PVP and citrate prevent AuNPs in clusters from severe agglomeration, thereby stabilizing the formed AuNPCs. After the completion of AuNPC formation, these stabilizing agents could be effectively removed by repeated washing with de-ionized water by centrifugation, while the assembly structure of AuNPCs was almost maintained (Fig. S13†). Finally, the use of a relatively large amount of Au precursors than Ag is critical to the production of AuNPCs. In the standard synthesis, we used the Au:Ag ratio of 50:1, which proved to be an optimum value. Although triangular NPCs were also prepared when the Au:Ag ratio was decreased gradually from 50:1 to 10:1 (average cluster size and NP size decreased to 77 ± 17 and 16 ± 4 nm, respectively), the Ag composition of the resultant NPCs, which was determined by ICP-AES, reached 17% (Fig. S14 and S15†). This indicates that Ag can be effectively replaced with Au when the Au:Ag ratio is around 50:1. Notably, when the Au:Ag ratio was less than 5:1, we could not obtain complete NPCs (Fig. S16†). In particular, unusual frame-like structures were generated when the Au:Ag ratio was less than 2.5:1, demonstrating that the galvanic replacement reaction occurs preferentially at the highly energetic sites of Ag nanoprisms, such as corners and edges, when an insufficient amount of Au precursors is present in the reaction medium.
23 and the pre-reduction of Au3+ to Au+ by AA is the key to the successful formation of AuNPCs. Analogous experiments in the absence of I− and/or without the pre-reduction process did not produce AuNPCs; instead ill-defined NPs were generated, where some of them had hollow features (Fig. S11†). This reveals that reducing the extent of galvanic replacement reaction by lowering the reduction potential of Au precursor accounts for the AuNPC formation. In previous studies, including ours, such control over the kinetics of galvanic replacement has been utilized to yield uncommon nanostructures.23–26 Second, employing suitable stabilizing agents, i.e., PVP and citrate, plays a crucial role in the formation of AuNPCs. In fact, the characteristic assembly structure was not formed when the reaction proceeded in the absence of PVP or citrate (Fig. S12†). Nanoparticle agglomerates or frame-like structures were obtained during the synthesis in the absence of PVP or citrate, respectively, which seem to originate from the extensive coalescence of constituent AuNPs. These observations imply that PVP and citrate prevent AuNPs in clusters from severe agglomeration, thereby stabilizing the formed AuNPCs. After the completion of AuNPC formation, these stabilizing agents could be effectively removed by repeated washing with de-ionized water by centrifugation, while the assembly structure of AuNPCs was almost maintained (Fig. S13†). Finally, the use of a relatively large amount of Au precursors than Ag is critical to the production of AuNPCs. In the standard synthesis, we used the Au:Ag ratio of 50:1, which proved to be an optimum value. Although triangular NPCs were also prepared when the Au:Ag ratio was decreased gradually from 50:1 to 10:1 (average cluster size and NP size decreased to 77 ± 17 and 16 ± 4 nm, respectively), the Ag composition of the resultant NPCs, which was determined by ICP-AES, reached 17% (Fig. S14 and S15†). This indicates that Ag can be effectively replaced with Au when the Au:Ag ratio is around 50:1. Notably, when the Au:Ag ratio was less than 5:1, we could not obtain complete NPCs (Fig. S16†). In particular, unusual frame-like structures were generated when the Au:Ag ratio was less than 2.5:1, demonstrating that the galvanic replacement reaction occurs preferentially at the highly energetic sites of Ag nanoprisms, such as corners and edges, when an insufficient amount of Au precursors is present in the reaction medium.
On the basis of the data presented, we can propose the two-step formation mechanism of the AuNPCs (Scheme 1). In the initial step, Au precursors, Au(I), are reduced by galvanic replacement reaction to rapidly form nuclei at the entire area of the Ag nanoprism. As previously mentioned, under the present synthesis conditions, the homogeneous nucleation of Au is unlikely to occur due to the decreased reduction potential of Au precursors. At the second step, these nuclei further grow into AuNPs until the complete consumption of the Au precursors with the help of ascorbic acid and citrate–H2O2, which can act as reducing agents for the reduction of Au precursors. However, since the reaction time is too fast (<10 s), we could not catch each step with microscopic techniques.
To investigate the efficacy of the synthesized AuNPCs as plasmonic platforms, we measured the SERS signals of various Raman dyes in aqueous solutions with the colloidal AuNPCs synthesized through the standard procedure. For SERS measurements, we prepared two types of Raman samples, distinguishable by the injection time of Raman dye: Raman dyes were adsorbed after and before the cluster formation for types I and II samples, respectively. For the preparation of type I samples, 0.5 mL of purified AuNPC solution and 0.5 mL of pre-diluted Raman dye solution were mixed to allow the uniform surface coverage of Raman dye molecules (“half–half dilution procedure”27). For preparing type II samples, pre-diluted Raman dyes were injected into the growth solution before the addition of reducing agents during the synthesis of AuNPCs. AuNPCs with similar assembly structures to those used in the preparation of type I samples were successfully generated in the presence of Raman dyes (Fig. S17†). In fact, types I and II methods have been typically employed in the preparation of Raman samples for environmental sensing and the fabrication of Raman probes for bioimaging applications, respectively. Fig. 2a–d show the SERS spectra of rhodamine 6G (R6G, 5 × 10−9 M), 4-nitrobenzenethiol (4-NBT, 5 × 10−8 M), crystal violet (CV, 1 × 10−10 M), and Nile blue (NB, 5 × 10−11 M) obtained with the AuNPCs by using 632.8 nm excitation. Apparently, characteristic SERS spectra could be obtained for such low-concentration of Raman dyes, which evidences the prominent SERS activity of the prepared AuNPCs. The above concentration of each Raman dye is the minimum molar concentration detectable by the AuNPCs. In the cases of the R6G and 4-NBT, both types of Raman samples gave comparable SERS signals, whereas the type II samples exhibited a higher SERS activity than the type I samples for the CV and NB. This can be attributed to the fact that the AuNPCs of type II samples have more chance of trapping Raman dyes at their hot spots compared to those of type I samples in the presence of low-concentration of Raman dyes. Notably, AuNPs with a similar average particle size (20 nm, Fig. S18†) to that of the particles in the AuNPCs gave no detectable SERS signal for all the Raman dyes under the same measurement conditions. This finding distinctly demonstrates the highly efficient SERS activity of the AuNPCs, as they are rich in interparticle gaps. To compare the SERS efficacies of the prepared AuNPCs and AuNPs, their surface areas were adjusted to a similar value (see the ESI† for details).
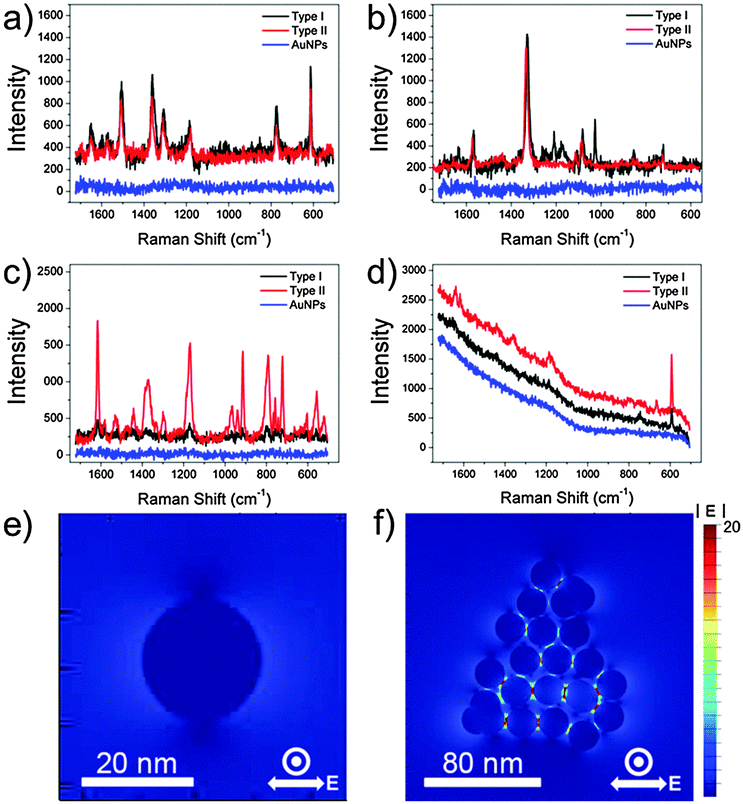 | ||
| Fig. 2 SERS spectra of (a) R6G (5 × 10−9 M), (b) 4-NBT (5 × 10−8 M), (c) CV (1 × 10−10 M), and (d) NB (5 × 10−11 M) obtained with a 632.8 nm excitation (2 mW, integration time = 10 s). FDTD-simulated |E| distributions of an (e) AuNP and (f) AuNPC. | ||
From the comparison of normal Raman signals of the aqueous solutions of Raman dyes, the analytical SERS enhancement factor (EF)6,28 of the AuNPCs (type II) was estimated to be 3.7 × 104, 1.7 × 104, and 1.2 × 105 for R6G, 4-NBT, and CV, respectively (see the ESI† for details). The analytical EF for NB could not be calculated due to the overwhelming fluorescence background of NB at the 632.8 nm excitation. Compared to some reported EFs, the measured analytical EFs are relatively low. This is because the analytical EFs are average values representing a broad distribution of EFs from the ensemble of dye molecules.28,29 As revealed by previous SERS studies, a subensemble of molecules residing at hot spots dominates the SERS signal.28–31 Accordingly, if we could count the exact number of dye molecules located at the hot spots of the AuNPCs, which is practically hard to evaluate with suitable reliability, much higher EFs could be obtained. Evidently, the present AuNPCs have an enough SERS activity to enable the detection of single molecules, as will be confirmed later.
To further verify the role of the interparticle gaps in the AuNPCs in enhancing their SERS activity, we performed the theoretical analysis of the field distribution around the nanostructures by using the finite-difference time domain (FDTD) method. As shown in Fig. 2e and f, the FDTD-simulated electric field amplitude (|E|) distribution of an AuNPC with the excitation wavelength of λex = 632.8 nm showed much stronger (35-fold) field localizations at the interparticle gaps relative to a spherical AuNP of the same NP size (21 nm). This clearly reveals that the interparticle gaps in the AuNPCs can act as SERS hot spots through the strong enhancement of the electromagnetic field. The calculated EF of the AuNPC obtained from the FDTD-simulated |E| at the gap was as high as 4 × 108.
In order to probe the single molecule-SERS capability of the prepared AuNPCs, an ultralow analyte concentration method was used with a suitable acquisition time for SERS measurements.32,33 Fig. S19a† shows the time-dependent SERS spectra of 1 × 10−7 M NB obtained with the AuNPCs (type II). The acquisition time for each spectrum was 100 ms, which typically allows the observation of single SERS event per colloidal particle in scattering volume.30 Apparently, stable SERS signals of NB were obtained. This can be attributed to the constant distribution of hot spots and NB molecules in the scattering volume, which could be achieved by the defined assembly structure of AuNPCs and the pre-dilution of Raman dyes before their adsorption on the AuNPCs, which leads to the uniform molecular coverage. SERS intensities of NB measured at 592 cm−1 showed a typical Gaussian distribution (Fig. S19b†), which is the common feature of multimolecule events. Notably, when the concentration of NB was decreased to 5 × 10−11 M (0.067 NB molecules per particle in the AuNPCs), SERS signals exhibited a strong fluctuation in intensity with spectral blinking (Fig. S19c†). As shown in Fig. S19d,† the distribution of NB SERS intensities was changed from Gaussian to Poisson distribution. These findings demonstrate that single-molecule SERS events could be detected using the AuNPCs.32,33 In fact, representative SERS spectra showed quantized signals for 1, 2, 3, 4, and 5 NB molecules (Fig. S19e and f†).
Besides the sensitivity, the stability of colloidal SERS platforms has been a critical issue in their applications for SERS sensing and imaging. In this regard, we investigated the SERS stability of the colloidal AuNPCs in water and under physiological conditions. Compared to individual AuNPs, the AuNPCs gave very stable SERS signals in water even for an analyte with multiple strong binding sites such as 1,4-phenylene diisocyanide (Fig. S20†), which has frequently induced the severe agglomeration of NPs in solution in an uncontrolled manner because they can act as a molecular linker between particles.14 Furthermore, the colloidal AuNPCs showed prominent SERS stability in PBS solution, while AuNPs exhibited irregular changes in SERS signals with time due to uncontrolled particle aggregation under high salt conditions (Fig. S21 and S22 for types I and II samples, respectively†). These results demonstrate that the AuNPCs have better SERS stability than AuNPs as they are already composed of assembled AuNPs; thus, they are less vulnerable to analyte-induced particle agglomeration.
In summary, we presented a facile aqueous synthetic method for the preparation of AuNPCs by the controlled galvanic replacement of AgNPs with Au precursors. The prepared AuNPCs showed greatly enhanced SERS activity and stability for various analytes in water and under physiological conditions. Furthermore, single molecule SERS signals could be detected using the AuNPCs. Since the colloidal suspension of AuNPCs has been used as an SERS platform in our work, we anticipate that this study will provide a new direction for the development of an efficient plasmonic platform for in vitro and in vivo biosensing and imaging. Further improvement of plasmonic performance and its application to the development of efficient plasmonic platforms are expected through better control over the size of the NP building blocks and their spacing in the final clusters. To address this issue, more precise tuning of the morphology of sacrificial templates as well as the kinetics of galvanic replacement reaction is currently underway.
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2010-0029149), and was also supported by Institute for Basic Science (IBS) [IBS-R004-D1] and KAIST RED&B project. S.L. acknowledges TJ Park Science Fellowship of POSCO TJ Park Foundation.
Notes and references
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2003, 107, 668 CrossRef CAS.
- J. A. Fan, C. Wu, K. Bao, J. Bao, R. Bardhan, N. J. Halas, V. N. Manoharan, P. Nordlander, G. Shvets and F. Capasso, Science, 2010, 328, 1135 CrossRef CAS PubMed.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494 CrossRef CAS PubMed.
- J. Kneipp, H. Kneipp, M. McLaughlin, D. Brown and K. Kneipp, Nano Lett., 2006, 6, 2225 CrossRef CAS PubMed.
- J.-L. Wu, F.-C. Chen, Y.-S. Hsiao, F.-C. Chien, P. Chen, C.-H. Kuo, M. H. Huang and C.-S. Hsu, ACS Nano, 2011, 5, 959 CrossRef CAS PubMed.
- J. W. Hong, S.-U. Lee, Y. W. Lee and S. W. Han, J. Am. Chem. Soc., 2012, 134, 4565 CrossRef CAS PubMed.
- N. J. Halas, S. Lal, W.-S. Chang, S. Link and P. Nordlander, Chem. Rev., 2011, 111, 3913 CrossRef CAS PubMed.
- D. R. Ward, N. K. Grady, C. S. Levin, N. J. Halas, Y. Wu, P. Nordlander and D. Natelson, Nano Lett., 2007, 7, 1396 CrossRef CAS PubMed.
- K. Kneipp, H. Kneipp and J. Kneipp, Acc. Chem. Res., 2006, 39, 443 CrossRef CAS PubMed.
- F. L. Yap, P. Thoniyot, S. Krishnan and S. Krishnamoorthy, ACS Nano, 2012, 6, 2056 CrossRef CAS PubMed.
- J. P. Camden, J. A. Dieringer, Y. Wang, D. J. Masiello, L. D. Marks, G. C. Schatz and R. P. V. Duyne, J. Am. Chem. Soc., 2008, 130, 12616 CrossRef CAS PubMed.
- C. Talley, J. Jackson, C. Oubre, N. Grady, C. Hollars, S. Lane, T. Huser, P. Nordlander and N. Halas, Nano Lett., 2005, 5, 1569 CrossRef CAS PubMed.
- M. Li, J. W. Kang, R. R. Dasari and I. Barman, Angew. Chem., Int. Ed., 2014, 53, 14115 CrossRef CAS PubMed.
- H. S. Kim, S. J. Lee, N. H. Kim, J. K. Yoon, H. K. Park and K. Kim, Langmuir, 2003, 19, 6701 CrossRef CAS.
- A. J. Mastroianni, S. A. Claridge and A. P. Alivisatos, J. Am. Chem. Soc., 2009, 131, 8455 CrossRef CAS PubMed.
- J.-H. Lee, G.-H. Kim and J.-M. Nam, J. Am. Chem. Soc., 2012, 134, 5456 CrossRef CAS PubMed.
- L. Xu, H. Kuang, C. Xu, W. Ma, L. Wang and N. A. Kotov, J. Am. Chem. Soc., 2012, 134, 1699 CrossRef CAS PubMed.
- J. Heo, D.-S. Kim, Z. H. Kim, Y. W. Lee, D. Kim, M. Kim, K. Kwon, H. J. Park, W. S. Yun and S. W. Han, Chem. Commun., 2008, 6120 RSC.
- Y. Bae, N. H. Kim, M. Kim, K. Y. Lee and S. W. Han, J. Am. Chem. Soc., 2008, 130, 5432 CrossRef CAS PubMed.
- G. S. Métraux, Y. C. Cao, R. Jin and C. A. Mirkin, Nano Lett., 2003, 3, 519 CrossRef.
- X. Hong, D. Wang, S. Cai, H. Rong and Y. Li, J. Am. Chem. Soc., 2012, 134, 18165 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892 CrossRef CAS PubMed.
- C. Gao, Z. Lu, Y. Liu, Q. Zhang, M. Chi, Q. Cheng and Y. Yin, Angew. Chem., Int. Ed., 2012, 51, 5629 CrossRef CAS PubMed.
- C. Gao, Q. Zhang, Z. Lu and Y. Yin, J. Am. Chem. Soc., 2011, 133, 19706 CrossRef CAS PubMed.
- E. González, J. Arbiol and V. F. Puntes, Science, 2011, 334, 1377 CrossRef PubMed.
- J. W. Hong, S. W. Kang, B.-S. Choi, D. Kim, S. B. Lee and S. W. Han, ACS Nano, 2012, 6, 2410 CrossRef CAS PubMed.
- B. L. Darby and E. C. Le Ru, J. Am. Chem. Soc., 2014, 136, 10965 CrossRef CAS PubMed.
- E. C. Le Ru, E. Blackie, M. Meyer and P. G. Etchegoin, J. Phys. Chem. C, 2007, 111, 13794 CAS.
- Y. Fang, N.-K. Seong and D. D. Dlott, Science, 2008, 321, 388 CrossRef CAS PubMed.
- E. C. Le Ru, J. Grand, I. Sow, W. R. C. Somerville, P. G. Etchegoin, M. Treguer-Delapierre, G. Charron, N. Félidj, G. Levi and J. Aubard, Nano Lett., 2011, 11, 5013 CrossRef CAS PubMed.
- N. Gandra, A. Abbas, L. Tian and S. Singamaneni, Nano Lett., 2012, 12, 2645 CrossRef CAS PubMed.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667 CrossRef CAS.
- A. M. Michaels, M. Nirmal and L. E. Brus, J. Am. Chem. Soc., 1999, 121, 9932 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4cc10377g |
| This journal is © The Royal Society of Chemistry 2015 |