
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Neptunyl(VI) centred visible LMCT emission directly observable in the presence of uranyl(VI)†
Sean D.
Woodall
a,
Adam N.
Swinburne
ab,
Nidhu
lal Banik
c,
Andrew
Kerridge‡
e,
Poppy
Di Pietro
e,
Christian
Adam
cd,
Peter
Kaden
c and
Louise S.
Natrajan
*ab
aThe Centre for Radiochemistry Research, The School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: louise.natrajan@manchester.ac.uk; Tel: +44 (0)1612751426
bThe Photon Science Institute, Alan Turing Building, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cKarlsruhe Institute of Technology (KIT), Institute for Nuclear Waste Disposal (INE), P.O. Box 3640, 76021 Karlsruhe, Germany
dUniversity of Heidelberg, Institute for Physical Chemistry, Im Neuenheimer Feld 253, 69120 Heidelberg, Germany
eDepartment of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK
First published on 16th December 2014
Abstract
Room temperature detection of neptunyl(VI) LMCT emission in a coordination compound and in the presence of uranyl(VI) is reported for the first time. Differences in the excitation profiles of the complexes enable spectral editing so either exclusively neptunyl(VI) or uranyl(VI) emission is observed or a sum of the two.
There is an urgent need to address the many environmental problems the nuclear age has brought about; in particular, the identification of (trace) radiotoxic actinide ions, their oxidation state and chemical form.1 In this regard, time resolved emission spectroscopy is becoming an invaluable tool with which to probe the electronic structure (oxidation state and coordination environment) of a given actinide ion in low concentrations that may represent those encountered in environmental and reprocessing conditions.2
The seminal work of Denning,3 amongst others, utilised the emissive properties of the uranyl(VI) ion to help construct the molecular orbital bonding diagram widely used today. However, corresponding studies on its periodic neighbour neptunyl(VI) are very limited.4 Currently, time-resolved spectroscopic studies of the uranyl(VI) ion are often used to provide valuable insight into the speciation of uranium species on minerals and sediments,5 particularly those present in geological disposal conditions, and to provide insight into the mechanisms behind promising bio-remediation techniques for the immobilisation of aquatically mobile uranyl(VI) species.2,6 Laser induced emission spectroscopy of uranyl(VI) is also being used for its identification in aqueous wastes, in future sustainable partitioning and transmutation closed fuel cycles and in situations where the concentration and volume of the sample is too small (and the activity too high to consider concentration) for other analytical techniques to be of use.
The lack of analogous reports involving neptunyl (and plutonyl) ions is due in part to the relative redox instability of these ions in aqueous solutions (cf. uranyl(VI)).7 There are very few neptunyl(VI) compounds that are redox stable in solution since neptunyl(V) is the most stable oxidation state, especially in aqueous (and therefore in most environmental and reprocessing) conditions. The relative proportion of each oxidation state (IV, V and VI) is heavily dependent on sample pH. In non-aqueous conditions, the +V oxidation state also predominates; the neptunyl(VI) chloride salt [NpO2Cl2(thf)]n undergoes partial reduction in thf solution over several days producing an isolable mixed oxidation state neptunyl(V)–(VI) salt,8 whereas in MeOH, rapid reduction to neptunyl(V) is observed. Clark reported that the addition of 18-crown-6 to perchloric or triflic acid solutions of NpO2(VI) ions resulted in the isolation of a NpO2(V) crown ether complex.9 Despite this, several pure oxidation state NpO2(VI) complexes have been isolated.10,11
Here, we report on a redox stable neptunyl(VI) coordination compound that is prepared from both neptunyl(V) and neptunyl(VI) precursors with the ligand TPIP (tetraphenylimidodiphosphinate),12 that may well serve to model solvated forms of neptunyl(VI), in that no redox active ligands are present (eqn (1)). Previously, we reported that TPIP reacts with uranyl(VI) salts to yield discrete monometallic, bimetallic and trimetallic complexes [UO2(TPIP)(thf)], [UO2(TPIP)(Cy3PO)], [UO2(TPIP)2]2 and [UO2(TPIP)2]3.13 Each uranyl(VI) ion possesses a unique luminescent fingerprint of emission maxima, spectral shape and radiative lifetimes to characterise the nuclearity and type of each complex. Limiting the absorption envelope of the chromophoric groups in the TPIP ligand to the UV (phenyl groups and relatively localized [N−–P–O] double bonds) gives rise to strongly emissive compounds in fluid solution at room temperature by inhibiting competitive back energy transfer processes from the emissive uranyl(VI) LMCT state. We reasoned that the same principles should apply for neptunyl(VI).
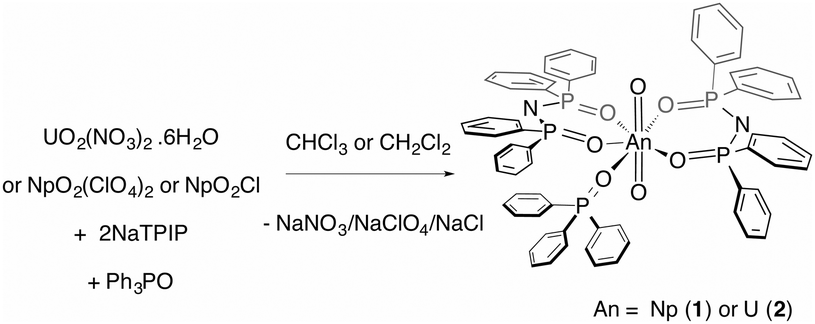 | (1) |
Treatment of neptunyl(V) chloride with two equivalents of NaTPIP and one of Ph3PO in mixtures of chloroform and methanol (10–50% methanol) resulted in quantitative conversion to the neptunyl(VI) complex [NpO2(TPIP)2(Ph3PO)] (1). The same complex can also be prepared from [NpO2(ClO4)2] in chloroform. The complete oxidation of [NpO2Cl] in the presence of TPIP and Ph3PO in organic solvents upon crystallisation is surprising and even in 50% methanol:chloroform solutions, 1 can be prepared and isolated. Moreover, 1 is stable with respect to reduction in 100% halogenated solvents for an indefinite time period (monitored for 9 months) as confirmed by UV-vis-nIR absorption spectroscopy. A principal f-centred absorption at 1232 nm characteristic of NpO2(VI) is observed and no significant absorptions at 980 nm that are typical for NpO2(V) are present.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8–11,14
8–11,14
The 31P solution NMR spectrum of 1 in CDCl3 shows a sharp resonance at 29 ppm and two broader resonances at 21 and −29 ppm (assigned as unbound Ph3PO, 29 ppm, and complexed Ph3PO and TPIP) indicating slow exchange of monodentate Ph3PO on the experimental timescale.† This chemical exchange was also corroborated by 2D 1H diffusion ordered spectroscopic (DOSY) measurements, where in 1, two species with diffusion coefficients of 6.7(2) × 10−10 m2 s−1 and ca. 14 × 10−10 m2 s−1 are discernible at 295 K.† The most broadened and downfield shifted proton resonances belong to a faster diffusing species (here, Ph3PO). The TPIP protons do not experience a large induced paramagnetic shift being more than three bonds away from the metal centre,12 but the longitudinal proton relaxation times (T1) are suggestive of a neptunyl 5f1 electronic configuration and range from 0.94 to 2.54 s at 300 K, providing further evidence of the assignment of the +VI oxidation state in 1.15 For comparison, resonances in uncomplexed Ph3PO (at 300 K) have relaxation times of 3.70–4.40 s.
Yellow single crystals of 1 for X-ray diffraction analysis were grown from slow evaporation of an NMR tube reaction of [NpO2Cl] plus two equivalents of TPIP and one of Ph3PO in an 8:1 v/v CDCl3:MeOD-d4 solution (Fig. 1). For comparative purposes, the uranyl(VI) analogue [UO2(TPIP)2(Ph3PO)]·CH2Cl2 (2) was prepared analogously.†
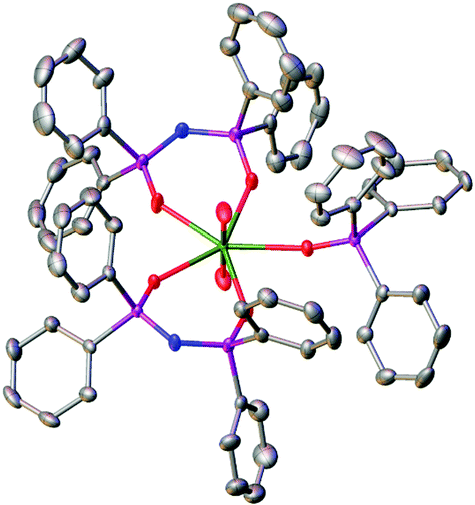 | ||
| Fig. 1 Solid state molecular structure of 1 with thermal ellipsoids set at the 50% probability level; H atoms are omitted for clarity. | ||
In the solid state, both complexes 1 and 2 are isostructural and the coordination geometries around the actinide cations are approximately pentagonal bipyramidal. Charge balancing together with optical and NMR data require a +VI oxidation state within the neptunium cation in 1. The neptunyl(VI) bond lengths of 1.7501(17) and 1.7470(17) Å (bond angle 179.21(8) Å) are in the range for previously seen neptunyl(VI) complexes,9,16 directly comparable to those in 2 at 1.767(3) and 1.764(3) Å, with the decrease in bond length of between 0.015 and 0.020 Å attributable to the actinide contraction. The Np–OTPIP bonds are between 2.3558(16) and 2.3584(17), shorter than the Np–OPh3PO bond length of 2.4099(16) Å. Whilst this indicates a stronger bond to the TPIP ligands, this is in contrast to 2 where all the equatorial bond lengths range 2.375(3) to 2.429(3) Å (O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) UO bond angle 179.10(15) Å).
UO bond angle 179.10(15) Å).
Previous studies of the photophysical properties of the neptunyl(VI) ion have shown that emission from several excited states in the near infrared region between 1452 and 1580 nm can be achieved in two ways: (i) by LMCT sensitization in room temperature aqueous solution in a polyoxometalate complex (via the O → W LMCT transitions);17 (ii) by direct excitation into neptunyl(VI) f-centered absorption bands in a frozen glass or the solid state in Cs2[NpO2Cl4].18 In both cases, the emission was assigned to interconfigurational transitions originating from excited states within the 5f1 manifold and no visible charge transfer emissions were reported.
In 1, excitation at these wavelengths (300–700 mm) in CH2Cl2 solution did not reveal any emission maxima in the near infrared region of the electromagnetic spectrum (using current instrumental set ups). However, excitation between 280 and 420 nm gave a vibrationally resolved visible emission band centred at 438 nm. The spectral shape of this emission band is independent of excitation wavelength suggesting that the emission originates from a common excited state. The fact that the excitation and emission profiles are broad indicates that the emission feature possesses considerable charge transfer character, which is confirmed by theoretical calculations (vide infra). The reconvoluted radiative lifetime of the emission, following 375 or 405 nm excitation is biexponential at 1.3 (95%) and 5.0 (5%) ns with the short lifetime difficult to measure accurately against a scatterer and the kinetic profile is monoexponential following tail fitting; τ = 1.5 ns. The lifetime is significantly shorter than its uranyl(VI) counterparts in fluid solution (1.66 μs in 2)† and also than that reported from the 5f1 uranyl(V) ion in the literature.19 All previous reports of emission from NpO2(V)20 and NpO2(VI) species are short-lived (<62 ns in solution),§ and emission from 1 is also expected to be short due to efficient non-radiative relaxational pathways through the 5f1 manifold. The energy spacings between the three maxima (Fig. 2) are 1349 and 1489 cm−1, are considerably higher than typical NpOyl Raman active symmetric stretch values (ca. 800 cm−1) (ref. 7, 9 and 21) and more likely to correspond to a P–N stretch from the TPIP ligand as measured experimentally in the UO2(VI) derivative 2 (range, 1163–1211 cm−1). This suggests that the emissive excited state possesses significant TPIP ligand character (vide infra). Interestingly, the excitation spectra show absorptions that correspond to previously reported UV neptunyl(VI) absorptions;7 these largely remain unassigned in the literature, but we presume these to be NpO ‘yl’ and ligand–Np (equatorial LMCT) transitions at ca. 340 nm and 380 nm in 1 respectively by analogy with uranyl(VI) TPIP complexes.13,22 Moreover, the excitation spectra show no transitions that correspond to TPIP π–π* absorptions, indicating that the emissive excited state is localised on the neptunyl unit itself. The possibility of the emission being phosphorescence from TPIP was also examined by recording the emission spectrum of the lanthanide analogue [Gd(TPIP)3] at 77 K,12 which showed similar features but did not correspond exactly to the emission profile seen in 1 and additionally was very weak.† The similarity of these spectra do however lend further weight to the conclusion that the emission in 1 possesses considerable TPIP character. By contrast, excitation between 320 and 420 nm in 2 resulted in a typical highly resolved uranyl(VI) emission spectrum centred at 522 nm that is 100% Oyl → U LMCT in character; the apparent electronic origin of the emission (E0–0) was determined as 20750 cm−1 and the average vibrational progression was measured as 809 cm1 which corresponds well to the ν1 Raman active symmetric uranyl stretch determined from experiment (825 cm−1) and no additional (P–N) vibrational coupling was apparent. The excitation spectrum of 2 recorded at the emission maxima is however a combination of TPIP → U and Oyl → U LMCT.13†
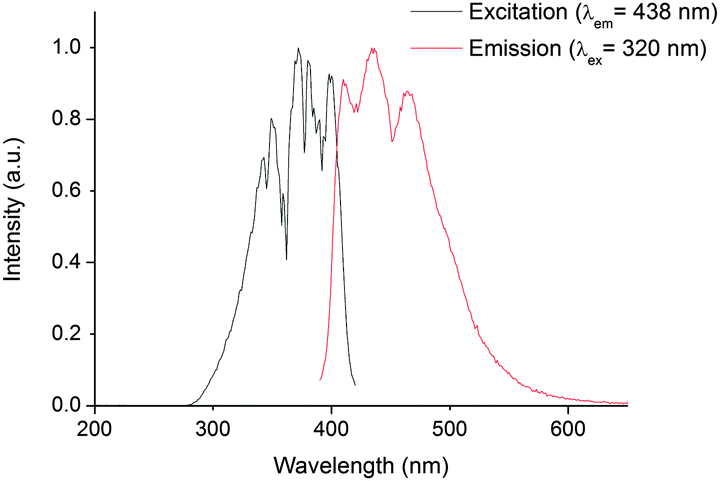 | ||
| Fig. 2 Steady state excitation spectrum (black trace, recorded at the emission maxima of 438 nm), and emission spectrum (red trace, recorded following 320 nm excitation), of 1 in CH2Cl2 at 295 K. | ||
In order to verify the origin of vibrational fine structure in the emission spectrum of 1, we performed a set of density functional theory (DFT) calculations on simplified structures in which phenyl groups were replaced with terminating hydrogens and solvation effects included via the COSMO implicit solvent model. These calculations, performed using version 6.4 of the TURBOMOLE code,23 employed the hybrid-GGA PBE0 (ref. 24) exchange correlation functional and Ahlrichs-style basis sets of polarised triple-ξ quality.25 Structural characterisation provided excellent agreement with crystallographic data, with An–Oyl bond lengths accurate to better than 0.01 Å and a calculated contraction of the An–Oyl bond of 0.026 Å when comparing the UO2(VI) and NpO2(VI) complexes. Equatorial coordination was also well reproduced, with an average Np–OTPIP bond length of 2.366 Å, shorter than the 2.443 Å Np–OPh3PO bond. Topological analysis via the quantum theory of atoms in molecules (QTAIM)† revealed a consistent increase in the magnitude of the electron density in the Np–OTPIP bonding region when compared to the Np–OPh3PO bond, commensurate with the stronger bonding determined experimentally.
Vibrational frequency analysis revealed the Np–Oyl stretch frequencies to occur at 913 cm−1 (symmetric) and 976 cm−1 (asymmetric), significantly lower than the spacings between maxima observed in the emission spectra, however two N–P stretching modes were calculated at 1281 cm−1 and 1282 cm−1. The absence of any other modes in the region of these vibrations led us to the possibility that these N–P modes are responsible for the observed vibrational fine structure. To test this hypothesis further, time-dependent (TD-) DFT simulations of the excited states of 1 were performed. These calculations revealed several excitations in the 344–397 nm region with similar oscillator strengths of the order 10−4. Of these, three excitations, at 363, 390, and 397 nm, had well-defined neptunyl LMCT character as well as substantial NTPIP(2p) → Np(5f) contributions of 55%, 22% and 20%, respectively. This finding lends strong support to our assertion that the origin of the observed fine structure is due to the TPIP N–P vibrational modes and that the emission is a combination of TPIP → Np and Oyl → Np charge transfer.
Compounds 1 and 2 have different excitation and emission profiles as well as radiative lifetime so we reasoned that it should be possible to discriminate between the two in solution. Indeed, addition of an equimolar dichloromethane solution of 2 to a dichloromethane solution of 1 (0.87 mM) resulted in an emission spectrum that is a sum of the individual components (following excitation at 320 and 420 nm). Interestingly, even in the presence of an excess of 2 (1.47 mM), both components are easily identifiable. Further, by changing the excitation wavelength, spectral editing can be achieved such as the response of purely NpO2(VI), (290 nm excitation), UO2(VI) (380 nm excitation) or a mixture of the two can be obtained (320 nm and 420 nm excitation, Fig. 3). In all spectra, the vibrational progression of each individual actinyl(VI) cation is evident suggesting no loss in structural integrity in 1 and 2.
 | ||
| Fig. 3 Steady state emission spectra of a mixture of 1 (0.87 mM) and 2 (1.47 mM) following 290 nm excitation (blue trace), 320 nm excitation (red trace) and 380 nm excitation (black trace) in CH2Cl2 at 295 K. | ||
The kinetic profile of both components in fluid solution at room temperature, following 420 nm excitation, is multiexponential and the major component (50%) possesses a radiative lifetime of 5 ns. The lifetimes of the minor components are 1 ns (23%) and 18 ns (24%).† Moreover, the time resolved emission spectra show the dominance of NpO2(VI) emission at short delay times (0 to 25 ns) and only UO2(VI) emission at longer delay times (30 to 100 ns), meaning that the longer lived component can be assigned to UO2(VI) emission.† The short lifetimes for the mixed solution of 1 and 2 suggests an efficient competitive pathway for emission quenching and the two complexes may be interacting with one another in solution. Overlap of the higher energy NpO2(VI) charge transfer bands with TPIP absorption in the UV-vis spectrum means that the absorption maxima corresponding to the emission are obscured. This precludes the determination of a quantum yield and experimentally, the exact origin of the emission.
The 31P, 1H NMR and 1H DOSY-NMR spectra also strongly suggest the two complexes may be aggregated in solution via loss of the Ph3PO ligands; the 1D spectra are broadened at 295 K when compared to the individual complexes with many overlapping resonances and several species are suggested by the 1H DOSY spectrum.† However, all attempts to isolate any intermediate (possible mixed metal) species were unsuccessful, producing in all cases single crystals of the most thermodynamically stable compounds 1 and 2 (and a solitary crystal of Ph3PO). This indicates that any aggregated species is only a transient in solution. It is interesting to note that even in the presence of uranyl(VI), no redox reactions of 1 in CH2Cl2 solution are observed; the possibility of forming intermetallic actinyl-oxo-actinide interactions between NpO2(VI) and UO2(VI)26 may be expected to be favourable, especially given that TPIP can promote such interactions with uranyl(VI).13
In conclusion, the neptunyl(VI) complex [NpO2(TPIP)2(Ph3PO)], (1, where TPIP = tetraphenylimidodiphosphinate) has been prepared from both NpO2(V) and NpO2(VI) precursors and has been found to be indefinitely redox stable in chlorinated solvent solutions. The ligand TPIP is not redox-active, lacks peripheral chromophores in the visible region, and therefore enables us to detect the vibrationally resolved visible emission of the NpO2(VI) cation in 1 for the first time. A combination of experimental evidence and DFT calculations has enabled the assignment of the emission to be a combination of Oyl → Np LMCT and TPIP → NpO2 LMCT emission (20 to 55%). Moreover, in an equimolar solution of 1 and its uranyl(VI) counterpart, 2, time resolved studies suggest that the two complexes may be aggregated in fluid solution and there is a cooperative pathway for emission quenching. Both broadband and selective excitation and time gating leads to spectral editing and this study illustrates that selective detection and discrimination of individual actinide cations is possible using time resolved emission techniques. We are currently investigating the effects of electron withdrawing and donating substituents in TPIP analogues to assess the relative contributions of the TPIP and NpO LMCT on the emission of NpO2(VI) compounds.
The authors gratefully acknowledge Simon Randall for help with the crystallographic data and Professor David Collison for helpful discussions. We thank the EPSRC for funding a Career Acceleration Fellowship (LSN and AK), postdoctoral funding (ANS), and studentships (SDW and PDP), The Royal Society for an equipment grant and The University of Manchester for support. The authors thank the European Commission Euratom FP7 funded project (no. 269923) EURACT-NMR for support.
Notes and references
- For recent reviews, see: M. Altmaier, X. Gaona and T. Fanghänel, Chem. Rev., 2013, 113, 901 CrossRef CAS PubMed; K. Maher, J. R. Bargar and J. E. Brown Jr., Inorg. Chem., 2013, 52, 510 CrossRef PubMed; L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Coord. Chem. Rev., 2014, 266–267, 171 CrossRef PubMed.
- L. S. Natrajan, Coord. Chem. Rev., 2012, 256, 1583 CrossRef CAS PubMed.
- R. G. Denning, Struct. Bonding, 1992, 79, 215 CrossRef CAS; R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125 CrossRef PubMed.
- J. Su, W. H. E. Schwarz and J. Li, Inorg. Chem., 2012, 51, 3231 CrossRef CAS PubMed.
- See for example: T. Arnold, S. Utsunomiya, G. Geipel, R. C. Ewing, N. Baumann and V. Brendler, Environ. Sci. Technol., 2006, 40, 4646 CrossRef CAS; E. J. Elzinga, C. D. Tait, R. J. Reeder, K. D. Rector, R. J. Donohoe and D. E. Morris, Geochim. Cosmochim. Acta, 2004, 68, 2437 CrossRef PubMed; K. F. Smith, N. D. Bryan, A. N. Swinburne, P. Bots, S. Shaw, L. S. Natrajan, J. F. W. Mosselmans, F. R. Livens and K. Morris, Geochim. Cosmochim. Acta, 2015, 148, 343 CrossRef PubMed.
- K. H. Williams, J. R. Bargar, J. R. Lloyd and D. R. Lovley, Curr. Opin. Biotechnol., 2013, 24, 489 CrossRef CAS PubMed; K. Grossman, T. Arnold, E. Krawczyk-Barsch, S. Diessner, A. Wobers, G. Benhard and R. Krawietz, Environ. Sci. Technol., 2007, 41, 6498 CrossRef.
- J. C. Hindman, L. B. Magnusson and T. J. LaChapelle, J. Am. Chem. Soc., 1949, 71, 687 CrossRef CAS; R. Sjoblom and J. C. Hindman, J. Am. Chem. Soc., 1951, 73, 1744 CrossRef; H. A. Friedman and L. M. Toth, J. Inorg. Nucl. Chem., 1980, 42, 1347 CrossRef; T. W. Newton, D. E. Hobart and P. D. Palmer, The Preparation and Stability of Pure Oxidation States of Neptunium, Plutonium, and Americium, Los Alamos National Laboratory, 1986, LA-UR-86-967 Search PubMed.
- S. M. Cornet, L. J. L. Häller, M. J. Sarsfield, D. Collison, M. Helliwell, I. May and N. Kaltsoyannis, Chem. Commun., 2009, 917 RSC.
- D. L. Clark, D. W. Keogh, P. D. Palmer, B. L. Scott and C. D. Tait, Angew. Chem., Int. Ed., 1998, 37, 64 Search PubMed.
- [NpO2(DPMMO)2Cl]2[NpO2Cl4] (DPPMO = bis(diphenyl-phosphino)methanedioxide) S. M. Cornet, M. P. Redmond, D. Collison, C. A. Sharrad, M. Helliwell and J. Warren, C. R. Chim., 2010, 13, 832 CrossRef CAS PubMed.
- Neptunyl(VI) bis(salicylidene)ethylenediamine (salen) is prepared from neptunyl(V) in methanol. D. G. Chuguryan, V. I. Dzyubenko, M. S. Grigoriev, A. I. Yanovski and Y. T. Struchkov, Radiokhimiya, 1988, 30, 41 CAS.
- S. W. Magennis, S. Parsons and Z. Pikramenou, Chem. – Eur. J., 2002, 8, 5761 CrossRef CAS.
- M. P. Redmond, S. M. Cornet, S. D. Woodall, D. Whittaker, D. Collison, M. Helliwell and L. S. Natrajan, Dalton Trans., 2011, 40, 3914 RSC.
- P. G. Hagan and J. M. Cleveland, J. Inorg. Nucl. Chem., 1966, 28, 2905 CrossRef CAS.
- In related neptunyl(V) TPIP solutions, the T1 values of the proton resonances are of millisecond order (10–110 ms). S. D. Woodall, PhD thesis, The University of Manchester, 2014.
- M. P. Wilkerson, H. J. Dewey, P. L. Gordon and B. L. Scott, J. Chem. Crystallogr., 2004, 34, 807 CrossRef CAS PubMed; N. W. Alcock, M. M. Roberts and D. Brown, J. Chem. Soc., Dalton Trans., 1982, 25 RSC.
- C. Talbot-Eeckelaers, S. J. A. Pope, A. J. Hynes, R. Copping, C. J. Jones, R. J. Taylor, S. Faulkner, D. Sykes, F. R. Livens and I. May, J. Am. Chem. Soc., 2007, 129, 2442 CrossRef CAS PubMed.
- M. P. Wilkerson and J. M. Berg, J. Phys. Chem. A, 2008, 112, 2515 CrossRef CAS PubMed; M. P. Wilkerson and J. M. Berg, Radiochim. Acta, 2009, 97, 223 CrossRef.
- R. Steudtner, T. Arnold, K. Grossman, G. Geipel and V. Brendler, Inorg. Chem. Commun., 2006, 9, 939 CrossRef CAS PubMed.
- R. Bradshaw, D. Sykes, L. S. Natrajan, R. J. Taylor, F. R. Livens and S. Faulkner, IOP Conf. Ser.: Mater. Sci. Eng., 2010, 9, 012047 CrossRef.
- C. Madic, G. M. Begun, D. E. Hobart and R. L. Hahn, Inorg. Chem., 1984, 23, 1914 CrossRef CAS.
- B. Drobot, R. Steudner, J. Raff, G. Geipel, V. Brendler and S. Tsushima, Chem. Sci. 10.1039/C4SC02022G.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165 CrossRef CAS.
- J. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS; C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- S. Wang, J. Diwu, E. V. Alekseev, L. J. Jouffret, W. Depmeier and T. E. Albrecht-Schmitt, Inorg. Chem., 2012, 51, 7016 CrossRef CAS PubMed; S. Skanthakumar, M. R. Antonio and L. Soderholm, Inorg. Chem., 2008, 47, 4591 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full experiemental, spectroscopic and computationad detaila. CCDC 1032118 and 1032119 for compounds 1 and 2. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08718f |
| ‡ Current address: Department of Chemistry, Lancaster University, Lancaster, LA1 4YB, UK. |
| § It is worth noting here that the nIR emisison of the NpO2(VI) aqua ion has not been dectected directly but the radiative lifetime has been estimated to be 7.6 ns (ref. 17). |
| This journal is © The Royal Society of Chemistry 2015 |