
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multicomponent low molecular weight gelators
Jaclyn
Raeburn
and
Dave J.
Adams
*
Department of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
First published on 28th November 2014
Abstract
Low molecular weight gelators (LMWG) self-assemble in solution into one-dimensional objects such as fibres or tapes. The entanglement of these fibres or tapes results in the formation of a network and a gel. In general, LMWG are investigated as single component systems. However, there are significant potential opportunities from mixed LMWG systems, which are rarely investigated. Here, we discuss the potential of multicomponent systems, and critically discuss the challenges.
 Dave J. Adams | Dave Adams joined the Department of Chemistry, University of Liverpool in 2008. He obtained his PhD from the University of York, working with Professor James Clark. After postdoctoral work at the Universities of York, Leeds and Leicester, he worked within Unilever for four years. His research interests lie in the areas of polymer and peptide self-assembly, low molecular weight gels, and porous materials. |
Introduction
Low molecular weight gelators (LMWG) are molecules that can self-assemble under specific conditions into structures that are able to immobilise the solvent.1–5 Typically, long one-dimensional objects such as fibres, tapes, tubes and helical structures can be formed. When these entangle, branch or otherwise interact, a network can be formed, which can result in a gel (Fig. 1). The gel properties are controlled by fibre widths, spatial distribution and number of entanglements or cross-links.1 | ||
| Fig. 1 Cartoon schematic showing the assembly of a LMWG into fibres, resulting in a network and gel formation. (a) LMWG dissolved in solution (b) self-assembly of LMWG starts to occur after application of a trigger (c) formation of the gel network by entanglement (d) a self-supporting bulk gel. | ||
LMWG are receiving significant current attention.6–11 There are potential applications in a wide range of areas, including cell culturing,12–14 sensing and catalysis,15–18 drug delivery,19–21 and optoelectronics.22–25 The range of molecular structures that can be used as LMWG is vast, including for example simple alkanes, sugars, modified amino acids, oligopeptides, dendrimers, bola-amphiphiles, peptide-amphiphiles, nucleobases, and C3-symmetric molecules.1,2,26–30 This diversity in molecular structure is coupled to the wide range of solvents that can be gelled. However, it should also be pointed out that design of a new LMWG based on knowledge of current systems is still a challenge, with even small structural variations often resulting in completely different behaviour. As a result, it is often stated that new LMWG are discovered by serendipity.4,31
Gels formed from LMWG are solid-like from a rheological perspective, although they can be formed at very low concentrations of gelator, sometimes as low as 0.1 wt%. These gels have a number of interesting properties. For example, the gels tend to be reversible, so application of a particular stimulus can result in a gel-to-liquid transition. Most LMWG gels are thermally reversible, with the melting temperature of the gel being nearly independent of the concentration of gelator. The gels can be stable for long periods of time (∼years). In other cases, interesting behaviour such as crystallisation of the LMWG from the gel phase can occur.32–36
The self-assembly into fibrous structures can also be affected strongly by how the self-assembly is carried out,37 properties by varying the process, not the LMWG itself. In the vast majority of cases, gels are typically formed thermally. The LMWG and solvent are heated until a solution is apparently formed. On cooling, a gel is formed. This can be thought of as an arrested crystallisation, where the LMWG assemble into one-dimensional structures as opposed to crystallising or precipitating. The rate of cooling here can affect the gel properties.38,39 Where the solvent is water, a range of other triggers can be used to form gels, including changes in pH, salt concentrations, or an enzymatic reaction with a suitable pre-gelator.8 For all of these cases, a rarely reported feature is also the concept of aging. It is not always the case that the materials remain the same over periods of time.40
In the vast majority of cases, LMWG systems are described for a single molecule. However, this does not need to be the case.41,42 There are a number of examples where two components are necessary to form a gel.41,43,44 Essentially here the two components form the LMWG in situ, interacting by non-covalent forces such as hydrogen bonding to form a complex that is the gelator. The structure of either of the two components can be adjusted, allowing in some cases a fine-tuning of the gel properties. Specific structural components can be pulled out of a mixture on the basis of interaction strengths.44 Whilst this is an extremely interesting area, we will not discuss this further here. Likewise, we do not discuss in detail the cases where a LMWG is assembled in the presence of something else, such as a surfactant,45,46 a liquid crystal,47 a gelling polymer,48–50 or a non-gelling additive such as a polymer, a clay, or graphene,51–58 although all of these potentially enable exciting new materials to be formed.
Instead, we focus on mixtures of LMWG where either component can form a gel alone. In this situation, three different scenarios can be envisaged: (i) the LMWG can randomly mix forming fibres that contain a statistical amount of each gelator; (ii) the LMWG can specifically associate such that fibres are formed that contain an exact ratio and order of gelators; (iii) the LMWG can self-sort such that fibres contain molecules of only one gelator or the other (Fig. 2(a)–(c)). Since in all cases the primary fibres can then entangle, we need to understand and control assembly of multiple components across multiple length-scales. In reality, there is the possibility for a combination of both to occur to a certain degree, although this is not generally acknowledged or discussed.
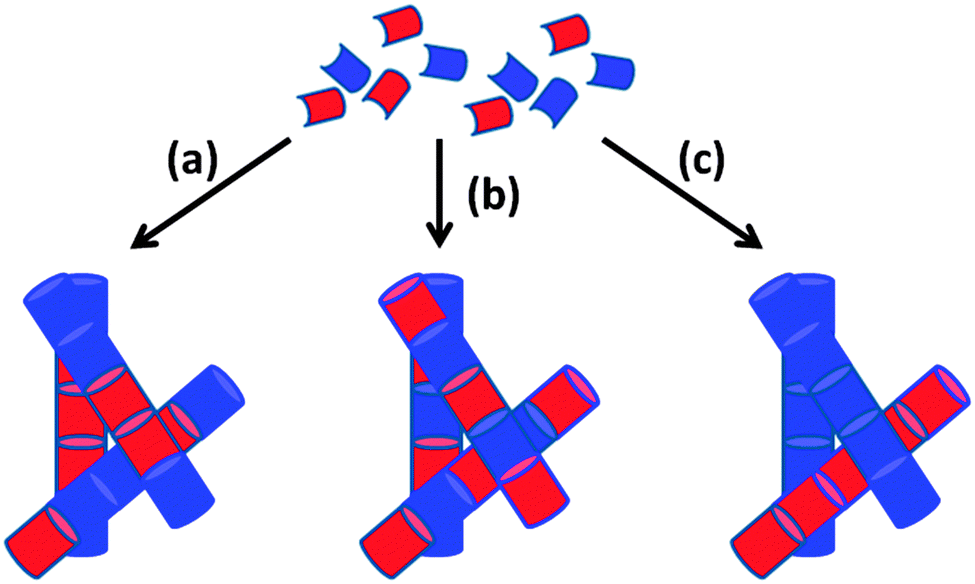 | ||
| Fig. 2 Cartoon schematic showing how the assembly of two LMWG (red and blue) can lead to (a) self-sorting; (b) random mixing; (c) specific co-assembly. | ||
Multicomponent LMWG systems open up opportunities to build exciting new materials. For example, self-sorting of gelators is potentially a powerful strategy to generate bulk heterojunctions (BHJ) for optoelectronic devices, assuming that the LMWG have the appropriate electronic properties. Entangled fibres of self-sorted gelators conceptually provide a system with a very high interfacial area between fibres. In 2008, Sugiyasu et al. showed that BHJ could be prepared from organogels by the thermal self-sorting of two specific LMWG incorporating suitable aromatic chromophores (Fig. 3).59 As another example of potential applications, Zhou et al. have shown that random mixing of LMWG into fibres can be used to decorate dipeptide fibres with a cell adhesive sequence present on a second LMWG.60
 | ||
| Fig. 3 The structures of Thio and Pery LMWG and a cartoon representation of the self-sorted network formed, which leads to p–n heterojunction points. Reprinted with permission from ref. 39. Copyright 2008 American Chemical Society. | ||
Here, we discuss the available literature on multicomponent LMWG systems, critically highlighting the state of the art, as well as discussing the barriers currently in place to future developments.
How can mixed LMWG systems be characterised?
When two LMWG are mixed, there are multiple levels of hierarchical assembly that need to be understood and controlled (Fig. 4). At the molecular level, how do the molecules pack? If the LMWG are mixing at the molecular level, does one LMWG dominate the packing or is the structure formed distinct from that formed by either component alone? For self-sorted systems, a key question is how does the presence of a network from one LMWG affect the assembly of the second LMWG? At the next length-scale, how do the fibres interact? One could envisage mixed systems having the potential for more or fewer cross-links than the analogous single component systems. For self-sorted systems, do the fibres prefer to entangle with fibres of the same LMWG, or is there no discrimination? For example, one could imagine that one network could grow on the fibres of the other, epitaxial growth could occur, intimate entanglement could occur or the networks could be entirely independent (two example hypothetical networks are shown schematically in Fig. 4 (left and right)). Finally, at a micrometer length scale, how are the fibres distributed in space? Are the fibres uniformly distributed throughout the gel, or are there local variations? Do these differ from the single component systems? | ||
| Fig. 4 Two hypothetical networks that could be formed from two self-sorted LMWG: (left) the fibres entangle around one another; (right) an interpenetrated network. | ||
Hence, to characterise these systems, information is needed across all these length scales.61,62 We should first acknowledge that this is difficult to do for single component LMWG systems. Indeed, how the LMWG pack into fibres is not well understood in most cases. There are some examples where information has been derived from X-ray scattering experiments, for examples using powder X-ray diffraction with appropriate background subtraction of the diffuse solvent peak.63 In many cases, information about the packing has been extracted from a crystal structure of the LMWG.31 However, at least in some cases, it is clear that the packing in the crystal structure does not correlate with the packing in the gel phase,35 raising questions as to how valid this approach is. Molecular packing information can also be extracted from fluorescence, circular dichroism (CD) and infra-red (IR) spectroscopy.62 At longer length scales, electron microscopy is often used to image the fibrous structures, although there can be issues here with drying artefacts. Small angle X-ray and neutron scattering approaches have also been used to characterise LMWG systems.64 Rheology is used to characterise the mechanical properties of the bulk gel.65 NMR can be used to monitor how much of the gelator has assembled since the LMWG becomes undetectable by solution state NMR on gelation.66,67
For mixtures of LMWG, the same techniques are used to characterise the systems, combined with comparing the data with the analogous single components. However, it is not always clear what results are expected. For example, if self-sorting occurs, it is likely that the rate of assembly of each component will be different. As a result, one component will be self-assembling in the presence of the other molecularly dispersed LMWG. There are a number of reports regarding how the assembly of a gelator can be affected by the presence of an additive,53–55,68 and as a result any differences seen in the assembly could be due to the effect of the additive, as opposed to being only due to the self-sorted network. Hence, in some cases, it is not clear whether co-assembly or self-sorting has occurred. For example, Jayawarna et al. have reported Fmoc-dipeptide LMWG, which are able to gel water on adding acid to a pH 8 solution.69 Fmoc-diphenylalanine was found to gel water at a pH of less than 8, but Fmoc-diglycine required that the pH was less than 4 for a gel to be formed. In a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 mol mol−1 mixed solution, a gel could be formed at pH 7, which was described as being more stable than for the pure materials alone. The fluorescence spectra showed the presence of a broad phosphorescence peak at 450 nm, which was not present in either of the single LMWG gels. A similar strong peak at 450 nm was observed elsewhere for a mixture of two Fmoc-amino acids on gel formation, which was not as strong for the individual components.70 One could interpret these data as being the result of formation of a co-assembled structure. Alternatively, this could be the result of the Fmoc-diglycine acting as a surfactant at pH 7 (it is known that the pKa of these materials scale with the relatively hydrophobicity and gelation occurs at the pKa (ref. 71 and 72)) and hence influencing and changing the self-assembly of the Fmoc-diphenylalanine. This situation is complicated by the later observations of the high sensitivity of the system to the mixing conditions during gel formation.73 Further work where a Fmoc-dipeptide LMWG was assembled using an enzymatic trigger in the presence of a Fmoc-amino acid or tripeptide surfactant-like structure suggested the formation of core–shell fibres, where the hydrophobic nanofibres of the LMWG are coated with the surfactant-like molecule.74 Depending on the choice of LMWG and surfactant-like molecule, different types of structure were suggested,75 where the two molecules could self-sort or co-assemble, either constructively or destructively. When the surfactant-like molecule is also a LMWG, it is unclear if the structures formed would then be “locked in”, or whether a structural change would occur on gelation.
:50 mol mol−1 mixed solution, a gel could be formed at pH 7, which was described as being more stable than for the pure materials alone. The fluorescence spectra showed the presence of a broad phosphorescence peak at 450 nm, which was not present in either of the single LMWG gels. A similar strong peak at 450 nm was observed elsewhere for a mixture of two Fmoc-amino acids on gel formation, which was not as strong for the individual components.70 One could interpret these data as being the result of formation of a co-assembled structure. Alternatively, this could be the result of the Fmoc-diglycine acting as a surfactant at pH 7 (it is known that the pKa of these materials scale with the relatively hydrophobicity and gelation occurs at the pKa (ref. 71 and 72)) and hence influencing and changing the self-assembly of the Fmoc-diphenylalanine. This situation is complicated by the later observations of the high sensitivity of the system to the mixing conditions during gel formation.73 Further work where a Fmoc-dipeptide LMWG was assembled using an enzymatic trigger in the presence of a Fmoc-amino acid or tripeptide surfactant-like structure suggested the formation of core–shell fibres, where the hydrophobic nanofibres of the LMWG are coated with the surfactant-like molecule.74 Depending on the choice of LMWG and surfactant-like molecule, different types of structure were suggested,75 where the two molecules could self-sort or co-assemble, either constructively or destructively. When the surfactant-like molecule is also a LMWG, it is unclear if the structures formed would then be “locked in”, or whether a structural change would occur on gelation.
In some cases, the mixtures of LMWG are used, but the type of assembly is not probed. For example, mixtures of Fmoc-dipeptides have been shown to form gels.76 There were changes in the rate of gelation and in the turbidity during assembly as compared to the individual LMWG, but this was ascribed either to the difficulties in forming more complex structures or diffusional effects involved in the co-formation of two separate populations of fibres (i.e. either co-assembly or self-sorting).
In other cases, the type of assembly is clear. For example, Smith and Smith have shown that self-sorting can be proven in a two component systems using NMR, differential scanning calorimetry (DSC) and SEM.40 They used NMR to show that one of the gelators was immobilised with the same profile at different temperatures in the mixed gel as when gelled alone (Fig. 5). This demonstrates that the assembly of the first LMWG is not being affected by the presence of the second gelator. DSC was used to show that the gel-to-sol temperatures for the individual LMWG were also observed in the mixed gel, showing again that the network of each LMWG was unaffected by the presence of the other. SEM was also consistent with these observations. Moffat and Smith have also shown very clear SEM data for a self-sorted system.77 Here, the two LMWG form fibres with very different diameters; fibres with both diameters could be clearly visualised in the mixed gel.
 | ||
| Fig. 5 NMR data showing that the immobilisation of one LMWG occurs with the same temperature profile in the self-sorted gels (squares) as in the single component gels (triangles). Reproduced from ref. 40. | ||
Elsewhere, Sugiyasu et al. have shown that the absorption spectrum of the gel formed from a mixture of two LMWG (structures shown in Fig. 3) overlapped with the sum of two spectra of individual gelators.59 The peaks attributed to the different LMWG changed independently on heating and the dissociation temperatures coincided with those of the pure gelators. Similarly, the CD spectrum of the mixed gel perfectly overlapped with the sum of the CD spectrum of each individual component. Hence, at a molecular level, this system was ascribed to a perfectly self-sorted system. SEM was less conclusive. Fibrous structures were imaged for both individual LMWG and also for the self-sorted system, but the diameters in all cases were very similar. This shows that although sorting at the molecular level is clear in this case, self-sorting at longer length scales is less certain. To demonstrate self-sorting at the fibre level, the properties of the gels were investigated, with the conductivity being used to infer self-sorting at the fibre level.
As an example of a co-assembling system, a phenothiazine-based LMWG was mixed with a naphthalene-based LMWG.78 In both cases, the LMWG formed a gel alone. The mixed gel was formed by a heat–cool cycle. Here, like the case above, the CD spectrum for the mixed system was very similar to that of the individual components. However, there were changes in the fluorescence spectra, and crucially the gel-to-sol temperature was higher for the mixed gel system compared to either of the individual component gels. This is all highly indicative of a co-assembled structure, presumably randomly co-assembled since there are no specific interactions present. On the fibre length scale, transmission electron microscopy (TEM) showed that the diameter of the fibres in the mixed gel was higher than for those in either individual gel. Hence, in this case, the assembly was attributed to a co-assembled system. As a result of this co-assembly, the rheological properties were higher than for either of the single component gels.
Energy transfer has been used to demonstrate effective co-assembly.79–81 For example, in an organogel, extremely effective energy transfer was found when fibres of an anthracene derivative were doped with a tetracene. Interestingly, the quenching and resulting spectrum was very similar whether the added tetracene was a gelator or a non-gelator, which implies that both were equally effectively assembled in the fibres of the anthracene LMWG.79
Foster et al. have shown how co-assembled gels can sometimes give rise to fibre morphologies that are not present in the gels of either individual component.82 A mixture of two bola-amphiphile amino acid based LMWG were found to form helical tapes, which were not formed by either of the individual LMWG. For other examples, separate fibres characteristic of individual LMWG were imaged, providing evidence of self-sorting. Interestingly, the co-assembly of a pyrene-based LMWG with an amino-acid based LMWG was found to lead to a co-assembled gel, as demonstrated by the absence of an excimer peak from the pyrene. This excimer, present in the pure pyrene–LMWG system arises from close packing of the pyrene units. Hence, the absence of this peak shows that the pyrenes must not be in close contact, implying co-assembly with the other LMWG.
Specifically co-assembled structures can be formed by mixing two molecules, one of which acts as a donor and the other as an acceptor. With appropriate donors and acceptors, charge transfer complexes are formed, which drives specific co-assembly. For example, Rao and George have described the co-assembly of a oligo(phenylvinylene) (OPV) and a perylene bisimide (PBI), acting as a donor and acceptor respectively.83 In water at a concentration of 3.3 mM, a gel is formed. The gel had a strong absorption at 700 nm, and was non-fluorescent, indicating charge transfer interactions are occurring (Fig. 6).
 | ||
| Fig. 6 (a) Absorption and (b) emission spectra for the donor (T-OPV) and acceptor (C-PBI) and the 1:1 mixture showing a strong charge transfer band and lack of fluorescence. Figure reproduced from ref. 80 with permission from John Wiley and Sons. | ||
The charge transfer band was strongest at a 1:1 stoichiometry of the two components, again suggesting specific co-assembly. In this particular case, the OPV and PBI are reported to assemble into one-dimensional structures alone; it is not clear whether the components can form gels alone, or whether only the mixture results in a gel. Das and Ghosh have also described a specifically assembled gel formed from a donor and acceptor LMWG.84 Here, both components gel individually. A strong charge transfer band was again observed. Interestingly, where a self-sorted analogue was formed, no charge transfer band was observed and the UV spectrum was instead equivalent to the simple mathematical addition of the spectra from the two components.
We have recently shown that self-sorted systems can be prepared and characterised these at multiple length scales.85 These self-sorted hydrogels were prepared using a controlled pH drop. Each LMWG gels at its respective pKa. Using NMR, we showed that the two LMWG (Fig. 7a) were immobilised at different pH and clearly were sequentially assembling (Fig. 7b).
 | ||
| Fig. 7 (a) Structures of LMWG used for pH-triggered self-sorting; (b) NMR data showing sequential assembly of 1 and 2 starting at their respective pKa; (c) schematic of the sequential assembly; (d) SANS data for (top) 1 alone; (middle) a mixture of 1 and 2, and (bottom), 2 alone. | ||
However, this does not necessarily prove that self-sorted structures are formed, since the disappearance of signal from the NMR spectrum is indicative of self-assembly, but does not provide any information as to the identity of the self-assembled structures. Fibre X-ray diffraction was used to show that the scattering in the mixed gel was the sum of the scattering from the individual components. Since co-assembly would be expected to lead to the disruption of the individual native lattice structures or the formation of a new unique structure, we used this data to demonstrate self-sorting. In this case, electron microscopy was less clear than for some other examples (for example the work of Moffat and Smith77), and it was not possible to identify fibres which were clearly from one LMWG or the other. However, small angle neutron scattering (SANS) could be used to show that the fibres were self-sorted (Fig. 7d). The scattering from one of the LMWG was very different from the other, with the first being consistent with a core–shell structure and the other with a worm-like structure. In the mixed gel, aspects of both structures were detected. However, we could not distinguish categorically between co-existing homomolecular assemblies and co-assembled average structures. Hence, we prepared an analogue of one of the LMWG with a deuterated naphthalene ring. This is expected to greatly reduce the contribution of this LMWG to the scattering, and hence for a self-sorted system, we would expect that the scattering would more resemble that of the other LMWG alone. This was the case, showing that self-sorting had occurred at the fibre length scale.
We later showed that this self-sorting lead to gels with higher rheological properties than the expected sum of the properties of a gel made from each individual component.86 We also found a system where a co-assembled gel was formed. In this case, the signals in the NMR for both LMWG started to disappear at the same pH. The fibre X-ray diffraction data and SANS data were less clear cut in this case, but we were able to show that the CD data was not a simple overlay of the expected data from both LMWG alone. Interestingly, the co-assembled gel also had higher rheological moduli than expected from the sum of the components. It therefore appears that one should treat the observation of stronger gels with caution in terms of linking to a specific structure. Once again, the gel properties arise from a number of parameters, including the fibre concentration, fibre dimensions and fibre moduli, the number of cross-links and type of cross-links. The relative importance of each of these parameters is unknown.37
From the above, it is clear that sometimes the type of assembly is clear and the data are unambiguous across all length scales. In other cases, the situation is less clear, or perhaps is only provided at one length scale. There are currently a relatively few examples of multicomponent systems and so it also not always obvious how to interpret the data as there are insufficient other examples from which to extrapolate.
How can mixed LMWG systems be designed?
As noted above, despite being a widely studied class of materials, the exact design rules for a LWMG are not fully understood. Many are discovered serendipitously, with further LMWG discovered by the synthesis of structural permutations. This is not always successful; subtle differences in both the molecular structure and the assembly conditions can be the difference between a gelator and a non-gelator. These nuances make it difficult to fully understand the gelation process, and this difficulty is inherently magnified in systems consisting of multiple gelators. This is further complicated by the need to understand how the two LMWG interact when in the presence of one another. There are not many examples where the assembly of a multicomponent gelator system has been designed and rationalised. Here, we discuss the data that is available, as a potential for design rules.In terms of designing multicomponent systems, it is plausible to expect that a system consisting of LMWG with very different structural motifs may encourage self-sorting. Conversely, when the two components are structurally similar, co-assembly may be encouraged. A number of examples have been reported where structurally dissimilar gelators have been used to form a self-sorted gel. These include the perylene and oligothiophene LMWG used to form BHJ (see Fig. 3 above).59 Here, the authors make the important point that the LMWG are sufficiently structurally similar to allow gelation of the same solvent, but the molecular lengths are slightly different and the number of hydrogen-bonding sites is different. Hence, self-sorting is preferred to mixing. Elsewhere, Smith’s group have shown that self-sorted systems can be formed from two very structurally dissimilar LMWG (A and B, Fig. 8a).40
 | ||
| Fig. 8 Molecular structure of LMWG used by Smith for preparing self-sorted and co-assembled structures. A and B self-sort;40 C and D self-sort;77 D and E co-assemble.77 | ||
However, such significant differences are not always required. From example Moffat and Smith showed that the LMWG C and D (Fig. 8b) formed self-sorted systems.77 Gelators D and E (Fig. 8b) formed co-assembled structures. It should be noted however that C and D individually form gels with very different melting temperatures, whilst D and E individually form gels with similar melting temperatures. It is therefore not absolutely clear whether it is the molecular structures that driving the different types of assembly or the thermal properties of the gels.
We have recently showed that a slow pH change could be used to prepare self-sorted gels using dipeptide based LMWG (e.g.Fig. 7a). Here, the apparent pKa of the dipeptide-based LMWG used scales with the hydrophobicity of the gelator. Hence, gelators with very different pKa can by synthesised. The self-assembly occurs at the pKa of the LMWG and so a slow pH change means that the pKa of one LMWG is reached before the other and hence this gelator assembles first. After this, the pH drops further and the second LMWG assembles. As discussed above, we have shown that this can be used to prepare systems that are self-sorted across multiple length scales. This self-sorting is therefore ‘programmed’ by the molecular structure, and even structurally quite similar molecules can have different pKa. This process works for a number of LMWG pairs. This approach has been shown to work in the bulk using the slow hydrolysis of glucono-δ-lactone to gluconic acid, which allows the slow pH change.85,86 We have also used a surface triggered approach, using electrochemistry to lower the pH.87 Self-sorting also occurs here. For this approach, the LMWG were structurally relatively similar, but with differences in gelation point (here pH, as opposed to the gelation temperature as used by Sugiyasu et al.59 or Moffat and Smith77). However, the concept of structural similarity does seem to be important; the one example we have found to date where co-assembly occurs is where the LMWG are very structurally similar.86
A relatively simple concept for designing multicomponent systems consisting of very structurally similar gelators is to use enantiomers. As chirality plays a vital role in an array of biological systems and can be expressed at not only the molecular level, but also in the helices of macro and supramolecular structures, it is a rational means to try to design co-assembling multicomponent LMWG systems. In many cases, individual enantiomers have been reported to be better gelators (individually) than the racemic mixture.88,89 Enantiomers can form aggregates that lead to fibril growth and network formation (and so gelation), but the use of racemates frequently results in precipitation90 and hence no gelation. However, in some cases racemic mixtures can lead to conglomerate formation, with the conglomerates resolving into separate aggregates resulting in gelation. The pure enantiomers would interact with enantiomers of the same conformation, leading to self-sorting at the molecular level as it may be expected that favourable packing occurs between similar molecules.
For example, Das et al. prepared gels from alkyl urethane based gelators using a heat–cool method, where they studied both enantiomeric forms of the LMWG, a version of the R-enantiomer that contained a longer chain substituent and the achiral version of the gelator.91 The longer chain length did not affect the individual gelation properties. When multicomponent gels were prepared by mixing the enantiomers (1R (or the longer chain equivalent 2R) and 1S), different properties were observed to those of the pure gels (Fig. 9).
 | ||
| Fig. 9 (a) Gel to sol transition temperatures in single component and mixed gels and (b) CD data obtained for xerogels (in isooctane) of mixed systems of 1R/1S with different ratios of enantiomer. Reprinted with permission from ref. 91. Copyright 2010 American Chemical Society. | ||
Individually, gels of 1R and 1S in isooctane showed the same thermal stability, as they both exhibited a Tgel of 65 °C. However, a racemic mixture of 1R and 1S resulted in a gel with lower thermal stability (Tgel of 55 °C). The solvent was found to play a role in the complexity of these mixed systems, as racemic mixtures could not form gels in dodecane, but an 80% enantiomeric excess (ee) of 1R mixed with 1S could form gels in this solvent, as could 1R and 1S individually. When these LMWG were mixed in dodecane, rheological measurements of the resulting gels showed the gels were less strong than for those formed from pure 1R gel. To rationalise these data, the CD spectra showed no linear dependence on the ee (Fig. 9b), which led to the suggestion of co-assembly occurring between enantiomers and hence, no conglomerate formation. On this basis alone, it was difficult to assign any exact rules for the design on multicomponent gels from these urethane gelators as chiral transcription to the macroscopic domain was not always guaranteed. Pure enantiomers could retain the chirality in the assembled state, even upon drying, but this was lost in mixed gels containing an achiral component. Furthermore, in the design of these systems the solvent choice is important. Individual enantiomers could form gels in dodecane but co-assembled racemates could not. Gels prepared in dodecane consisting of a chiral–achiral mixture showed no chiral amplification, unlike the gel in isooctane, indicating further the importance of solvent interaction when designing these systems. Other than just chiral recognition at the molecular level being crucial to the design of these materials, other key features of the gelator molecules can be tuned to try to influence the interaction between gelators in a mixed system. For instance, of the chiral–achiral systems described by Das et al., the system where the lengths of side chain alkyl groups were similar resulted in gels with larger fibres, better thermal stability and larger yield stress than the mixed gel with differing side chain lengths. This demonstrates the important role of molecular recognition (not just molecular chirality) leading to favourable packing in multiple gelators with similar structures. No one property can be considered in isolation when attempting to design these systems.
Pochan and Schneider’s groups have shown an example of synergistic design of mixed gels of racemic mixtures of their 20 amino acid β-hairpin gelator, MAX1.92 Gels were prepared by increasing the ionic strength (using NaCl) to screen the positive charges, leading to decreased electrostatic repulsion between protonated lysine side chains and intramolecular folding precedes assembly. The D-enantiomer of MAX1 (DMAX1) can undergo folding under the same conditions as that for the L-enantiomer (MAX1) and produces hydrogels with virtually the same mechanical properties. The molecular packing of the LMWG in the gels appears to be the same as the CD data for DMAX1 is the mirror image of MAX1 (Fig. 10a), as expected for the opposite enantiomer, and shows that β-sheet structures are formed. In a mixed system, altering the ratio of MAX1 to DMAX1 affects the mechanical rigidity of the gels. Using 1:3 and 3:1 mixtures of MAX1:DMAX1 more than doubles the G′ values of the gels as compared to that of the single enantiomers (Fig. 10b).
 | ||
| Fig. 10 (a) CD wavelength spectra of pure MAX1 (□), 3:1 MAX1:DMAX1 (▲), 1:1 MAX1:DMAX1 (●), 1:3 MAX1:DMAX1 (▼) and pure DMAX1 (◆). (b) Storage moduli of gels with varying mole fractions of enantiomer. Reprinted with permission from ref. 92. Copyright 2011 American Chemical Society. | ||
An equal ratio of MAX1:DMAX1 further enhances the rheological properties. TEM shows that the LMWG assembles into fibrils with a width of ∼3 nm, the same as individual MAX1 LMWG. These synergistic effects were rationalised as being due to favourable interactions between enantiomers, although it is unclear whether these are on the molecular or the network level. Although the packing of the LMWG was found to be the same, there still could be some interactions between enantiomers at the molecular level that may guide the self-assembly and result in co-assembly of enantiomers to give homogeneous fibrils. Conversely, if the favourable interactions are having an effect at a longer length level, perhaps in the network formation, then it could be possible that segregation of enantiomers in racemic mixtures may occur and lead to self-sorting of enantiomers. From the CD data collected, the chirality in this case is dominated by majority rule but it is unclear if this aspect has any influence on the macroscopic properties of the mixed gels. As the mixed gels bear no resemblance in bulk properties to either gelled individually, it would appear that the design of these gels cannot be fully rationalised on the understanding that self-recognition of enantiomers leads to more favourable packing and better gels. Here, either self-sorting or co-assembly at the molecular level may be suggested, but it is not fully understood what happens beyond this length scale. This system highlights the hierarchical nature of the assembly process as differing chiral properties of the supposed aggregates can react synergistically. This was not observed in the previous example. Chirality alone cannot always be a sound rationale for the design of mixed gelator systems, it would seem.
Swanekamp et al. tried to elucidate the design rules of enantiomeric multicomponent gels from studies of amphipathic peptides.93 When the two gelators were labeled with donor and quencher molecules, respectively, co-assembled gels displayed no fluorescence. This appeared to confirm co-assembled structures and a hybrid ‘rippled’ β-sheet co-assembled structure was proposed – slightly different to the packing proposed for the gels of the individual components. Here, the basic rule of design does not rely on enantiomeric recognition – as this may be expected to produce enantiomerically distinct, self-sorted gel networks, but relies on the recognition of structurally similar molecules favourably interacting with one another when mixed together. Interestingly, Swanekamp et al. drew comparison of their system with that of Pochan and Schneider mentioned above, suggesting that their multicomponent gels could also consist of co-assembled, alternating enantiomers due to the similar alternating hydrophobic and hydrophilic patterning features of both systems.
Although not strictly a mixed gel system, Adhikari et al. have shown systems whereby the chirality of the LMWG in the presence of an amino acid additive is translated into the supramolecular helicity of the resulting fibres of the gel.94 CD data collected for a gel of Fmoc-L-Glu (the LMWG) and L-Lys, – where the gelator and amino acid additively co-assemble in equimolar quantities – was an exact mirror image of the CD spectrum collected for a gel of Fmoc-D-Glu and D-Lys, with left-handed and right-handed helices respectively. When all four components were mixed to form a multicomponent gel, the LMWG/amino acid pairings with the same conformations (L or D) still appeared to co-assemble; left and right-handed helices were both observed in the final gel (Fig. 11). Interestingly, these opposing helices were observed in (almost) equal amounts, which suggests that self-sorting of the gelators (and their amino acid ‘partner’) occurs.
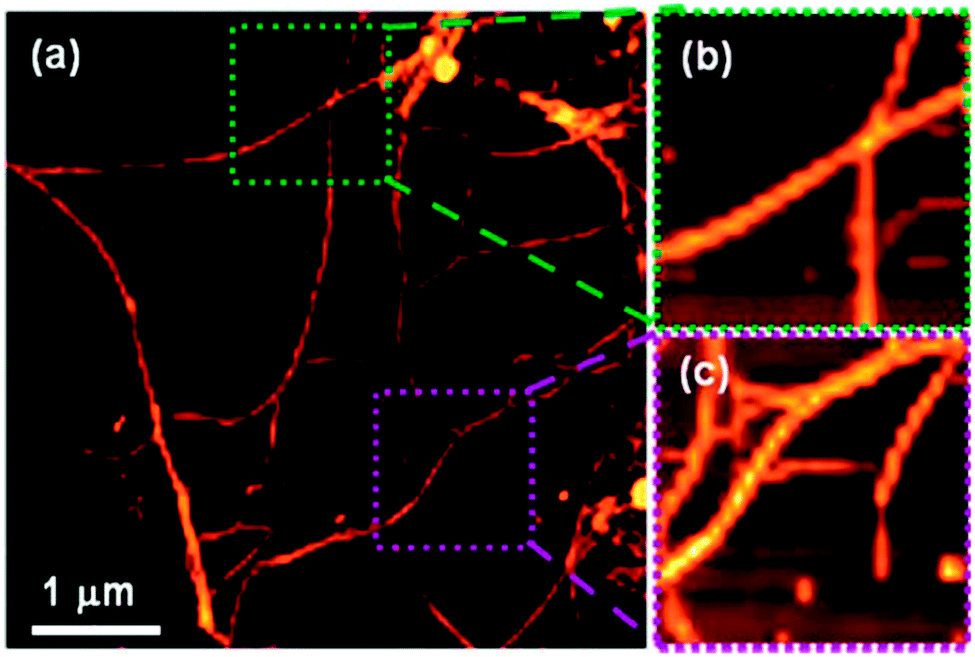 | ||
| Fig. 11 (a) AFM image of racemic mixture of multi-component (four components) hydrogel [Fmoc-(L + D)Glu + (L + D)Lys] showing the presence of both left- and right-handed helical fibres; (b) zoomed image showing a right-handed helical fibre (green region) and (c) shows zoomed image of a left-handed helical fibre (purple region). Reproduced from ref. 94. | ||
Cicchi et al. also showed that enantiomeric discrimination leads to self-sorting.95 They noted a linear correlation between CD maximum and enantiomeric composition when they varied the enantiomeric ratios in organogels of long chain carbamate-based materials. When opposing helices are mixed, it may be plausible to assume that supramolecular self-sorting of these helices will result, due to dissimilar modes of packing. It is clear that opposing enantiomers do not always favour packing with like enantiomers, and cooperative interactions between components can be just as important as chirality to the packing of gelator molecules in a mixed system.
Clearly chirality can play a pivotal role in the design of mixed gelator systems, with enantiomeric recognition governing the supramolecular arrangement preceding gelation in some cases. However, it does not control the design explicitly. By extrapolation of this concept, self-recognition can be utilised to control assembly by designing gelators with similar structural motifs rather than the same chirality.
The majority of self-sorted systems are prepared by a heat–cool cycle. The gelling temperature of a LMWG is hard to predict in advance. Hence, there is a requirement for control over the structural properties of the gelator. Conceptually, preparing self-sorted systems requires the assembly of structurally distinct LMWG. As one example, Ghosh’s group has designed a self-sorted organogel system.84,96 Donor (D) and acceptor (A) based LMWG were used. To avoid co-assembly, the LMWG were designed carefully by the placement of amide functionality on the gelators. The design of this subset of materials can be manipulated to allow a co-assembled supramolecular structure to form. NDI-1 forms a self-sorted gel with the DAN-1 donor (Fig. 12).
 | ||
| Fig. 12 Structures of two NDI-based (blue) gelators where the position of the amide functionalities (red) differ and a dialkoxy-naphthalene (blue) gelator possessing symmetrical amide functionality (red). | ||
By altering the structure of NDI-1 to give a gelator where the two amide groups are in close proximity on a single arm of the acceptor chromophore (Fig. 12) allowed for stronger H-bonding interactions between the two LMWG. The latter system leads to intense red gels due to charge transfer (CT) interactions between donor and acceptor chromophores. The occurrences of CT interactions are inherently dictated by the propensity for H-bonding interactions between DA pairs, and the introduction of amide functionality on the opposing chromophores of the D and A allows the degree of H-bonding to be tuned (to an extent). CT interactions are an attribute of a co-assembled system in DA pairs with related structures. NDI-2–DAN-1 gels form alternate DA stacks where H-bonding is geometrically allowed in the CT state of the DA pairs. The DA system of NDI-1 and DAN-1, relies on a self-complementary H-bonding system consisting of symmetrical amide (and so H-bonding) functionality on the donor and unsymmetrical amide functionality on the acceptor chromophore. Here, not all amides can participate in the H-bonding of the alternate co-stacking structure. This work shows that subtle changes in very similar materials can be the difference between a self-sorted and co-assembled system, making the design of multicomponent systems a real challenge. However, self-recognition between gelators seems to be an efficient rule for the basis of initial design of these systems.
Another example of self-recognition was demonstrated by Afrasiabi and Kraatz.97 They prepared a series of Boc-L-Phe-L-Lys(Z)-OMe (Z = carboxybenzyl) analogues with pyrene or ferrocene groups attached at the N-terminus. The addition of these groups was to provide some stimuli responsiveness to the hydrogel systems. A variety of mixtures showed gelation (in a variety of organic solvents) under both thermal (heating–cooling) and sonication (heating–sonicating) conditions. Here, the basic rule of multicomponent gelator design is amino acid sequence manipulation; where the amino acid sequence of a peptide-based gelator can be tuned to promote either co-assembled or self-sorting structures. When the amino acid sequence on opposing gelators was the same, co-assembly occurred. Conversely, when the amino acid sequence was not the same, self-sorting between gelators in the mixed system occurred. In the self-sorted networks, distinct regions of nanofibres and nanotapes were imaged, showing the self-sorting was also prevalent at this length scale.
Self-recognition motifs appear to be an effective rule for the design of many multicomponent gelator systems, however, systems based on other design rules have been developed. Nilsson’s group proposed that the design of multicomponent gels could be influenced by the addition of various halogen substituents to a benzyl group of one of the gelators in their mixed gels.98 The addition of a halogen affects the π–π stacking interactions between assembled gelators. They have shown that the choice and position of the halogen substituent can affect the mechanical properties of the resulting multicomponent gel. Co-assembly was favoured due to offset π–π interactions between the halogenated substituents on the benzyl ring of one gelator and the non-substituted benzyl of the other gelator. Complementary quadrupoles were not a requirement for co-assembly. To try to clarify the design rules for their multicomponent gels, Nilsson’s group looked at a system consisting of an Fmoc-Phe gelator containing a perfluorinated ring co-assembled with Fmoc-Phe. Using a solvent-mediated trigger, the perfluorinated gelator could assemble (and gel) alone. Fmoc-Phe formed a precipitate under the same conditions. However, a 1:1 mixture of the two components formed gels with no evidence of precipitate, suggesting that Fmoc-Phe was incorporated into the fibres and co-assembly had occurred. Replacing Fmoc-Phe with Fmoc-D-Phe in the mixed system resulted in no gelation and therefore gave an indication that the interactions dictating co-assembly are selective and complementary. Furthermore, no gels were formed when Fmoc-Leu was used instead of Fmoc-Phe. The importance here is that these two gelators have similar hydrophobicities, implying that hydrophobic interactions are not sufficient to promote assembly. On this basis, manipulation of the complementary π–π interactions via substitution on one of the benzyl rings is a design rule that provides the option for future development and design.
Conclusions, challenges and opportunities for multi-component systems
It is clear from the above that there are limited design rules for the formation of self-sorted, or co-assembled multicomponent gels. In general, it appears that structural diversity between the LMWG tends to favour self-sorting and specific co-assembly can be driven by charge transfer interactions. However, the non-covalent interactions between molecules are subtle and sometimes hard to understand. In some cases, the difficulty in being able to distinguish between where a self-sorted or co-assembled gel has been formed complicates the understanding.A further issue for all of these design concepts revolves around the veracity of the cartoon in Fig. 2. In this cartoon, we imply that the starting point is a molecular solution of the two LMWG. This may not be the case. For example, in heat–cool triggered gels, it may be that the LMWG have not molecularly dissolved, but are rather dispersed as aggregates. For the pH triggered systems, it is clear that self-assembled aggregates exist at high pH before the pH is lowered to form a gel. The importance of the pre-gelling structures is not clear, but we expect that it is likely that these are important and perhaps template the assembly in some way.
Finally, we note that there is undoubtedly a significant importance to how the self-assembly is carried out.37 Even within a single process, it may well be that the kinetics of assembling the mixture may have an effect. For example, one can imagine in a temperature-triggered gel that the rate of cooling could be critical. This is often over-looked and rarely discussed, but should be considered.
As mentioned, LMWG are receiving significant attention at the moment. They are being used for a number of applications and there are many exciting developments. Alongside this, there is a growing awareness of the complexities in understanding and controlling the self-assembly processes. The vast majority of examples are for single component systems. However, by using multiple components, there is the opportunity to prepare exciting materials. This approach is of course more difficult, since now it is necessary to control the assembly of multiple components across different length scales and potentially at different times.
There have been a number of examples where mixed LMWG systems provide gels with improved properties compared to single component systems. For example, a mixture of two oxalamide-based LMWG has been shown to be able to gel significantly more solvent than either gelator alone.99 An increase in the gel-to-sol temperature was also reported. There are a number of examples for both mixed and self-sorted systems where the rheological properties are improved compared to what might be expected from the individual components.78,86,91,100,101 Even from these relatively few examples however, it is clear that there is not a simple link between the rheological data and the type of assembly.
There are of course a number of other challenges. As has been described above, a number of techniques can be used to probe the nature of the mixed systems. However, there are not many examples where the type of assembly has been proved at both the molecular level and also at the fibre and network length scale. For self-sorted systems, a number of examples show self-sorting at the molecular level, but self-sorting at the fibre length scale is rarely demonstrated. In rare occasions, it is possible to differentiate between fibres by microscopy.77,86 However, many LMWG are observed to assemble into fibres with similar dimensions, so differentiating between them in a mixed system is difficult. This is on top of potential issues with drying artefacts.
To further this area, it is clear that we need to develop a significantly better understanding as to how co-assembled or self-sorted gels can be designed, and critically how to truly understand the assembly across multiple length scales. Gelation is often described as a kinetically controlled process, and it is not clear that the gel state is the thermodynamic minimum. In mixed systems, this becomes even more complex. Additionally, it is not clear whether all of the mixed systems described above are 100% self-sorted or 100% co-assembled. It could be that there is a mixture of both types of assembly in either or both, and the methods used to probe the assembly are simply more sensitive to one type of aggregate.
Despite this, there are clear opportunities in materials with improved and designed rheological properties (it is probably easier to mix two known LMWG to design specific properties than design a new LMWG from first principles), as well as for materials with specific properties in optoelectronics, cell culturing etc. For co-assembled structures, it may be possible to continuously tune properties by the ratio of LMWG. It may also be possible to add specific amounts of a LMWG containing a specific functional or addressable group without affecting the mechanical properties, allowing enhanced properties; this would be especially true if the spatial location of the added second LMWG could be controlled. As such, the current relatively restricted number of examples is only just starting to scratch the surface of the possible mixed structures.
Acknowledgements
We thank the EPSRC for funding (EP/L021978/1).Notes and references
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 2005, 3615–3631 CrossRef.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1218 CrossRef CAS PubMed.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821–836 RSC.
- C. Tomasini and N. Castellucci, Chem. Soc. Rev., 2013, 42, 156–172 RSC.
- M. D. Segarra-Maset, V. J. Nebot, J. F. Miravet and B. Escuder, Chem. Soc. Rev., 2013, 42, 7086–7098 RSC.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- M. A. Rogers and R. G. Weiss, New J. Chem., 2015 10.1039/C4NJ01439A.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- M. C. Branco and J. P. Schneider, Acta Biomater., 2009, 5, 817–831 CrossRef CAS PubMed.
- J. B. Matson, R. H. Zha and S. I. Stupp, Curr. Opin. Solid State Mater. Sci., 2011, 15, 225–235 CrossRef CAS PubMed.
- D. M. Ryan and B. L. Nilsson, Polym. Chem., 2012, 3, 18–33 RSC.
- B. Escuder, F. Rodriguez-Llansola and J. F. Miravet, New J. Chem., 2010, 34, 1044–1054 RSC.
- J. F. Miravet, B. Escuder and C. Berdugo, Org. Biomol. Chem., 2014 10.1039/C4OB02003K.
- J. F. Miravet and B. Escuder, Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 117–156 Search PubMed.
- D. Diaz Diaz, D. Kuhbeck and R. J. Koopmans, Chem. Soc. Rev., 2011, 40, 427–448 RSC.
- X. Li, Y. Wang, C. Yang, S. Shi, L. Jin, Z. Luo, J. Yu, Z. Zhang, Z. Yang and H. Chen, Nanoscale, 2014, 6, 14488–14494 RSC.
- K. J. Skilling, F. Citossi, T. D. Bradshaw, M. Ashford, B. Kellam and M. Marlow, Soft Matter, 2014, 10, 237–256 RSC.
- M. Rodrigues, A. C. Calpena, D. B. Amabilino, M. L. Garduno-Ramirez and L. Perez-Garcia, J. Mater. Chem. B, 2014, 2, 5419–5429 RSC.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- J. D. Tovar, Acc. Chem. Res., 2013, 46, 1527–1537 CrossRef CAS PubMed.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644–656 CrossRef CAS PubMed.
- J. Puigmarti-Luis and D. B. Amabilino, Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 195–254 Search PubMed.
- P. Chen, Colloids Surf., A, 2005, 261, 3–24 CrossRef CAS PubMed.
- N. Zweep and J. H. van Esch, in Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 1–29 Search PubMed.
- D. K. Smith, Chem. Commun., 2006, 34–44 RSC.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5133–5138 CrossRef CAS PubMed.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- Y. Wang, L. Tang and J. Yu, Cryst. Growth Des., 2008, 8, 884–889 CAS.
- G. O. Lloyd and J. W. Steed, Soft Matter, 2011, 7, 75–84 RSC.
- D. Braga, S. d'Agostino, E. D'Amen and F. Grepioni, Chem. Commun., 2011, 47, 5154–5156 RSC.
- K. A. Houton, K. L. Morris, L. Chen, M. Schmidtmann, J. T. A. Jones, L. C. Serpell, G. O. Lloyd and D. J. Adams, Langmuir, 2012, 28, 9797–9806 CrossRef CAS PubMed.
- D. J. Adams, K. Morris, L. Chen, L. C. Serpell, J. Bacsa and G. M. Day, Soft Matter, 2010, 6, 4144–4156 RSC.
- J. Raeburn, A. Zamith Cardoso and D. J. Adams, Chem. Soc. Rev., 2013, 42, 5143–5156 RSC.
- X. Huang, P. Terech, S. R. Raghavan and R. G. Weiss, J. Am. Chem. Soc., 2005, 127, 4336–4344 CrossRef CAS PubMed.
- J. Cui, A. Liu, Y. Guan, J. Zheng, Z. Shen and X. Wan, Langmuir, 2009, 26, 3615–3622 CrossRef PubMed.
- M. M. Smith and D. K. Smith, Soft Matter, 2011, 7, 4856–4860 RSC.
- L. E. Buerkle and S. J. Rowan, Chem. Soc. Rev., 2012, 41, 6089–6102 RSC.
- M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
- A. R. Hirst, J. F. Miravet, B. Escuder, L. Noirez, V. Castelletto, I. W. Hamley and D. K. Smith, Chem. – Eur. J., 2009, 15, 372–379 CrossRef CAS PubMed.
- W. Edwards and D. K. Smith, J. Am. Chem. Soc., 2014, 136, 1116–1124 CrossRef CAS PubMed.
- A. Heeres, C. van der Pol, M. Stuart, A. Friggeri, B. L. Feringa and J. van Esch, J. Am. Chem. Soc., 2003, 125, 14252–14253 CrossRef CAS PubMed.
- A. Brizard, M. Stuart, K. van Bommel, A. Friggeri, M. de Jong and J. van Esch, Angew. Chem., Int. Ed., 2008, 47, 2063–2066 CrossRef CAS PubMed.
- T. Kato, Y. Hirai, S. Nakaso and M. Moriyama, Chem. Soc. Rev., 2007, 36, 1857–1867 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Soft Matter, 2013, 9, 8730–8736 RSC.
- D. J. Cornwell, B. O. Okesola and D. K. Smith, Angew. Chem., Int. Ed., 2014, 126, 12669–12673 CrossRef.
- J. Wang, Z. Wang, J. Gao, L. Wang, Z. Yang, D. Kong and Z. Yang, J. Mater. Chem., 2009, 19, 7892–7896 RSC.
- N. Javid, S. Roy, M. Zelzer, Z. Yang, J. Sefcik and R. V. Ulijn, Biomacromolecules, 2013, 14, 4368–4376 CrossRef CAS PubMed.
- G. Pont, L. Chen, D. G. Spiller and D. J. Adams, Soft Matter, 2012, 8, 7797–7802 RSC.
- L. Chen, S. Revel, K. Morris, D. G. Spiller, L. C. Serpell and D. J. Adams, Chem. Commun., 2010, 46, 6738–6740 RSC.
- J.-L. Li and X.-Y. Liu, Adv. Funct. Mater., 2010, 20, 3196–3216 CrossRef CAS.
- Y. J. Adhia, T. H. Schloemer, M. T. Perez and A. J. McNeil, Soft Matter, 2012, 8, 430–434 RSC.
- J. L. Li, X. Y. Liu, C. S. Strom and J. Y. Xiong, Adv. Mater., 2006, 18, 2574–2578 CrossRef CAS.
- Q. Wang, X. Xiao, Y. Hu, H. Wang and Y. Yang, RSC Adv., 2014, 4, 22380–22386 RSC.
- Y. Ohsedo, M. Oono, K. Saruhashi, H. Watanabe and N. Miyamoto, RSC Adv., 2014, 4, 44837–44840 RSC.
- K. Sugiyasu, S.-i. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- F. M. Menger and K. L. Caran, J. Am. Chem. Soc., 2000, 122, 11679–11691 CrossRef CAS.
- V. J. Nebot and D. K. Smith, in Functional Molecular Gels, The Royal Society of Chemistry, 2014, pp. 30–66 Search PubMed.
- E. Ostuni, P. Kamaras and R. G. Weiss, Angew. Chem., Int. Ed. Engl., 1996, 35, 1324–1326 CrossRef CAS.
- J.-B. Guilbaud and A. Saiani, Chem. Soc. Rev., 2011, 40, 1200–1210 RSC.
- C. Yan and D. J. Pochan, Chem. Soc. Rev., 2010, 39, 3528–3540 RSC.
- B. Escuder, M. Llusar and J. F. Miravet, J. Org. Chem., 2006, 71, 7747–7752 CrossRef CAS PubMed.
- D. C. Duncan and D. G. Whitten, Langmuir, 2000, 16, 6445–6452 CrossRef CAS.
- R. Huang, W. Qi, L. Feng, R. Su and Z. He, Soft Matter, 2011, 7, 6222–6230 RSC.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- L. Chen, S. Revel, K. Morris, L. C. Serpell and D. J. Adams, Langmuir, 2010, 26, 13466–13471 CrossRef CAS PubMed.
- C. Tang, R. Ulijn and A. Saiani, Eur. Phys. J. E: Soft Matter Biol. Phys., 2013, 36, 1–11 CrossRef PubMed.
- W. Helen, P. de Leonardis, R. V. Ulijn, J. Gough and N. Tirelli, Soft Matter, 2011, 7, 1732–1740 RSC.
- Y. M. Abul-Haija, S. Roy, P. W. J. M. Frederix, N. Javid, V. Jayawarna and R. V. Ulijn, Small, 2014, 10, 973–979 CrossRef CAS PubMed.
- S. Fleming, S. Debnath, P. W. J. M. Frederix, N. T. Hunt and R. V. Ulijn, Biomacromolecules, 2014, 15, 1171–1184 CrossRef CAS PubMed.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- J. R. Moffat and D. K. Smith, Chem. Commun., 2009, 316–318 RSC.
- D. Li, Y. Shi and L. Wang, Chin. J. Chem., 2014, 32, 123–127 CrossRef CAS.
- A. Del Guerzo, A. G. L. Olive, J. Reichwagen, H. Hopf and J.-P. Desvergne, J. Am. Chem. Soc., 2005, 127, 17984–17985 CrossRef CAS PubMed.
- J.-P. Desvergne, A. G. L. Olive, N. M. Sangeetha, J. Reichwagen, H. Hopf and A. Del Guerzo, Pure Appl. Chem., 2006, 78, 2333–2339 CAS.
- L. Chen, S. Revel, K. Morris and D. J. Adams, Chem. Commun., 2010, 46, 4267–4269 RSC.
- J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem. – Eur. J., 2014, 20, 279–291 CrossRef CAS PubMed.
- K. V. Rao and S. J. George, Chem. – Eur. J., 2012, 18, 14286–14291 CrossRef CAS PubMed.
- A. Das and S. Ghosh, Chem. Commun., 2011, 47, 8922–8924 RSC.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O’Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- C. Colquhoun, E. R. Draper, E. G. B. Eden, B. N. Cattoz, K. L. Morris, L. Chen, T. O. McDonald, A. E. Terry, P. C. Griffiths, L. C. Serpell and D. J. Adams, Nanoscale, 2014, 6, 13719–13725 RSC.
- J. Raeburn, B. Alston, J. Kroeger, T. O. McDonald, J. R. Howse, P. J. Cameron and D. J. Adams, Mater. Horiz., 2014, 1, 241–246 RSC.
- M. de Loos, J. van Esch, R. M. Kellogg and B. L. Feringa, Angew. Chem., Int. Ed., 2001, 40, 613–616 CrossRef CAS.
- Z. Yang, G. Liang, M. Ma, Y. Gao and B. Xu, J. Mater. Chem., 2007, 17, 850–854 RSC.
- J. H. Fuhrhop and C. Boettcher, J. Am. Chem. Soc., 1990, 112, 1768–1776 CrossRef CAS.
- R. K. Das, R. Kandanelli, J. Linnanto, K. Bose and U. Maitra, Langmuir, 2010, 26, 16141–16149 CrossRef CAS PubMed.
- K. J. Nagy, M. C. Giano, A. Jin, D. J. Pochan and J. P. Schneider, J. Am. Chem. Soc., 2011, 133, 14975–14977 CrossRef CAS PubMed.
- R. J. Swanekamp, J. T. M. DiMaio, C. J. Bowerman and B. L. Nilsson, J. Am. Chem. Soc., 2012, 134, 5556–5559 CrossRef CAS PubMed.
- B. Adhikari, J. Nanda and A. Banerjee, Soft Matter, 2011, 7, 8913–8922 RSC.
- S. Cicchi, G. Ghini, L. Lascialfari, A. Brandi, F. Betti, D. Berti, P. Baglioni, L. Di Bari, G. Pescitelli, M. Mannini and A. Caneschi, Soft Matter, 2010, 6, 1655–1661 RSC.
- A. Das, M. Molla and S. Ghosh, J. Chem. Sci., 2011, 123, 963–973 CrossRef CAS PubMed.
- R. Afrasiabi and H.-B. Kraatz, Chem. – Eur. J., 2013, 19, 15862–15871 CrossRef CAS PubMed.
- D. M. Ryan, T. M. Doran and B. L. Nilsson, Langmuir, 2011, 27, 11145–11156 CrossRef CAS PubMed.
- Z. Dzolic, K. Wolsperger and M. Zinic, New J. Chem., 2006, 30, 1411–1419 RSC.
- D. G. Velázquez and R. Luque, Chem. – Eur. J., 2011, 17, 3847–3849 CrossRef PubMed.
- S. Boothroyd, A. Saiani and A. F. Miller, Biopolymers, 2014, 101, 669–680 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |