
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Polymeric biomaterials for the delivery of platinum-based anticancer drugs
Jihoon
Kim†
a,
Swapan
Pramanick†
a,
Duhwan
Lee
a,
Hansoo
Park
*b and
Won Jong
Kim
*a
aCenter for Self-assembly and Complexity, Institute for Basic Science and Department of Chemistry, Pohang University of Science and Technology (POSTECH), 77 Cheongam-ro, Nam-gu, Pohang 790-784, Republic of Korea
bSchool of Integrative Engineering, Chung-Ang University, Seoul 156-751, Korea. E-mail: wjkim@postech.ac.kr; Tel: +82-54-279-2104; Fax: +82-54-279-3399
First published on 20th April 2015
Abstract
Since cisplatin, cis-diamminedichloroplatinum(II), received FDA approval for use in cancer treatment in 1978, platinum-based drugs have been one of the most widely used drugs for the treatment of tumors in testicles, ovaries, head and neck. However, there are concerns associated with the use of platinum-based anticancer drugs, owing to severe side effects and drug resistance. In order to overcome these limitations, various drug-delivery systems have been developed based on diverse organic and inorganic materials. In particular, the versatility of polymeric materials facilitates the tuning of drug-delivery systems to meet their primary goals. This review focuses on the progress made over the last five years in the application of polymeric nanoparticles for the delivery of platinum-based anticancer drugs. The present article not only describes the fundamental principles underlying the implementation of polymeric nanomaterials in platinum-based drug delivery, but also summarizes concepts and strategies employed in the development of drug-delivery systems.
 Jihoon Kim | Dr Jihoon Kim received his BSc from the Department of Life Sciences and obtained a double major from the Department of Chemistry of Pohang University of Science and Technology (POSTECH) in 2009. He obtained his PhD from the Department of Chemistry of POSTECH under the supervision of Prof. Won Jong Kim in 2014. Currently, he is a postdoctoral fellow and focuses on developing various systems for the delivery of nitric oxide/gene/drug. |
 Swapan Pramanick | Dr Swapan Pramanick received his BSc in Chemistry from Calcutta University. He obtained his MSc in Organic Chemistry from Jadavpur University, India in 2000. He obtained his PhD in the area of Medicinal Chemistry from Indian Institute of Chemical Biology (IICB), Kolkata, India in 2008. He joined Chembiotek, a branch of TCG Lifesciences in India, and worked as a Senior Research Scientist in the Synthetic Organic Chemistry research group for about 4 years. In 2011, he was appointed as a Research Associate in the research group of Prof. Robert P. Borris in the University of Hawaii at Hilo (UHH), Hawaii, USA. Since 2013 he has been a Research Associate in Prof. Won Jong Kim's group for biomedical polymer research in the Department of Chemistry of POSTECH, South Korea. |
 Duhwan Lee | Duhwan Lee obtained his BSc from the Department of Chemistry of Hanyang University in 2010. He is a graduate student pursuing his PhD under the supervision of Prof. Won Jong Kim in the Department of Chemistry, POSTECH. He has focused on the development of bioreducible gene/drug delivery systems with various polymers and nanomaterials. |
 Hansoo Park | Hansoo Park is an associate professor at the School of Integrative Engineering of Chung-Ang University. He has obtained an undergraduate degree from the Department of Chemical Engineering of Hanyang University, a master degree from the Department of Chemical Engineering of Korea Advanced Institute of Science and Technology, and a Ph.D. degree from the Department of Bioengineering of Rice University. He joined Stanford University as a post-doc researcher. His current research interests include biomaterials for stem cell engineering, tissue engineering, drug delivery systems, and cancer treatment. |
 Won Jong Kim | Prof. Won Jong Kim received his BSc from Hanyang University in 1998, and his M.S. and Ph.D. in Biomolecular Engineering in 2004 from Tokyo Institute of Technology. During his graduate studies with Profs T. Akaike and A. Maruyama, he developed a polymer-mediated DNA detection system. From 2004 to 2007, he was a postdoctoral fellow at the University of Utah under the supervision of Prof. Sung Wan Kim. Currently, he is an associate professor at the Department of Chemistry, POSTECH and a group leader of the Center for Self-assembly and Complexity, Institute for Basic Science (IBS). |
1. Introduction
Chemotherapy, i.e., the treatment of diseases by administration of chemical compounds, is currently one of the most effective ways to treat cancer. Among a variety of anticancer agents, cisplatin, a leading platinum (Pt)-based anticancer drug, has been used for more than three decades, either as a single therapeutic agent or in combination with other agents.1,2 Following the success of cisplatin, various Pt-based drugs (Fig. 1) have been developed, although only a small subset has received US Food and Drug Administration (FDA) approval. Nowadays, they play a crucial role in the treatment of various cancers, such as cancers in testicles, ovaries, head, and neck.3–5 However, the use of Pt-based anticancer drugs is associated with drug resistance and inactivation by vascular and cellular components, and is associated with severe side effects incurred by the systemic delivery, such as nephrotoxicity, ototoxicity, neurotoxicity, and emetogenesis.6–9 Therefore, various delivery systems have been developed to prevent the shortcomings of Pt-based chemotherapy and to increase its efficacy.10–12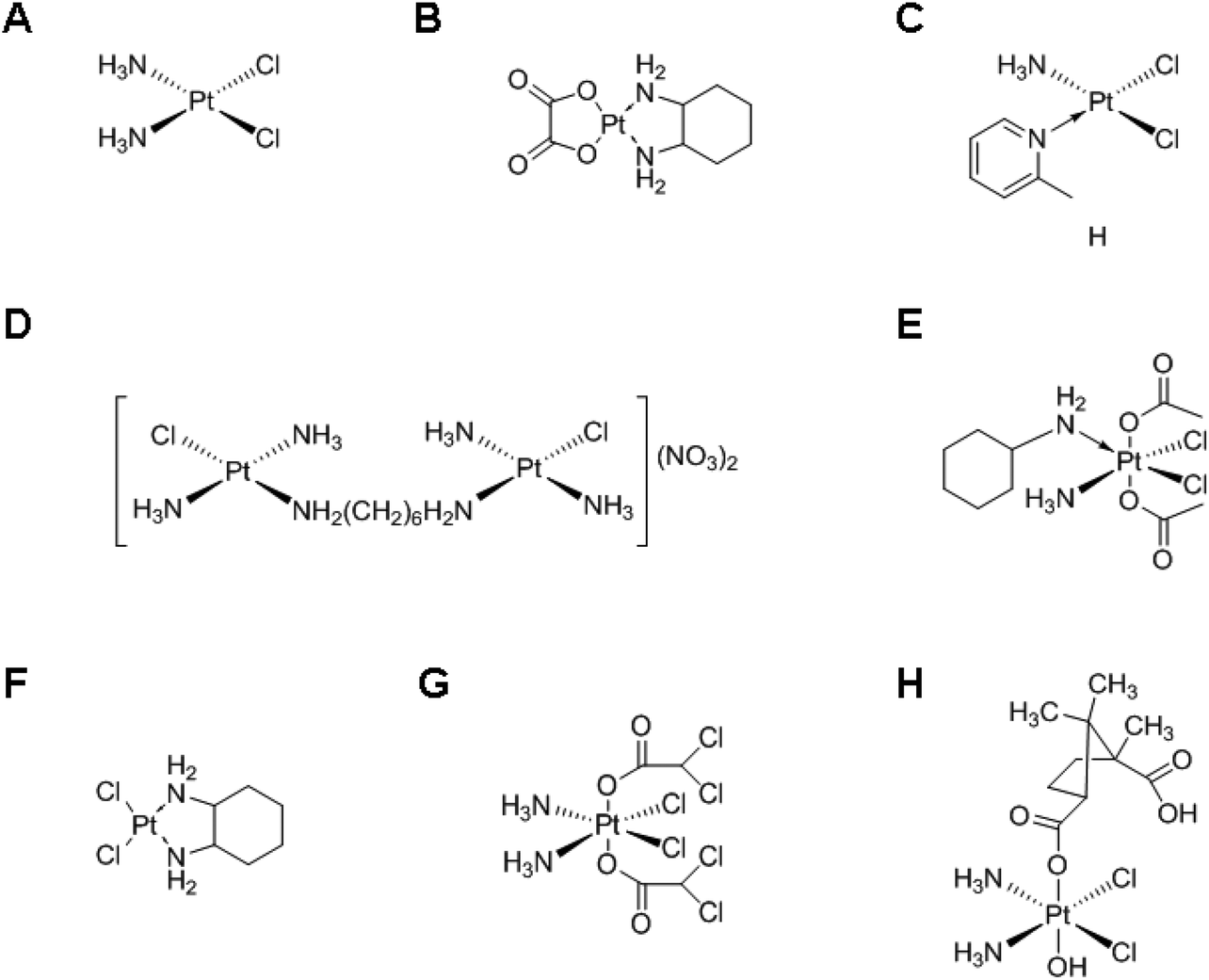 | ||
| Fig. 1 Chemical structures of various Pt-based drugs. (A) Cisplatin, (B) oxaliplatin, (C) ZD0473, (D) BBR 3005, (E) satraplatin, (F) DACHPt, (G) mitaplatin and (H) camplatin. | ||
Approaches like carrier-based drug delivery generally exploit the difference between normal and tumor tissues for increasing the efficacy and selectivity of a drug towards the target tissue. More specifically, the enhanced permeability and retention (EPR) effect is based on the increased permeability of nano-sized macromolecules coupled with the poor lymphatic clearance and slow venous return in the tumor tissues.13–17 Long-circulating nanocarriers have greater chances to be exposed to a tumor site, compared to low-molecular weight drugs which are rapidly cleared from the circulation. Thus, the EPR effect results in passive targeting of drug-loaded nanoparticles to the tumor tissues. In some cases, this can be further enhanced by active targeting, the use of nanoparticles functionalized with ligands for selectively binding to tumor-specific moieties exposed at the target cells.16–19 Such moieties generally include transporters, antigens, or receptors whose quantity or functionality is higher in tumors compared to normal tissues. Drug-delivery systems based on nanocarriers include micelles,20,21 liposomes,22 dendrimers,23 silica nanoparticles,24 and organic/inorganic hybrid nanoparticles.25 The most fascinating features of polymer-based delivery systems arise from the versatility of the polymer sources and their combinatorial synthesis. This versatility enables an easy tuning of the properties of the nanoparticles to meet the purpose of using delivery systems.21
For the successful drug delivery, drug encapsulation efficiency is one of the important indexes, especially in the case of poorly bioavailable drugs. To maximize the efficacy of anticancer drugs, the developed drug delivery systems must contain large amounts of drugs as much as possible. The polymeric nanoparticles facilitate efficient encapsulation of drugs into the nanocarriers by exploiting the compatibility between drugs and matrixes of polymeric nanoparticles or by employing the chemical conjugation between the polymeric backbone and drugs. In addition, the nanoparticles must release the drugs at a controlled rate. The controlled release of drugs has been achieved through the utilization of stimuli-responsive chemical bonds between drugs and the polymeric backbone or among polymeric blocks comprising the nanoparticles. Therefore, the ultimate goal of this review is to discuss the strategies for loading Pt-based drugs into the polymeric nanoparticles efficiently and for modulating their release in an appropriate manner, which affect significantly the anticancer efficacy.
In this review, we discuss the progress in the field of polymeric delivery systems of Pt-based anticancer drugs over the past five years. The article is organized into the following sections: first, we briefly introduce the characteristics of each structurally different Pt-based anticancer drug, involving both Pt(II)- and Pt(IV)-based drugs; then, we systematically discuss the encapsulation needed for delivering the Pt-based drugs; finally, we highlight the conjugation methods used for delivering Pt(II)- and Pt(IV)-based drugs.
2. Structure and cellular mechanism of Pt(II) and Pt(IV) compounds
Although numerous Pt-based drugs have been developed, most of them can be categorized into two groups, based on Pt(II) and Pt(IV), according to the molecular structure. As a background reference for the subsequent sections, here we provide a comprehensive summary of the structure and cellular mechanism of Pt(II) and Pt(IV)-based drugs.The most representative Pt(II)-based drug is cisplatin, which is the cis isomer of the square planar configuration (Fig. 1A).26,27 According to the HSAB (Hard–Soft Acid–Base) theory, Pt can be defined as a soft metal with high affinity for soft non-metal sulfur.26,27 Therefore, during blood circulation, unintended deactivation of cisplatin may be caused by biomolecules containing thiol groups, such as cysteine and human serum albumin.26–29 Some studies have demonstrated that about 65–98% of Pt becomes bound to blood proteins one day after cisplatin administration.28,29 Therefore, only a small number of Pt(II)-based drugs can maintain their activity during blood circulation and enter cancer cells mainly by passive diffusion and/or by the copper transporter CTR1 (Fig. 2).3,26–30 The relatively low concentration of chloride in the cells facilitates the replacement of chloro ligands in Pt(II)-based drugs by water, resulting in the mono-aquated Pt complex. The aquated form is reactive to guanine and adenine in DNA. The resulting intra- and inter-strand crosslinks distort the DNA duplex, causing the apoptosis of cancer cells.3,26,27,31,32 However, in some cases, the efficacy of cisplatin may be limited by inherent or acquired cisplatin resistance. The major mechanisms of resistance can be classified into four categories: inactivation by increased thiol-containing molecules, an increased DNA-repair capacity, reduced uptake/enhanced efflux, and failure of apoptotic pathways.3,26,30,31 Oxaliplatin (Fig. 1B) is a successful alternative drug to overcome the limitations of cisplatin in the treatment of cisplatin-resistant tumors. It has been reported that the bulky diaminocyclohexane (DACH) ligand of oxaliplatin contributes to overcoming the drug resistance arising from the mechanism of DNA mismatch repair recognition.3,30,33–35 In addition, cis-ammine(2-methylpyridine)dichloroplatinum(II) (ZD0473) is another alternative drug candidate for overcoming the cisplatin resistance induced by increased thiol-containing cellular components (Fig. 1C). It has been proposed that the steric hindrance of ZD0473 prevents the approach of glutathione (GSH). This strategy is not only able to maintain the cytotoxic ability of Pt-based drugs, but is also able to reduce deactivation by GSH.3,30,35 Another strategy to avoid cisplatin resistance involves BBR3005, a multinuclear Pt(II)-based drug (Fig. 1D). By inducing the irreversible B → Z transition in DNA structures with long range intra- and inter-strand crosslinks, multinuclear Pt(II)-based drugs prevent the detection of damaged DNA by DNA damage-recognition proteins.35,36
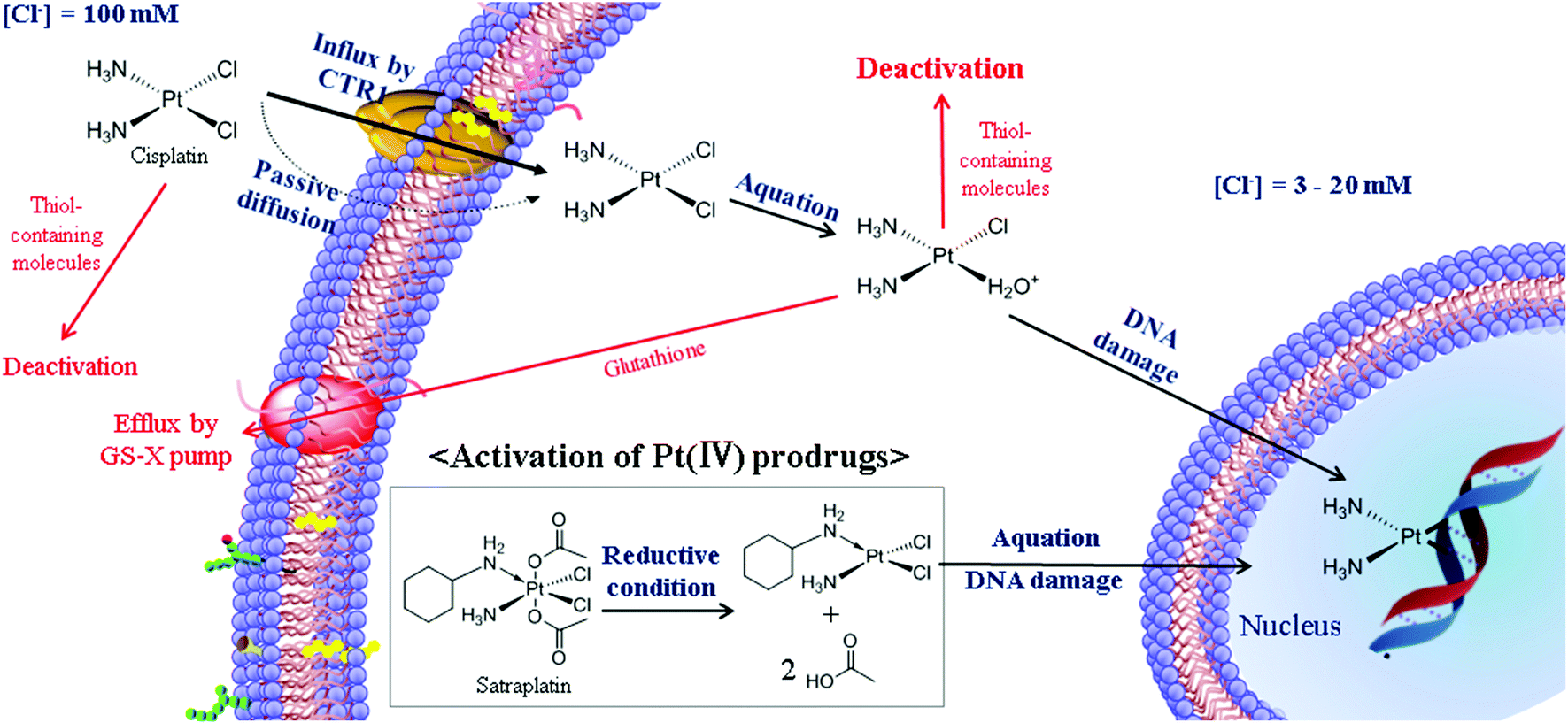 | ||
| Fig. 2 Schematic diagram of the cytotoxic pathway of Pt-based drugs. Cisplatin is a representative. After entering the cell by copper transferase receptor (CTR1) or passive diffusion, cisplatin was activated by replacing a chloride ion with water molecules in low concentration of chloride ions (aquation). The aquated cisplatin binds to DNA. Distortion of DNA activates the programmed cell death. Pt(IV) prodrugs were activated by reduction of two axial positions, resulting in the release of active Pt(II) complexes. After this, the mechanism is similar to cisplatin. | ||
Although a Pt(IV) complex was firstly identified together with cisplatin in Rosenberg's original work, its potential as an anticancer drug has been recently recognized.26,32,37–39 Pt(IV)-based drugs have a d6 octahedral geometry, generally believed to be less reactive towards nucleophiles than Pt(II) complexes. The axial ligands of Pt(IV) complexes have an important role in making them relatively inert compared to the Pt(II) complexes. The enhanced stability of Pt(IV) complexes is expected to improve the stability of Pt-based drugs and increase their half-life in the blood stream. In addition, the axial ligands ideally can exhibit functional moieties that can be modified by targeting ligands to achieve active targeting, by other components in order to overcome drug resistance, and by polymers for developing drug-delivery systems.32,37–39 The Pt(IV)-based drugs undergo reduction to Pt(II) inside the cells, which eliminates the two axial ligands of the octahedral Pt(IV)-based drug (Fig. 2). The reactive Pt(II) analogs produced by the reduction process exert the anticancer activity.26,32,37–39 Because of the reactivity change before and after reduction, Pt(IV)-based drugs are generally considered to be prodrugs.38 However, only a few Pt(IV) complexes such as satraplatin (Fig. 1E) have been involved in significant clinical trials, despite the rational design of Pt(IV) prodrugs.37–39 In order to proceed to further clinical practice, it is highly necessary to establish new synthetic chemistry, optimize the kinetics of reduction, and develop well-designed drug delivery systems.38,39
3. Physical encapsulation of Pt(II)
3.1. Traditional techniques
Traditional strategies for the delivery of Pt-based drugs are based on physical encapsulation—an approach that has been widely used in general drug delivery systems. These strategies have the merits of easy and simple procedures for loading drugs into the nanoparticles; however, they have been lamented for their low loading stability which causes the unintended loss of drugs during blood circulation.40 Several techniques have been developed for the potential use of encapsulation methods.41–44 Desolvation is one of the most frequently used strategies for fabricating nanoparticles. Protein nanoparticles developed by the desolvation method represent ideal candidates for the delivery of anticancer drugs because of their biodegradable, less immunogenic, and non-toxic characteristics.41,42 Very recently, a folic acid-conjugated gelatin nanoparticle (Cis-GN-FA) for cisplatin delivery has been developed through a double desolvation method.45 In the case of folate-negative A549 lung cancer cells, it was difficult to distinguish Cis-GN-FA from Cis-GNs. In contrast, the Cis-GN-FA nanoparticles show a significant decrease of the half maximal inhibitory concentration (IC50) (8.3 mM) in folate receptor-active HeLa cells, compared to Cis-GNs (15.1 mM) and cisplatin (40.2 mM), implying that further in vivo investigations of Cis-GN-FA are needed.The emulsion technique is another common encapsulation method, which is based on the compatibility between a drug and a matrix.41–43 In the case of poorly soluble and lipophilic drugs, the oil-in-water (o/w) single emulsion method has been utilized for physical encapsulation of the drugs. In addition, double emulsion techniques like water-in-oil-in-water (w/o/w), solid-in-oil-in-water (s/o/w), water-in-oil-in-oil (w/o/o), and solid-in-oil-in-oil (s/o/o) have been employed for compounds with high solubility in water, including proteins, peptides, and drugs like cisplatin.41–43 These common techniques have been frequently applied in cisplatin-delivery systems. A study illustrating the development of cisplatin-encapsulated poly(lactic-co-glycolic acid) (PLGA) nanoparticles (NP) by the double emulsion technique has also been reported recently.46 The developed PLGA NPs showed a sustained cisplatin release, with a cumulative release of 50% over 3 weeks, highlighting the potential of this system for avoiding multiple administrations of cisplatin in cancer treatment. In another study, cisplatin was emulsified into methoxy poly(ethylene glycol)-block-poly(lactic-co-glycolic acid)-block-poly(L-lysine) (mPEG-b-PLGA-b-PLL) nanoparticles (CDDP-NPs) using the w/o/w double emulsion method, followed by conjugation of an epidermal growth factor (EGF) ligand to the CDDP-NPs via EDC/NHS chemistry.47 The developed CDDP-NP-EGF not only showed sustained drug release profiles in vitro, but also showed lower nephrotoxicity and improved anticancer effects on ovarian carcinoma in vivo. This simple and universal emulsion technique has been used to develop novel and complex delivery systems as well. In 2014, Chen et al. reported an interesting nanocarrier system which releases both Pt(II)-based drugs and O2 in response to the local H2O2 concentration typical of the tumor environment (Fig. 3).48 In this system, Pt(II)-based drugs ([PtLCl]Cl) and catalase were incorporated into the aqueous core of PLGA nanoparticles via the double emulsion method. The catalase generated O2 from intracellular H2O2, resulting in the rupture of the nanoparticle shell by the increased internal pressure. In addition, the Pt(II)-based drug and O2 exhibited anticancer effects and the ability to overcome hypoxia-induced multidrug resistance (MDR),49 respectively. Although further investigations are necessary, this novel system could enhance the anticancer efficiency of cisplatin at the in vitro level.
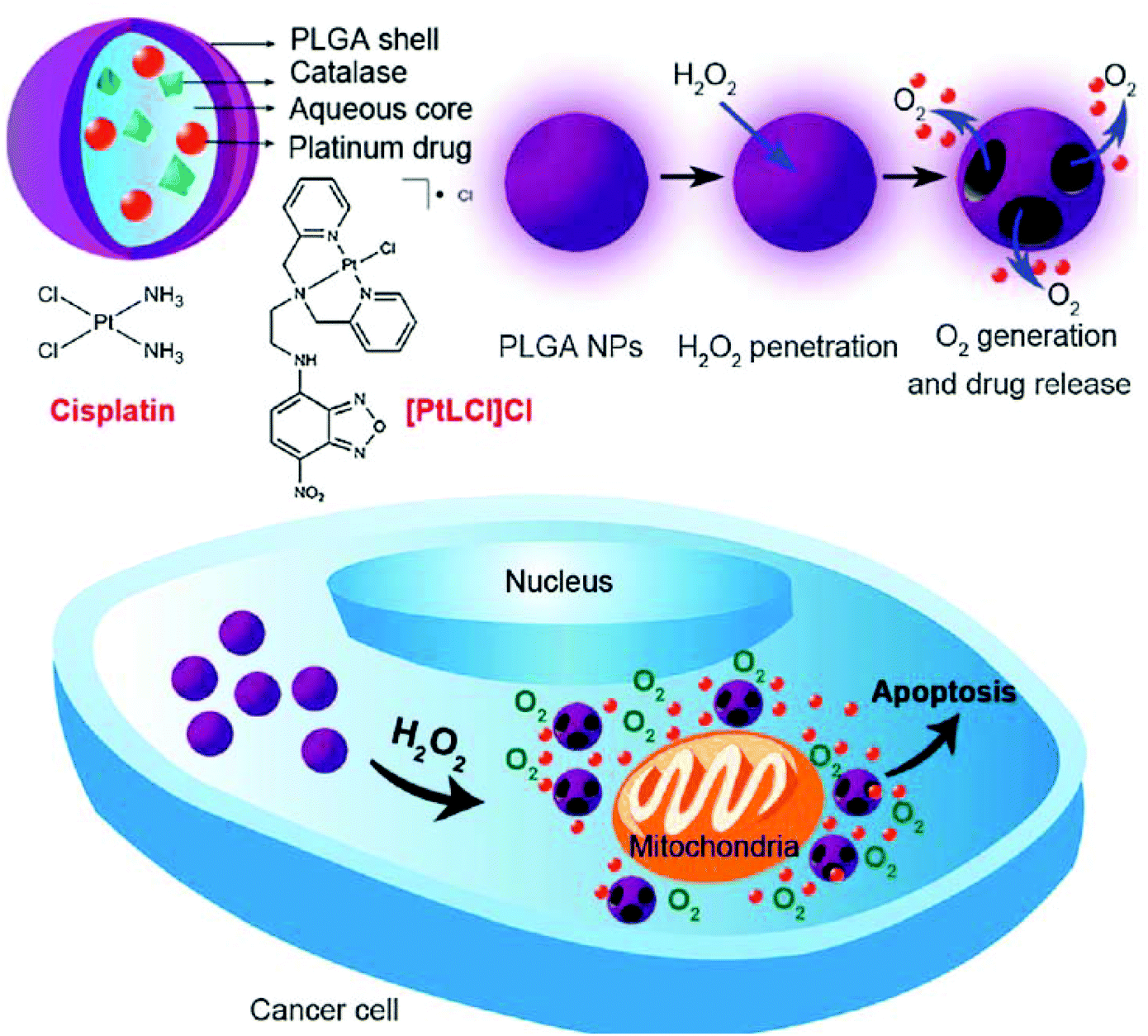 | ||
| Fig. 3 Schematic illustration of H2O2-responsive PLGA nanoparticles containing Pt(II) drugs and O2-generating catalase and the mechanism of drug release by H2O2 (reproduced from ref. 48 with permission from the Royal Society of Chemistry). | ||
Nanoprecipitation, based on the interfacial deposition, is a convenient and commonly used method for the preparation of polymeric nanoparticles.43,44 Recently, a biodegradable multifunctional nanoparticle (MNP) system for co-delivery of cisplatin and genes has been developed. Cisplatin-loaded poly(lactic acid) (PLA) nanoparticles were prepared via nanoprecipitation, and the surface was then coated with chitosan for electrostatic complexation with genes.50 The MNP is thus composed of a PLA inner core loaded with cisplatin and a cationic chitosan (CS) outer layer with P62 siRNA (siP62) and/or β5-expressing plasmid DNA (pβ5) (Fig. 4). The co-delivered genes could knock down P62 and restore β5 expression, resulting in a remarkable reduction in the IC50 value of cisplatin in cisplatin-resistant ovarian cancer cells. This simple co-delivery system has thus shown a potential ability to overcome the problems arising in the treatment of cisplatin-resistant tumors. Huang et al. have recently reported another co-delivery system capable of changing the tumor microenvironment for enhanced anticancer effects.51 To improve the encapsulation efficiency, cisplatin was coated with dioleoyl phosphatidic acid (DOPA). The DOPA-coated cisplatin cores facilitated the co-encapsulation of cisplatin and rapamycin, an mTOR inhibitor with antiangiogenic properties, into the PLGA via nanoprecipitation. The developed PLGA nanoparticles not only suppressed the tumor growth, but also altered the microenvironment of the tumor, inhibiting angiogenesis, reducing tumor-associated fibroblasts and collagen expression, and improving EPR effects of the nanoparticles in tumors. These findings have raised the expectations in the development of novel theranostic systems based on multifunctional nanoparticles such as quantum dots, superparamagnetic iron oxide nanoparticles, gold nanoparticles, and so on.
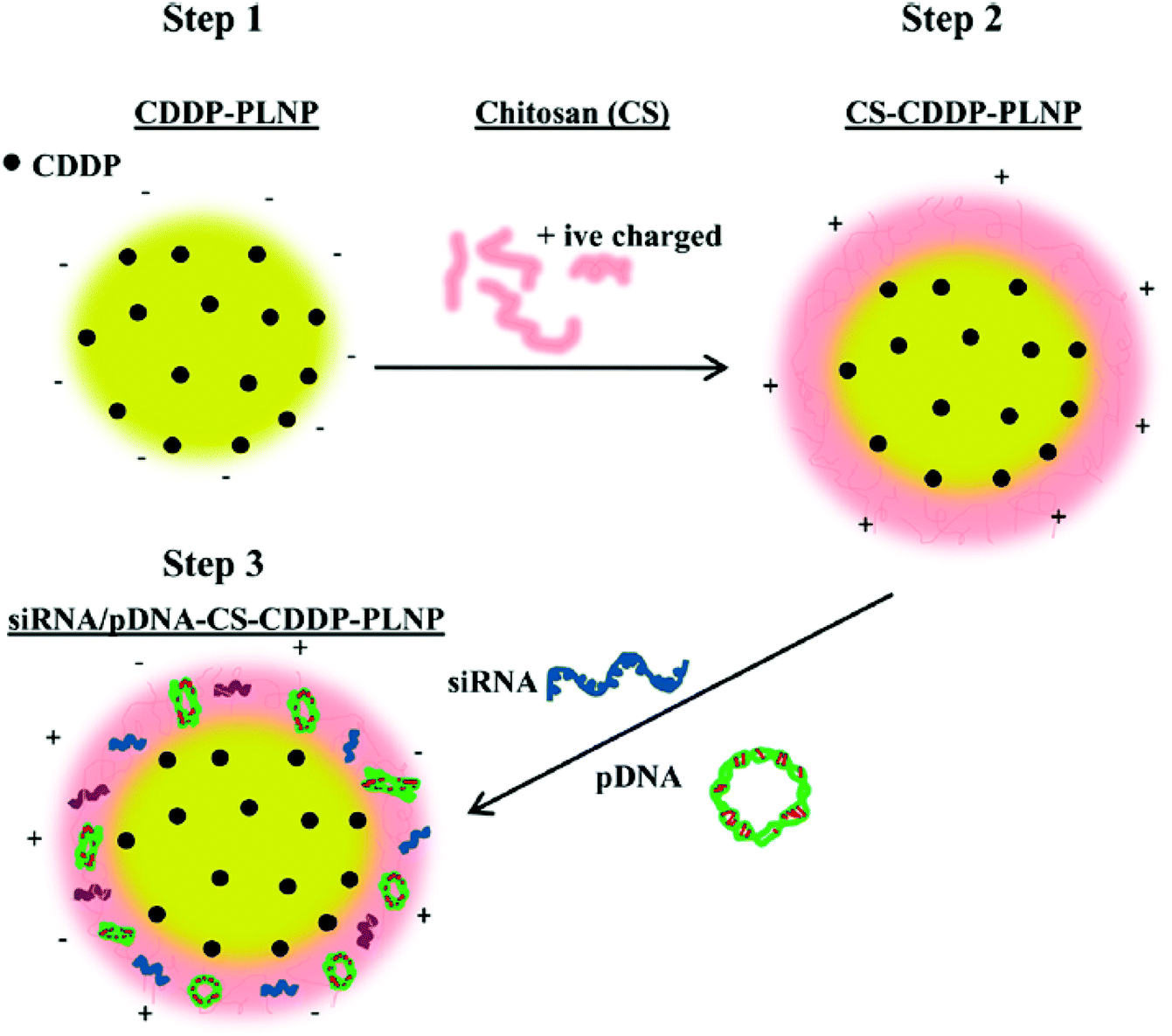 | ||
| Fig. 4 Schematic diagram of the preparation of siRNA/pDNA-CS-CDDP-PLNP. CDDP-PLNP was prepared by the nanoprecipitation method (Step 1), which was subsequently coated with CS layers (Step 2). The siRNA/pDNA-CS-CDDP-PLNP complex was formed via electrostatic interactions between siRNA/pDNA and CS-CDDP-PLNP (Step 3) (reproduced from ref. 50 with permission from the American Chemical Society). | ||
3.2. Micelles
Polymeric micelles are nanoscopic core–shell structures formed by amphiphilic block copolymers. They are generated when the hydrophobic portions of the polymers are driven to the interior of the structure, while the hydrophilic portions are exposed to the aqueous environment. These micelles have been widely used for general drug-delivery systems, as the hydrophobic interior has the capacity to hold drugs that are poorly soluble in aqueous solution.12,20,52,53 However, as far as cisplatin delivery is concerned, the hydrophilic nature of cisplatin makes it difficult to obtain cisplatin-encapsulated micelles. The successful encapsulation of a hydrophobic Pt(II) compound, cis-(cha)2Pt(NO3)2, into the amphiphilic cyclotriphosphazene [NP(MPEG750) (GlyPheLeu)2Et]3 (CP750) to form the micelle was recently reported.54 Compared to free cis-(cha)2Pt(NO3)2, the micelle not only exhibited prolonged blood circulation and large systemic exposure (AUC), but also showed excellent tumor accumulation and low acute toxicity. In particular, the micelle showed significant anticancer effects on the SNU638 cell line, a drug-resistant stomach carcinoma cell.4. Physical encapsulation of Pt(IV)
The fundamental principles for physical encapsulation of a Pt(IV) prodrug in a nanoparticle are in line with those of Pt(II)-based drugs. The primary difference is that a Pt(IV) prodrug has two more ligands at its axial positions, and these additional ligands can be useful to encapsulate the Pt(IV)-based drug into the nanoparticle. For instance, a Pt(IV)-based drug can be easily encapsulated into the hydrophobic parts of a nanoparticle if its axial ligands are modified by hydrophobic groups.55 Recently, Farokhzad and Lippard's group reported the development of targeted-delivery systems with reduced side effects by using simple nanoprecipitation and hydrophobic Pt(IV) prodrugs.56,57 Hydrophobic aliphatic chains at the axial position of Pt(IV) drugs helped in the efficient encapsulation of the drug into the hydrophobic core of methoxy poly(ethylene glycol)-block-poly(lactic-co-glycolic acid) (PEG-b-PLGA) nanoparticles via nanoprecipitation.55–57 In addition, the inertness of Pt(IV) is expected to mitigate side effects, like nephrotoxicity, compared to Pt(II)-based drugs, because the protection of the axial position of the drug can reduce the reactivity of Pt towards biomolecules. In particular, the conjugation of the cyclic arginine-glycine-aspartate (cRGD) ligand or the prostate-specific membrane antigen (PSMA) aptamer onto the nanocarriers facilitated the active targeting of drugs to the corresponding receptor on a tumor. In fact, cRGD-tethered and hydrophobic Pt(IV) drug-encapsulated mPEG-b-PLGA nanoparticles exhibited favorable anticancer effects on αvβ3 integrin-overexpressed cancer cell lines in vitro.56 Furthermore, PSMA-tethered and hydrophobic Pt(IV) drug-encapsulated mPEG-b-PLGA nanoparticles showed significant anticancer effects on PSMA-overexpressed LNCaP cells in xenograft mouse models, as well as a remarkable pharmacokinetic index without nephrotoxicity and changes in body weight.57 These basic principles have been extended to the development of several co-delivery systems. In 2013, Farokhzad and Walker's group modified the procedures for preparing mPEG-b-PLGA nanoparticles containing hydrophobic Pt(IV) prodrugs, in order to simultaneously load siRNA.58 By adding a cationic lipid-like molecule (G0-C14) in the double emulsion process, they were able to obtain a nanoparticle whose structure involves an aqueous core, a cationic and hydrophobic layer formed by G0-C14 and PLGA, and a hydrophilic outer shell composed of PEG. The G0-C14 has a pivotal role in the fabrication of the nanoparticle, because it provides flexible hydrophobic tails for self-assembly into the PLGA matrix, as well as efficient binding with the negatively-charged siRNA. These formulations enable the fabrication of nanoparticles with a dense structure, resulting in a sustained release of siRNA and Pt(IV) drug payloads. In addition, the nanoparticles exhibit a significant inhibition of tumor growth in vivo owing to the synergy of antitumor effects by Pt(IV) prodrugs and sensitization by siREV1/siREV3L. Moreover, hydrophobicity can be introduced by conjugating hydrophobic anticancer drugs at the axial position of a Pt(IV) prodrug. Zhang's group reported the synthesis of lipid–polymer hybrid nanoparticles for the delivery of a drug conjugate composed of a hydrophilic Pt(IV) prodrug and the hydrophobic paclitaxel (Ptxl) (Fig. 5).59 The drug conjugate can be loaded into the lipid–polymer hybrid nanoparticles via nanoprecipitation. The loading of a drug conjugate system within a polymeric nanocarrier is expected not only to provide a route for improving loading of Pt-based drugs, but also to prevent limitations in anticancer effects arising from the variation of pharmacokinetics and biodistribution of different drugs in cocktail chemotherapy strategies.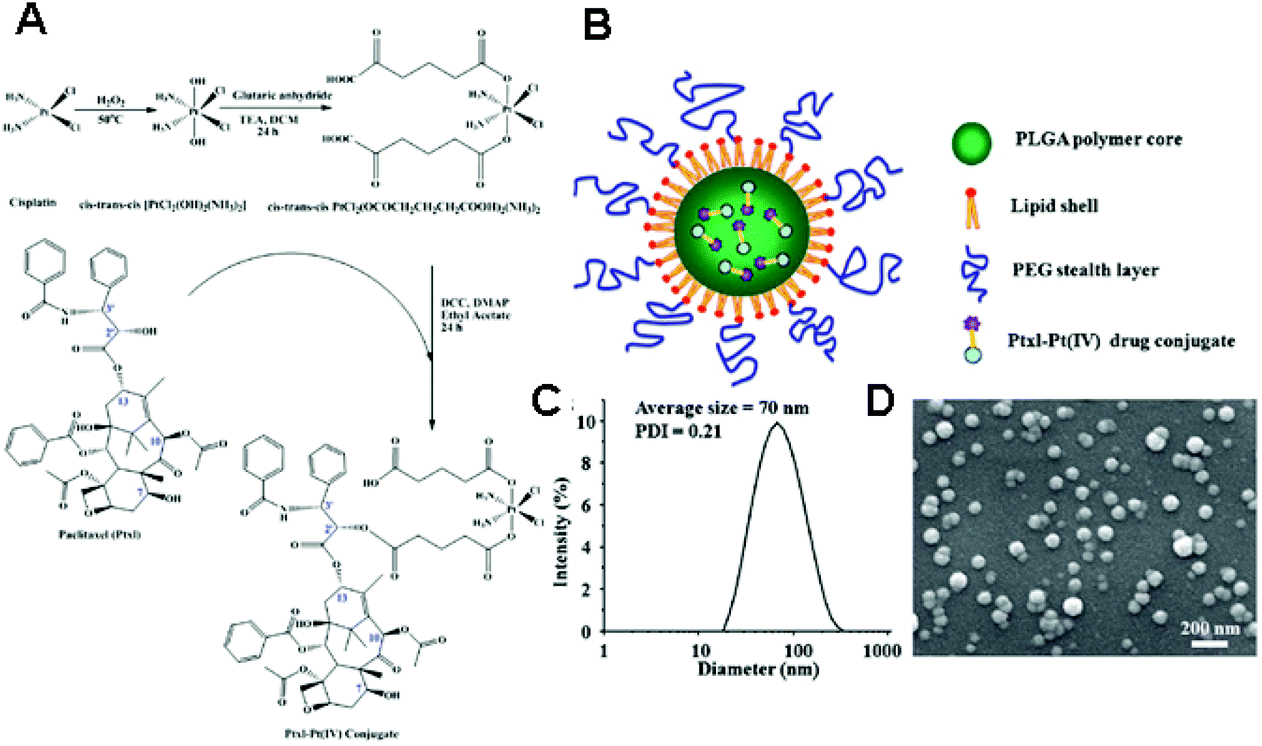 | ||
| Fig. 5 (A) Synthesis of a drug conjugate composed of Ptxl and Pt(IV) prodrugs. (B) Schematic illustration of a Ptxl–Pt(IV) conjugate loaded polymeric nanoparticles. (C) Dynamic light scattering (DLS) analysis and (D) scanning electron microscopy (SEM) images of the nanoparticles containing a Ptxl–Pt(IV) conjugate (reproduced from ref. 59 with permission from the Royal Society of Chemistry). | ||
5. Conjugation of Pt(II)
5.1. Pt(II)-conjugated micelles via labile leaving ligands
Pt(II)-based drugs typically consist of two permanently bound ligands, usually amines, and two labile leaving ligands like chlorides or carboxylate functional groups. The conjugation of Pt(II)-based drugs to a polymeric carrier has been accomplished either via leaving ligands or via permanent ligands. Conjugation via leaving ligands is widely used, as inspired by carboplatin which carries carboxylate as a leaving ligand. It is relatively straightforward to obtain polymer–Pt(II) conjugates structurally similar to carboplatin by conjugating a Pt(II)-based drug to a polymer having pendant carboxyl functionalities.10,37 In addition, monofunctional conjugation, bifunctional conjugation, and/or crosslinking of polymer chains can be obtained upon coordinating a Pt(II)-based drug to a polymer containing carboxylate groups.60,61There have been several notable reports on the development and evaluation of micelles incorporating Pt(II)-based drugs via coordination between carboxylate functional groups of a block copolymer and leaving ligands of the drugs.62–77 For example, Sengupta et al. developed self-assembled micelles by complexation via mono- and di-carboxylate linkages between poly(ethylene glycol)-poly(isobutylene-alt-maleic anhydride) (PEG-PIMA) and diaquated cisplatin. The nano-sized particles exhibited superior antitumor efficiency compared with free cisplatin, owing to the EPR effect.65 Moreover, tumor-targeted micelles for the delivery of Pt(II)-based drugs against specific receptor-positive cell lines have been frequently reported.66–68 For instance, Zhang and Yu's group developed micelles fabricated from folate-conjugated poly(ethylene glycol)-graft-α,β-poly [(N-amino acidyl)-aspartamide] (FA-PEG-g-PAAsp) and Pt(II)-based drugs, and demonstrated their favorable antitumor effects.68 In addition, as a representative example of co-delivery, we discuss the polymeric co-delivery system inspired by the GEMOX chemotherapy regimen, i.e., co-administration of gemcitabine and oxaliplatin.69 Gemcitabine is a nucleoside derivative of deoxycytidine and is involved in the repair and synthesis of DNA. It has been demonstrated that gemcitabine is effective on non-small cell lung cancer, pancreatic cancer, bladder cancer, colon, and breast cancer.69,70 However, the low stability of gemcitabine in vivo requires the administration of high doses, which causes adverse side effects. To overcome these problems, Jing's group developed micelles containing oxaliplatin–polymer (P(Pt)) and gemcitabine–polymer (P(Gem)) conjugates.69 Due to the identical polymer backbones, poly(ethylene glycol)-block-poly(L-lactide-co-2-methyl-2-carboxyl-propylene carbonate) (mPEG-b-P(LA-co-MCC)), P(Pt) and P(Gem) could co-assemble to form hybrid micelles with controlled gemcitabine/oxaliplatin ratios. The co-assembled micelle exhibited enhanced drug accumulation in the tumor site, resulting in the efficient inhibition of tumor growth compared to single-drug administration and co-administration.
Traditionally, poly(aspartic acid), poly(glutamic acid), and poly(methacrylic acid) were employed for generating polymer–Pt(II) complexes. However, these common polymers coordinate to the Pt(II)-based drugs in a non-specific geometry, resulting in uncontrolled crosslinking and mixed configurations such as monodentate or bidentate coordination.71,72 Therefore, Stenzel's group aimed to develop a structurally defined Pt(II)-based drug carrier by investigating the effects of polymer architectures or by using neighboring carboxylate functional groups.71,72 Their analysis confirmed that polymers that form 7-membered rings with Pt enhance the solubility of polymer–Pt(II) conjugates, while those that form 11-membered rings led to low solubility due to inter-polymer crosslinking.71 In addition, it was revealed that statistical copolymers showed complete drug release, in contrast to block copolymers that released around 40% of a Pt-based drug.71 However, it was found that the cytotoxicity was not closely related to the drug release, but was related to the cellular uptake.71 Stenzel's group also tried to develop polymeric micelles with defined geometry by using bidentate carboxylate ligands.72 They demonstrated that the formation of 6-membered rings with Pt provided stability to the complex, and that drug release was increasingly more effective as the hydrophobic block with cisplatin became shorter.72
A major disadvantage of micelles is their instability during blood circulation, due to the unintended contact with blood components and the dilution to below critical micellar concentration (CMC). Covalent crosslinking has attracted significant interest as one of the options for improving the stability of micelles.73,74 This methodology has also been applied to the delivery of Pt(II)-based drugs using micelles.71,75–77 For example, Stenzel's group used 2,2′-(ethylenedioxy)bis(ethylamine) as a crosslinker of micelles, not only to stabilize the micelles, but also to improve the cytotoxic effects of cisplatin by inducing efficient cellular uptake.71 However, permanently stable crosslinkers have a potential risk to limit the drug release and interrupt the renal clearance of the polymeric carriers.76,77 Therefore, Huynh et al. utilized an acid-degradable acetal crosslinker to prepare cisplatin-conjugated micelles composed of poly(oligo(ethylene glycol) methylether methacrylate)-block-poly(N-hydroxysuccinic methacrylate)-block-poly(1,1-di-tert-butyl 3-(2-(methacryloyloxy)ethyl) butane-1,1,3-tricarboxylate) (POEGMEMA-b-PNHSMA-b-PMAETC).76 POEGMEMA occupies the hydrophilic shell, and the PMAETC block is responsible for the complex formation with cisplatin. The hydroxyl succinimide groups in the PNHSMA block react with the diamine of the acetal crosslinker to produce the crosslinked micelle. Qiao's group employed the same strategy to develop pH-sensitive micelles for cisplatin delivery (Fig. 6).77 The linear-brush diblock copolymer contains anhydride groups and grafted PEG, which self-assemble into the micelle. Subsequently, the micelle was crosslinked by the reaction between anhydride groups of diblock copolymers and diamine of the acetal crosslinkers. These two types of pH-sensitive micelles showed a faster drug release under acidic conditions and a higher cellular uptake compared to free drugs and uncrosslinked micelles, thus resulting in enhanced anticancer effects.
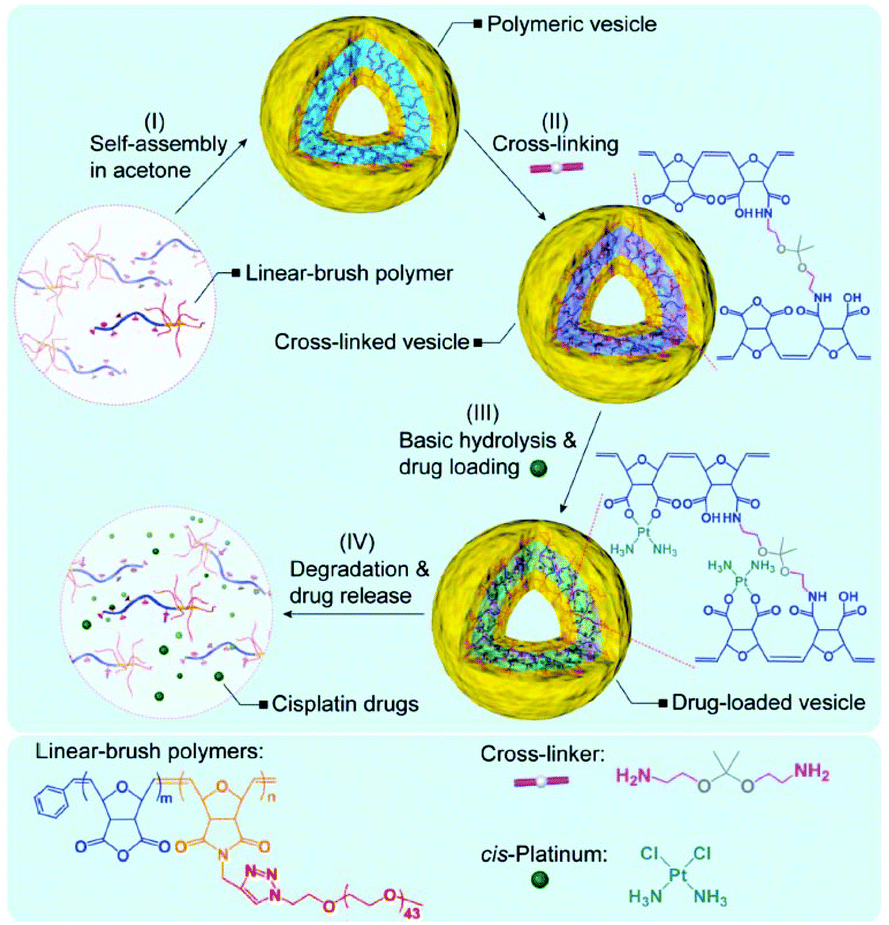 | ||
| Fig. 6 Schematic diagram of the preparation of a Pt(II)-loaded polymeric vesicle with an acid-degradable crosslinker (reproduced from ref. 77 with permission from the Royal Society of Chemistry). | ||
5.2. Metallosomes
Besides the regular spherical micelles, amphiphilic block copolymers can self-assemble into diverse ordered structures, such as worm-like micelles, multilayer structures, and polymersomes. It has been reported that the structure of amphiphilic block copolymers in aqueous solutions is determined by the ratio of hydrophilic block to total polymer mass. The polymersome can be defined as a polymer-based liposome formed from synthetic polymers, rather than lipids or natural polymers, via supramolecular assembly.78–84 Polymersomes are typically hollow spheres consisting of an aqueous core and a polymeric bilayer shell. The shells are composed of three parts: an inner hydrophilic region, a middle hydrophobic part, and an outer hydrophilic part. The polymersomes have attracted considerable attention in the field of drug-delivery systems owing to their superior properties. First, they can encapsulate hydrophilic agents in their aqueous core and integrate hydrophobic drugs within the hydrophobic regions of the shells. In addition, the dense structure of the hollow shell not only provides structural stability to the polymersomes, but also reduces the permeability of the inner components. This reduced permeability can enhance the drug-loading efficiency and reduce the leakage of the drugs during blood circulation. Furthermore, the diverse synthetic routes for block copolymers provide an easy way to tune the properties of the polymersomes. In 2012, Kataoka's group reported a polymersome with a remarkable structure for the delivery of Pt(II)-based drugs (Fig. 7).85 In contrast with general polymersomes formed by hydrophobic or electrostatic interactions, they developed metallosomes by exploiting the coordination of Pt(II)-based drugs. The complex between (1,2-diaminocyclohexane)platinum(II) (DACHPt) (Fig. 1F) and the carboxylic functional groups of Y-shaped block copolymer (PEGasus-b-PLGA-Chole) composed of ω-cholesteroyl-poly(L-glutamic acid) and two-armed poly(ethylene glycol) triggered the metallosome formation. The metallosome showed sustained release of active Pt(II)-based drugs and effective suppression of tumor growth. In addition, by showing the feasibility of loading water-soluble fluorescent molecules into the metallosomes, this strategy shows potential applications to the co-delivery of other therapeutic agents.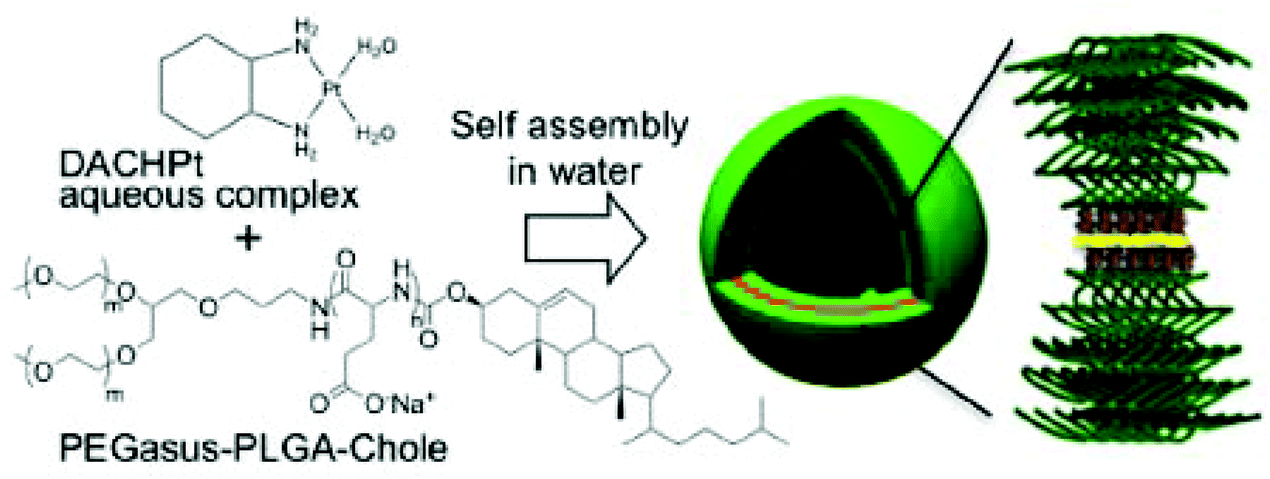 | ||
| Fig. 7 Schematic diagram of the formation of metallosomes via self-assembly of the polymer–metal complex. A Pt in the DACHPt was located between two polymer chains by coordinating with carboxylates in the poly(L-glutamic acid) (PLGA) segment (reproduced from ref. 85 with permission from the American Chemical Society). | ||
5.3. Micelles crosslinked by Pt(II)-based drugs
The ability of Pt(II)-based drugs to coordinate to two carboxylic groups provides a route to crosslink nanoparticles composed of polymers with carboxylic functional groups.62–64,66,67,86–89 That is, conjugation via leaving ligands of Pt(II)-based drugs can perform an important role in stabilizing the nanoparticles, as well as in conjugating the drug to the carrier.60–64,66,67,86–89 Song et al. developed a polymeric micelle using the biocompatible triblock copolymer, methoxy poly(ethylene glycol)-block-poly(L-glutamic acid)-block-poly(L-phenylalanine) (mPEG-b-P(Glu)-b-P(Phe)), for the co-delivery of Ptxl and cisplatin.87 The triblock copolymer was rationally designed: the hydrophobic P(Phe) block is responsible for the Ptxl encapsulation, the P(Glu) segment performs the conjugation of cisplatin and the crosslinking of the micelles, and mPEG in the outer corona can prolong the blood circulation time. It was demonstrated that the crosslinks created via coordination of cisplatin contributed to alleviate the initial burst release of Ptxl from the micelles. In addition, the micelles showed an enhanced anti-tumor efficacy with reduced side effects in vivo. Kim et al. reported an interesting strategy to form micelles containing cisplatin and photodynamic agents.88 They employed cisplatin as a crosslinker among components containing carboxylic functional groups, such as phthalocyanine (DPc) and poly(ethylene glycol)-block-poly(L-aspartic acid) (PEG-b-PLAn). The developed polymer–metal complex micelles (PMCM) formed unimodal nano-sized particles that exhibited high stability in buffer solution. Although the generation of singlet oxygen was confirmed, the synergistic effects of cisplatin and singlet oxygen still need to be investigated. Koseva's group exploited the crosslinking ability of cisplatin to develop a reversible PEGylated strategy.89 A residual leaving ligand of PEG-cisplatin was used as a glue between PEG and the carboxylic functional groups in star-type block copolymers. This strategy, capable of simultaneously loading Pt(II)-based drugs, is expected to control the pharmacological behavior of the drugs and the nanocarriers.5.4. Pt(II)-conjugated nanoparticles via permanently bound ligands
Chemical conjugation via permanently bound ligands has been occasionally employed to obtain polymer–Pt(II) conjugates because of the difficulty in modification of permanent ligands.10,37 In particular, the biodegradability of Pt(II)-conjugated nanoparticles is important in the case of conjugation via permanently bound ligands. According to various studies, it has been hypothesized that the stable macromolecular structures of polymer–Pt(II) conjugates formed via permanent ligands suppress the chemotherapeutic efficiency of the Pt(II)-based drugs.12 Therefore, many research studies have employed acid-degradable linkers for developing polymer–Pt(II) conjugates via permanently bound ligands.In the process of developing and exploiting polymer-caged nanobins (PCNs) formed by crosslinking polymers which are incorporated into bare liposomes, Lee et al. evaluated the ability of PCNs to load and deliver two types of cytotoxic agents, doxorubicin and Pt(II)-based drugs (Fig. 8A).90–92 The doxorubicin could be loaded into the liposomes by hydrophobic interactions. By conjugating the pH-sensitive C6-amine moiety of a cisplatin derivative (PtII(NH3)2(N-AcLys)) (Fig. 8B) to the carboxylic acid in the polymeric shell composed of cholesterol-terminated poly(acrylic acid), the developed PCN also contains a cisplatin derivative in the shell.92 This co-delivery system exhibited higher synergistic cytotoxicity than the combination of either the free drugs or the separately nano-packaged drugs. These results clearly demonstrate that the PCN platform can offer new means for building synergy into combination chemotherapy. Binauld et al. reported a strategy to introduce acid-degradable linkages between the polymer backbone and the permanently bound amine ligands of Pt(II)-based drugs (Fig. 9).93 Their methodology exploited the high stability of the bonds between Pt and permanently bound ligands to enhance the pH responsiveness. The micelle formed by self-assembly of pH-sensitive polymer–Pt(II) conjugates showed an acid-accelerated drug release behavior.
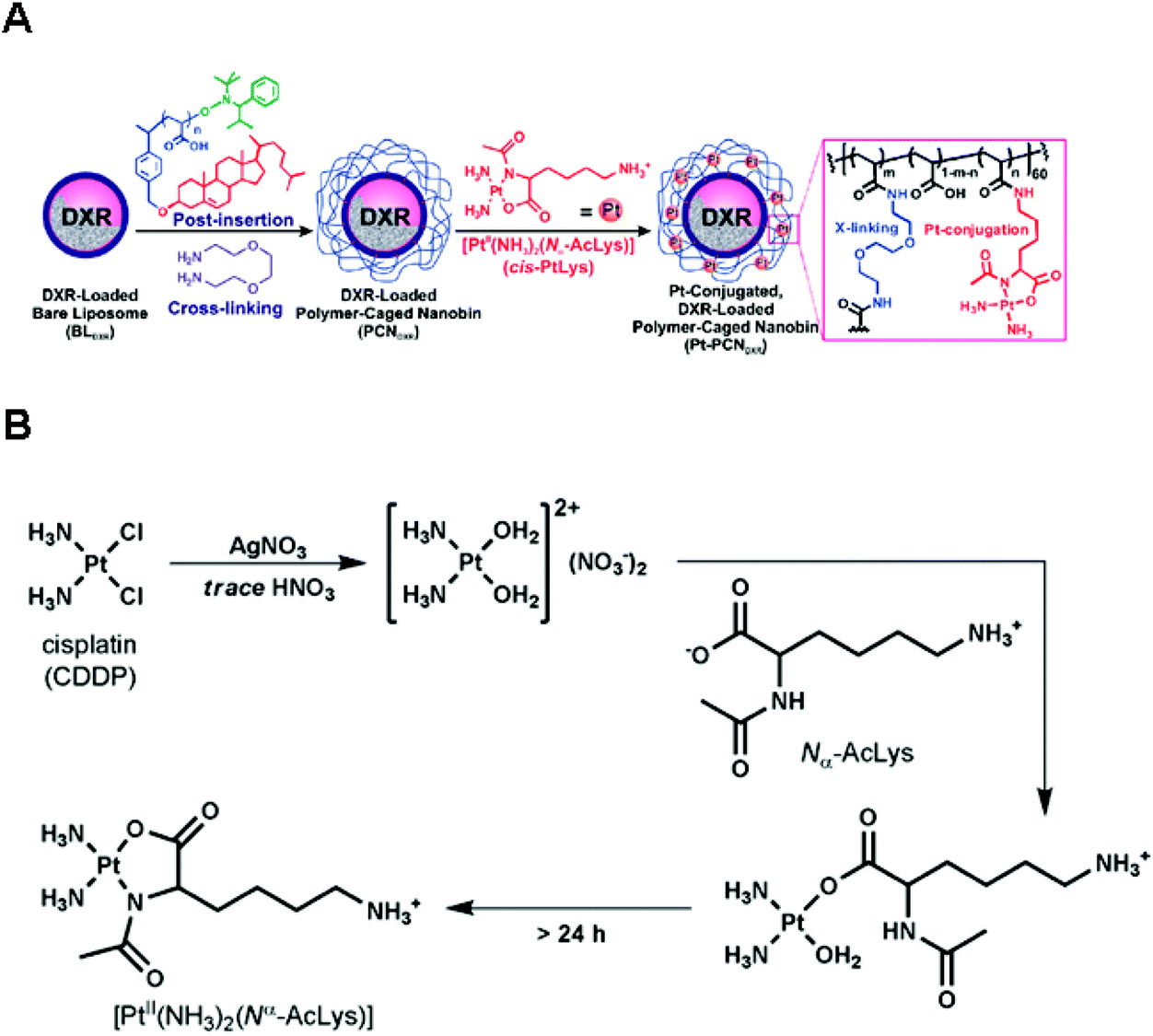 | ||
| Fig. 8 (A) Synthesis of Pt(II)-conjugated and doxorubicin (DXR)-loaded Polymer-Caged Nanobins (PCNs). The DXR-loaded liposomes were coated with polymers which could act as a reservoir for Pt(II) drugs. Polymer shells provide structural stability to liposomes and controlled release of DXR and Pt(II) drugs. (B) Synthetic scheme of [PtII(NH3)2(Nα-AcLys)] (reproduced from ref. 92 with permission from the American Chemical Society). | ||
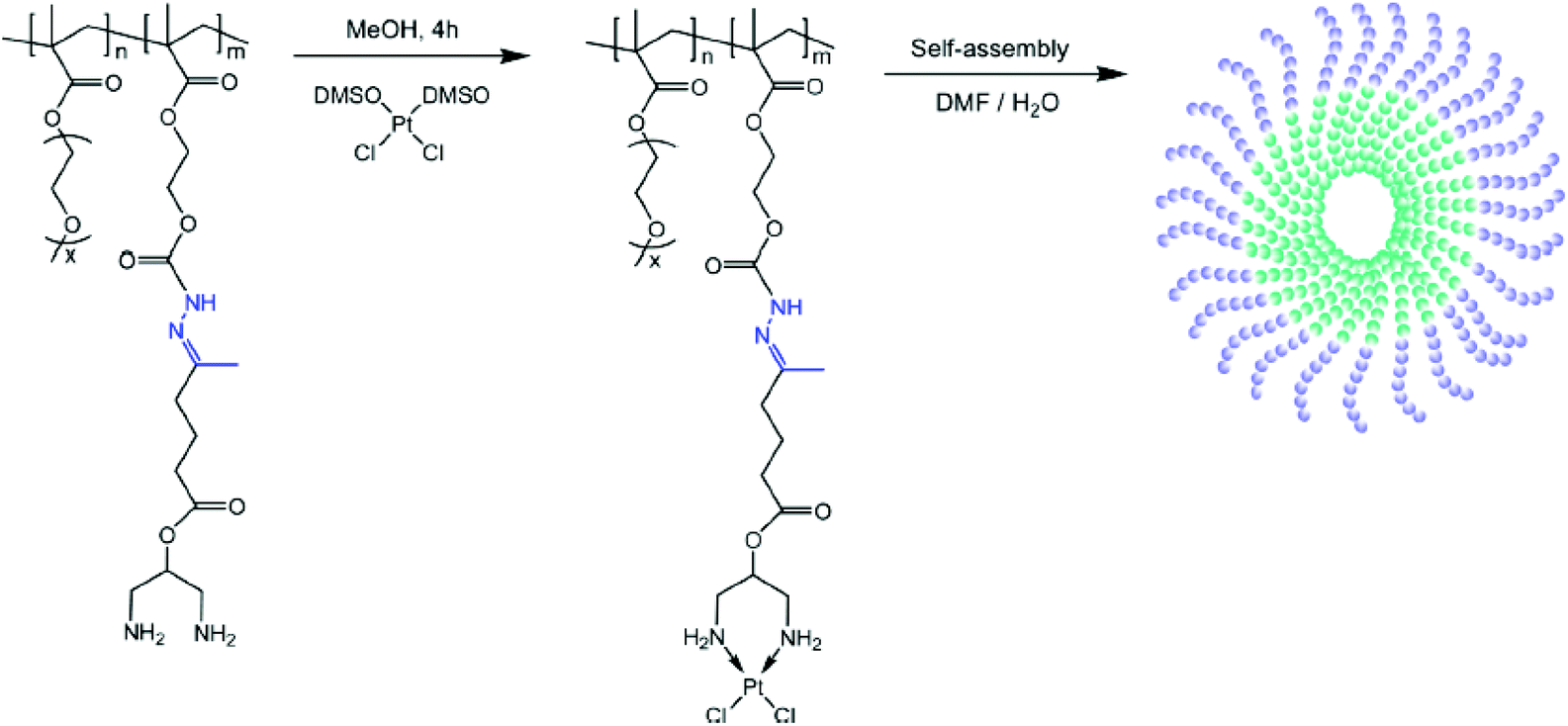 | ||
| Fig. 9 Preparation of the hydrazone copolymer–Pt(II) complex and illustration of micelles through self-assembly of polymer–drug conjugates. The hydrazone bond was cleavable under acidic conditions, resulting in the release of Pt(II) (reproduced from ref. 93 with permission from the American Chemical Society). | ||
6. Conjugation of Pt(IV)
6.1. Traditional strategies with Pt(IV)–polymer conjugates
The axial ligands of Pt(IV) complexes provide the drugs an opportunity to be encapsulated efficiently into the nanoparticles via traditional and simple methods.32,37–39,55 In addition, combination chemotherapy has emerged as a possible solution for drug resistance, due to its ability to interrupt one or more cellular mechanisms related to apoptosis and survival.94,95 In particular, a prodrug comprised of Pt(IV)-based drugs and polymers can contribute to effectively encapsulate other anticancer agents, as well as Pt(IV)-based drugs, into the nanoparticles developed by nanoprecipitation. Mi et al. developed a strategy for targeted co-delivery of a Pt(IV) prodrug and docetaxel.94D-α-Tocopheryl-co-poly(ethylene glycol) 1000 succinate (TPGS), which has a water-soluble amphiphilic structure, was employed to incorporate a Pt(IV) prodrug. TPGS-COOH, the TPGS-Pt(IV) prodrug, and PLA-TPGS were nanoprecipitated with docetaxel to form the nanoparticles (HTCP-NP) for combination chemotherapy. Herceptin, which targets the HUR-2 receptor overexpressed in breast cancer, could react with carboxylic groups exposed on the HTCP-NP surface. This approach can be applied to the targeted co-delivery of various hydrophilic and hydrophobic drugs. Kolishetti et al. illustrated a platform technology for combination chemotherapy involving a Pt(IV)–polymer conjugate and the microfluidic nanoprecipitation method.95 The Pt(IV)-based drugs were tethered to the pendant hydroxyl group of a hydrophobic poly(lactide) derivative, followed by the formation of nanoparticles with docetaxel and the PEG-b-PLGA block copolymer through the microfluidic nanoprecipitation method. This platform technology provides excellent drug encapsulation efficiency and reproducibility. In addition, the introduction of the A10 aptamer supplemented the nanoparticles with the ability to target PSMA-overexpressed prostate cancer cells. PSMA-targeted nanoparticles encapsulating a Pt(IV) prodrug and docetaxel showed enhanced cytotoxicity compared to nanoparticles containing the individual single drugs. This approach showed significant potential for providing conventional methods capable of developing uniform nanoparticles for a targeted co-delivery system incorporating Pt-based drugs.6.2. Pt(IV)-conjugated micelles
In addition to providing hydrophobicity to Pt-based drugs, the axial ligands of Pt(IV) complexes can enable the conjugation of the drug directly with the nanoparticles,32,37–39 which would allow the successful and efficient incorporation of Pt-based drugs into the micelles. Stenzel's group demonstrated the ability of a polymeric micelle containing oxoplatin and folate ligands to deliver the Pt(IV)-based drug into folate receptor-overexpressed ovarian cancer cells (Fig. 10).96 In this system, oxoplatin was successfully loaded into the micelles via an ester linkage between the carboxyl group of hydrophobic segments of the amphiphilic block copolymer and the hydroxyl group of oxoplatin. The micelle was designed to release cisplatin by hydrolysis of the ester linkage and subsequent reduction of oxoplatin in the cytosol. Folate ligands were introduced to the ends of hydrophilic segments through the interaction between phenylboronic acid and dopamine. This simple chemistry enables easy control of the amount of folate ligands on the micelle, which affects the IC50 value of oxoplatin. In addition, the micellar core was crosslinked to provide structural stability to the micelle, allowing the drugs to enter the cell efficiently.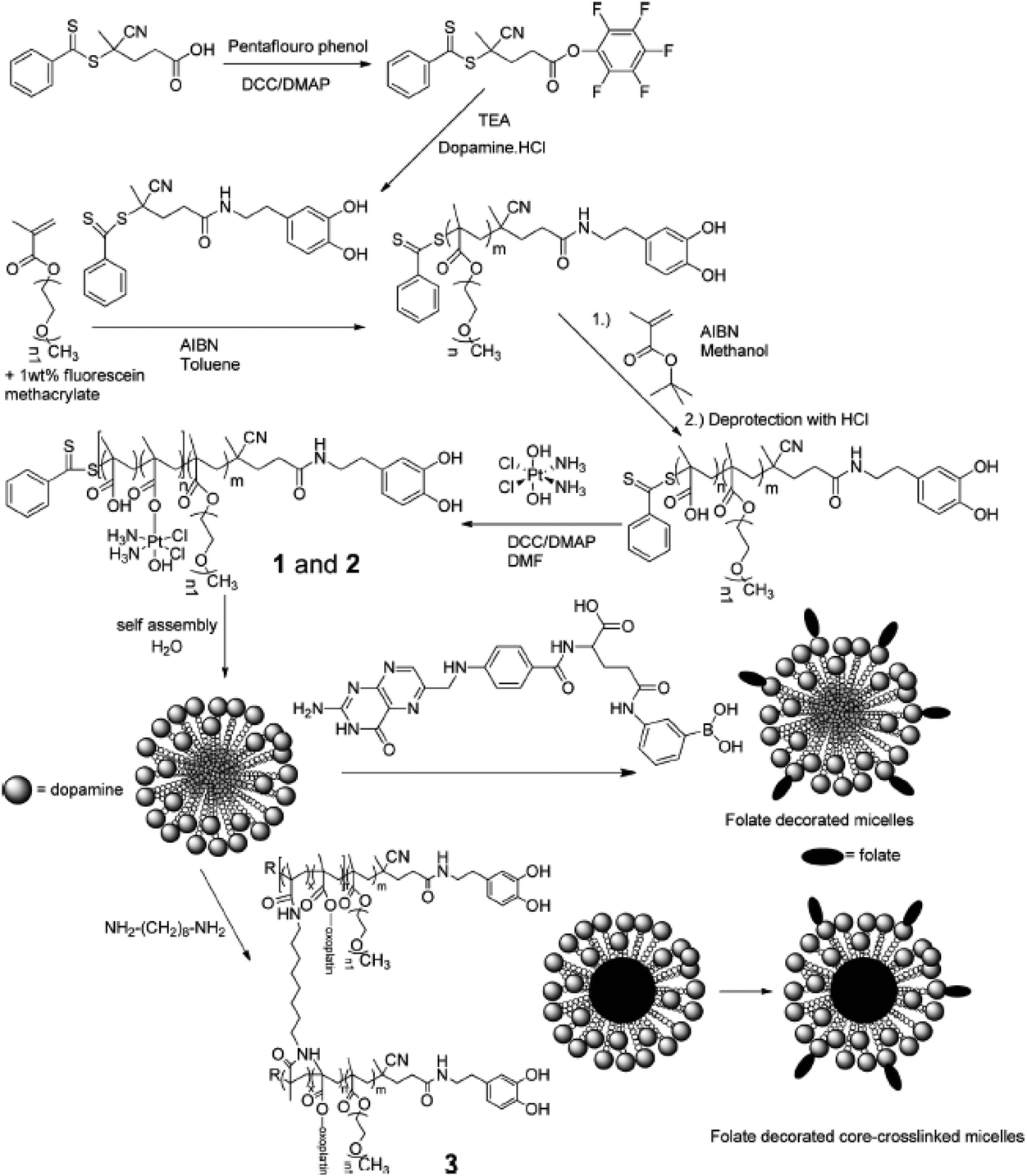 | ||
| Fig. 10 Synthesis of Pt(IV) prodrug attached poly(oligo(ethylene glycol) methyl ether methacrylate)-b-poly(methacrylic acid) block copolymers (p(OMGMEMA)-b-p(MMA)) with a pendant dopamine group. After self-assembling, the folate ligands were introduced on the surface of micelles via dopamine–phenylboronic ester interaction and the cores of micelles were crosslinked for structural stability (reproduced from ref. 96 with permission from the American Chemical Society). | ||
Although it has been reported that Pt(IV)-based drugs are activated by the cellular environments, the drug efficacy can be limited due to the dependency of the drug release on indirect processes such as acid hydrolysis and/or reduction.97–100 Following a high demand for systems capable of releasing Pt(II) complexes directly, Sadler and co-workers reported a photosensitive Pt(IV)–diazido complex which is converted to Pt(II) species under mild UV irradiation.97,98 The UV irradiation breaks the Pt–N3 bond, resulting in the release of the Pt(II) complex.97,98 Based on the photosensitive characteristics of the Pt(IV)–diazido complex, polymeric micelles which showed light-responsive release of active Pt(II)-based drugs have been developed by conjugating the Pt(IV)–diazido complex to polymers via an ester or amide linkage.99,100 The micelles showed rapid release of Pt(II) species under UV irradiation, whereas they were stable under dark conditions. These micelles also exhibited enhanced cytotoxicity under UV irradiation, as well as inhibition of tumor growth in vivo, with reduced side effects.
Li et al. reported multi-stage Pt(IV) prodrug-delivery systems mimicking the common polymeric gene-delivery systems.101 The mono-carboxylated Pt(IV) complexes were conjugated with cationic dendrimers, and the as-synthesized cationic prodrugs formed polyplex micelles with the anionic diblock copolymer. According to the design, the cationic Pt(IV)–dendrimer conjugates are dissociated from the polyplex micelles due to the charge conversion of anionic segments of the diblock copolymer at tumoral acidic pH (∼6.0). Subsequently, cisplatin can be released from Pt(IV)–dendrimer conjugates by the reductive environment in the cytosol. The polyplex micelles showed an ability for deep penetration in tumor tissue and enhanced anticancer effects compared to cisplatin.
6.3. Pt(IV)-conjugated micelles for co-delivery
Approaches involving drug-conjugated micelles have attracted considerable attention for the development of co-delivery systems. Unlike the physical encapsulation strategies, conjugated micelle systems provide Pt(IV)-based drugs and other combinatorial drugs with structural stability and ability of controlled release. Xiao et al. investigated micelles for co-delivery of daunomycin (DRB) and oxaliplatin.102 The DRB and the prodrug of oxaliplatin were attached to a biodegradable amphiphilic polymer, and the resulting two types of drug–polymer conjugates were co-assembled into the micelle. The different linkages between each drug and the polymer have a different effect on the drug release. The polymer–Pt(IV) conjugate exhibited rapid release of the Pt(II) complex obtained by reduction, while the polymer–DRB conjugate showed sustained release due to acid hydrolysis. In addition, the micelles showed reduced systemic toxicity and synergistic effects both in vivo and in vitro.The two axial ligands of Pt(IV)-based drugs can be used for incorporating additional functionalities. For example, if one ligand contains agents for combination therapy and the other is conjugated with the nanocarriers, a co-delivery system with synergistic effects, enhanced stability, and passive targeting ability can be developed. Inspired by mitaplatin (Fig. 1G), which contains two dichloroacetate (DCA) ligands in the axial positions of cisplatin, Xiao et al. developed micelles containing a Pt(IV) prodrug with a DCA ligand and a carboxylic group in its axial positions.103 DCA is a mitochondria-targeting small molecule which inhibits glycolysis, enhancing the mitochondrial apoptosis in cancer and suppressing tumor growth. The carboxylic group at the axial positions of a DCA–Pt(IV) conjugate was linked to an amphiphilic biodegradable block copolymer (methoxy poly(ethylene glycol)-block-poly(caprolactone)-block-poly(L-lysine), mPEG-b-PCL-b-PLL) for the formation of micelles. The enhanced anticancer effects of this polymeric delivery system might be attributed to the individual pharmacological mechanism of each anticancer drug. Qi et al. described a similar Pt(IV) prodrug delivery system.104 Camphor anhydride is a derivative of camphor that can down-regulate the intracellular levels of Bcl-2 expression, enabling to overcome the resistance to cisplatin. Conjugation of camphor anhydride with one of the axial positions of Pt(IV) prodrugs generated camplatin (Fig. 1H) and the other axial position was further linked to biocompatible mPEG-b-PCL-b-PLL. The as-prepared polymer–camplatin conjugate could self-assemble into micelles. The micelles exhibited enhanced cytotoxicity on both cisplatin-sensitive and cisplatin-resistant cell lines compared to free cisplatin, because of the synergistic effects of the cytotoxic Pt-based drug and the camphoric acid chemosensitizer.
6.4. Micelles crosslinked by Pt(IV) prodrugs
In addition to providing functional moieties that can be modified with other therapeutic agents, the two axial ligands can act as crosslinkers among polymers, leading to the formation and stabilization of the micelle structures. By taking advantage of this characteristic, Aryal et al. reported a pH-responsive delivery system, with the Pt(IV) prodrug employed as both an anticancer agent and a crosslinker between diblock copolymers.105 In this system, the Bi(PEG-PLA)–Pt(IV) conjugate was obtained by crosslinking two PEG-b-PLA block copolymers by one Pt(IV) prodrug via pH-responsive hydrazone bonds (Fig. 11). The amphiphilic Bi(PEG-PLA)–Pt(IV) could self-assemble into micelles. This pH-responsive micelle showed a rapid release of cisplatin in acidic pH, which contributed to the increased cytotoxicity compared to free cisplatin. The strategy involving the use of a Pt(IV) prodrug as a crosslinker was extended to develop shell- or core-crosslinked micelles for stabilizing the micelle structure and preventing the premature release of the drug. Song et al. reported shell-crosslinked mPEG-b-PCL-b-PLL micelles based on a dicarboxyl Pt(IV) prodrug.106 Two axial succinic moieties of the Pt(IV) prodrug could react via EDC/NHS chemistry with free amine groups in the PLL segment occupying the hydrophilic shell of the micelles. Compared with free cisplatin and prodrugs, shell-crosslinked micelles not only showed a sustained release of cisplatin with enhanced cytotoxicity in vitro, but also exhibited selective accumulation into tumor sites in vivo.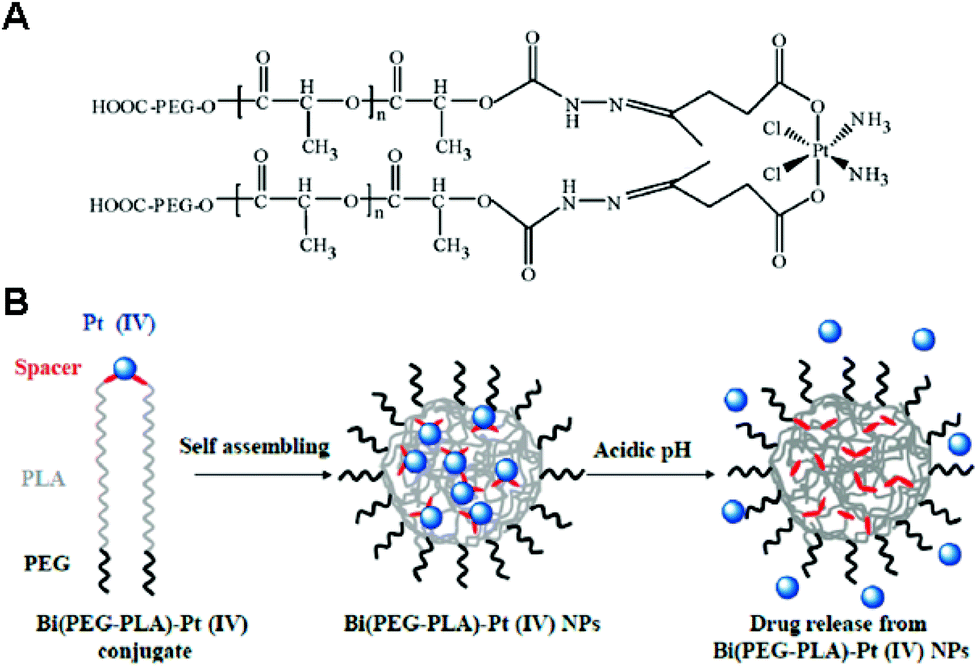 | ||
| Fig. 11 (A) Structure of the Bi(PEG-PLA)-Pt(IV) polymer–prodrug conjugate. (B) Schematic illustration of the preparation and operation of the pH-responsive Pt(IV) delivery system. Bi(PEG-PLA)-Pt(IV) NPs were formed through self-assembly of polymer–drug conjugates. Under acidic conditions, the hydrazine bonds were hydrolyzed, resulting in the rapid release of cisplatin (reproduced from ref. 105 with permission from the American Chemical Society). | ||
7. Conclusions
Since the successful clinical transfer of cisplatin and oxaliplatin, various Pt-based drugs have been developed and they have shown significant antitumor effects in vitro. Nevertheless, most free Pt-based drugs have failed to enter clinical use due to the severe side effects and lower activity than cisplatin. Polymeric nanoparticles have emerged as one of the alternative strategies for overcoming the disadvantages of bare Pt-based drugs. For example, the NC-6004 micelle comprised of cisplatin-conjugated PEG-b-poly(glutamic acid) (PEG-b-P(Glu)) was developed by Kataoka's group and has shown reduced nephrotoxicity and improved antitumor efficacy in pancreatic cancer. This is currently in a Phase III clinical trial.10–12,107 Inspired by the successful development of NC-6004, NC-4016 was also developed by conjugating DACHPt with the PEG-b-P(Glu). NC-4016 has also shown potential therapeutic efficacy in various tumor models and is currently in a Phase I clinical trial.10–12,108,109 ProlindacTM (AP5346) is one of the polymer-Pt(II) conjugates, which contains DACHPt conjugated with N-(2-hydroxypropyl)methacrylamide (HPMA) via a pH-sensitive coordinating group. This is currently in Phase II clinical studies for advanced ovarian cancer.10,12,110 In addition to the polymeric delivery systems, various liposomal nanoparticles containing Pt-based drugs have been reported for the clinical trials.10,111–113 The increasing number of Pt-based drug delivery systems under clinical evaluation have motivated many researchers to develop a more advanced strategy for effective anticancer therapy.In this review, we have summarized and discussed the progress attained in the development of polymeric nanoparticles for effective delivery of Pt-based drugs, focusing on drug configurations and incorporation methods. Whereas micelles have been the most employed systems in the past five years, standard techniques such as desolvation, emulsion, and nanoprecipitation have also been frequently used for the delivery of Pt-based drugs. Crosslinking strategies and integration of targeting ligands have also been employed to improve the loading efficiency and the tumor accumulation of drugs. Co-delivery of other therapeutic agents or chemosensitizers has provided a method for overcoming the resistance to cisplatin. In particular, unique polymeric nanoparticles like metallosomes and PCN have been developed as potential routes to improve anticancer effects and mitigate side effects. Most strategies described in this review offer rational alternatives to existing conventional methods. These efforts are expected to pave the way for the practical advancement and clinical use of Pt-based anticancer therapy.
Acknowledgements
This work was supported by the Research Center Program of IBS (Institute for Basic Science) (CA1203-02) and the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2013R1A1A1076136) in Korea.Notes and references
- A. S. Abu-Surrah and M. Kettunen, Curr. Med. Chem., 2006, 13, 1337–1357 CrossRef CAS.
- X. Wang and Z. Guo, Dalton Trans., 2008, 1521–1532 RSC.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573–584 CrossRef CAS PubMed.
- M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2261 CrossRef CAS.
- S. P. Fricker, Dalton Trans., 2007, 4903–4917 RSC.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9–23 CrossRef CAS PubMed.
- A. A. Argyriou, P. Polychronopoulos, G. Iconomou, E. Chroni and H. P. Kalofonos, Cancer Treat. Rev., 2008, 34, 368–377 CrossRef CAS PubMed.
- S. R. McWhinney, R. M. Goldberg and H. L. McLeod, Mol. Cancer Ther., 2009, 8, 10–16 CrossRef CAS PubMed.
- X. Yao, K. Panichpisal, N. Kurtzman and K. Nugent, Am. J. Med. Sci., 2007, 334, 115–124 CrossRef PubMed.
- H. S. Oberoi, N. V. Nukolova, A. V. Kabanov and T. K. Bronich, Adv. Drug Delivery Rev., 2013, 65, 1667–1685 CrossRef CAS PubMed.
- H. Cabral and K. Kataoka, J. Controlled Release, 2014, 190, 465–476 CrossRef CAS PubMed.
- M. Callari, J. R. Aldrich-Wright, P. L. de Souza and M. H. Stenzel, Prog. Polym. Sci., 2014, 39, 1614–1643 CrossRef CAS PubMed.
- K. Maruyama, Adv. Drug Delivery Rev., 2011, 63, 161–169 CrossRef CAS PubMed.
- A. K. Iyer, G. Khaled, J. Fang and H. Maeda, Drug Discovery Today, 2006, 11, 812–818 CrossRef CAS PubMed.
- V. Torchilin, Adv. Drug Delivery Rev., 2011, 63, 131–135 CrossRef CAS PubMed.
- T. Lammers, W. E. Hennink and G. Storm, Br. J. Cancer, 2008, 99, 392–397 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Preat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- J. D. Byrne, T. Betancourt and L. Brannon-Peppas, Adv. Drug Delivery Rev., 2008, 60, 1615–1626 CrossRef CAS PubMed.
- F. Marcucci and F. Lefoulon, Drug Discovery Today, 2004, 9, 219–228 CrossRef CAS.
- K. Kataoka, A. Harada and Y. Nagasaki, Adv. Drug Delivery Rev., 2001, 47, 113–131 CrossRef CAS.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1–20 CrossRef CAS.
- T. M. Allen and P. R. Cullis, Adv. Drug Delivery Rev., 2013, 65, 36–48 CrossRef CAS PubMed.
- E. R. Gillies and J. M. J. Frechet, Drug Discovery Today, 2005, 10, 35–43 CrossRef CAS.
- B. Ahn, J. Park, K. Singha, H. Park and W. J. Kim, J. Mater. Chem. B, 2013, 1, 2829–2836 RSC.
- T. L. Doane and C. Burda, Chem. Soc. Rev., 2012, 41, 2885–2911 RSC.
- R. A. Alderden, M. D. Hall and T. W. Hambley, J. Chem. Educ., 2006, 83, 728–734 CrossRef CAS.
- D. Gibson, Dalton Trans., 2009, 10681–10689 RSC.
- A. I. Ivanov, J. Christodoulou, J. A. Parkinson, K. J. Barnham, A. Tucker, J. Woodrow and P. J. Sadler, J. Biol. Chem., 1998, 273, 14721–14730 CrossRef CAS PubMed.
- R. C. DeConti, B. R. Toftness, R. C. Lange and W. A. Creasey, Cancer Res., 1973, 33, 1310–1315 CAS.
- D. Wang and S. J. Lippard, Nat. Rev. Drug Discovery, 2005, 4, 307–320 CrossRef CAS PubMed.
- M. Ohmichi, J. Hayakawa, K. Tasaka, H. Kurachi and Y. Murata, Trends Pharmacol. Sci., 2005, 26, 113–116 CrossRef CAS PubMed.
- M. D. Hall and T. W. Hambley, Coord. Chem. Rev., 2002, 232, 49–67 CrossRef CAS.
- D. Fink, S. Nebel, S. Aebi, H. Zheng, B. Cenni, A. Nehme, R. D. Christen and S. B. Howell, Cancer Res., 1996, 56, 4881–4886 CAS.
- E. Raymond, S. Faivre, S. Chaney, J. Woynarowski and E. Cvitkovic, Mol. Cancer Ther., 2002, 1, 227–235 CAS.
- Y.-P. Ho, S. C. F. Au-Yeung and K. K. W. To, Med. Res. Rev., 2003, 23, 633–655 CrossRef CAS PubMed.
- P. Perego, C. Caserini, L. Gatti, N. Carenini, S. Romanelli, R. Supino, D. Colangelo, I. Viano, R. Leone, S. Spinelli, G. Pezzoni, C. Manzotti, N. Farrell and F. Zunino, Mol. Pharmacol., 1999, 55, 528–534 CAS.
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451–2466 CrossRef CAS PubMed.
- M. D. Hall, H. R. Mellor, R. Callaghan and T. W. Hambley, J. Med. Chem., 2007, 50, 3403–3411 CrossRef CAS PubMed.
- E. Gabano, M. Ravera and D. Osella, Dalton Trans., 2014, 43, 9813–9820 RSC.
- S. R. Croy and G. S. Kwon, Curr. Pharm. Des., 2006, 12, 4669–4684 CrossRef CAS.
- C. P. Reis, R. J. Neufeld, A. J. Ribeiro and F. Veiga, Nanomedicine, 2006, 2, 8–21 CrossRef CAS PubMed.
- S. Sundar, J. Kundu and S. C. Kundu, Sci. Technol. Adv. Mater., 2010, 11, 014104 CrossRef.
- S. Vrignaud, J.-P. Benoit and P. Saulnier, Biomaterials, 2011, 32, 8593–8604 CrossRef CAS PubMed.
- S. Hornig, T. Heinze, C. R. Becer and U. S. Schubert, J. Mater. Chem., 2009, 19, 3838–3840 RSC.
- N. Dixit, K. Vaibhav, R. S. Pandey, U. K. Jain, O. P. Katare, A. Katyal and J. Madan, Biomed. Pharmacother., 2015, 69, 1–10 CrossRef CAS PubMed.
- A. C. Jayasuriya and A. J. Darr, J. Biomed. Sci. Eng., 2013, 6, 586–592 CrossRef CAS.
- Y. Wang, P. Liu, L. Qui, Y. Sun, M. Zhu, L. Gu, W. Di and Y. Duan, Biomaterials, 2013, 34, 4068–4077 CrossRef CAS PubMed.
- H. Chen, W. He and Z. Guo, Chem. Commun., 2014, 50, 9714–9717 RSC.
- A. L. Harris, Nat. Rev. Cancer, 2002, 2, 38–47 CrossRef CAS PubMed.
- A. Babu, Q. Wang, R. Muralidharan, M. Shanker, A. Munshi and R. Ramech, Mol. Pharmaceutics, 2014, 11, 2720–2733 CrossRef CAS PubMed.
- S. Guo, C. M. Lin, Z. Xu, L. Miao, Y. Wang and L. Huang, ACS Nano, 2014, 8, 4996–5009 CrossRef CAS PubMed.
- J. Gong, M. Chen, Y. Zheng, S. Wang and Y. Wang, J. Controlled Release, 2012, 159, 312–323 CrossRef CAS PubMed.
- K. Miyata, R. J. Christie and K. Kataoka, React. Funct. Polym., 2011, 71, 227–234 CrossRef CAS PubMed.
- V. B. Jadhav, Y. J. Jun, J. H. Song, M. K. Park, J. H. Oh, S. W. Chae, I.-S. Kim, S.-J. Choi, H. J. Lee and Y. S. Sohn, J. Controlled Release, 2010, 147, 144–150 CrossRef CAS PubMed.
- S. Dhar, F. X. Gu, R. Langer, O. C. Farokhzad and S. J. Lippard, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17356–17361 CrossRef CAS PubMed.
- N. Graf, D. R. Bielenberg, N. Kolishetti, C. Muus, J. Banyard, O. C. Farokhzad and S. J. Lippard, ACS Nano, 2012, 6, 4530–4539 CrossRef CAS PubMed.
- S. Dhar, N. Kolishetti, S. J. Lippard and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1850–1855 CrossRef CAS PubMed.
- X. Xu, K. Xie, X.-Q. Zhang, E. M. Pridgen, G. Y. Park, D. S. Cui, J. Shi, J. Wu, P. W. Kantoff, S. J. Lippard, R. Langer, G. C. Walker and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 18638–18643 CrossRef CAS PubMed.
- S. Aryal, C.-M. Jack Hu, V. Fu and L. Zhang, J. Mater. Chem., 2012, 22, 994–999 RSC.
- N. Nishiyama, S. Okazaki, H. Cabral, M. Miyamoto, Y. Kato, Y. Sugiyama, K. Nishio, Y. Matsumura and K. Kataoka, Cancer Res., 2003, 63, 8977–8983 CAS.
- L. L. Komane, E. H. Mukaya, E. W. Neuse and C. E. J. van Rensburg, J. Inorg. Organomet. Polym., 2008, 18, 111–123 CrossRef CAS.
- Y. Xiong, W. Jiang, Y. Shen, H. Li, C. Sun, A. Ouahab and J. Tu, Biomaterials, 2012, 33, 7182–7193 CrossRef CAS PubMed.
- Z. Ahmad, Z. Tang, A. Shah, S. Lv, D. Zhang, Y. Zhang and X. Che, Macromol. Biosci., 2014, 14, 1337–1345 CrossRef CAS PubMed.
- F. Zhang, M. Elsabahy, S. Zhang, L. Y. Lin, J. Zou and K. L. Wooley, Nanoscale, 2013, 5, 3220–3225 RSC.
- A. Paraskar, S. Soni, S. Basu, C. J. Amarasiriwardena, N. Lupoli, S. Srivats, R. S. Roy and S. Sengupta, Nanotechnology, 2011, 22, 265101 CrossRef PubMed.
- N. V. Nukolova, H. S. Oberoi, Y. Zhao, V. P. Chekhonin, A. V. Kabanov and T. K. Bronich, Mol. Pharmaceutics, 2013, 10, 3913–3921 CAS.
- M. Li, Z. Tang, Y. Zhang, S. Lv, H. Yu, D. Zhang, H. Hong and X. Chen, J. Mater. Chem. B, 2014, 2, 3490–3499 RSC.
- Y. Xue, X. Tang, J. Huang, X. Zhang, J. Yu, Y. Zhang and S. Gui, Colloids Surf., B, 2011, 85, 280–288 CrossRef CAS PubMed.
- H. Song, H. Xiao, M. Zheng, R. Qi, L. Yan and X. Jing, J. Mater. Chem. B, 2014, 2, 6560–6570 RSC.
- C. M. Galmarini, J. R. Mackey and C. Dumontet, Lancet Oncol., 2002, 3, 415–424 CrossRef CAS.
- V. T. Huynh, G. Chen, P. de Souza and M. H. Stenzel, Biomacromolecules, 2011, 12, 1738–1751 CrossRef CAS PubMed.
- V. T. Huynh, P. de Souza and M. H. Stenzel, Macromolecules, 2011, 44, 7888–7900 CrossRef CAS.
- A. Rosler, G. W. M. Vandermeulen and H. A. Klok, Adv. Drug Delivery Rev., 2012, 64, 270–279 CrossRef PubMed.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- J. Peng, T. Qi, J. Liao, B. Chu, Q. Yang, W. Li, Y. Qu, F. Luo and Z. Qian, Biomaterials, 2013, 34, 8726–8740 CrossRef CAS PubMed.
- V. T. Huynh, S. Binauld, P. L. de Souza and M. H. Stenzel, Chem. Mater., 2012, 24, 3197–3211 CrossRef CAS.
- Q. Fu, J. Xu, K. Ladewig, T. M. A. Henderson and G. G. Qiao, Polym. Chem., 2015, 6, 35–43 RSC.
- B. M. Discher, Y.-Y. Won, D. S. Ege, J. C.-M. Lee, F. S. Bates, D. E. Discher and D. A. Hammer, Science, 1999, 284, 1143–1146 CrossRef CAS.
- D. E. Discher and F. Ahmed, Annu. Rev. Biomed. Eng., 2006, 8, 323–341 CrossRef CAS PubMed.
- D. E. Discher, V. Ortiz, G. Srinivas, M. L. Klein, Y. Kim, D. Christian, S. Cai, P. Photos and F. Ahmed, Prog. Polym. Sci., 2007, 32, 838–857 CrossRef CAS PubMed.
- D. H. Levine, P. P. Ghoroghchian, J. Freudenberg, G. Zhang, M. J. Therien, M. I. Greene, D. A. Hammer and R. Murali, Methods, 2008, 46, 25–32 CrossRef CAS PubMed.
- O. Onaca, R. Enea, D. W. Hughes and W. Meier, Macromol. Biosci., 2009, 9, 129–139 CrossRef CAS PubMed.
- H. K. Cho, I. W. Cheong, J. M. Lee and J. H. Kim, Korean J. Chem. Eng., 2010, 27, 731–740 CrossRef CAS PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto, Y. Anraku, A. Kishimura, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2012, 134, 13172–13175 CrossRef CAS PubMed.
- R. Wang, X. Hu, H. Xiao, Z. Xie, Y. Huang and X. Jing, J. Mater. Chem. B, 2013, 1, 744–748 RSC.
- W. Song, Z. Tang, M. Li, S. Lv, H. Sun, M. Deng, H. Liu and X. Chen, Acta Biomater., 2014, 10, 1392–1402 CrossRef CAS PubMed.
- J. Kim, H.-J. Yoon, S. Kim, K. Wang, T. Ishii, Y.-R. Kim and W.-D. Jang, J. Mater. Chem., 2009, 19, 4627–4631 RSC.
- E. Stoyanova, V. Mitova, P. Shestakova, A. Kowalczuk, G. Momekov, D. Momekova, A. Marcinkowski and N. Koseva, J. Inorg. Biochem., 2013, 120, 54–62 CrossRef CAS PubMed.
- S.-M. Lee, H. Chen, C. M. Dettmer, T. V. O'Halloran and S. T. Nguyen, J. Am. Chem. Soc., 2007, 129, 15096–15097 CrossRef CAS PubMed.
- S.-M. Lee, H. Chen, T. V. O'Halloran and S. T. Nguyen, J. Am. Chem. Soc., 2009, 131, 9311–9320 CrossRef CAS PubMed.
- S.-M. Lee, T. V. O'Halloran and S. T. Nguyen, J. Am. Chem. Soc., 2010, 132, 17130–17138 CrossRef CAS PubMed.
- S. Binauld, W. Scarano and M. H. Stenzel, Macromolecules, 2012, 45, 6989–6999 CrossRef CAS.
- Y. Mi, J. Zhao and S.-S. Feng, J. Controlled Release, 2013, 169, 185–192 CrossRef CAS PubMed.
- N. Kolishetti, S. Dhar, P. M. Valencia, L. Q. Lin, R. Karnik, S. J. Lippard, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 17939–17944 CrossRef CAS PubMed.
- W. Scarano, H. T. T. Duong, H. Lu, P. L. De Souza and M. H. Stenzel, Biomacromolecules, 2013, 14, 962–975 CrossRef CAS PubMed.
- P. J. Bednarski, F. S. Mackay and P. J. Sadler, Anticancer Agents Med. Chem., 2007, 7, 75–93 CrossRef CAS.
- F. S. Mackay, J. A. Woods, P. Heringová, J. Kašpárková, A. M. Pizarro, S. A. Moggach, S. Parsons, V. Brabec and P. J. Sadler, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20743–20748 CrossRef CAS PubMed.
- H. Xiao, G. T. Noble, J. F. Stefanick, R. Qi, T. Kiziltepe, X. Jing and B. Bilgicer, J. Controlled Release, 2014, 173, 11–17 CrossRef CAS PubMed.
- R. Du, H. Xiao, G. Guo, B. Jiang, X. Yan, W. Li, X. Yang, Y. Zhang, Y. Li and X. Jing, Colloids Surf., B, 2014, 123, 734–741 CrossRef CAS PubMed.
- J. Li, Y. Han, Q. Chen, H. Shi, S. ur Rehman, M. Siddiq, Z. Ge and S. Liu, J. Mater. Chem. B, 2014, 2, 1813–1824 RSC.
- H. Xiao, W. Li, R. Qi, L. Yan, R. Wang, S. Liu, Y. Zheng, Z. Xie, Y. Huang and X. Jing, J. Controlled Release, 2012, 163, 304–314 CrossRef CAS PubMed.
- H. Xiao, L. Yan, Y. Zhang, R. Qi, W. Li, R. Wang, S. Liu, Y. Huang, Y. Li and X. Jing, Chem. Commun., 2012, 48, 10730–10732 RSC.
- R. Qi, H. Xiao, S. Wu, Y. Li, Y. Zhang and X. Jing, J. Mater. Chem. B, 2015, 3, 176–179 RSC.
- S. Aryal, C.-M. J. Hu and L. Zhang, ACS Nano, 2010, 4, 251–258 CrossRef CAS PubMed.
- H. Song, R. Wang, H. Xiao, H. Cai, W. Zhang, Z. Xie, Y. Huang, X. Jing and T. Liu, Eur. J. Pharm. Biopharm., 2013, 83, 63–75 CrossRef CAS PubMed.
- P. Plummer, R. H. Wilson, H. Calvert, A. V. Boddy, M. Griffin, J. Sludden, M. J. Tilby, M. Eatock, D. G. Pearson, C. J. Ottley, Y. Matsumura, K. Kataoka and T. Nishiya, Br. J. Cancer, 2011, 104, 593–598 CrossRef PubMed.
- T. Ueno, K. Endo, K. Hori, N. Ozaki, A. Tsuji, S. Kondo, N. Wakisaka, S. Murono, K. Kataoka, Y. Kato and T. Yoshizaki, Int. J. Nanomedicine, 2014, 9, 3005–3012 CrossRef CAS PubMed.
- H. Wu, H. Cabral, K. Toh, P. Mi, Y.-C. Chen, Y. Matsumoto, N. Yamada, X. Liu, H. Kinoh, Y. Miura, M. R. Kano, H. Nishihara, N. Nishiyama and K. Kataoka, J. Controlled Release, 2014, 189, 1–10 CrossRef CAS PubMed.
- D. P. Nowotnik and E. Cvitkovic, Adv. Drug Delivery Rev., 2009, 61, 1214–1219 CrossRef CAS PubMed.
- M. J. A. de Jonge, M. Slingerland, W. J. Loos, E. A. C. Wiemer, H. Burger, R. H. J. Mathijssen, J. R. Kroep, M. A. G. den Hollander, D. van der Biessen, M.-H. Lam, J. Verweij and H. Gelderblom, Eur. J. Cancer, 2010, 46, 3016–3021 CrossRef CAS PubMed.
- G. P. Stathopoulos, D. Antoniou, J. Dimitroulis, J. Stathopoulos, K. Marosis and P. Michalopoulou, Cancer Chemother. Pharmacol., 2011, 68, 945–950 CrossRef CAS PubMed.
- N. Seetharamu, E. Kim, H. Hochster, F. Martin and F. Muggia, Anticancer Res., 2010, 30, 541–545 CAS.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |