
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Enhancement of deoxyribozyme activity by cationic copolymers†
Jueyuan
Gao
a,
Naohiko
Shimada
b and
Atsushi
Maruyama
*b
aGraduate School of Engineering, Kyushu University, 744 CE11 Motooka, Nishi, Fukuoka 819-0395, Japan
bDepartment of Biomolecular Engineering, Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259-B57 Nagatsuta, Midori, Yokohama, 226-8501, Japan. E-mail: amaruyama@bio.titech.ac.jp
First published on 3rd October 2014
Abstract
Deoxyribozymes, or DNAzymes, are DNA molecules with enzymatic activity. DNAzymes with ribonuclease activity have various potential applications in biomedical and bioanalytical fields; however, most constructs have limited turnover despite optimization of reaction conditions and DNAzyme structures. A cationic comb-type copolymer accelerates DNA hybridization and strand exchange rates, and we hypothesized that the copolymer would enhance deoxyribozyme activity by promoting turnover. The copolymer did not change DNAzyme activity under single-turnover conditions, suggesting that the copolymer affects neither the folding structure of DNAzyme nor the association of a divalent cation, a catalytic cofactor, to DNAzyme. The copolymer enhanced activity of the evaluated DNAzyme over a wide temperature range under multiple-turnover conditions. The copolymer increased the DNAzyme kcat/KM by fifty-fold at 50 °C, the optimal temperature for the DNAzyme in the absence of the copolymer. The acceleration effect was most significant when the reaction temperature was slightly higher than the melting temperature of the enzyme/substrate complex; acceleration of two orders of magnitude was observed. We concluded that the copolymer accelerated the turnover step without influencing the chemical cleavage step. In contrast to the copolymer, a cationic surfactant, CTAB, strongly inhibited the DNAzyme activity under either single- or multiple-turnover conditions.
Introduction
Deoxyribozymes (DNAzymes) are DNA molecules with enzymatic activity. Compared with protein enzymes and ribozymes, the DNAzymes are easier to synthesize, active under a wider range of temperature and buffer conditions, and more readily modified. The first DNAzyme was demonstrated by Breaker and Joyce in 1994.1 Since then DNAzymes with diverse enzyme activities have been reported.2,3 DNAzymes with RNA cleaving (ribonuclease) activity have been most extensively studied. RNA-cleaving DNAzymes have been demonstrated to inactivate target RNAs,4 and detect divalent metal ions5 and have been used in construction of computational device elements.6 Sensitive detection of DNA/RNA sequences has also been reported.7RNA-cleaving DNAzymes are composed of a catalytic core and substrate-recognizing domains. DNAzyme-catalyzed reactions involve several steps. The first is the formation of a substrate/DNAzyme (ES) complex. The next step is the chemical cleavage of the substrate with the aid of a divalent cation. This step is followed by the release of the cleavage products. This generates a DNAzyme ready to load a fresh substrate for turnover. DNAzyme activity has been enhanced by screening core sequences under different selection pressures,8–13 use of modified nucleotides,14–17 conjugation with intercalators,18 and optimization of divalent metal cations.19–21 Both the chemically cleaving step and the turnover step should be improved for enhancing overall activity of DNAzyme. DNAzymes designed to form a stable complex with substrates showed higher catalytic activity under single-turnover conditions, but occasionally resulted in low turnover efficacy owing to a slower product release from the enzymes.17,21 Similarly, at low reaction temperatures the release rate of the product is limited, but at high temperature slow substrate/DNAzyme association limits the reaction rate. Conditions that increase dissociation and association dynamics of nucleotide hybrids are considered to improve turnover efficacy.
We have explored interactions between DNA strands with a cationic comb-type copolymer composed of a polycationic backbone and water soluble graft chains.22,23 The copolymer with more than 80 weight% graft chains forms a soluble inter-polyelectrolyte complex with DNA without inducing the coil–globule transition of the DNA.24–26 The copolymer considerably stabilizes double-stranded DNA (dsDNA)23,27 and triple-stranded DNA structures.22,28 Interestingly, the copolymer stabilized these structures by increasing the association rates rather than decreasing dissociation rates.29,30 This kinetic effect of the copolymer is unique because decrease in the dissociation rate was generally accounted for hybrid stabilization by other methods including the use of chemically modified nucleotides.31,32 More notably, the copolymer facilitates a strand exchange between a dsDNA and a homologous strand.33,34 The copolymer likely increases dissociation and association dynamics of nucleotide hybrids. The activity of the copolymer was utilized to refine a DNA analytical method35 and nanodevices.36,37
We hypothesized that the turnover efficacy of DNAzyme can be improved by the copolymer, if the copolymer satisfies the following requirements: the copolymer disorders neither (1) association of a divalent cation to DNAzyme nor (2) active structures of DNAzymes. In the present study, we examined the effect of the copolymer on the activity of the 10–23 DNAzyme,8,38 one of the most active RNA-cleaving DNAzymes, under single- and multiple-turnover conditions.
Experiment
Materials
Sodium poly(vinyl sulfonate) (PVS, 25 wt% aq.) and poly(L-lysine hydrobromide) (PLL·HBr, MW = 7.5 × 103) were obtained from Sigma-Aldrich Co. LLC (St. Louis, USA), and dextran (Dex, MW = 8 × 103–1.2 × 104) was obtained from Funakoshi Co. (Tokyo, Japan). Sodium hydroxide, sodium chloride, magnesium sulfate, manganese(II) chloride tetrahydrate, and acetic acid were purchased from Wako Pure Chemical Industries (Osaka, Japan). Sodium tetraborate decahydrate, urea, (2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid (HEPES), and ethylenediaminetetraacetic acid tetrasodium salt (EDTA·4Na) were purchased from Nacalai Tesque, Inc. (Kyoto, Japan). HPLC-grade oligonucleotides with the sequences summarized in Fig. 1 were purchased from Fasmac Co., Ltd (Atsugi, Japan) and used without further purification. A cationic comb-type copolymer poly(L-lysine)-graft-Dextran (PLL-g-Dex) was prepared according to the previously published procedure.22,23 Briefly, PLL-g-Dex was prepared by a reductive amination reaction of dextran with PLL. The resulting copolymer was isolated through an ion exchange column, dialyzed, and lyophilized. The product was characterized by 1H NMR and GPC. PLL-g-Dex of 90 wt% dextran (11.5 mol% of lysine units of PLL were modified with dextran) was used in this study.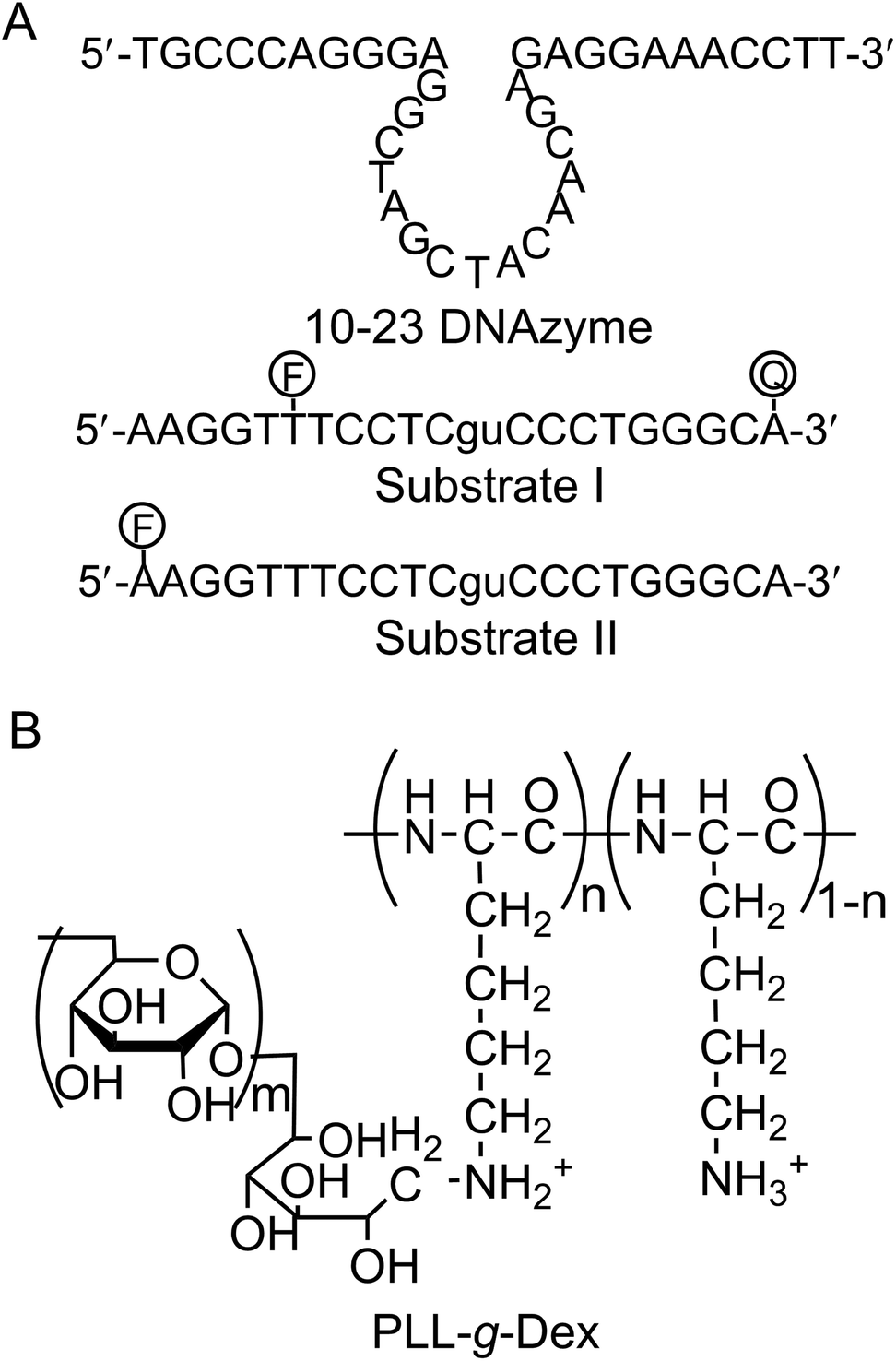 | ||
| Fig. 1 (A) Sequences of 10–23 DNAzyme and substrates. Substrate I was labeled with both a fluorophore (F, FITC) and a quencher (Q, BHQ-1), and substrate II was labeled with only a fluorophore. (B) The structural formula of the cationic comb-type copolymer PLL-g-Dex. Mw of PLL backbone: 7.5 × 103, Mw of dextran grafts: 1.0 × 104, grafting degree of dextran: 11.5 mol%. | ||
FRET analysis of the DNAzyme cleavage reaction
Substrate I (final concentration: 200 nM) was dissolved in a reaction buffer consisting of 50 mM HEPES (pH 7.3), 150 mM NaCl, and 5.0 mM Mg2+ or Mn2+ in the absence or presence of PLL-g-Dex. Unless otherwise noted, the ratio of [positively charged amino groups]copolymer/[negatively charged phosphate groups]DNA (N/P ratio) was 2. The substrate was pre-incubated with PLL-g-Dex at the reaction temperature for 5 min in a quartz cell. After the pre-incubation, DNAzyme solution (final concentration 6.7 nM) was injected into the cell to initiate the reaction. The fluorescence intensity was measured using a FP-6500 spectrofluorometer (Jasco, Tokyo, Japan) at an excitation wavelength, λex, of 494 nm and an emission wavelength, λem, of 520 nm. The fluorescence intensity curve over time was used to fit the equation It = I0 + (I∞ − I0) (1 − e−kobst), where It was the fluorescence intensity at any reaction time t, I∞ was the fluorescence intensity of the synthesized product, I0 was the initial fluorescence intensity (background). The kobs was obtained by fitting the initial 20% of the cleavage reaction curve.Gel electrophoresis analysis of DNAzyme reaction progress
Substrate II (500 nM) was mixed with DNAzyme (750 nM for single-turnover or 16.7 nM for multiple-turnover reactions) in 50 mM HEPES buffer (pH = 7.3) containing 0.5 mM Mg2+ and 150 mM NaCl with or without PLL-g-Dex (N/P = 2). The reaction mixtures were incubated at 25 °C. Aliquots (3 μL) were taken at various reaction times, and 0.5 μL of 0.1 M EDTA was added to stop the reaction. After addition of 3 μL formamide and 0.5 μL 5 mg mL−1 PVS, reaction products were separated on a denaturing polyacrylamide gel (16%) run at 25 °C for 75 min. The fluorescence was monitored using a Bio-Rad molecular imager pharosFX at λex 388 nm and λem 532 nm.Determination of DNAzyme kinetic constants
The Michaelis–Menten kinetic parameters, kcat and KM, were determined from the y-intercept and the negative slope, respectively, of the best-fit line to the Eadie–Hofstee plot of V0versus V0/[S].21 Each plot consisted of five data points for values of [S] that ranged from KM to 10-fold KM, with [S] always ≥10-fold excess over [E]. V0 was determined from the initial 10% of the curve. All the reactions were carried out in the presence of 5 mM Mn2+, in the absence or presence (N/P = 2) of PLL-g-Dex in 50 mM HEPES (pH 7.3) and 150 mM NaCl, and were initiated by injecting the DNAzyme solution.Results
Influence of the cationic comb-type copolymer on multiple- and single-turnover reactions
Firstly, we investigated the effect of the copolymer PLL-g-Dex on the ribonuclease activity of the 10–23 DNAzyme on substrate I (Fig. 1) with 0.5 mM Mg2+ as a cofactor under multiple turnover conditions, [S]/[E] = 30 (Fig. 2A). Whereas in the absence of the copolymer kobs was 1.3 × 10−3, in the presence of the copolymer at an N/P ratio of 2, kobs was 3-fold greater.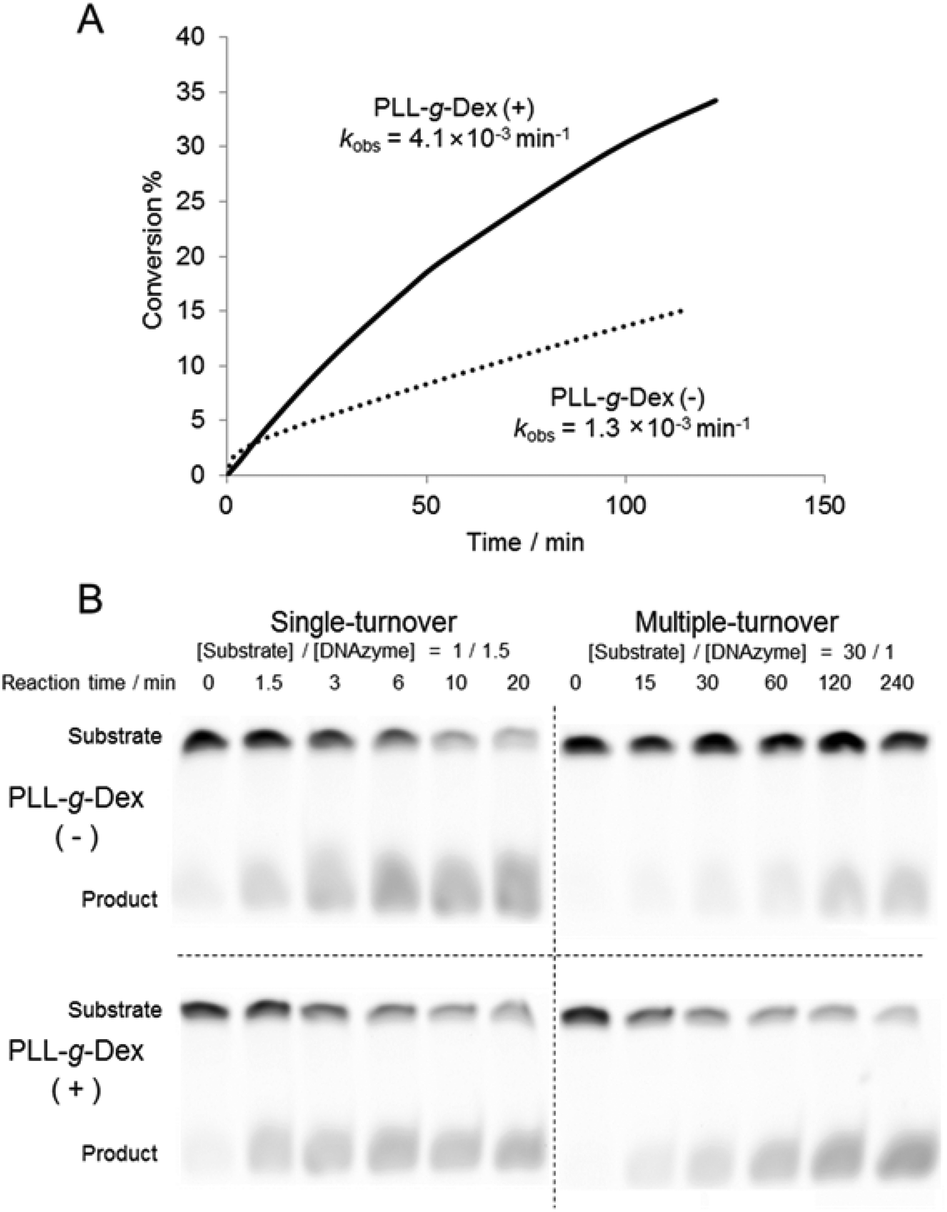 | ||
| Fig. 2 Influence of PLL-g-Dex on single- and multiple-turnover reaction of DNAzyme at 0.5 mM Mg2+ and 25 °C. (A) DNAzyme multiple-turnover reactions in the absence and presence of PLL-g-Dex (N/P = 2) were monitored by analysis of labeled substrate I ([substrate I]0 = 200 nM, [DNAzyme] = 6.7 nM). (B) DNAzyme reactions under single- and multiple-turnover conditions in the absence and presence (N/P = 2) of PLL-g-Dex were monitored by analysis of substrate II. The samples contained 500 nM substrate II and either 750 nM (single-turnover) or 16.7 nM (multiple-turnover) DNAzyme. | ||
To estimate the role of the copolymer in the observed acceleration, the DNAzyme reactions were carried out under either single- or multiple-turnover conditions. Substrate I (500 nM) was mixed with 750 nM or 16.7 nM of DNAzyme in a buffer containing 0.5 mM MgSO4 with or without PLL-g-Dex. After incubation at 25 °C the reaction mixtures were separated by denatured gel electrophoresis (Fig. 2B). As the reaction time increased the intensity of the upper band, which corresponds to an uncleaved substrate, decreased and the intensity of the lower band, which corresponds to products, increased. In the reaction under the single-turnover conditions no difference in cleavage rates was observed regardless of the presence of PLL-g-Dex. The results indicated that the copolymer did not significantly influence the chemical cleavage activity of the DNAzyme. The active folding structure of the DNAzyme and the association of a divalent metal ion to DNAzyme were likely not disordered by the copolymer. In contrast, under multiple-turnover reaction conditions faster cleavage was observed in the presence of PLL-g-Dex, implying that the copolymer promoted turnover. Calculated thermal melting temperatures (Tms) at 0.5 mM Mg2+ were 39.8 °C for the enzyme complex with the 5′ product and 54.0 °C for the enzyme complex with the 3′ product when the product concentration was 500 nM and DNAzyme concentration was 16.7 nM.39 This calculation suggests that dissociation of the products from the DNAzyme should be slow at 25 °C. The copolymer likely facilitates the strand exchange of EP for ES at 25 °C.
The effect of the copolymer was compared with that of cethyltrimethylammonium bromide (CTAB), a cationic surfactant known to promote DNA hybridization.40 CTAB significantly decreased the rate of the DNAzyme reaction under both single- and multiple-turnover conditions (Fig. 3A and B). We observed turbidity in the reaction mixture in the presence of CTAB (Fig. 3C). CTAB likely inhibited the DNAzyme activity by DNA compaction and aggregation.41
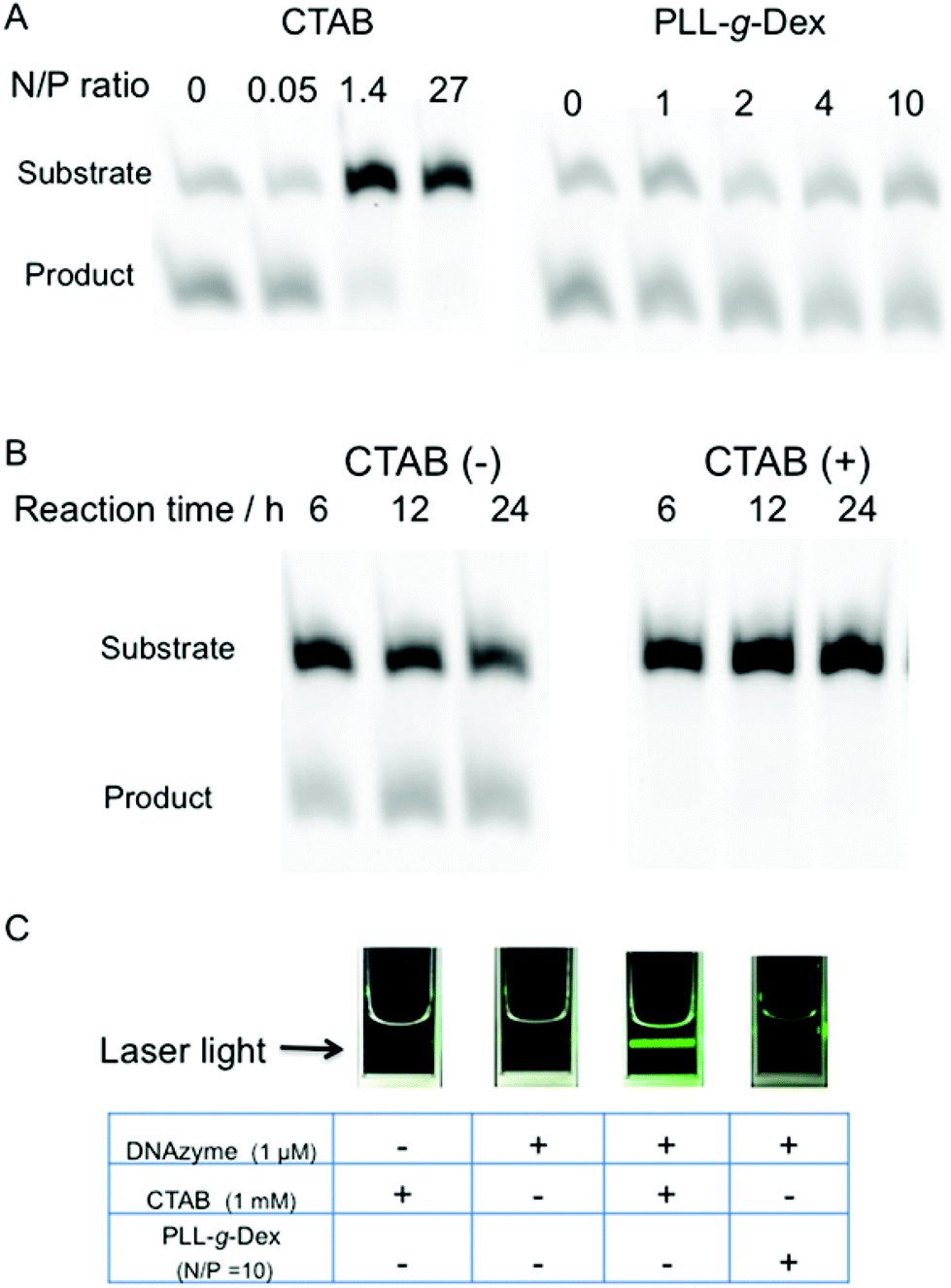 | ||
| Fig. 3 (A) Comparison of PLL-g-Dex and CTAB on DNAzyme reactions under single-turnover reaction conditions (20 min reaction at 25 °C, 0.5 mM MgSO4, 0.5 μM substrate II, and 0.75 μM DNAzyme). (B) The influence of CTAB (N/P = 27, 1 mM) on multiple-turnover DNAzyme reaction (reaction at 25 °C, 0.5 mM MgSO4, 0.5 μM substrate II, and 0.017 μM DNAzyme). (C) Solubility of DNA complex with either CTAB or PLL-g-Dex. | ||
Rate-determining step shifted by the copolymer
To investigate further the effect of the copolymer on the DNAzyme multiple-turnover reaction, the temperature dependence of the reaction was assessed. Cleavage activity with or without the copolymer increased as the reaction temperature was increased from 25 °C to 60 °C (Fig. 4 and 5D). When the DNAzyme reaction was carried out at 60 °C, a significant loss in the activity was observed in the absence of the copolymer. As the calculated value of Tm39 of the DNAzyme/substrate hybrid is about 53 °C, this loss in the DNAzyme reactivity likely resulted from a decrease in the association rate of the substrate with the DNAzyme. In the presence of the copolymer, no loss in activity was observed until 60 °C (Fig. 5D), indicating that the copolymer increased the association rate of a substrate with a DNAzyme. At 60 °C, the copolymer enhanced the DNAzyme activity by 30-fold relative to the activity in the absence of the copolymer. | ||
| Fig. 4 Temperature dependence of DNAzyme multiple-turnover reactions in 5 mM Mg2+ with or without PLL-g-Dex. Samples containing 200 nM substrate I and 6.7 nM DNAzyme were incubated in the absence or presence of PLL-g-Dex (N/P = 2) at (A) 25 °C, (B) 50 °C, and (C) 60 °C. | ||
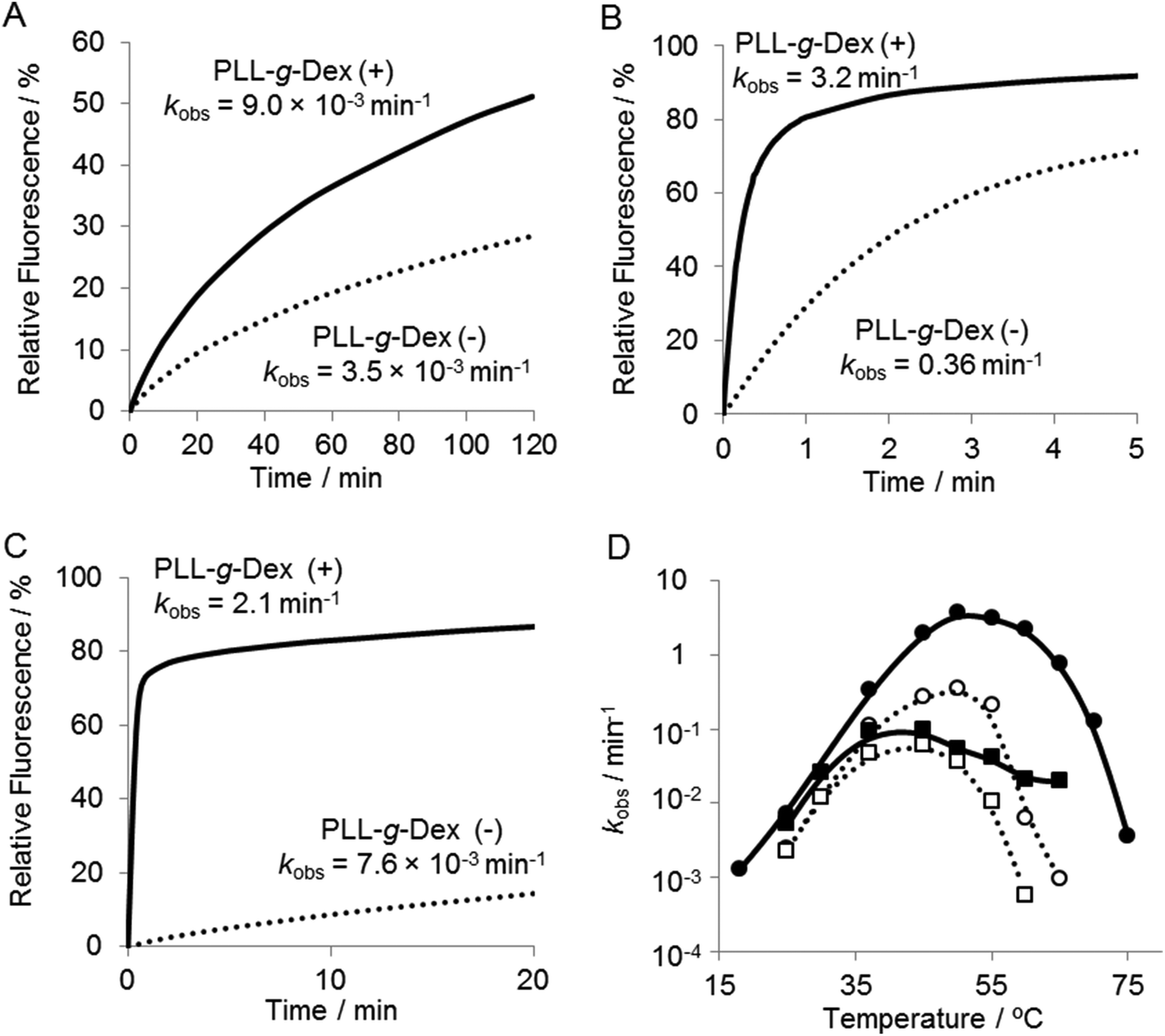 | ||
| Fig. 5 Temperature dependence of DNAzyme multiple-turnover reactions at 5 mM Mn2+ with or without PLL-g-Dex. Samples containing 200 nM substrate I and 6.7 nM DNAzyme were incubated in the absence or presence of PLL-g-Dex (N/P = 2) at (A) 25 °C, (B) 50 °C, and (C) 60 °C. (D) Temperature dependence of rate constants, kobs, estimated in 5 mM Mg2+ (square) and in Mn2+ (circle) in the absence (dotted lines) and presence (solid lines) of the copolymer. | ||
It should be noted that the reaction in the presence of the copolymer at 50 °C or higher obeyed the zero-order reaction kinetics (Fig. 4B and C). This result indicated that the reaction rate was limited by the chemical cleavage process and not by the turnover process involving product release and substrate binding. This supports our hypothesis that the copolymer facilitates the turnover process. Moreover, this observation suggested that further acceleration of the DNAzyme reaction should be possible by replacing Mg2+ with a more active metal ion.
The multiple-turnover reaction accelerated in the presence of Mn2+ and the copolymer
Various divalent metal ions have been evaluated as cofactors of the 10–23 DNAzyme. Among the metal ions investigated, Mn2+ is one of the most active cofactors.21 Hence, we carried out the reaction with Mn2+ either in the absence or presence of the copolymer (Fig. 5). At temperatures below 30 °C only slight differences were observed in the DNAzyme activity in Mn2+ and Mg2+ regardless of the absence or presence of the copolymer (Fig. 4Avs. Fig. 5A, D). This is reasonable because under 30 °C the rate-determining step is not the chemical cleavage process but the turnover process. At higher temperatures a more significant enhancement by Mn2+ was observed in the presence of the copolymer than in the absence. The DNAzyme activity at 50 °C was increased nearly 60-fold by replacing Mg2+ for Mn2+ in the presence of the copolymer, whereas it was increased only 10-fold in the absence of the copolymer. As described above, the DNAzyme reaction with the copolymer in Mg2+ at higher temperature was limited by the chemical cleavage step; the Mn2+ ion effectively increased the overall reaction rate by facilitating this step. At 60–65 °C the DNAzyme was two orders of magnitude more active in the presence of the copolymer than without. The observed rate constants were higher over the range of temperature from 35 °C to 65 °C in the presence than in the absence of the copolymer (Fig. 5D). To confirm the role of the copolymer in the 10–23 DNAzyme reactions, the catalytic parameters were determined under multiple-turnover conditions and are summarized in Table 1. The copolymer did not affect kcat but considerably decreased KM. Hence, the copolymer did not affect the chemical cleavage step but facilitated ES complex formation. As a result the copolymer increased kcat/KM, a measure of the overall enzymatic activity, by 50-fold at 50 °C.| Temperature | PLL-g-Dex | V max (nM min−1) | k cat (min−1) | K M (M) | k cat/KM (M−1 min−1) |
|---|---|---|---|---|---|
| a N/P = 2. | |||||
| 37 °C | − | 6.6 × 101 | 6.6 | 1.5 × 10−7 | 4.4 × 107 |
| + | 5.8 × 10−1 | 5.8 | 4.5 × 10−9 | 1.3 × 109 | |
| 50 °C | − | 1.0 × 102 | 5.2 × 10 | 8.5 × 10−7 | 6.1 × 107 |
| + | 1.5 × 102 | 7.8 × 10 | 2.4 × 10−8 | 3.2 × 109 | |
Influence of the copolymer on the ES complex
To determine the effect of the copolymer on the stability of the ES complex UV melting experiments were performed in the absence of divalent metal ions. As shown in Fig. 6, the Tm of the complex was 47 °C in the absence of the copolymer. The Tm was increased to 60 °C in the presence of the copolymer, indicating that the copolymer stabilized the ES complex. The effect of the copolymer on the ES complex was also evaluated by circular dichroismic (CD) spectroscopy using substrates in which ribonucleotides were replaced with deoxyribonucleotides to prevent cleavage during the CD measurement. As shown in Fig. 7A, the CD profile characteristics of B-DNA with 240 nm negative and 260 nm positive bands were obtained for a mixture of DNAzymes and substrates. A slight difference in the CD signal was observed with Mn2+. The copolymer did not change the CD profiles regardless of the presence of Mn2+. Though CD spectroscopy is not sensitive enough for us to be able to discuss the local structures of the complex, the result implied that interactions of the copolymer did not significantly influence the conformation of the ES complex. Also, the copolymer did not influence the Mn2+-induced conformational change of the complex, indicative of inertness of the copolymer to Mn2+/DNA interactions. The stabilizing effect of the copolymer on the ES complex was also shown by this CD analysis. At 65 °C the ES complex was dissociated in the absence of the copolymer but was stable in the presence of the copolymer (Fig. 7B). | ||
| Fig. 6 UV melting profile of the DNAzyme/substrate complex in the presence or absence of the PLL-g-Dex copolymer. Samples contained 1.5 μM substrate I and 1.5 μM DNAzyme in the absence (dotted line) or presence (solid line, N/P = 2) of PLL-g-Dex in 50 mM HEPES (pH = 7.3), 150 mM NaCl. Temperature was increased 1 K min−1, and spectra were recorded on a V-630 spectrophotometer (Jasco). | ||
 | ||
| Fig. 7 CD spectra of complexes between the DNAzyme and the DNA-substituted substrate (ribo gu nucleotides of the substrate were replaced with deoxy GT nucleotides to avoid cleavage) at (A) 25 °C and (B) 65 °C in the absence or presence of the divalent ion and PLL-g-Dex. Samples containing DNAzyme (2.5 μM) were mixed with DNA substrate (2.5 μM) in 50 mM HEPES (pH = 7.3), 150 mM NaCl in the absence (dotted line) or presence (N/P = 2, solid line) of PLL-g-Dex and in the absence (blue line) or presence (red line) of 5 mM Mn2+. | ||
The dependence of the DNAzyme reaction on the concentration of the copolymer was estimated in the presence of Mn2+ at 50 °C. As shown in Fig. 8, the cleavage rate increased with an increase in polymer concentration until a plateau at an N/P ratio of about 0.7. This indicates that an electrostatically equivalent amount of the copolymer was sufficient for maximum rate enhancement.
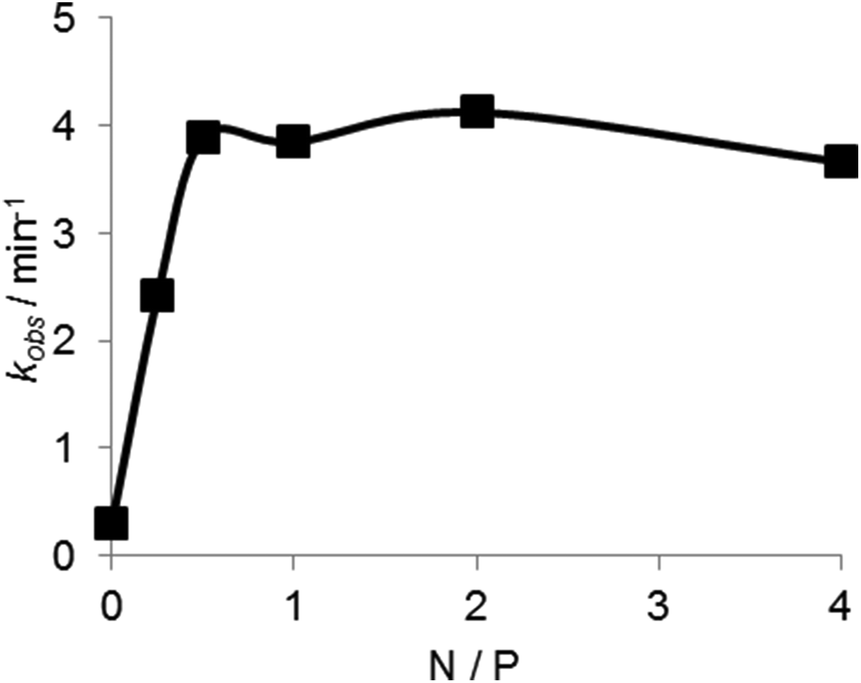 | ||
| Fig. 8 PLL-g-Dex concentration dependence of the DNAzyme multiple-turnover reaction rate. Reactions were performed at 50 °C, 5 mM Mn2+, 6.7 nM DNAzyme, and 200 nM substrate I with various concentrations of PLL-g-Dex. | ||
Discussion
We have shown that cationic comb-type copolymers accelerate DNA hybridization and stabilize DNA structures through inter-polyelectrolyte complex formation. In this study we examined how PLL-g-Dex influenced the ribonuclease activity of the 10–23 DNAzyme. As shown in Fig. 2B, the copolymer did not influence the DNAzyme activity under single-turnover conditions. This result indicated that association with the copolymer did not affect the chemical cleavage activity of the DNAzyme. This was also supported by the fact that the copolymer did not influence kcat (Table 1). The association of the copolymer did not alter the shape of the CD spectrum of the ES complex suggesting that the copolymer does not significantly alter the secondary structure of the ES complex (Fig. 7). These results also implied that the association of the polycationic copolymer did not markedly influence the interaction of a divalent metal ion with the ES complex. This inertness of the copolymer to the interaction between the divalent metal ion and the DNA is interesting. We have previously observed that the copolymer did not interfere with K+-dependent DNA quadruplex formation.42Under multiple-turnover reaction conditions the copolymer enhanced the activity of the DNAzyme (Fig. 2 and 4). The enhancement was not large when Mg2+ was used as a cofactor. The zero-order reaction kinetics observed in the presence of the copolymer (Fig. 4B and C) implied that the rate-determining step was shifted to the chemical cleavage step by the presence of the copolymer. The result clearly revealed that the copolymer increased the turnover rate. Indeed, in the presence of Mn2+, which results in higher cleavage activity than Mg2+, the copolymer more significantly enhanced the DNAzyme activity (Fig. 3 and 4).
In addition to the increase in the DNAzyme reaction rate, the copolymer also increased the effective temperature range of the DNAzyme reaction. In the absence of the copolymer, the DNAzyme activity began to decrease at 55 °C and the enzyme was almost completely inactive at 60 °C (Fig. 5D). In the presence of the copolymer, substantial activity was retained up to 60 °C. At 60 °C the copolymer accelerated the reaction by 250-fold relative to the reaction in the absence of the copolymer (Fig. 5C) by stabilizing the ES complex. At the optimal temperature, 50 °C, for the DNAzyme in the absence of the copolymer, the copolymer increased the kcat/KM value by 50 times (Table 1). The increase in the kcat/KM value was largely caused by the decrease in the KM value by the presence of the copolymer. The copolymer facilitated association of DNAzyme to its substrate.
Interestingly the copolymer enhanced the DNAzyme reaction even at lower temperatures, 20–27 °C, where product release is the rate-determining step, though the magnitude of the enhancement was not as large as at 50 °C. As previously reported, the copolymer accelerates a strand exchange reaction between the DNA duplex and its complementary single strand.33,34 The copolymer likely facilitates exchange of a product with a substrate on the DNAzyme by a similar mechanism. This enhancement of DNAzyme activity was also observed as a decrease in the KM value in Michaelis–Menten parameters (Table 1). Michaelis–Menten kinetics assumes that the enzyme is regenerated soon after the product is produced. But this is not the case because scission products are bound to binding arms and inhibit the reaction. An accelerated turnover can be evaluated as an increase in the dissociation constant Ki of the enzyme-inhibitor complex. According to this inhibition model, kcat does not change but apparently KM decreases, suggesting that the copolymer increased the turnover efficacy.
As shown in Fig. 8, an electrostatically equivalent amount (N/P = 1) of the copolymer was sufficient to produce the maximum efficacy. The copolymer at N/P = 2 increased kcat/KM values by 50-fold compared to the reaction in the absence of the copolymer (Table 1). The copolymer concentration was only 150 nM (in strand) (or 8.9 μM in ionic unit) at this N/P ratio, suggesting stable association of the copolymer with DNA. Regardless of the strong association of the copolymer, inertness of the copolymer to DNAzyme conformation and its interaction with a divalent cation should be noted. Abundant dextran grafts of the copolymer play a pivotal role in this strong but gentle interaction. The copolymer may be used to refine DNAzyme-based biomedical and nanomechanical devices.
Conclusions
The cationic comb-type copolymer, PLL-g-Dex, was shown to enhance the DNAzyme cleavage reaction. The copolymer accelerated the turnover process without affecting the chemical cleavage step and increased the active temperature window of DNAzyme reaction.Acknowledgements
Parts of this work were supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Robotics” (no. 24104003), “Nanomedicine Molecular Science” (no. 2306) and the Cooperative Research Program of “Network Joint Research Center for Materials and Devices” from the Ministry of Education, Culture, Sports, Science and Technology, by Center of Innovation (COI) Program, Japan Science and Technology Agency (JST), and by KAKENHI (no. 23240074, 25350552) from Japan Society for the Promotion of Science.Notes and references
- R. R. Breaker and G. F. Joyce, A DNA enzyme that cleaves RNA, Chem. Biol., 1994, 1, 223–229 CrossRef CAS
.
- D. A. Baum and S. K. Silverman, Deoxyribozymes: useful DNA catalysts in vitro and in vivo, Cell. Mol. Life Sci., 2008, 65, 2156–2174 CrossRef CAS PubMed
- S. K. Silverman, Catalytic DNA (deoxyribozymes) for synthetic applications-current abilities and future prospects, Chem. Commun., 2008, 3467–3485 RSC
- F. S. Santiago, H. C. Lowe, M. M. Kavurma, C. N. Chesterman, A. Baker, D. G. Atkins and L. M. Khachigian, New DNA enzyme targeting Egr-1 mRNA inhibits vascular smooth muscle proliferation and regrowth after injury, Nat. Med., 1999, 5, 1264–1269 CrossRef CAS PubMed
- J. Li and Y. Lu, A Highly Sensitive and Selective Catalytic DNA Biosensor for Lead Ions, J. Am. Chem. Soc., 2000, 122, 10466–10467 CrossRef CAS
- M. N. Stojanovic, T. E. Mitchell and D. Stefanovic, Deoxyribozyme-based logic gates, J. Am. Chem. Soc., 2002, 124, 3555–3561 CrossRef CAS PubMed
- D. M. Kolpashchikov, A Binary Deoxyribozyme for Nucleic Acid Analysis, ChemBioChem, 2007, 8, 2039–2042 CrossRef CAS PubMed
- S. W. Santoro and G. F. Joyce, A general purpose RNA-cleaving DNA enzyme, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4262–4266 CrossRef CAS
- K. Schlosser and Y. Li, Diverse evolutionary trajectories characterize a community of RNA-cleaving deoxyribozymes: a case study into the population dynamics of in vitro selection, J. Mol. Evol., 2005, 61, 192–206 CrossRef CAS PubMed
- S. H. Mei, Z. Liu, J. D. Brennan and Y. Li, An efficient RNA-cleaving DNA enzyme that synchronizes catalysis with fluorescence signaling, J. Am. Chem. Soc., 2003, 125, 412–420 CrossRef CAS PubMed
- W. Chiuman and Y. Li, Revitalization of six abandoned catalytic DNA species reveals a common three-way junction framework and diverse catalytic cores, J. Mol. Biol., 2006, 357, 748–754 CrossRef CAS PubMed
- J. C. Lam, S. O. Kwan and Y. Li, Characterization of non-8–17 sequences uncovers structurally diverse RNA-cleaving deoxyribozymes, Mol. BioSyst., 2011, 7, 2139–2146 RSC
- W. Chiuman and Y. Li, Evolution of high-branching deoxyribozymes from a catalytic DNA with a three-way junction, Chem. Biol., 2006, 13, 1061–1069 CrossRef CAS PubMed
- J. He, D. Zhang, Q. Wang, X. Wei, M. Cheng and K. Liu, A novel strategy of chemical modification for rate enhancement of 10–23 DNAzyme: a combination of A9 position and 8-aza-7-deaza-2′-deoxyadenosine analogs, Org. Biomol. Chem., 2011, 9, 5728–5736 CAS
- Q. Wang, D. Zhang, Y. Liu, M. Cheng, J. He and K. Liu, A structure-activity relationship study for 2′-deoxyadenosine analogs at A9 position in the catalytic core of 10–23 DNAzyme for rate enhancement, Nucleic Acid Ther., 2012, 22, 423–427 CAS
- B. Nawrot, K. Widera, M. Wojcik, B. Rebowska, G. Nowak and W. J. Stec, Mapping of the functional phosphate groups in the catalytic core of deoxyribozyme 10–23, FEBS J., 2007, 274, 1062–1072 CrossRef CAS PubMed
- S. Schubert, D. C. Gul, H. P. Grunert, H. Zeichhardt, V. A. Erdmann and J. Kurreck, RNA cleaving ‘10–23’ DNAzymes with enhanced stability and activity, Nucleic Acids Res., 2003, 31, 5982–5992 CrossRef CAS PubMed
- H. Asanuma, H. Hayashi, J. Zhao, X. Liang, A. Yamazawa, T. Kuramochi, D. Matsunaga, Y. Aiba, H. Kashida and M. Komiyama, Enhancement of RNA cleavage activity of 10–23 DNAzyme by covalently introduced intercalator, Chem. Commun., 2006, 5062–5064 RSC
- N. Sugimoto, Y. Okumoto and T. Ohmichi, Effect of metal ions and sequence of deoxyribozymes on their RNA cleavage activity, J. Chem. Soc., Perkin Trans. 2, 1999, 1381–1386 RSC
- M. Bonaccio, A. Credali and A. Peracchi, Kinetic and thermodynamic characterization of the RNA-cleaving 8–17 deoxyribozyme, Nucleic Acids Res., 2004, 32, 916–925 CrossRef CAS PubMed
- S. W. Santoro and G. F. Joyce, Mechanism and utility of an RNA-cleaving DNA enzyme, Biochemistry, 1998, 37, 13330–13342 CrossRef CAS PubMed
- A. Maruyama, M. Katoh, T. Ishihara and T. Akaike, Comb-type polycations effectively stabilize DNA triplex, Bioconjugate Chem., 1997, 8, 3–6 CrossRef CAS PubMed
- A. Maruyama, H. Watanabe, A. Ferdous, M. Katoh, T. Ishihara and T. Akaike, Characterization of interpolyelectrolyte complexes between double-stranded DNA and polylysine comb-type copolymers having hydrophilic side chains, Bioconjugate Chem., 1998, 9, 292–299 CrossRef CAS PubMed
- Y. Sato, Y. Kobayashi, T. Kamiya, H. Watanabe, T. Akaike, K. Yoshikawa and A. Maruyama, The effect of backbone structure on polycation comb-type copolymer/DNA interactions and the molecular assembly of DNA, Biomaterials, 2005, 26, 703–711 CrossRef CAS PubMed
- N. Sonda, M. Hirano, N. Shimada, A. Kano, S. Kidoaki and A. Maruyama, Cationic Comb-type Copolymers Do Not Cause Collapse but Shrinkage of DNA Molecules, Chem. Lett., 2011, 40, 250–251 CrossRef CAS
- A. Maruyama, N. Sonda, K. Yamasaki, S. Kidoaki, N. Shimada, M. Maeshiro and M. Miyazaki, Cationic comb-type copolymer excludes intercalating dye from DNA without inducing DNA condensation, Curr. Nanosci., 2014, 10, 185–188 CrossRef CAS
- A. Maruyama, Y.-I. Ohnishi, H. Watanabe, H. Torigoe, A. Ferdous and T. Akaike, Polycation comb-type copolymer reduces counterion condensation effect to stabilize DNA duplex and triplex formation, Colloids Surf., B: Biointerfaces, 1999, 16, 273–280 CrossRef CAS
- A. Ferdous, H. Watanabe, T. Akaike and A. Maruyama, Poly(L-lysine)-graft-dextran copolymer: Amazing effect on triplex stabilization under physiological relevant conditions (in vitro), Nucleic Acids Res., 1998, 26, 3949–3954 CrossRef CAS PubMed
- H. Torigoe, A. Ferdous, H. Watanabe, T. Akaike and A. Maruyama, Poly(L-lysine)-graft-dextran copolymer promotes pyrimidine motif triplex DNA formation at physiological pH. Thermodynamic and kinetic studies, J. Biol. Chem., 1999, 274, 6161–6167 CrossRef CAS PubMed
- L. Wu, N. Shimada, A. Kano and A. Maruyama, Poly(l-lysine)-graft-dextran copolymer accelerates DNA hybridization by two orders, Soft Matter, 2008, 4, 744–747 RSC
- H. Torigoe, Thermodynamic and Kinetic Effects of N3′→P5′ Phosphoramidate Modification on Pyrimidine Motif Triplex DNA Formation, Biochemistry, 2001, 40, 1063–1069 CrossRef CAS PubMed
- H. Torigoe, Y. Hari, M. Sekiguchi, S. Obika and T. Imanishi, 2′-O,4′-C-methylene bridged nucleic acid modification promotes pyrimidine motif tripler DNA formation at physiological pH-Thermodynamic and kinetic studies, J. Biol. Chem., 2001, 276, 2354–2360 CrossRef CAS PubMed
- W. J. Kim, T. Ishihara, T. Akaike and A. Maruyama, Comb-type cationic copolymer expedites DNA strand exchange while stabilizing DNA duplex, Chemistry, 2001, 7, 176–180 CrossRef CAS
- W. J. Kim, T. Akaike and A. Maruyama, DNA strand exchange stimulated by spontaneous complex formation with cationic comb-type copolymer, J. Am. Chem. Soc., 2002, 124, 12676–12677 CrossRef CAS PubMed
- W. J. Kim, Y. Sato, T. Akaike and A. Maruyama, Cationic comb-type copolymers for DNA analysis, Nat. Mater., 2003, 2, 815–820 CrossRef CAS PubMed
- S. W. Choi, N. Makita, S. Inoue, C. Lesoil, A. Yamayoshi, A. Kano, T. Akaike and A. Maruyama, Cationic comb-type copolymers for boosting DNA-fueled nanomachines, Nano Lett., 2007, 7, 172–178 CrossRef CAS PubMed
- J. Du, L. Wu, N. Shimada, A. Kano and A. Maruyama, Polyelectrolyte-assisted transconformation of a stem-loop DNA, Chem. Commun., 2013, 49, 475–477 RSC
- J. Nowakowski, P. J. Shim, G. S. Prasad, C. D. Stout and G. F. Joyce, Crystal structure of an 82-nucleotide RNA-DNA complex formed by the 10–23 DNA enzyme, Nat. Struct. Biol., 1999, 6, 151–156 CrossRef CAS PubMed
- M. Zuker, Mfold web server for nucleic acid folding and hybridization prediction, Nucleic Acids Res., 2003, 31, 3406–3415 CrossRef CAS PubMed
- B. W. Pontius and P. Berg, Rapid renaturation of complementary DNA strands mediated by cationic detergents: a role for high-probability binding domains in enhancing the kinetics of molecular assembly processes, Proc. Natl. Acad. Sci. U. S. A., 1991, 88, 8237–8241 CrossRef CAS
- J. Carlstedt, D. Lundberg, R. S. Dias and B. Lindman, Condensation and decondensation of DNA by cationic surfactant, spermine, or cationic surfactant–cyclodextrin mixtures: Macroscopic phase behavior, aggregate properties, and dissolution mechanisms, Langmuir, 2012, 28, 7976–7989 CrossRef CAS PubMed
- R. Moriyama, N. Shimada, A. Kano and A. Maruyama, DNA assembly and re-assembly activated by cationic comb-type copolymer, Biomaterials, 2011, 32, 2351–2358 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: % Cleavage vs. reaction time plots for Fig. 2B. See DOI: 10.1039/c4bm00256c |
| This journal is © The Royal Society of Chemistry 2015 |