
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a UHPLC method for the detection of organic gunshot residues using artificial neural networks
Regina Verena
Taudte
,
Claude
Roux
,
David
Bishop
,
Lucas
Blanes
,
Philip
Doble
and
Alison
Beavis
*
Centre for Forensic Science, University of Technology, PO Box 123 Broadway, Sydney, NSW 2007, Australia. E-mail: Alison.Beavis@uts.edu.au
First published on 5th May 2015
Abstract
The introduction of lead and heavy-metal free ammunition to the market challenges the current protocol for gunshot residue (GSR) investigations, which focuses on the inorganic components. Future proofing GSR analysis requires the development and implementation of new methods for the collection and analysis of organic GSR (OGSR) into operational protocols. This paper describes the development and optimisation of an ultra high performance liquid chromatography method for the analysis of 32 compounds potentially present in OGSR. An artificial neural network was applied to predict the retention times of the target analytes for various gradients for rapid determination of optimum separation conditions. The final separation and analysis time for the 32 target analytes was 27 minutes with limits of detection ranging from 0.03 to 0.21 ng. The method was applied to the analysis of smokeless powder and samples collected from the hands of a shooter following the discharge of a firearm. The results demonstrate that the method has the potential for use in cases involving GSR.
1. Introduction
The increasing number of firearm related incidents requires accurate and reliable analytical methods of investigation.1 Gunshot residues (GSR) consist of burnt and unburnt particles released from a firearm that originate from the primer, propellant powder, or other particle sources in the firearm and represent an important evidence in firearm related events.2,3 GSR analyses can support findings such as the presence of a suspect at the crime scene, an estimate of the shooting distance,4–7 the discharge time,8–10 and the linkage of a specific weapon and/or ammunition to the incident.11–16The internationally accepted method for detection and analysis of GSR is scanning electron microscopy/energy dispersive X-ray spectroscopy (SEM/EDX).3,17 This method depends on the presence of particles that are characteristic of GSR incorporating lead, antimony and barium, which originate from the primer. Due to the capability to selectively identify a single GSR particle based on morphology and elemental composition, SEM/EDX continues to be the method of choice for GSR identification in forensic casework and is unequalled by any other bulk analysis method.18,19 In the 1970s lead free (LF) and heavy-metal free (HMF) ammunitions were introduced to the market to decrease the exposure of frequent shooters to toxic gases and hazardous particulates released from the primer and the bullet itself.20 Although these ammunitions have been available for a few decades, recent legislative changes in some US states (e.g. California) prohibiting lead ammunition for hunting21 and the shift of other groups, most notable the US military, to lead free ammunition22 accentuate the increasing trend towards LF and HMF ammunition. The replacement of lead and/or heavy-metals has been accomplished by different processes, such as enclosing parts or the entire projectile with brass, copper or gilding metal, using a non-lead containing sintered metal for the fabrication of the projectile,23 and substituting lead, barium and antimony with other components such as diazodinitrophenol (DDNP).24,25 So far, different elemental profiles of spherical particles produced by LF and/or HMF ammunitions have been elucidated.20,23,26,27 These studies suggest that elemental profiles of LF and HMF GSR particulates are in consistency with the composition of the respective primers. However, identification of GSR particles formed by LF and HMF ammunition is problematic. The composition is not exclusive to firearm handling25 and IGSR particles from LF and HMF ammunition could potentially derive from other sources than firearms. Therefore, SEM/EDX analysis of IGSR from LF and HMF ammunition could possibly lead to false-negative results.23,26–28 Moreover, interpretation issues correlating to memory effects,29 GSR-like particles from environmental and occupational sources,30–32 and secondary or tertiary33–36 constitute additional challenges.
It is important to point out, that particle analysis by SEM/EDX continues to be the method of choice for GSR analysis as LF and HMF only constitutes a relatively small proportion of the ammunition on the market at present. However, the increasing trend towards LF and HMF ammunition and the associated limitations call for an adjustment of current Standard Operation Procedures (SOPs) for GSR investigation.
The analysis of organic gunshot residues (OGSR) originating from the propellant powder and primer provides additional information, complementary to that obtained by SEM-EDX analysis. Thus, incorporation of OGSR analysis to existing SOPs is beneficial. This incorporation must give consideration to the necessary sequencing of the organic and inorganic analyses, ensuring one does not impact on or preclude the subsequent testing.
When developing an analytical method, it is important to include both, OGSR from propellant powder as well as from primer mixtures to target a majority of the compounds potentially present.
OGSRs may be analysed using a variety of analytical methods including liquid chromatography (LC),37,38 gas chromatography (GC),39,40 micellar electrokinetic capillary electrophoresis (MECE),41 time-of-flight secondary ion mass spectrometry (TOF-SIMS),42 and desorption electrospray ionisation mass spectrometry (DESI-MS).43,44
Traditional LC method development consists of changing individual parameters one at a time, while keeping all others constant. This is a time consuming and challenging process given the large number of parameters and their possible interactions. An additional difficulty can be posed when a large number of compounds require separation, as in the case of OGSR analysis. An alternative route for rapid method development is the application of artificial neural networks (ANNs) which are predictive data-processing programs that mimic the way a human brain processes information. The processing units in ANNs consist of neurons, units, and nodes arranged in several interconnected layers.45 Multilayer perceptron (MLP) ANNs are constructed with three layers; the input layer, hidden layer and output layer.46 Each node of the input layer is associated with an experimental factor. The data is processed in the hidden layer by an activation function, whilst each node in the output layer is associated to a response.45 An advantage of ANNs against other predictive statistical network approaches is the capacity of an ANN to learn from a set of training examples that contain both the input and output data.45 A potential deficiency of ANNs is the possibility to over-learn or over-fit the network. In such cases, the ANN functions well with the training data points; however, its predictive capacity for other data points is sub optimal. Overlearning can be minimised by monitoring the error of predictions with a verification data set. Verification error that is greater than the training indicates over-fitting.47 ANNs have been applied to the separations of herbicides,48 cosmetic preservatives,49 benzodiazepines,46 organic explosives,50 peptides,51 and fatty acids methyl esters.52
This paper presents the rapid development and optimisation of a UHPLC method for the analysis of 32 OGSR target compounds with the aid of an ANN. To the best of our knowledge, this is the first publication showing the application of an ANN to OGSR.
2. Experimental
2.1 Reagents and standards
A summary of the target OGSR compounds is provided in Table 1. The internal standard 2-naphthol (99.0% certified purity) was obtained from Dr Ehrenstorfer (Augsburg, Bavaria, Germany). HPLC grade acetonitrile (ACN) and methanol (MEOH) were supplied by ChemSupplies Pty Ltd (Gillman, SA, Australia). Ultrapure grade water (18.2 MΩ cm−1) was obtained from a Sartorius 611 water purification system.| Compound | Abbreviation | Function | Standard concentration | Source |
|---|---|---|---|---|
| a Ref. 2, 13, 23, 27, 37 and 57. b Cerilliant – Round Rock, TX, USA; AccuStandard – New Hssaven, CT, USA; ChemService – West Chester, PA, USA. Dr Ehrenstorfer GmbH – Augsburg, Bavaria, Germany. | ||||
| Nitroglycerin | NG | Propellant | 1000 μg mL−1 in ACN | Cerilliant |
| 1,2-Dinitroglycerin | 1,2-DNG | Explosive | 100 μg mL−1 in ACN | AccuStandard |
| 1,3-Dinitroglycerin | 1,3-DNG | Explosive | 100 μg mL−1 in ACN | AccuStandard |
| Nitroguanidine | NGU | Flash suppressor | 100 μg mL−1 in MeOH | AccuStandard |
| 2,4,6-Trinitrotoluene | TNT | Explosive, sensitiser | 1000 μg mL−1 in ACN | ChemService |
| 2,3-Dinitrotoluene | 2,3-DNT | Flash suppressor | 99.5% certified purity | Dr Ehrenstorfer GmbH |
| 2,4-Dinitrotoluene | 2,4-DNT | Flash suppressor | 1000 μg mL−1 in ACN | ChemService |
| 2,6-Dinitrotoluene | 2,6-DNT | Flash suppressor | 1000 μg mL−1 in ACN | ChemService |
| 3,4-Dintirotoluene | 3,4-DNT | Flash suppressor | 1000 μg mL−1 in MeOH | ChemService |
| 4-Amino-2,6-dinitrotoluene | 4-A-2,6-DNT | Flash suppressor | 1000 μg mL−1 in ACN | ChemService |
| 3-Nitrotoluene | 3-NT | Explosive, flash suppressor | 1000 μg mL−1 in ACN | ChemService |
| 1,3,5-Trinitrobenzene | TNB | Explosive | 1000 μg mL−1 in ACN | ChemService |
| 1,3-Dinitrobenzene | 1,3-DNB | Explosive | 1000 μg mL−1 in ACN | ChemService |
| Nitrobenzene | NB | Explosive | 1000 μg mL−1 in ACN | ChemService |
| N,N′-Diphenylurea | N,N′-DPU | Stabiliser, plasticiser | 97.5% certified purity | Dr Ehrenstorfer GmbH |
| Methyl centralite | MC | Stabiliser, plasticiser | 100 μg mL−1 in 50% ACN | AccuStandard |
| Ethyl centralite | EC | Stabiliser, plasticiser | 500 μg mL−1 in ACN | AccuStandard |
| 1,3-Benzenediol | Resorcinol | Stabiliser, plasticiser | 100 μg mL−1 in MeOH | AccuStandard |
| Dimethyl phthalate | DMP | Plasticiser | 1000 μg mL−1 in MeOH | AccuStandard |
| Diethyl phthalate | DEP | Plasticiser | 100 μg mL−1 in MeOH | AccuStandard |
| Dibutyl phthalate | DBP | Plasticiser | 99% certified purity | ChemService |
| Diphenylamine | DPA | Stabiliser | 1000 μg mL−1 in MeOH | AccuStandard |
| 2-Nitrodiphenylamine | 2-NDPA | Stabiliser (DPA derivative) | 100 μg mL−1 in ACN | AccuStandard |
| 4-Nitrosodiphenylamine | 4-nDPA | Stabiliser (DPA derivative) | 99% certified purity | ChemService |
| N-Nitrosodiphenylamine | N-nDPA | Stabiliser (DPA derivative) | 1000 μg mL−1 in MeOH | AccuStandard |
| 2,4-Dinitrodiphenylamine | 2,4-DNDPA | Stabiliser (DPA derivative) | 97.5% certified purity | Dr Ehrenstorfer GmbH |
| Ethylene glycol dinitrate | EGDN | Explosive | 100 μg mL−1 in ACN | AccuStandard |
| Pentaerythritol tetranitrate | PETN | Explosive, sensitiser | 1000 μg mL−1 in ACN | ChemService |
| Octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine | HMX | Explosive | 1000 μg mL−1 in ACN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (1:1) :MeOH (1:1) |
AccuStandard |
| Hexahydro-1,3,5-trinitro-1,3,5-triazine | RDX | Explosive | 1000 μg mL−1 in ACN | ChemService |
| 2,4,6-Trinitrophenylmethyl nitramine | Tetryl | Sensitiser | 1000 μg mL−1 in ACN | ChemService |
| Diazodinitrophenol | DDNP | Initiating explosive | 100 μg mL−1 in ACN | AccuStandard |
2.2 Instrumentation
An Agilent 1290 ultra high performance liquid chromatography (UHPLC) system (Agilent Technologies) was used for all analyses. The system incorporated a binary pump, vacuum degasser, standard autosampler, thermostats for the column and sample compartments, and a UV detector. UV was monitored at 214 nm and 254 nm. ChemStation software (Version B 04.02, Agilent Technologies) was used for instrument control, data acquisition and analysis. Standards and samples were analysed on a Zorbax RRHD Eclipse XDB-C18 3 × 100 mm, 1.8 μm (Agilent Technologies) using a flow rate of 0.8 mL min−1 and a 1 μL injection volume. A 1290 Infinity in-line filter (0.2 μm, Agilent Technologies) was installed to prolong column lifetime.The identities of the compounds detected by UHPLC analysis with UV detection were confirmed by mass spectrometric detection using an Agilent Technologies 6490 triplequad mass spectrometer controlled by MassHunter software version B.06.00 (Agilent Technologies). The mass spectrometer was connected to an atmospheric pressure chemical ionisation (APCI) ion source (G1947 A/B) from Agilent Technologies. Multiple reaction monitoring (MRM) mode was used, which provides a high degree of certainty in identifying compounds based on their precursor-to-product transitions. The conditions for the MRM of the compounds were optimised either using the Optimizer Software (Agilent Technologies) or manually and are shown in Table 2. The most abundant and specific ions were chosen.
| Compound | Ionization mode | Precursor ion [m/z] | Product ions [m/z] |
|---|---|---|---|
| 2-Naphthol | APCI+ | 145.07 | 104.0, 62.9, 60.1 |
| DPA | APCI+ | 170.1 | 152.1, 93.0, 78.0 |
| N-nDPA | APCI+ | 199.0 | 181.0, 128.0, 77.0 |
| 4-nDPA | APCI+ | 199.09 | 182.0, 128.0, 126.9 |
| 2-NDPA | APCI+ | 215.08 | 197.0, 180.9, 180.0 |
| EC | APCI+ | 269.16 | 148.1, 119.9, 92.1 |
| MC | APCI+ | 241.14 | 134.1, 106.0, 93.1 |
| NG | APCI− | 226.0 | 195.9, 133.9, 75.9 |
| DNG | APCI− | 181.01 | 85 |
| DNG | APCI+ | 183.03 | 77.0, 51.2 |
2.3 Experimental design
Preliminary scouting experiments were conducted to determine independent mobile phase parameters and to define the experimental space. A mixture of the compounds was injected and different selectivity variables tested. It was determined that mobile phase strength and temperature had the greatest influence on the resolution of the OGSR mix which is in agreement with previous studies.53–55 These two factors were compared in their influence on the resolution of the compounds of interest, whereby mobile phase and temperature were changed and the shift of the retention time and peak width was measured. The gradient was the most significant factor affecting resolution, followed by the temperature. MeOH/water was selected as the mobile phase as it has been previously applied to GSR analysis. Additionally, MeOH presents a smaller environmental hazard than ACN.56 No buffer was used. A starting concentration of 30% MeOH and 70% water was chosen as the initial concentration for all gradients as this ensured that all compounds eluted after the void time. Linear gradient slopes of 0.6, 1.2, 1.8, 3.0 and 6.0% MeOH per min at 23 °C were examined. A schematic diagram of the experimental space is shown in Fig. 1.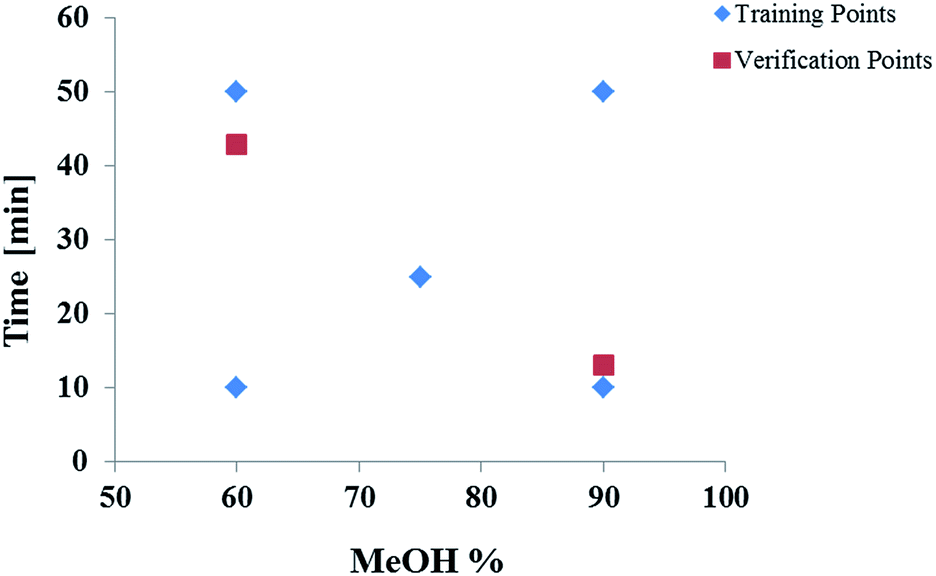 | ||
| Fig. 1 Representation of gradients defining the experimental space for input data to the ANN. Five gradients were used as training points to train the network, two gradients were used as verification data to mitigate overlearning. | ||
All 33 compounds (32 OGSR and one internal standard) were run individually in duplicates at the five different gradients to provide the training data (average retention times) for the ANN. Aliquots of the stock standards were diluted for UHPLC analysis in MeOH:ACN (1:1) to generate solutions at working concentrations of 20 ppm with 20 ng injected. Two gradients at 0.7% per min and 4.6% per min were used to provide verification data in order to examine the suitability of the ANN for the prediction of the average retention times.
2.4 Artificial neural networks
Trajan Neural Networks, Version 6.0 (Trajan Software Ltd.), was used for simulating the ANNs and predicting optimised experimental conditions. The slope of the five different gradients were presented to the software as input data and the average retention times of the 33 compounds respectively for each gradient were used as output values. An automated heuristic approach was applied to determine the optimal architecture of the ANN. During this process, the number of nodes in the hidden layer in the architecture was varied, and the network with the lowest error selected. This network was used to predict gradients between 0.6% per min and 6% per min with increments of 0.1% per min.2.5 Additional separation optimisation
The best separation by the ANN identified from the shortest time and highest resolution was further optimised. This involved assessing additional variables such as temperature and the initial methanol concentration. The temperatures evaluated were 23 °C, 35 °C, 37 °C, 39 °C, 41 °C, 43 °C, and 45 °C, while the initial concentration of MeOH was optimised by monitoring peak shape and number of peaks at 30%, 25%, 20%, 15%, and 10% of MeOH. Isocratic intervals were also introduced to the gradient profile to further separate the structural isomers and improve low resolution areas of the chromatogram.2.6 Method validation
Working standard solutions in the range of 0.1–100 ng were injected in seven replicates to construct calibration curves. The limits of detection (LODs) were calculated using (3.3 × σ)/S, where σ is the standard deviation of the slope and S the slope of the calibration curve. The limits of quantification (LOQs) were based on 3.3 × LOD. Intra and inter-day variations were examined by analysing the standards on three randomly chosen days with n = 7 in the morning and n = 7 in the evening.2.7 Ammunitions, firearms and sample preparation
| Ammunition number | Caliber | Ammunition manufacturer (country of origin) | Firearm model (country of origin) |
|---|---|---|---|
| 1 | WinClean 45 (LF) | Winchester (Australia) | Colt (USA) |
| 2 | 45 Auto CP | Winchester (Australia) | Colt (USA) |
| 3 | 44 Rem Magnum | PMC (USA) | Smith & Wesson model 629-4 (USA) |
| 44 Rem Magnum | Winchester (Australia) | Smith & Wesson model 629-4 (USA) | |
| 4 | 9 mm Parabellum | Blazer, CCI (USA) | Beretta model 902FS (Italy) |
| 5 | 357 Magnum | Winchester (Australia) | Smith & Wesson revolver model 686-3 (USA) |
| 357 Magnum | PMC (USA) | Smith & Wesson revolver model 686-3 (USA) | |
| 6 | 22 LR high velocity | Remington (USA) | Sport King (USA) |
| 22 LR high velocity | Winchester (Australia) | Sport King (USA) | |
| 22 LR high velocity | Remington (USA) | Rifle 70 (Marlin, USA) | |
| 22 LR high velocity | Winchester (Australia) | Rifle 70 (Marlin, USA) | |
| 7 | 40 S&W WinClean (LF) | Winchester (Australia) | Glock (Austria) |
| 8 | 40 S&W | Winchester (Australia) | Glock (Austria) |
| 9 | 12 gauge (SuperX) | Winchester (Australia) | Shotgun, model 870 (Remington, USA) |
5 mL MTBE were added to the swab in a scintillation vial. After a 5 min sonication, the solvent was removed under a steady stream of nitrogen gas and the sample reconstituted in 196 μL of mobile phase and 4 μL internal standard was added. The sample was filtered using a 0.2 μm syringe filter prior to analysis by UHPLC.
3. Results and discussion
3.1 ANN training
The separation of the large number of explosives possibly present in OGSR is problematic due to the significant number of isomers, such as DNT and/or amino-DNT isomers. The use of an ANN is an efficient means to separate the greatest number of compounds with a linear gradient. Further small refinements may then be made such as increasing temperature; varying MeOH starting concentration; and introduction of isocratic conditions in regions of poor separation, particularly where the structural isomers elute.Initially, the ANN training data consisted of five linear gradient slopes as the independent input variable, whilst the average of the duplicate retention times for each of the 33 standards was used as the dependent output variables. These 165 experimental points were used to construct a suitable ANN architecture to adequately model the response surface. An iterative heuristic process resulted in a multi-layer perceptron network with one input node, 19 nodes in the hidden layer, and 33 nodes in the output layer representing the retention times of the 33 compounds (Fig. 2).
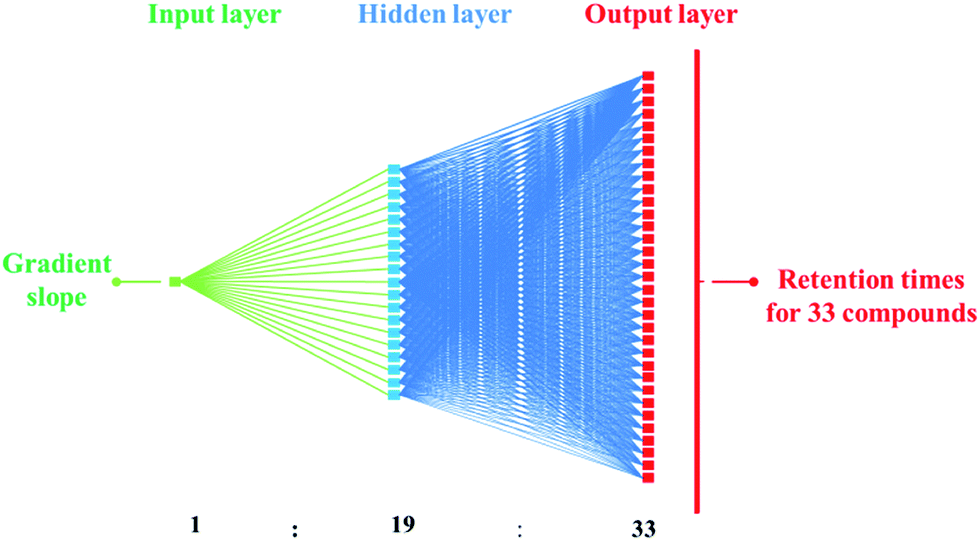 | ||
| Fig. 2 Schematic diagram of the 1:1–19–33:33 MLP network providing the smallest error for the prediction of the retention times of the 33 compounds of interest. The gradient slope represents the input data, the retention times are given through the output data. | ||
This network was used to predict retention times for all compounds within the experimental space with gradient increments of 0.1% per min. The minimum peak pair difference (the retention time difference between two closest peaks) was calculated and a response resolution plot constructed (Fig. 3). The highest point on the response resolution plot (0.7% per min MeOH) represented the best performing gradient in terms of resolution. However, the predicted analysis time was 54.0 min, which is of limited practical use. Therefore a compromise between resolution and runtime was made, with the gradient at 4.6% per min offering a suitable outcome in terms of run time and resolution. This gradient separated 22 compounds, whilst 11 compounds co-eluted.
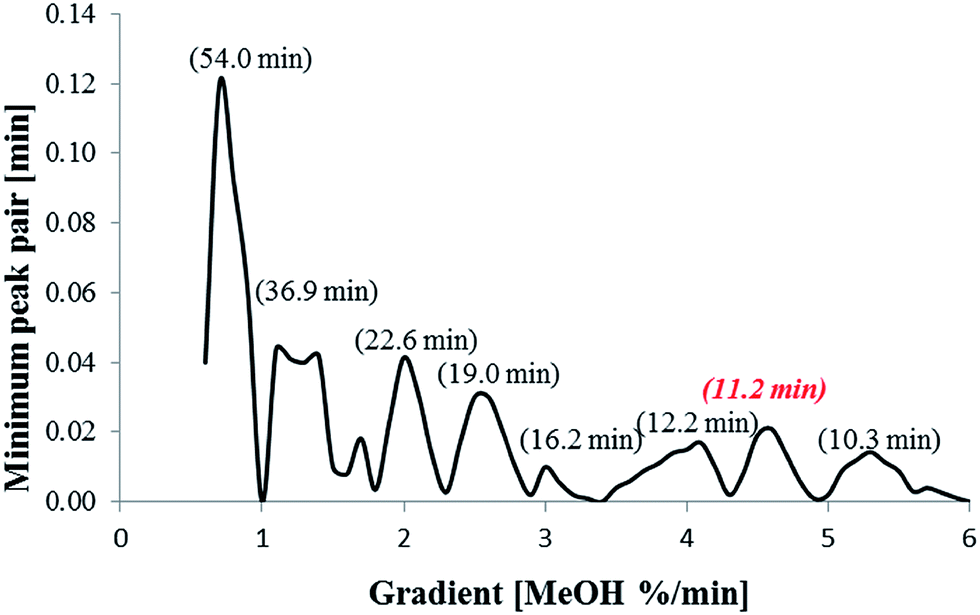 | ||
| Fig. 3 Response resolution plot. The minimum peak pair is plotted versus the gradient (MeOH% per min). The run times of the maxima of the minimum peak pairs (representing the best resolution) are shown in the brackets. The gradient with 4.6% per min MeOH increase was used as it provided efficient resolution and short analysis time. | ||
The possibility of overfitting was eliminated by running the individual standards at gradients of 0.7% per min and 4.6% per min for use as verification data points for retraining of the ANN. Using the same process as described before, the ANN with the smallest error was again determined to be a MLP network with 1:1–19–33:33 architecture. This together with the high correlation (R2 = 0.999) between observed and predicted retention times demonstrated that the ANN adequately described the response surface. Differences between measured and predicted retention times were in the range between 0–7.44%.
3.2 Additional optimisation
The gradient slope identified using ANN was excellent, except for isomers which required addition optimisation, and was applied throughout additional optimisation process which consisted of modifications to the initial concentration of MeOH%; increasing the column temperature; and incorporation of isocratics steps.The initial MeOH concentration of the mobile phase had the strongest influence on the first eight peaks; RDX and 1,3-DNG coeluted at 5% MeOH; whilst EGDN and HMX coeluted at 15% MeOH. All of the first eight peaks were baseline separated and clearly distinguishable at 10% MeOH. Increasing the temperature to 43 °C further increased the resolution due to improved mass transfer with 27 peaks separated. Two isocratic steps were then introduced to separate tetryl, TNT, DMP and the DNT isomers. Separation of the 33 target analytes is shown in Fig. 4.
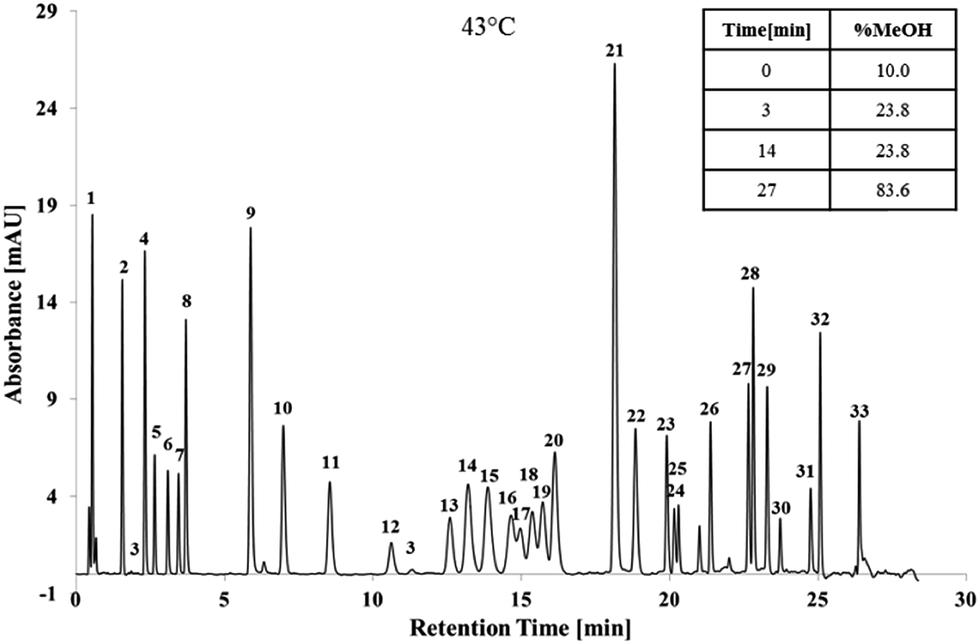 | ||
| Fig. 4 Optimised separation of 32 organic gunshot residue compounds under 214 nm. 1 = NGU, 2 = resorcinol, 3 = DDNP, 4 = RDX, 5 = 1,3-DNG, 6 = 1,2-DNG, 7 = EGDN, 8 = HMX, 9 = TNB, 10 = 1,3-DNB, 11 = NB, 12 = NG, 13 = tetryl, 14 = TNT, 15 = 4-A-2,6-DNT, 16 = 3,4-DNT, 17 = DMP, 18 = 2,4-DNT, 19 = 2,6-DNT, 20 = 2,3-DNT, 21 = 2-naphthol (IS), 22 = m-NT, 23 = DEP, 24 = N,N′-DPU, 25 = PETN, 26 = 4-nDPA, 27 = MC, 28 = N-nDPA, 29 = DPA, 30 = 2,4-DNDPA, 31 = 2-NDPA, 32 = EC, 33 = DBP. | ||
3.3 Method validation
The analytical figures of merit are shown in Table 4. Relative standard deviation (RSD) values of retention times were between 0.010% (EC, MC) and 1.2% (1,2-DNG), area RSD values between 0.18% (MC) and 1.6% (2,4-DNDPA), and k′ RSD values between 0.010% (DBP) and 1.5% (1,2-DNG). The method showed high linearity with correlation coefficients (R2) between 0.988 (DDNP) and 0.999 (2,3-DNT, m-NT, 4-A-2,6-DNT, 2-NDPA, DMP, DBP, N,N′-DPU), y-intercepts between 0.00023 (NG) and 0.041 (DDNP), and slopes between 0.00048 (2,4-DNDPA) and 0.025 (4-A-2,6-DNT); standard errors of the calibration curve ranged between 4.3 × 10−3 (resorcinol) and 3.4 × 10−5 (2,4-DNDPA). The lowest RSD of the capacity factors was 0.0100% (DBP); whilst the highest was for NGU at 4.00%. Peak area RSDs ranged between 0.201% (1,3-DNG) and 1.55% (2,4-DNDPA). The LODs were between 0.03 ng (N,N′-DPU, 2-NDPA) and 0.21 ng (DDNP) at 214 nm; comparable to values previously reported using UHPLC-UV detection.38 LOQs were between 0.10 ng (N,N′-DPU, 2-NDPA) and 0.71 ng (2,4-DNDPA). These LODs indicate that the method is suitable for casework application.2,37,60,61 Intra and interday variations were determined over a three day period by comparing the peak area of the seven replicate analyses. The mean intraday variation across the three day period was between 0.22% RSD (NGU) and 9.7% RSD (2-NDPA). The interday variation ranged from 0.017% RSD (N,N′-DPU) to 12% RSD (2,4-DNDPA).| Compound | Average retention time [min] | Capacity factor, k′ | Sensitivity | |
|---|---|---|---|---|
| LOD [ng] | LOQ [ng] | |||
| a Internal standard. | ||||
| NG | 10.55 | 18.9 | 0.14 | 0.43 |
| 1,2-DNG | 3.134 | 4.91 | 0.085 | 0.26 |
| 1,3-DNG | 2.693 | 4.08 | 0.068 | 0.21 |
| NGU | 0.5430 | 0.0200 | 0.11 | 0.33 |
| TNT | 13.26 | 23.9 | 0.075 | 0.23 |
| 2,3-DNT | 16.07 | 29.3 | 0.041 | 0.12 |
| 2,4-DNT | 16.16 | 29.5 | 0.13 | 0.40 |
| 2,6-DNT | 15.60 | 28.4 | 0.069 | 0.21 |
| 3,4-DNT | 14.51 | 26.34 | 0.062 | 0.18 |
| m-NT | 18.81 | 34.5 | 0.041 | 0.12 |
| 4-A-2,6-DNT | 13.76 | 25.0 | 0.044 | 0.13 |
| TNB | 5.913 | 10.1 | 0.078 | 0.24 |
| m-DNB | 7.018 | 12.2 | 0.065 | 0.20 |
| NB | 8.593 | 15.1 | 0.068 | 0.20 |
| DPA | 23.30 | 42.9 | 0.091 | 0.28 |
| 2-NDPA | 24.77 | 45.7 | 0.035 | 0.10 |
| 4-nDPA | 20.99 | 38.6 | 0.059 | 0.18 |
| N-nDPA | 22.67 | 41.7 | 0.080 | 0.24 |
| 2,4-DNDPA | 23.94 | 44.2 | 0.23 | 0.71 |
| Resorcinol | 1.548 | 1.92 | 0.11 | 0.32 |
| EC | 25.05 | 46.3 | 0.11 | 0.32 |
| MC | 22.80 | 42.0 | 0.079 | 0.24 |
| RDX | 2.323 | 3.37 | 0.079 | 0.24 |
| HMX | 3.706 | 5.97 | 0.075 | 0.23 |
| PETN | 20.17 | 37.0 | 0.10 | 0.31 |
| Tetryl | 12.51 | 22.6 | 0.054 | 0.16 |
| EGDN | 3.435 | 5.48 | 0.087 | 0.26 |
| DMP | 14.91 | 27.1 | 0.039 | 0.12 |
| DEP | 21.51 | 39.6 | 0.13 | 0.40 |
| DBP | 26.38 | 48.8 | 0.034 | 0.11 |
| N,N′-DPU | 19.86 | 36.5 | 0.034 | 0.10 |
| DDNP | 1.848 | 2.48 | 0.21 | 0.65 |
| 2-Naphthola | 18.16 | 33.2 | N/A | N/A |
3.4 Simulated case samples
The method was applied to a real case scenario using smokeless powder samples and hand swabs taken after shooting a firearm. A total of 78 test fires were conducted in triplicate. The sample preparation of the hand swabs was previously developed and tested on samples taken from skin spiked with OGSR. The mean recovery for targeted OGSR collected from hands was 18%, ranging from 9% (TNB) to 24% (NG). Possible interferences were only found for DBP. Example chromatograms for ammunition 8 (lead containing 40 S&W) before and after shooting using a 22 Glock (Austria) are shown in Fig. 5.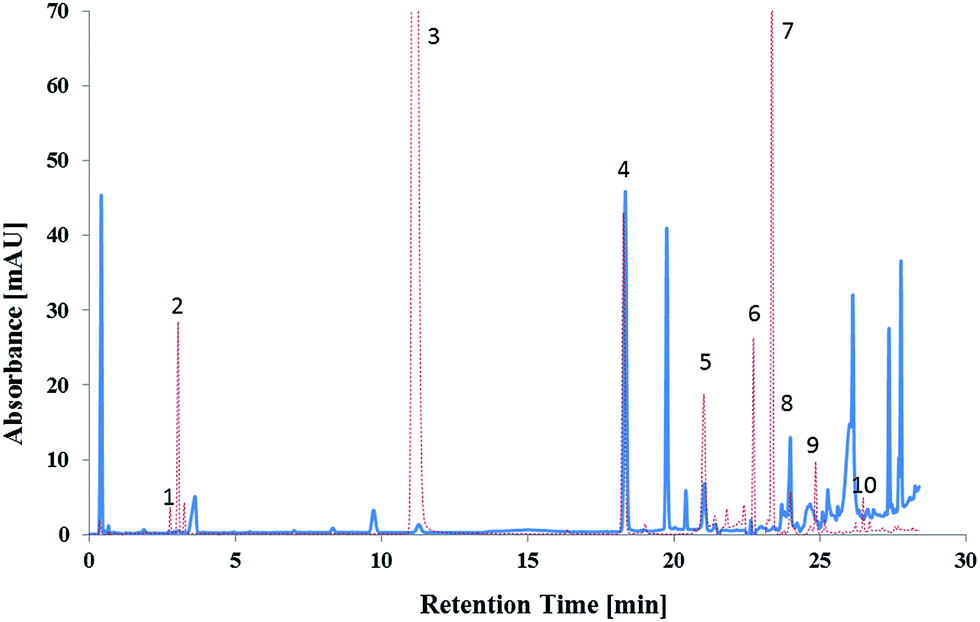 | ||
| Fig. 5 Overlaid chromatograms of smokeless powder before shooting (40 S&W, Winchester, Australia; red dashed line) and the gunshot residues collected from the hands of a shooter after discharge using a 22 Glock pistol (blue line). 1 = 1,2-DNG, 2 = 1,3-DNG, 3 = NG, 4 = 2-naphthol (IS), 5 = DEP, 6 = MC, 7 = DPA, 8 = 2-NDPA, 9 = EC, 10 = DBP. | ||
Various OGSR were detected in the unburned smokeless powders and hand swab samples. All UHPLC-UV detections were confirmed by tandem mass spectrometry using the above outlined method. Each of the tested powders included NG, thus was a double based powder. NG was found in 62.2% of the hand swab samples, 1,2-DNG in 66.7% and 1,3-DNG in 34.6%. The stabilisers DPA, EC and MC were detected in 47.4%, 73.5%, and 72.9% of the hand swab samples respectively. The fact that EC and MC were detected in approximately 3/4 of the hand swab samples after firing ammunition is of high importance since MC and EC are considered to be the most characteristic compounds for OGSR.37,62 DPA alone is not characteristic for OGSR. However, when detected in combination with its derivates, it is considered as indicative for OSGR.63 DPA derivates detected included N-nDPA (5.1%), 4-nDPA (6.4%), and 2-NDPA (75.6%). The results show, that the developed method can provide vital information in the investigation of a firearm related event. Additionally, the combined detection of the various compounds increases the evidential value of the developed method for OSGR investigation.
Moreover, the method was able to quantify the compounds of interest present in smokeless powder samples and hand swabs after only a single cartridge was discharged. For the different powders tested before and after shooting, differences in the chemical composition could be found and profiles for each powder established. The method has therefore the potential to distinguish between different ammunitions based on their varying composition and potentially link ammunition to the OGSR found on the hands of a shooter.
The concentrations of the detected compounds were relatively low (in the low ng range), which is in consistency with previous reported concentration2,64 and underlines the significance of a sensitive method as developed here.
It is important to consider, that swabs were used in order to collect OGSR from the hands of a shooter. Studies comparing swabbing of OGSR to other commonly applied collection techniques can inform on which technique is most suitable for OGSR collection. Applying the most suitable collection technique might ultimately improve the collection efficiency and increase the levels of OGSR detected. Since police commonly uses GSR stubs in order to collect IGSR from hands, it should be tested whether OGSR extracted from GSR stubs can be detected using the developed method.
In order to implement such a method in routine casework additional research is required along with individual laboratory validation studies in order to ensure that quality standards for OGSR investigations are met.
4. Conclusion
A gradient UHPLC method was developed for the quantitative analysis of 32 OGSR using an ANN for rapid optimisation. The ANN was trained with average retention times and provided excellent correlation between observed and predicted retention times with errors between 0 and 7.44%. The ANN predicted a gradient at 4.6% per min MeOH providing the best compromise between resolution and run time, with 22 compounds baseline separated. Further optimisations of the initial MeOH concentration, temperature and implementation of two isocratic steps resulted in separation of all 32 OGSR and the internal standard. The final method separated all of the target analytes in under 27 min with detection limits between 0.03 and 0.21 ng at 214 nm. The LODs were lower than previously reported OGSR concentrations in simulated scenarios.2,37,60,61 The relatively large number of target compounds and low LODs indicate the method is applicable for forensic investigations of firearm related incidents. OGSR were identified in a real case scenario whereby samples were taken from shooters hands at a shooting range. Further research will focus on the collection and extraction protocols for GSR samples in order to establish a new protocol for GSR collection allowing the analysis of both, IGSR and OGSR.Acknowledgements
This project is supported by a Premier's Research and Industry Fund grant provided by the South Australian Government Department of Further Education, Employment, Science and Technology.References
- U. N. O. O. D. A. Crime, Global Study on Homicide – Trends, Contexts, Data, UNODC, Vienna, 2011 Search PubMed.
- H.-H. Meng and B. Caddy, J. Forensic Sci., 1997, 42(4), 553–570 CAS.
- O. Dalby, D. Butler and J. W. Birkett, J. Forensic Sci., 2010, 55(4), 924–943 CrossRef PubMed.
- V. J. M. Di Maio, Gunshot Wounds – Practical Aspects of Firearms, Ballistics and Forensic Techniques, CRC Press, Florida, Boca Raton, 2nd edn, 1999 Search PubMed.
- E. Turillazzi, et al. , Forensic Sci. Int., 2013, 231(1–3), 142–149 CrossRef CAS PubMed.
- M. López-López and C. García-Ruiz, Forensic Sci. Int., 2014, 239, 79–85 CrossRef PubMed.
- A. Santos, et al. , Forensic Sci. Int., 2015, 247, 62–68 CrossRef CAS PubMed.
- C. Andersson and J. Andrasko, J. Forensic Sci., 1999, 44(1), 211–213 Search PubMed.
- J. Andrasko and S. Ståhling, J. Forensic Sci., 1999, 44(3), 487–495 Search PubMed.
- J. Andrasko and S. Ståhling, J. Forensic Sci., 2000, 45(6), 1250–1255 CAS.
- W. A. MacCrehan, et al. , J. Forensic Sci., 2001, 46(1), 57–62 CAS.
- W. A. MacCrehan, M. R. Reardon and D. L. Duewer, J. Forensic Sci., 2002, 47(2), 260–266 CAS.
- J. Bueno, V. Sikirzhytski and I. K. Lednev, Anal. Chem., 2012, 84(10), 4334–4339 CrossRef CAS PubMed.
- M. Lopez-Lopez, J. L. Ferrando and C. Garcia-Ruiz, Anal. Chem., 2012, 84, 3581–3585 CrossRef CAS PubMed.
- Z. Brożek-Mucha and A. Jankowicz, Forensic Sci. Int., 2001, 123(1), 39–47 CrossRef.
- J. Yañez, et al. , Microchem. J., 2012, 101, 43–48 CrossRef PubMed.
- Z. Brożek-Mucha, BioMed Res. Int., 2014, 2014, 11 Search PubMed.
- H. H. Meng and H.-C. Lee, J. Forensic Sci., 2007, 6(1), 39–54 Search PubMed.
- A. M. O'Mahony, et al. , Anal. Chem., 2014, 86(16), 8031–8036 CrossRef PubMed.
- A. Martiny, et al. , Forensic Sci. Int., 2008, 177(1), e9–e17 CrossRef PubMed.
- Wildlife C. D. O. F. A., cited 2014 22.10., Available from, http://www.dfg.ca.gov/wildlife/hunting/lead-free/.
- Chiaramonte P., 2013, cited 2014 22.10., Available from, http://www.foxnews.com/us/2013/12/21/end-line-for-lead-bullet-regulations-bans-force-switch-to-green-ammo/.
- Z. Oommen and S. M. Pierce, J. Forensic Sci., 2006, 51(3), 509–519 CrossRef CAS PubMed.
- S. Hales, Improving Forensic Casework Analysis and Interpretation of Gunshot Residue (GSR) Evidence, PhD Thesis, Center of Forensic Science, University of Technology, Sydney, 2011.
- Z. Abrego, et al. , Analyst, 2014, 139(23), 6232–6241 RSC.
- L. Gunaratnam and K. Himberg, J. Forensic Sci., 1994, 39(2), 532–536 CAS.
- A. Harris, J. Forensic Sci., 1995, 40(1), 27–30 CAS.
- W. A. MacCrehan, K. D. Smith and W. F. Rowe, J. Forensic Sci., 1998, 43(1), 119–124 CAS.
- S. Charles, B. Nys and N. Geusens, Forensic Sci. Int., 2011, 212(1–3), 22–26 CrossRef PubMed.
- C. Torre, et al. , J. Forensic Sci., 2002, 47(3), 494–504 CAS.
- P. V. Mosher, et al. , Can. Soc. Forensic Sci. J., 1998, 31, 157–168 CrossRef CAS PubMed.
- J. S. Wallace and J. McQuillan, J. Forensic Sci. Soc., 1984, 24(5), 495–508 CrossRef CAS.
- R. E. Berk, et al., An Empirical Study, J. Forensic Sci., 2007, 52(4), 838–841 CrossRef PubMed.
- S. Charles and N. Geusens, Forensic Sci. Int., 2012, 216(1–3), 78–81 CrossRef PubMed.
- J. French and R. Morgan, Forensic Sci. Int., 2015, 247(0), 14–17 CrossRef CAS PubMed.
- J. French, R. Morgan and J. Davy, X-Ray Spectrom., 2014, 43(1), 56–61 CrossRef CAS PubMed.
- D. Laza, et al. , J. Forensic Sci., 2007, 52(4), 842–850 CrossRef CAS PubMed.
- J. L. Thomas, D. Lincoln and B. R. McCord, J. Forensic Sci., 2013, 58(3), 609–615 CrossRef CAS PubMed.
- D. Muller, et al. , J. Forensic Sci., 2007, 52(1), 75–78 CrossRef CAS PubMed.
- A. Zeichner, et al. , J. Forensic Sci., 2003, 48(5), 961–972 CAS.
- D. M. Northrop, J. Forensic Sci., 2001, 46(3), 549–559 CAS.
- C. M. Mahoney, G. Gillen and A. J. Fahey, Forensic Sci. Int., 2006, 158(1), 39–51 CrossRef CAS PubMed.
- M. Morelato, et al. , Forensic Sci. Int., 2012, 217(1–3), 101–106 CrossRef CAS PubMed.
- R. V. Taudte, et al. , BioMed Res. Int., 2014, 2014, 965403 Search PubMed.
- L. V. Fausett, Fundamentals of Neural Networks, ed. D. Fowley, Prentice-Hall, Inc, New Jersey, US, 1994 Search PubMed.
- R. Webb, P. Doble and M. Dawson, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877(7), 615–620 CrossRef CAS PubMed.
- P. Doble, et al. , Forensic Sci. Int., 2003, 132(1), 26–39 CrossRef CAS.
- A. T. K. Tran, et al. , Talanta, 2007, 71(3), 1268–1275 CrossRef CAS PubMed.
- E. Marengo, et al. , J. Chromatogr. A, 2004, 1029(1–2), 57–65 CrossRef CAS PubMed.
- S. Casamento, et al. , J. Forensic Sci., 2003, 48(5), 1075–1083 CAS.
- K. Petritis, et al. , Anal. Chem., 2003, 75(5), 1039–1048 CrossRef CAS.
- V. K. Gupta, et al. , Talanta, 2011, 83(3), 1014–1022 CrossRef CAS PubMed.
- J. W. Dolan, J. Chromatogr. A, 2002, 965(1–2), 195–205 CrossRef CAS.
- P. L. Zhu, et al. , J. Chromatogr. A, 1996, 756(1–2), 21–39 CrossRef CAS.
- P. L. Zhu, J. W. Dolan and L. R. Snyder, J. Chromatogr. A, 1996, 756(1–2), 41–50 CrossRef.
- T. Borch and R. Gerlach, J. Chromatogr. A, 2004, 1022(1–2), 83–94 CrossRef CAS PubMed.
- B. Paull, et al. , J. Forensic Sci., 2004, 49(6), 1–6 CrossRef.
- J. A. Mathis and B. R. McCord, J. Chromatogr. A, 2003, 988(1), 107–116 CrossRef CAS.
- C. E. Wissinger and B. R. McCord, J. Forensic Sci., 2002, 47(1), 168–174 CAS.
- Z. P. Wu, et al. , Analyst, 1999, 124(11), 1563–1567 RSC.
- M. Zhao, et al. , J. Forensic Sci., 2008, 53(4), 807–811 CrossRef CAS PubMed.
- M. H. Mach, A. Pallos and P. F. Jones, J. Forensic Sci., 1978, 23(3), 446–455 CAS.
- Y. Tong, et al. , Analyst, 2001, 126, 480–484 RSC.
- Z. Wu, et al. , J. Forensic Sci., 2001, 46(3), 495–501 CAS.
| This journal is © The Royal Society of Chemistry 2015 |