
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Simultaneous intracellular redox potential and pH measurements in live cells using SERS nanosensors†
L. E.
Jamieson
a,
A.
Jaworska
bc,
J.
Jiang
a,
M.
Baranska
bc,
D. J.
Harrison
d and
C. J.
Campbell
*a
aEaStCHEM, School of Chemistry, University of Edinburgh, Edinburgh, EH9 3JJ, UK. E-mail: colin.campbell@ed.ac.uk
bFaculty of Chemistry, Jagiellonian University, 3 Ingardena Str., 30-060 Krakow, Poland
cJagiellonian Centre for Experimental Therapeutics (JCET), Jagiellonian University, 14 Bobrzynskiego Str., 30-348 Krakow, Poland
dMedical and Biological Sciences Building, University of St Andrews, North Haugh, St Andrews, Fife, KY16 9TF, UK
First published on 20th February 2015
Abstract
Intracellular redox potential is a highly regulated cellular characteristic and is critically involved in maintaining cellular health and function. The dysregulation of redox potential can result in the initiation and progression of numerous diseases. Redox potential is determined by the balance of oxidants and reductants in the cell and also by pH. For this reason a technique for quantitative measurement of intracellular redox potential and pH is highly desirable. In this paper we demonstrate how surface enhanced Raman scattering (SERS) nanosensors can be used for multiplexed measurement of both pH and redox potential in live single cells.
Introduction
Intracellular redox potential is a measure of the oxidising or reducing potential of the environment within a cell. It is a delicate balance between a number of species including reactive oxygen species (ROS), antioxidant enzymes and small molecules that exist as redox couples.1,2 These numerous intracellular species interact with each other to maintain a tightly regulated redox potential.3 Cellular redox potential and its regulation is critically important for maintaining cellular health and integrity and is involved in the regulation of numerous essential processes.4–7 When the delicate balance of intracellular redox potential is disrupted the resulting consequences include the initiation and progression of numerous diseases including neurodegeneration,8 cardiovascular diseases9 and cancer.8,10The importance of redox potential in vital aspects of cell function and the implication of disruption of redox potential on disease makes establishing an effective method for measuring intracellular redox potential an important goal. Techniques such as the glutathione recycling assay allow an estimate of redox potential to be made in a population of cells, but lack any information about dynamics or cell-to-cell variability.11 Fluorescent dyes such as DCFH12 allow the measurement of ROS, but not a quantitative measurement of potential. More recently dyes such as Redoxfluor-1 (RF1)13 have been used to report reversibly on intracellular redox potential. These dyes have the disadvantage that they act as ‘switches’ only determining whether the redox potential is above or below a certain value, the half-cell potential of the dye, thus failing to give quantitative determination of intracellular redox potential. The current ‘gold standard’ technique for measuring intracellular redox potential is the use of redox active Green Fluorescent Proteins (roGFPs).14–16 Various roGFPs have been engineered with specific cysteine residues, which allow the protein to exist as a redox couple either with two free thiols or a disulfide (cystine). Depending on the oxidation state of the residues the fluorescence excitation maxima of the roGFP changes and therefore the ratio of reduced to oxidised roGFP can be determined and thus the redox potential is calculated. This technique allows in vivo real time and reversible monitoring of intracellular redox potential. It also has the advantage that the engineered roGFPs can be designed to target particular organelles and therefore report on intracellular variations.14 While this technique has many advantages over the previous techniques developed roGFPs have been reported to primarily respond to glutathione concentration17 and thus the range over which they can report is relatively small and, while recent developments have seen improvements in this area, there is a need for a complementary approach that allows quantification of a larger range of potentials.
Our group has developed a technique which involves using surface enhanced Raman scattering (SERS) nanosensors that report on intracellular redox potential in live cells. We used redox sensitive probe molecules attached to gold nanoparticles (NPs) which are delivered to cells and whose SERS spectra report in a ratiometric manner on the redox potential of their intracellular environment.18,19
When making a redox potential measurement it is also necessary to measure pH as the calculation of redox potential is pH dependent.19 Since SERS has previously been used to make intracellular pH measurements20–23 we consider that there is an opportunity to combine these nanosensors to make a multiplexed measurement of redox potential and pH and therefore gain a more accurate measure of the microenvironment within the cell.
Results and discussion
Nanosensor calibration
MBA has been well documented as a pH sensitive SERS reporter.20–24 MBA nanosensors (NS) (Fig. 1A) have pH responsive signals in their SERS spectra, most significantly signals at approximately 1400 cm−1 and approximately 1700 cm−1, assigned to COO− stretching and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching respectively. The COO− stretching mode signal intensity increases with increasing pH, due to increased MBA deprotonation and the CO stretching mode signal intensity increases with decreasing pH. The strong signals at approximately 1080 cm−1 and approximately 1590 cm−1 are attributed to pH independent aromatic ring vibration and can therefore be used as reference signals.
O stretching respectively. The COO− stretching mode signal intensity increases with increasing pH, due to increased MBA deprotonation and the CO stretching mode signal intensity increases with decreasing pH. The strong signals at approximately 1080 cm−1 and approximately 1590 cm−1 are attributed to pH independent aromatic ring vibration and can therefore be used as reference signals.
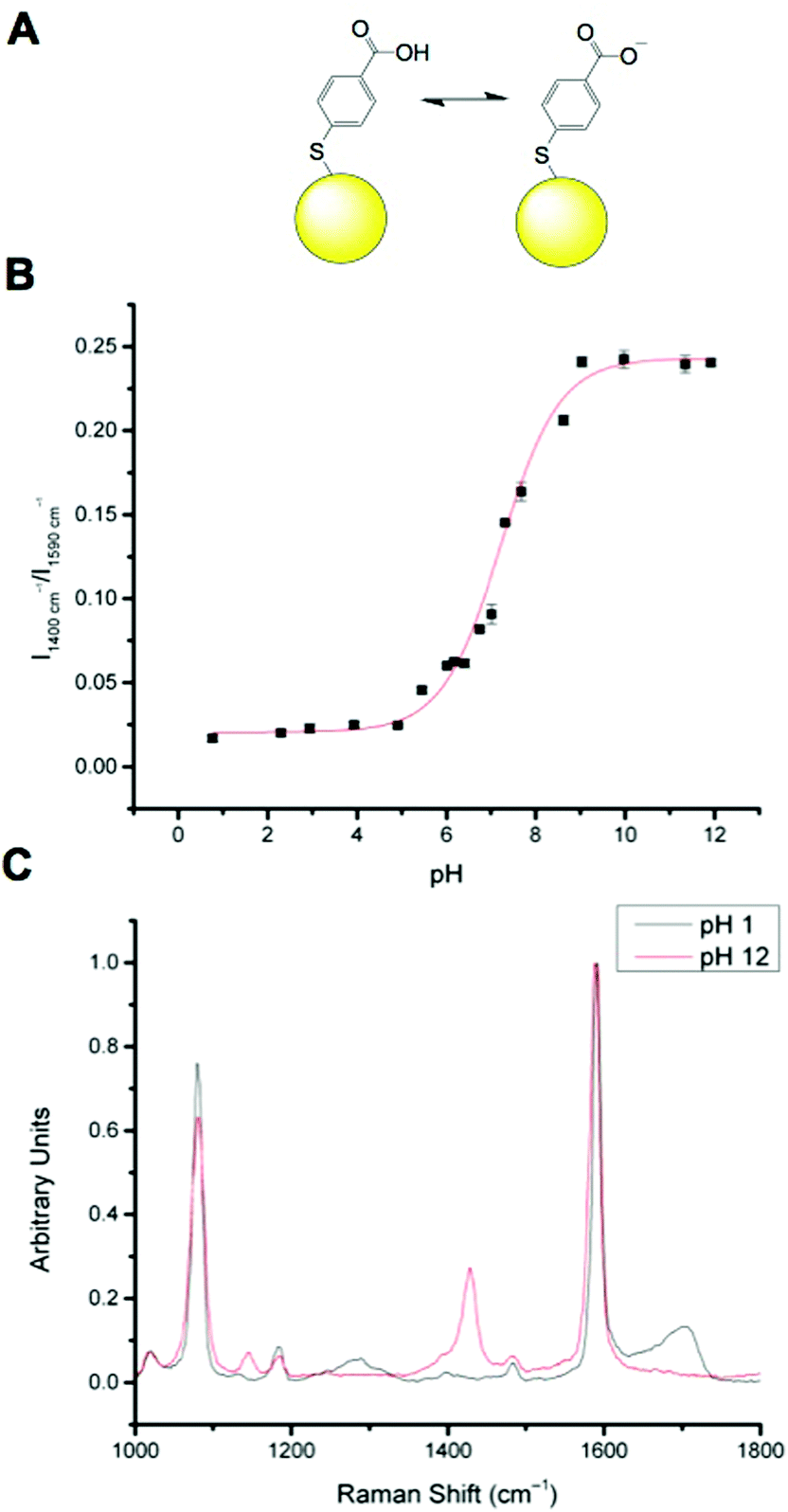 | ||
| Fig. 1 MBA-NP nanosensor. (A): Equilibrium between protonated and deprotonated MBA attached to a gold NP (yellow sphere, not to scale). (B): Calibration curve for MBA-NP nanosensors (R2 = 0.98687). (C): Representative spectra of fully protonated (pH 1) and fully deprotonated (pH 12) MBA-NPs. | ||
Calibration of MBA nanoparticles (MBA-NPs) is generated by plotting pH vs. relative intensity of selected signals. The expected sigmoid is centred around the pKa of the MBA and demonstrates that MBA-NPs are most sensitive to changes in pH between 6 and 8 (Fig. 1B). This shape of curve is to be expected since the transition between acid and base is expected to follow the Henderson–Hasselbach equation. The pH range offered by the MBA-NPs is well suited to the measurement of intracellular pH and we have previously demonstrated that MBA-NPs can accurately report on changes to the cytosolic pH.23 Representative spectra of fully protonated and deprotonated MBA-NPs are shown in Fig. 1C.
We have previously reported that N-[2-({2-[(9,10-dioxo-9,10-dihydroanthracen-2-yl)formamido]ethyl}disulfanyl)ethyl]-9,10-dioxo-9,10-dihydroanthracene-2-carboxamide (referred to as AQ) can be used as a redox sensitive SERS reporter when conjugated to gold nanoparticles (Fig. 2A).19 The signal at 1666 cm−1, corresponding to CO stretching reports on the oxidation state of the reporter and can be plotted relative to the non redox sensitive signal at 1606 cm−1 corresponding to CC stretching. Calibration of AQ-NPs is described in the Experimental section and the resultant calibration plot is given in Fig. 2B. The resultant calibration is a stretched sigmoid centred around the E1/2 of AQ. The stretched nature of the plot has been seen before25 and is probably the result of hindered electron or proton transfer between the solution and the nanosensor. We have demonstrated that AQ can be used to report on redox potentials that span from normoxia to hypoxia.18,19 Representative spectra of fully oxidised and fully reduced AQ-NPs are shown in Fig. 2C.
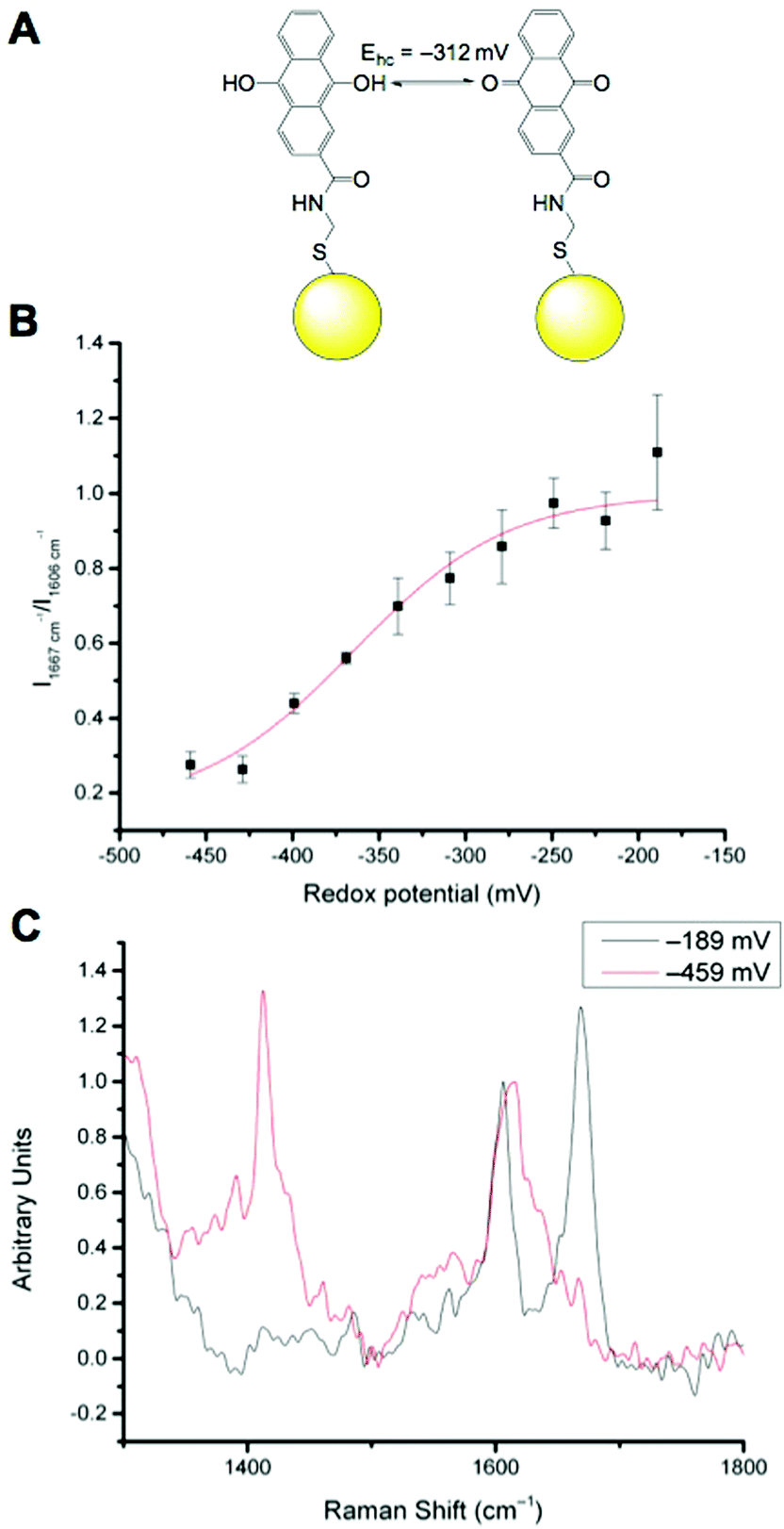 | ||
| Fig. 2 AQ-NP nanosensor. (A): Equilibrium between oxidised and reduced AQ attached to a gold NP (yellow sphere, not to scale). (B): Calibration curve for AQ-NP nanosensors (R2 = 0.96855). (C): Representative spectra of fully oxidised (−189 mV) and fully reduced (−459 mV) AQ-NPs. | ||
Intracellular SERS measurements
In previous work MBA-NPs have been used to monitor intracellular pH23,24 and AQ-NPs have been used to monitor intracellular redox potential.19 Our study investigates the data processing necessary to combine these two measurements. Cells were incubated with MBA-NPs and AQ-NPs as described in the Experimental section. Cells were located and imaged and then Raman mapping was performed with acquisitions in 1 μm steps in x and y. Hierarchical k-means cluster analysis was performed on the resultant maps (Fig. 3). This grouped similar spectra together into a single cluster represented by a colour on the false colour images and identified regions that contained NPs. The colours chosen do not indicate the magnitude of pH or redox potential only that a group of spectra have similar features. An average spectrum representative of the cluster was generated for each colour. Cluster spectra were processed manually by subtracting a baseline (8 points) in order to give an average spectrum for the cell. Of all spectra collected signals from MBA-NPs were predominant, with fewer AQ-NP signals in comparison. All cluster spectra with AQ-NP signals were multiplexed with MBA-NP signals while other clusters gave singleplex MBA-NP signals. For this reason, data analysis was simplified by determining only redox potential from multiplexed spectra and using the abundant singleplex MBA-NP signals for simplified processing for pH determination. This approach allowed intracellular redox potential and pH predominantly to be simultaneously measured on a whole cell basis. From the 4 mapped cells, cluster analysis for Cell 1 and 2 gave 2 clusters. Analysis of the spectra revealed that one cluster gave a singleplex spectrum with only MBA-NP signals and the other gave a multiplex spectrum with evidence of both MBA-NP and AQ-NP signals. The remaining cells (3 and 4) revealed 3 clusters. In each case one cluster contained a singleplex spectrum with MBA-NP signals in isolation and the other 2 contained multiplexed signals at different ratios.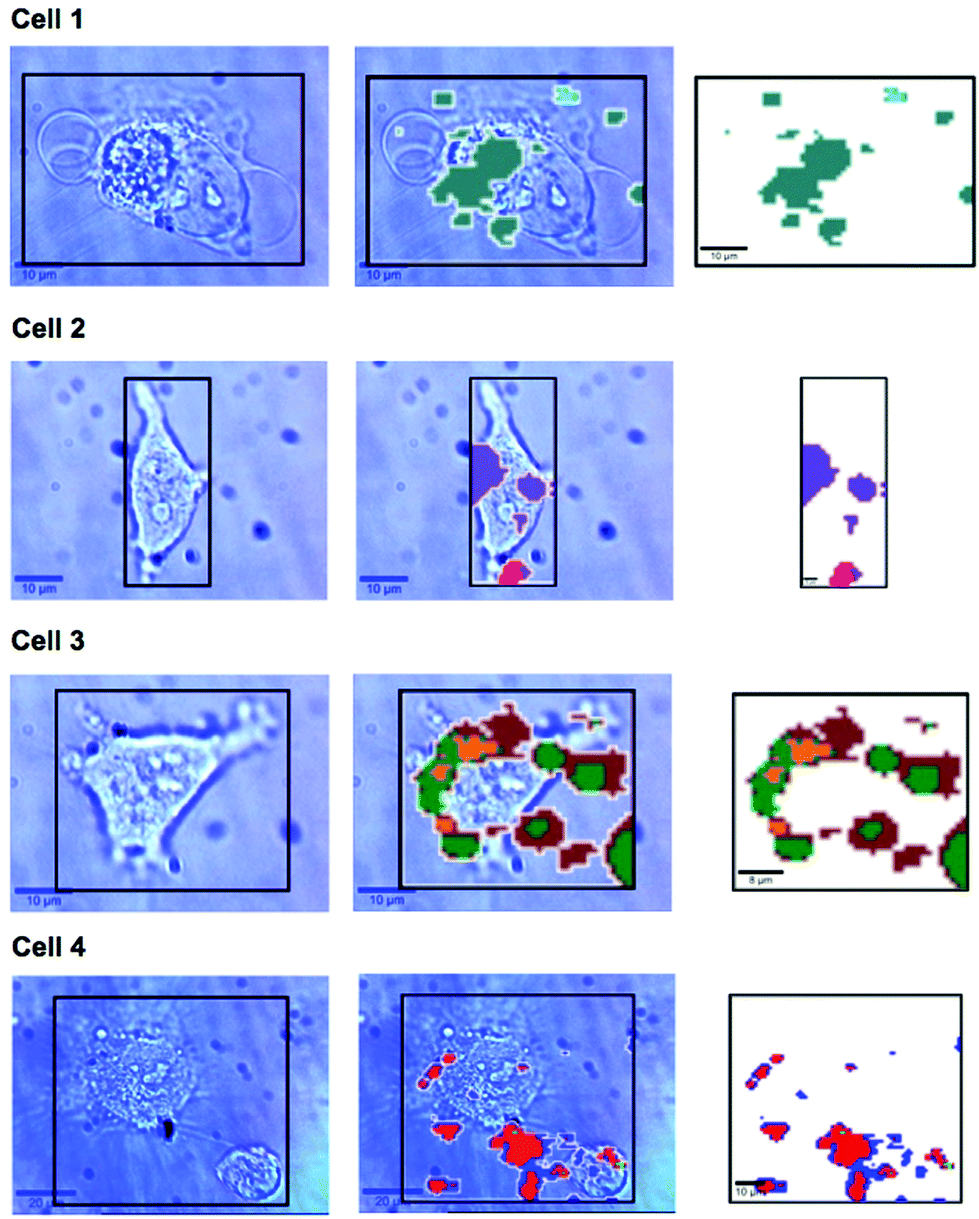 | ||
| Fig. 3 Photographs of SERS mapped cells (left column), area highlighted with black box corresponds to the Raman mapped area represented by the corresponding k-means cluster map (right hand column). Photographs and cluster maps are overlaid in the middle column. Each cluster from k-means cluster analysis is represented by a different colour. Cell 1 – turquoise cluster corresponds to a singleplex spectrum of MBA-NPs giving pH 7.1, cyan cluster corresponds to a multiplex spectrum of MBA-NPs and AQ-NPs from which redox potential −304 mV was extracted by peak fitting (pH-adjusted to −297 mV). Cell 2 – purple cluster corresponds to a singleplex spectrum of MBA-NPs giving pH 6.9 and the pink cluster corresponds to a multiplex spectrum of MBA-NPs and AQ-NPs from which redox potential −258 mV was extracted by peak fitting (pH-adjusted to −241 mV). Cell 3 – green cluster corresponds to a singleplex spectrum of MBA-NPs giving pH 7.0 and the yellow and brown clusters correspond to multiplex spectra of MBA-NPs and AQ-NPs from which redox potentials of −276 mV and −345 mV were extracted respectively by peak fitting (pH-adjusted to −272 mV and −332 mV respectively). Cell 4 – light green cluster corresponds to a singleplex spectrum of MBA-NPs giving pH 7.1 and the blue and red clusters correspond to multiplex spectra of MBA-NPs and AQ-NPs from which redox potentials of −337 mV and −306 mV were extracted respectively by peak fitting (pH-adjusted to −320 mV and −300 mV respectively). | ||
For spectra with only MBA-NP signals peak finding was performed to determine the intensity of the peak at 1400 cm−1 relative to the peak at 1590 cm−1 and subsequently determine pH from the calibration data. For spectra with multiplexed signals, peak fitting, as described in the Experimental section, was performed to extract the intensity of the peak at 1667 cm−1 relative to that at 1606 cm−1 and subsequently determine redox potential from the calibration data. Fig. 4 illustrates one example of peak fitting for the blue cluster in cell 4. A GaussAmp function was used with fixed y0, peak widths and peak centres and peak intensities of the fitted peaks from AQ-NPs were obtained. All singleplex cluster spectra analysed for pH measurements are overlaid in Fig. 5A and all multiplex cluster spectra analysed for redox measurements are overlaid in Fig. 5B.
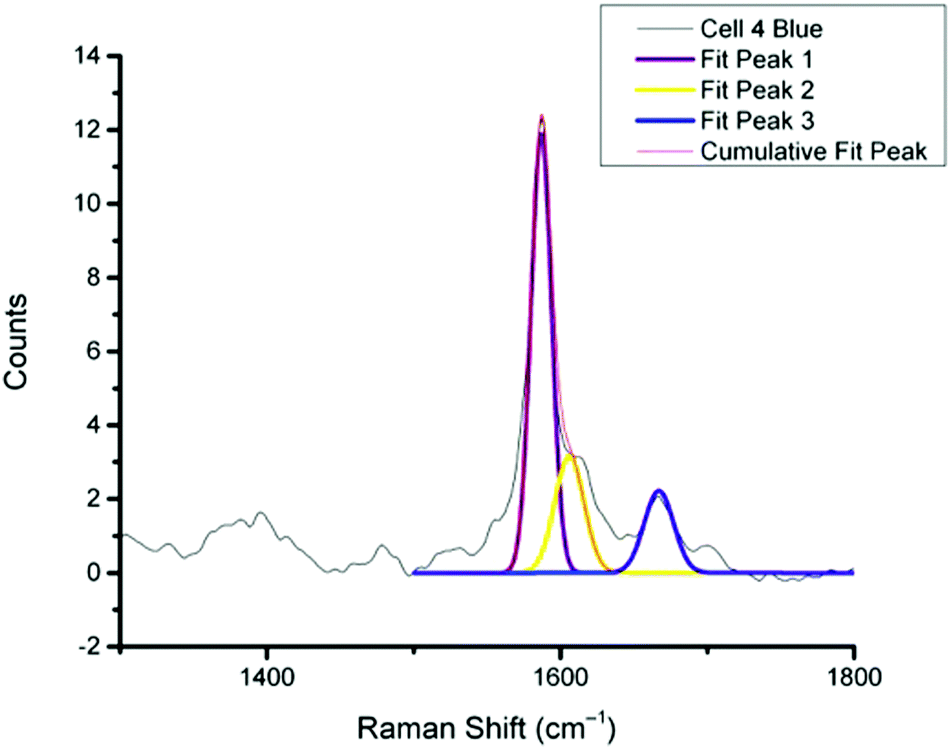 | ||
| Fig. 4 Representative example of peak fitting for a multiplex spectrum generated from k-means cluster analysis (cell 4 blue cluster). Peaks were fitted using a GaussAmp function with constrained parameters resulting in the 3 individual peak fits, Fit 1, 2 and 3 and the overall resultant Cumulative Fit Peak shown overlaid on the original spectrum (black line). | ||
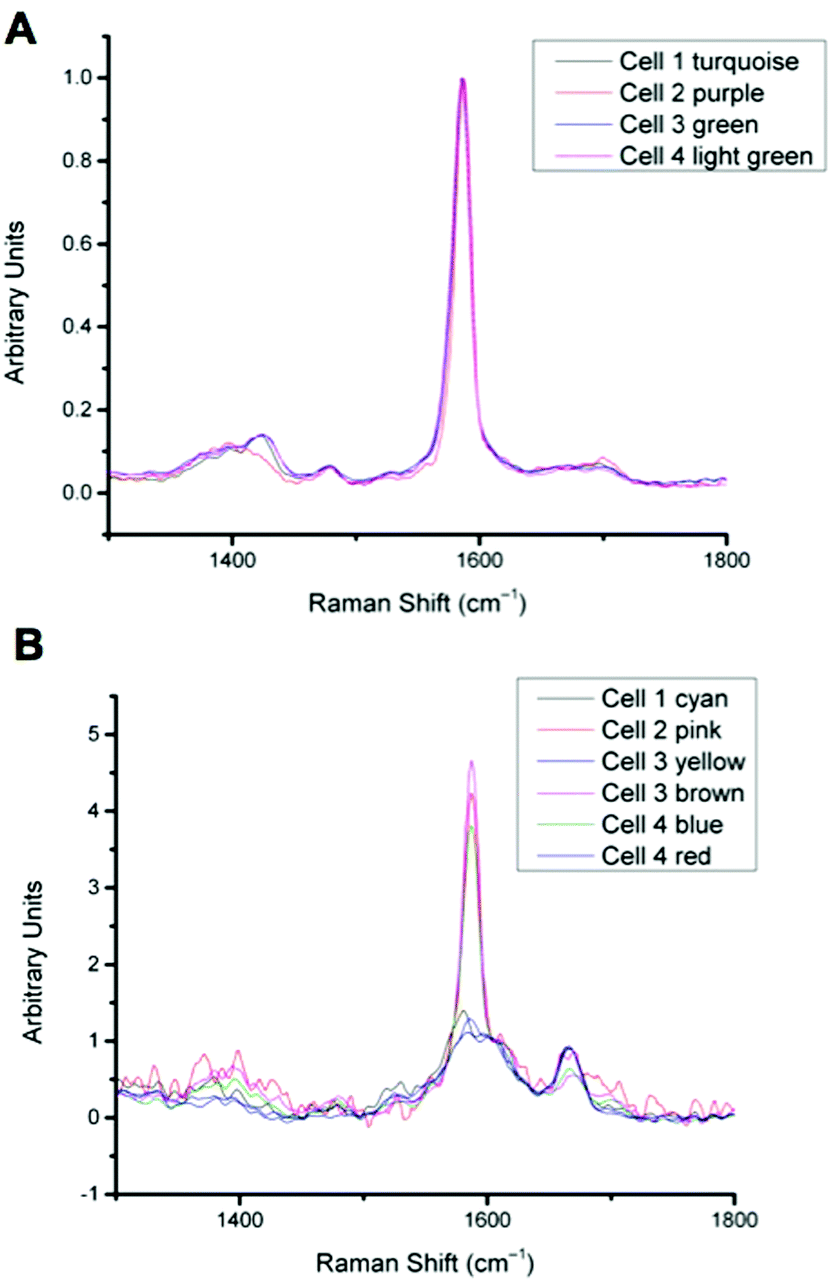 | ||
| Fig. 5 A: Overlay of all singleplex spectra from cells 1–4 corresponding to the clusters indicated. Spectra show MBA-NP signals corresponding to the pH values given in Table 1, calculated by taking the intensity ratio of the peaks around 1400 cm−1 and 1590 cm−1. B: Overlay of all multiplex spectra from cells 1–4 corresponding to the clusters indicated. Spectra show both MBA-NP and AQ-NP signals overlapping. Peaks were fitted to extract the intensity ratio of peaks at 1670 cm−1 and 1600 cm−1 to give redox potential values given in Table 1. | ||
Table 1 documents pH and redox potential values determined from each cluster from each cell in Fig. 3. The significance of multiplexing pH and redox potential measurements is to allow for redox potential values to be corrected relative to pH. Jiang et al.19 demonstrated that the half-cell potential of AQ relative to pH changed as expected from theory, by 60 mV per pH unit (Fig. S1†). Therefore the redox potential values determined from each cell could be corrected based on the pH determined from the same cell to give both pH and pH-corrected redox potential on a whole cell basis.
| Cell | pH (colour) | Redox potential (mV) (colour) | pH-adjusted redox potential (mV) (colour) |
|---|---|---|---|
| 1 | 7.1 (turquoise) | −304 (cyan) | −297 (cyan) |
| 2 | 6.9 (purple) | −258 (pink) | −241 (pink) |
| 3 | 7.0 (green) | −276 (yellow) | −272 (yellow) |
| −345 (brown) | −332 (brown) | ||
| 4 | 7.1 (light green) | −337 (blue) | −320 (blue) |
| −306 (red) | −300 (red) |
A more detailed analysis of Fig. 3. reveals that in some cases the colours representing clusters from NPs do not always overlay cell location. It is likely that this is a result of drift during measurements of live cells and not signals from extracellular particles as we used well established NP delivery protocols and the cells were washed multiple times before map acquisition.18,23,24,26–29 It is unlikely but possible that some NP remained outside cells after washing which could introduce error to cluster measurements however if this was the case the contribution from NP signals outside cells should have been minimal to the overall cluster. Both pH and redox potential differences between cells are apparent and in the case of cell 3 and cell 4 intracellular redox potential differences are also apparent. Intercellular pH variations were minimal, as expected for healthy cells, which maintain a pH around 7.2. Calculated pH varied by 0.2 pH units between 6.9 and 7.1. This is in agreement with measurements assigned to cytosolic pH when the same MBA-NP reporter was used independently.23 Redox variations were more substantial, with adjusted potential varying by 91 mV from −241 mV to −332 mV. This is in line with the normoxic range in A549 cells determined using AQ-NP19 and the large range could be attributed to varying stages of cell life-cycle. In the future this technique could be used to investigate more significant changes in response to, for example, disease and drug treatments.
Our results are the first demonstration of simultaneous measurement of redox potential and pH and this greatly improves our ability to analyse intracellular microenvironments. Cluster analysis provides a simple way of condensing large volumes of information for analysis of average redox potential and pH on a cell by cell basis, providing a platform for detailed analysis of whole cell differences and response to treatment. Continued improvements in reporter design will allow for improved multiplexing of spectra with potential to include markers for other biochemical parameters.
Experimental
Nanoparticle synthesis
Mercaptobenzoic acid (MBA), gold(III) chloride trihydrate, sodium citrate tribasic dehydrate and methanol were purchased from Sigma and were of analytical grade. Gold nanoparticles (GNPs) were prepared according to the Frens procedure.30 In this synthesis Au ions are reduced by citrate ions. A stock solution of MBA at the concentration of 1 mM was prepared by diluting an appropriate mass of the solid in ethanol. A stock solution of AQ at the concentration of 100 μM was prepared by diluting an appropriate mass of the solid in DMSO to a concentration of 10 mM and diluting the resultant solution with PBS to 100 μM.MBA-NP and AQ-NP solutions were prepared by mixing 100 μl of the 1 mM solution of MBA and 400 μl of 100 μM solution of AQ respectively with 1 ml of the gold colloid. After 5 min, mixtures were centrifuged for 10 min at 3000 rpm. For MBA-NPs supernatant was removed and NPs were re-dissolved in PBS.
Nanosensor calibration AQ
SERS spectroelectrochemistry was carried out as described by Jiang et al.19 Analysis was performed using Origin 9. After smoothing (9 points, Savitzky–Golay) and manual baseline subtraction (8 points), two peaks were fitted in the region from 1500 cm−1 to 1800 cm−1 using the GaussAmp function. Peak centres were fixed to approximately 1606 cm−1 (CC stretching) and 1667 cm−1 (CO stretching), y0 was fixed to 0, and peak widths were fixed to 10 (for representative peak fitting see Fig. S2†). The relative intensity of the fitted peaks at 1667 cm−1 and 1606 cm−1 were plotted against redox potential and Origin 9 was used to fit a Boltzmann equation to the resulting calibration curve.
Nanosensor calibration MBA
Solutions of MBA-NPs were mixed with buffers in the pH range 0–12 (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) and solutions were placed in a 96 well plate. Multiple spectra for each pH were acquired directly from the wells. The relative intensity of bands corresponding to COO− stretching (approximately 1400 cm−1) and aromatic ring stretching (approximately 1590 cm−1) were calculated using OPUS software and plotted against pH. Origin 9 was used to fit a Boltzmann equation to the calibration curve.
:1 v/v) and solutions were placed in a 96 well plate. Multiple spectra for each pH were acquired directly from the wells. The relative intensity of bands corresponding to COO− stretching (approximately 1400 cm−1) and aromatic ring stretching (approximately 1590 cm−1) were calculated using OPUS software and plotted against pH. Origin 9 was used to fit a Boltzmann equation to the calibration curve.
Buffers were prepared by firstly dissolving appropriate masses of solid analytes in deionised water to make 0.1 M solutions of sodium hydroxide, sodium phosphate, disodium hydrogen phosphate and phosphate acid purchased from Sigma-Aldrich and of analytical grade. Appropriate volumes of each of these solutions were mixed to prepare buffer solutions and pH was measured using a pH meter.
pH dependence of AQ half-wave potential
The half-wave potential of AQ at different pH values was determined by cyclic voltammetry as described by Jiang et al.19 and agreed with theoretical predications (Fig. S1†).Cellular delivery
Experiments were conducted using the EAhy926 cell line. Cells were grown in plastic wells with glass bottom in DMEM supplemented with 10% fetal bovine serum, 2 mM L-glutamine, penicillin, streptomycin, and 2% HAT. Cells were incubated at 37 °C and 5% CO2 in a humidified incubator. 24 h after passaging medium was replaced by medium without serum and left for 1 hour. Then, 200 μl of MBA-NP solution and 400 μl of AQ-NP solution were added to the serum-free medium in the wells and left for approximately 2 hours. Media was removed, cells washed with PBS and Raman measurements were carried out immediately.Instrumentation
The electronic absorption spectra of the gold colloid, the gold colloid mixed with MBA and gold colloid mixed with AQ were recorded with a UV-Vis-NIR Perkin Elmer spectrophotometer (model Lambda 35) in the range of 190–1100 nm with a resolution of 2 nm. Quartz cells of 1 cm were used.SERS mapping of cells was carried out by using a WITec system, equipped with an immersive objective with magnification of 60× and a He–Ne laser (632.8 nm). For all measurements, integration time was 0.03 s with a single accumulation, laser power of 10 mW and grating 600 g mm−1. SERS spectra of MBA-NPs and AQ-NPs as a reference for cell mapping were recorded by placing a sample in a glass cuvette and using an air objective (20×). Three SERS spectra were acquired for each freshly prepared sample. The latter was prepared by mixing 500 μL of the Au colloid with 5 μL of a 1 × 10−3 M MBA/AQ solution in a glass cuvette. For Raman mapping, raster scans over single living cells were carried out with a computer-controlled x,y-stage. The mapping step was 1 μm.
Data processing for intracellular measurements
Hierarchical k-means cluster analysis of SERS maps was performed by using WITec Project 2.06 software. The spectra were analysed in the region of 400–1800 cm−1 after a routine procedure for cosmic rays removal and smoothing (13 points) using a Savitzky–Golay algorithm.Spectra of resultant clusters were analysed using Origin 9 software. An 8 point baseline was subtracted from all spectra in the region 1000–1800 cm−1. For spectra with only MBA signals, peak heights were determined using the peak finding function and the 1st derivative method. For spectra with both MBA and AQ signals, three peaks were fitted in the region from 1500 cm−1 to 1800 cm−1 using the GaussAmp function. Peak centres were fixed to approximately 1587 cm−1 and width 7 (MBA ring breathing), 1606 cm−1 and width 10 (AQ CC stretching) and 1667 cm−1 and width 10 (AQ CO stretching) and y0 was fixed to 0. Relative intensities of the signals at 1667 cm−1 and 1606 cm−1 were used to determine redox potential.
Conclusions
In this paper we have presented the first example of multiplexing pH and redox responsive SERS nanosensors for intracellular live single cell measurement on a cell by cell basis. This provides quantitative information on the cell microenvironment, superior to previous techniques used for intracellular redox potential or pH measurements independently or together. MBA-NPs were calibrated and shown to be responsive in the pH range 6–8. AQ-NPs were calibrated and shown to be responsive in the range from −250 mV to −400 mV. Analysis of SERS maps of single cells incubated with both SERS reporters by k-means cluster analysis followed by spectral processing revealed detailed quantitative information on intracellular pH and redox potential on a whole cell basis. Redox potential measurements could be accurately corrected for pH and both inter- and intra- cellular differences were observed.Acknowledgements
The authors gratefully acknowledge the School of Chemistry at the University of Edinburgh, a Neil Campbell Travel Award, the Faculty of Chemistry at Jagiellonian University and Jagiellonian Centre for Experimental Therapeutics (JCET). A. J.'s work was supported by National Center of Science (grant PRELUDIUM DEC-2012/05/N/ST4/00218) and by the European Union from the resources of the European Regional Development Fund under the Innovative Economy Programme (grant coordinated by JCET-UJ, No POIG.01.01.02-00-069/09).Notes and references
- M. Kemp, Y.-M. Go and D. P. Jones, Free Radicals Biol. Med., 2008, 44, 921–937 CrossRef CAS PubMed.
- Y.-M. Go and D. P. Jones, Biochim. Biophys. Acta, 2008, 1780, 1273–1290 CrossRef CAS PubMed.
- F. Q. Schafer and G. R. Buettner, Free Radicals Biol. Med., 2001, 30, 1191–1212 CrossRef CAS.
- G. Filomeni, G. Rotilio and M. R. Ciriolo, Cell Death Differ., 2005, 12, 1555–1563 CrossRef CAS PubMed.
- D. P. Jones, J. Intern. Med., 2010, 268, 432–448 CrossRef CAS PubMed.
- H. Liu, R. Colavitti, I. I. Rovira and T. Finkel, Circ. Res., 2005, 97, 967–974 CrossRef CAS PubMed.
- V. Mallikarjun, D. J. Clarke and C. J. Campbell, Free Radical Biol. Med., 2012, 53, 280–288 CrossRef CAS PubMed.
- G. Waris and H. Ahsan, J. Carcinog., 2006, 5, 14 CrossRef PubMed.
- Y.-M. Go and D. P. Jones, Free Radicals Biol. Med., 2011, 50, 495–509 CrossRef CAS PubMed.
- M. W. Dewhirst, Y. Cao and B. Moeller, Nat. Rev. Cancer, 2008, 8, 425–437 CrossRef CAS PubMed.
- I. H. Shaik and R. Mehvar, Anal. Bioanal. Chem., 2006, 385, 105–113 CrossRef CAS PubMed.
- R. P. Rastogi, S. P. Singh, D.-P. Häder and R. P. Sinha, Biochem. Biophys. Res. Commun., 2010, 397, 603–607 CrossRef CAS PubMed.
- E. W. Miller, S. X. Bian and C. J. Chang, J. Am. Chem. Soc., 2007, 129, 3458–3459 CrossRef CAS PubMed.
- C. T. Dooley, T. M. Dore, G. T. Hanson, W. C. Jackson, S. J. Remington and R. Y. Tsien, J. Biol. Chem., 2004, 279, 22284–22293 CrossRef CAS PubMed.
- G. T. Hanson, R. Aggeler, D. Oglesbee, M. Cannon, R. A. Capaldi, R. Y. Tsien and S. J. Remington, J. Biol. Chem., 2004, 279, 13044–13053 CrossRef CAS PubMed.
- H. Ostergaard, A. Henriksen, F. G. Hansen and J. R. Winther, EMBO J., 2001, 20, 5853–5862 CrossRef CAS PubMed.
- M. Gutscher, A. Pauleau, L. Marty, T. Brach, G. H. Wabnitz, Y. Samstag, A. J. Meyer and T. P. Dick, Nat. Methods, 2008, 5, 553–559 Search PubMed.
- C. A. R. Auchinvole, P. Richardson, C. McGuinnes, V. Mallikarjun, K. Donaldson, H. McNab and C. J. Campbell, ACS Nano, 2012, 6, 888–896 CrossRef CAS PubMed.
- J. Jiang, C. Auchinvole, K. Fisher and C. J. Campbell, Nanoscale, 2014, 6, 12104–12110 RSC.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, J. Phys. Chem. C, 2010, 114, 7421–7426 CAS.
- J. Kneipp, H. Kneipp, B. Wittig and K. Kneipp, Nano Lett., 2007, 7, 2819–2823 Search PubMed.
- S. W. Bishnoi, C. J. Rozell, C. S. Levin, M. K. Gheith, B. R. Johnson, D. H. Johnson and N. J. Halas, Nano Lett., 2006, 6, 1687–1692 CrossRef CAS PubMed.
- A. Jaworska, L. E. Jamieson, K. Malek, C. J. Campbell, J. Choo, S. Chlopicki and M. Baranska, Analyst, 2015 10.1039/c4an01988a.
- M. A. Ochsenkuhn, P. R. T. Jess, H. Stoquert, K. Dholakia and C. J. Campbell, Nano Lett., 2009, 3, 3613–3621 CAS.
- W. J. Albery, M. G. Boutelle, P. J. Colby and A. R. Hillman, J. Electroanal. Chem. Interfacial Electrochem., 1982, 133, 135–145 CrossRef CAS.
- M. Ekkapongpisit, A. Giovia, C. Follo, G. Caputo and C. Isidoro, Int. J. Nanomed., 2012, 7, 4147–4158 CAS.
- R. R. Sathuluri, H. Yoshikawa, E. Shimizu, M. Saito and E. Tamiya, PLoS One, 2011, 6, e22802 Search PubMed.
- J. Huang, C. Zong, H. Shen, M. Liu, B. Chen, B. Ren and Z. Zhang, NanoSmallMicro, 2012, 10, 2577–2584 Search PubMed.
- A. Jaworska, T. Wojcik, K. Malek, U. Kwolek, M. Kepczynski, A. A. Ansary, S. Chlopicki and M. Baranska, Microchim. Acta, 2014, 8–11 Search PubMed.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an02365j |
| This journal is © The Royal Society of Chemistry 2015 |