
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Conformational dynamics of α-synuclein: insights from mass spectrometry†
Ashley S.
Phillips
a,
Alexandre F.
Gomes
b,
Jason M. D.
Kalapothakis
c,
Jay E.
Gillam
c,
Jonas
Gasparavicius
d,
Fabio C.
Gozzo
b,
Tilo
Kunath
e,
Cait
MacPhee
c and
Perdita E.
Barran
*a
aManchester Institute of Biotechnology, University of Manchester, Manchester, M1 7DN, UK. E-mail: Perdita.barran@manchester.ac.uk
bDalton Mass Spectrometry Laboratory, University of Campinas – UNICAMP, Campinas, São Paulo, Brazil
cSchool of Physics and Astronomy, University of Edinburgh, Edinburgh, EH9 3FD, UK
dSchool of Chemistry, University of Edinburgh, Edinburgh, EH9 3JJ, UK
eMRC Centre for Regenerative Medicine, University of Edinburgh, Edinburgh, EH16 4UU, UK
First published on 25th February 2015
Abstract
The aggregation and deposition of α-synuclein in Lewy bodies is associated with the progression of Parkinson's disease. Here, Mass Spectrometry (MS) is used in combination with Ion Mobility (IM), chemical crosslinking and Electron Capture Dissociation (ECD) to probe transient structural elements of α-synuclein and its oligomers. Each of these reveals different aspects of the conformational heterogeneity of this 14 kDa protein. IM-MS analysis indicates that this protein is highly disordered, presenting in positive ionisation mode with a charge state range of 5 ≤ z ≤ 21 for the monomer, along with a collision cross section range of ∼1600 Å2. Chemical crosslinking applied in conjunction with IM-MS captures solution phase conformational families enabling comparison with those exhibited in the gas phase. Crosslinking IM-MS identifies 3 distinct conformational families, Compact (∼1200 Å2), Extended (∼1500 Å2) and Unfolded (∼2350 Å2) which correlate with those observed in solution. ECD-Fourier Transform-Ion Cyclotron Resonance Mass Spectrometry (ECD-FT-ICR MS) highlights the effect of pH on α-synuclein structure, identifying the conformational flexibility of the N and C termini as well as providing evidence for structure in the core and at times the C terminus. A hypothesis is proposed for the variability displayed in the structural rearrangement of α-synuclein following changes in solution pH. Following a 120 h aggregation time course, we observe an increase in the ratio of dimer to monomer, but no gross conformational changes in either, beyond the significant variations that are observed day-to-day from this conformationally dynamic protein.
Introduction
Mass Spectrometry (MS) and the hybrid technique, Ion Mobility Mass Spectrometry (IM-MS) have emerged as useful tools to investigate the early stages of aggregation of proteins implicated in protein misfolding diseases, and the multiple conformational families that co-exist during the process.1 IM-MS can be used to measure heterogeneous samples (both in terms of stoichiometry and conformation), requires a low sample volume and allows a relatively short transfer time from bench to instrument. There are different forms of ion mobility instruments, however in this work we use drift tube based measurements (DT-IM-MS). DT-IM-MS has been described in detail elsewhere,2 but briefly, it is an analytical technique that measures the time it takes a given m/z selected ion to pass through a drift cell filled with an inert buffer gas, under the influence of a weak electrostatic field. Under low field conditions, injected ions drift through the drift cell and are retarded as well as thermalized by collisions with the buffer gas. The mobility of an ion (K) is defined as the ratio between the drift velocity (vd), which is measured by measuring the arrival time and the applied electric field (E), and then converting to the reduced mobility (K0) by normalising the temperature and pressure of the drift gas. The mobility of any given ion is related to its rotationally averaged Collision Cross Section (CCS) (Ω) according to the Mason-Schamp equation3 where N is the number gas density, μ is the reduced mass of the ion and the buffer gas, and z is the charge on the ion.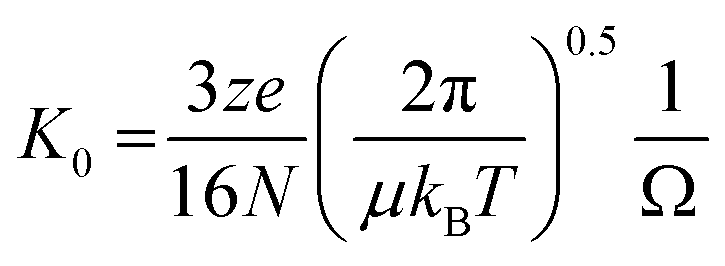 | (1) |
The aggregation of and the pre-fibrillar aggregates formed by β-2-microglobulin has been studied using IM-MS approaches by the Vachet,4 Radford5,6 and Ashcroft groups.7–10 Bowers and co-workers have extensively studied amyloid-β11–16 with contributions from Robinson.17 Other work has examined amylin,18–24 Tau25 and a peptide from the amyloid disease associated protein Transthyretin or TTR.26 These constitute several of the 30 different proteins which have been implicated as causal factors in human disease due to their inability to adopt or remain in their native conformational state.27 Such diseases are referred to as protein misfolding diseases due to the change in shape of the precursor protein, and most neurodegenerative diseases fall into this category. The mechanism of dysfunction varies however, a common identifier is that normally soluble proteins form organised, insoluble fibrillar and oligomeric aggregates.27 Parkinsons Disease (PD) is one of these diseases and the protein involved is α-synuclein, a major constituent of the characteristic Lewy bodies, where it presents in an organised fibrillar state.28,29
α-synuclein is a member of a subset of proteins known as, intrinsically disordered proteins (IDPs), defined as lacking (or having regions lacking) tertiary fold observable on the timescale of an NMR experiment. α-synuclein is a 140 amino acid protein encoded by a single gene, SNCA and in neurons it is localised predominantly in the presynaptic nerve terminals and nucleus.30 The exact function of α-synuclein is unknown however, it is theorised to be involved in synaptic function and neurotransmitter release.31 There is a large body of evidence indicating that α-synuclein exists as an unstructured monomer under a range of biophysical assays31–34 and even in cells.35 However, the α-synuclein found in Lewy bodies has condensed to fibrillar aggregates rich in β-sheet structure. The aggregation pathway has been described as containing multiple intermediate oligomer structures including spherical36 and ring-shaped31,37 forms and is susceptible to pertubation, through changes to environmental factors. These factors include agitation, temperature and pH.38 The transition from native conformation to these amyloid deposits is therefore not a single step, nor a single pathway, and hence in order to understand the mechanisms at play it is critical to identify the species present at each step. Oligomers formed in the early stages of aggregation are most applicable as drug targets since they are reported to be the most toxic species that mediate neurodegeneration.7,39
The Bowers,40 Kaltashov41 and Grandori42 have previously demonstrated the conformational plasticity of α-synuclein using mass spectrometry based approaches. All groups have reported the presence of multiple co-existing α-synuclein conformers under native conditions, including tightly folded and extended conformers.40–42 These studies have demonstrated the importance of solution conditions on the conformers presented by α-synuclein, in particular pH dependence. Bernstein et al. and Frimpong et al. independently reported a shift to lower charged, more compact species at low pH,40,41 whereas Natalello et al. showed a shift to higher charged, more extended conformers at basic pH.42 Bernstein et al. also noted the presence of dimer species at pH 2.5 and their absence at pH 7.40 In comparison, Frimpong et al. described the presence of dimer species at pH 2.5 to pH 8. The selective stabilisation of α-synuclein conformers via different alcohols has also been displayed via MS.41,42 Taking the data of these studies alone, it is clear that α-synuclein is extremely susceptible to environmental perturbations and even under tightly controlled conditions, significant variation in the data produced occurs.
MS approaches have also been applied to study other aspects of α-synuclein for example to study the effect of fragments on aggregation43,44 and to characterise the influence of metal ion binding, on conformation and aggregation.42,45–47 MS has been used to examine post-translational modifications in α-synuclein, identifying phosphorylated species in CSF48 and to investigate the effect of such post translational modifications on the behaviour of α-synuclein.49,50 Naturally occurring in vivo crosslinking of α-synuclein has been investigated with MS and implicated as both inducing and preventing aggregation.51,52 Photo-induced crosslinking of α-synuclein in vitro has been used to probe the properties and cellular toxicity of oligomers and the di-tyrosine cross links were identified with mass spectral analysis.53
Hydrogen-Deuterium Exchange (HDX) can be used in conjunction with MS to monitor protein conformational dynamics54,55 and has been applied to the study of monomeric and low order oligomeric species of α-synuclein.56–59 These studies have confirmed that the α-synuclein monomer is an unstructured solvent accessible protein, as a result of the rapid exchange which is seen across the entire sequence length.56,57 Following the induction of an amyloid state, HDX has helped uncover structural changes experienced by α-synuclein,56,58 but apparent inconsistencies in results from different groups highlights the dynamic nature of α-synuclein. Furthermore HDX has also allowed the designation of two distinct oligomers with different levels of protection, which may represent the intermediates of two different aggregation pathways.59 HDXs ability to define structural information is backed by evidence from Lee et al., whose work on the binding of α-synuclein to lipids, highlights regions of protection in the N terminus, which correlates with the induction of α-helical structure in the α-synuclein N terminus following binding to a SLAS micelle, described previously.57,60
MS has also enabled the study of the binding interactions of α-synuclein with a number of ligands including potential drug candidates, and in vivo partners,61,62 including spermine,63 and dopamine.64 In this study MS and IM-MS are applied; to investigate the conformational plasticity of α-synuclein, to detect low order oligomeric species and to follow the early stages of aggregation. In vitro chemical crosslinking is used here in conjunction with IM-MS to capture transient conformational populations of monomeric α-synuclein. ECD-FT-ICR MS is also applied to investigate the structure of monomeric and dimeric species from different solution conditions. Our data is compared to data reported in previous studies. We use transmission electron microscopy to determine fibril formation under the solution conditions employed in MS studies.
Experimental methods and materials
Materials
AR grade ammonium acetate, sodium phosphate, was purchased from Fisher Scientific (Loughborough, UK). BS3 crosslinker, sodium carbonate, sodium acetate, HEPES, potassium hydroxide and acetic acid were purchased from Sigma Aldrich (UK). Water was purified with an Arium 611 water purification system (Sartorius, Göttingen, Germany).α-synuclein preparation
α-synuclein was expressed recombinantly, from a pT7–7 vector containing human α-synuclein gene, kindly provided by Dr Jean-Christophe Rochet, Purdue University and purified as described previously;65 a Resource Q column (GE Healthcare Life Sciences, UK) was used. α-synuclein used for crosslinking, IM-MS and ECD-FT-ICR-MS analysis, was purified as described, concentrated using Vivaspin 6 centrifugal sample concentrators (MWCO 10 kDa) (GE Healthcare Life Sciences, UK) and applied to a HiPrep 26/10 desalting column (GE Healthcare Life Sciences, UK) pre-equilibrated with 100 mM ammonium acetate, flow rate 10 mL min−1. The eluent was lyophilised and stored at −80 °C prior to use. Prior to analysis, if required, samples were thawed and dialysed into a 50 mM ammonium acetate buffer solution (pH 7) overnight at 4 °C using 3.5 kDa MWCO Slide-A-Lyzer Dialysis cassettes (Thermo Scientific, USA). Post-dialysis concentration was determined by a BCA assay (Pierce, Thermo Scientific, USA) prior to aliquoting.Aggregation procedure
α-synuclein (70 μM, 50 mM ammonium acetate, pH 7) was incubated at 37 °C with agitation at 200 rpm in an incubator shaker, a method adapted from the work of Hashimoto et al. and Fink et al.66,67 Samples were incubated individually and at specified time points were removed and analysed immediately. The time taken to transfer the sample from incubation to nano-spraying tip was never more than 3 minutes. The samples were incubated for up to 120 hours and analysed at t = 0, 24, 48, 72, 96 and 120 hours.Crosslinking procedure
For α-synuclein crosslinking experiments, the conditions were based on the manufacturers recommended conditions and the previously published work of Chen et al.68 and primarily, Iglesias et al.69 Here, four buffer conditions, each at 50 mM, sodium acetate (pH 4), sodium phosphate (pH 6), HEPES (pH 8) and sodium carbonate (pH 10) were used. The pH of the HEPES buffer was modified with KOH. Since each crosslinking reagent has optimal operating conditions in terms of pH for maximum reactivity, by altering the pH during the crosslinker incubation, we are able to modulate the number of modifications. Lyophilised α-synuclein was reconstituted in each buffer solution to which BS3 at 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar excess was added, final α-synuclein concentration, 100 μM. Crosslinking reactions were incubated at room temperature for 1 hour. Reactions were quenched by the addition of 1 M ammonium acetate. Samples were desalted via buffer exchange into 50 mM ammonium acetate (pH 6.8) using Amicon Ultra centrifuge filters, MWCO 10 kDa (Millipore).
:1 molar excess was added, final α-synuclein concentration, 100 μM. Crosslinking reactions were incubated at room temperature for 1 hour. Reactions were quenched by the addition of 1 M ammonium acetate. Samples were desalted via buffer exchange into 50 mM ammonium acetate (pH 6.8) using Amicon Ultra centrifuge filters, MWCO 10 kDa (Millipore).
Nano-Electrospray Ionisation (nESI) was used for all MS and IMMS experiments, with the exception of ECD-FT-ICR MS. nESI tips were fabricated in-house using thin wall capillaries (ID 0.9 mm) (World Precision Instruments, Inc, USA) using a Fleming/Brown micropipette puller (Sutter Instruments Co., USA). Samples were ionised via the insertion of platinum wire into the nESI tip to which a voltage was applied.
Mass spectrometry
MS experiments were conducted on an Ultima API-US instrument (Micromass, Manchester, UK). Desalted α-synuclein was diluted with 50 mM ammonium acetate to a concentration of 70 μM. MS was conducted in positive mode. Source conditions were kept as similar as possible to enable comparison: capillary voltage ∼1.6 kV, cone voltage 165 V and source temperature 80 °C. For pH variation experiments, the solution pH was modified by the addition of acetic acid.Drift Tube Ion Mobility Mass Spectrometry (DT-IM-MS)
Drift Time Ion Mobility Mass Spectrometry measurements were performed on an in-house modified Q-ToF instrument (Micromass, Manchester, UK). The instrument has been modified via the addition of a 5.1 cm long copper drift cell and additional post source ion optics to carry out gas phase separation of protein ions based on the ion mobility, further instrument specifics are detailed elsewhere.70 Each experiment in the time course was performed in triplicate. Measurements were made at eight different drift voltages between 60 V and 10 V. For aggregation and crosslinking IM-MS experiments, tuning conditions were kept as similar as possible: capillary voltage ∼1.6 kV, cone voltage ∼98 V and source temperature 80 °C. The temperature and pressure of helium in the drift cell were on average: 302.4 K and 3.8 Torr, respectively, but are recorded and used to calculate mobilities for each experiment.Ion arrival time distributions (ATD) are recorded by synchronising the entry of each ion pulse into the drift cell with mass spectral acquisition. The mobility of the ion of interest was obtained from a linear plot of the average arrival time versus pressure/temperature, from which the rotationally-averaged Collision Cross Section (CCS) for each resolvable species at a given charge state was found using eqn (1). Collision Cross Section Distributions (CCSD) were created by plotting the average CCS against peak intensity values and charge. Data analysis was conducted using Masslynx v4.1 (Waters Corporation, USA), Origin v8–9.5 (OriginLab Corporation, USA) and Microsoft Excel (Microsoft, USA).
ECD-FT-ICR-MS
Lyophilised α-synuclein was reconstituted in 50 mM ammonium acetate to a concentration of 30 μM. Samples were prepared at pH 3.5 and pH 6.8. pH was adjusted by addition of acetic acid.Protein samples were ionised and introduced to the mass spectrometer using a Triversa Nanomate (Advion, New York, USA). Spectra were acquired with a Bruker Solarix 12 T FT-ICR Mass Spectrometer (Bruker Daltronics, Bremen, Germany). ECD-FT-ICR MS was performed after first acquiring a native mass spectrum by tuning source optics. Specific ion species were then isolated using the mass resolving quadrupole prior to MS/MS via ECD. For ECD, 1.7 A was applied to the cathode filament, 22 V to the lens, 1.2 V to the bias, and a pulse of between 15 and 20 ms was employed. Fragmentation data is the sum of 70 acquisitions. Data analysis was performed using DataAnalysis (Bruker Daltronics, Bremen, Germany). The SNAP 2.0 algorithm was used for peak picking and the matching of fragment peaks to calculated fragment masses was conducted by Prosight PTM (v1.0).71
Results and discussion
Mass spectrometry experiments, contrasting day to day variations, and the effect of lowered pH with data from aggregation time courses
Fig. 1A and B show MS spectra obtained from identically prepared samples taken under highly similar source conditions on the same instrument, approximately one month apart. In each case the spectra are predominantly due to the monomeric form of α-synuclein with a mass of 14453 Da (cf. theoretical average mass, 14460 Da). Both spectra also show evidence of higher order aggregates at low intensity. Comparison of the two spectra demonstrates the aforementioned conformational flux as well as the difficulty in acquiring reproducible data from this highly disordered protein. The most obvious change is seen in the relative population of different charge states as well as the distribution between monomer and higher order aggregates. In both cases, the protein exhibits a wide CSD, Fig. 1B shows a CSD of 5 ≤ z ≤ 19 for the monomeric species [aSyn + zH]z+, cf. a distribution of 5 ≤ z ≤ 20 in Fig. 1A. However, the most intense species in Fig. 1A is at z = 17 (m/z = 851.6) and in Fig. 1B is z = 14 (m/z = 1033.9). Other differences in the relative intensities of ions can be observed: for example the z = 10 ion (m/z = 1447) has significant intensity in Fig. 1A but is weaker in 1B, and here the z = 11 species (m/z = 1315.5) is much more abundant. Fig. 1A has multimodal distribution with at least three modes, z = 17, z = 14 and z = 11, by contrast, the data in Fig. 1B has a predominantly monomodal distribution, centred on z = 14. Fig. 1B shows less intensity in peaks assigned as dimeric species. This can be clearly identified by comparison of the relative abundance of monomer and dimer peaks of Fig. 1A and B and the less distinct multimodal distribution of Fig. 1B. This change is accompanied by lowered intensities of the observed trimeric and tetrameric species.
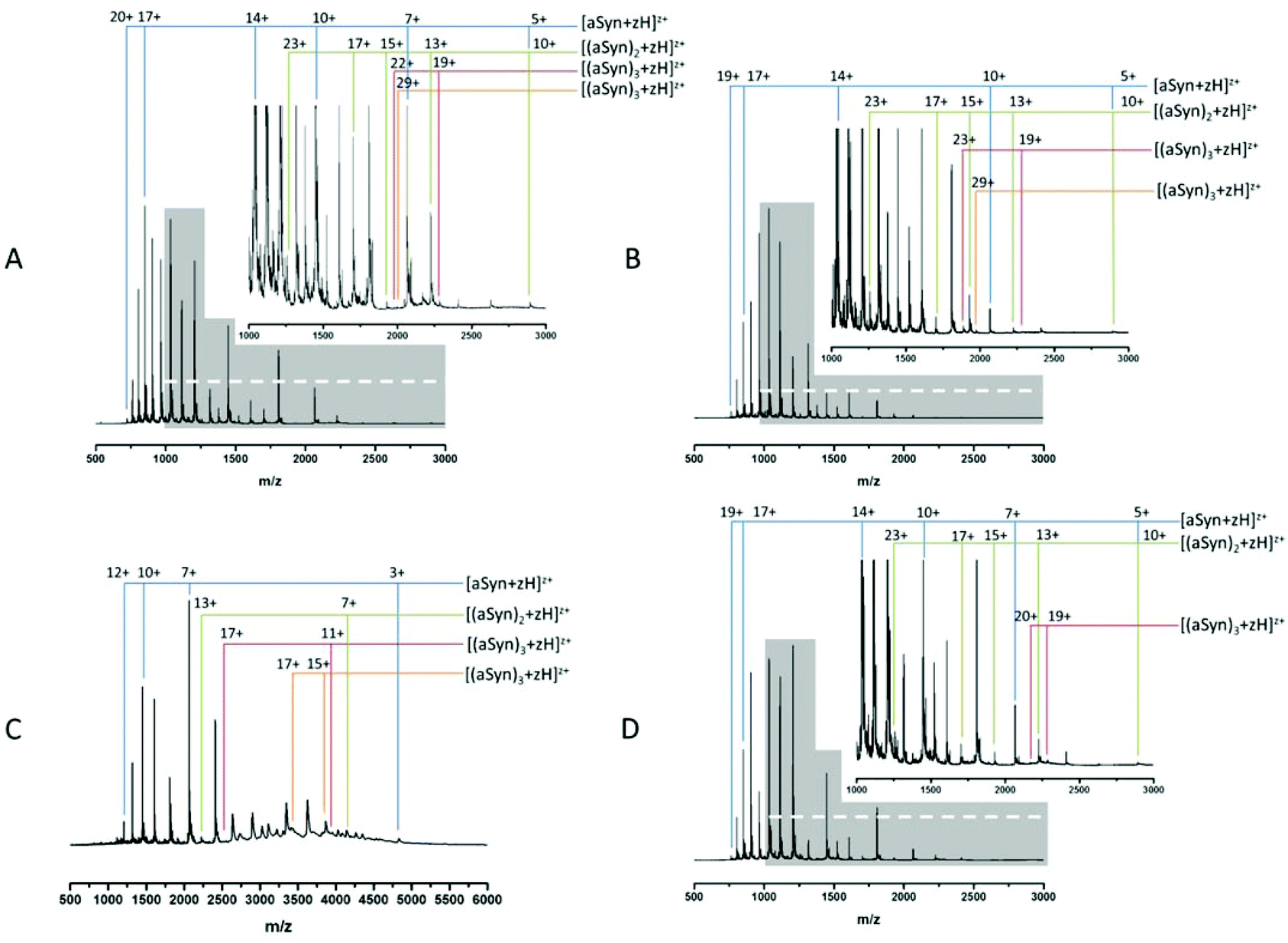 | ||
| Fig. 1 Comparison of nESI-MS data of α-synuclein (1 mg mL−1) (50 mM ammonium acetate). A & B. α-synuclein pH 6.8, spectra taken under similar instrumental and identical sample conditions. C. α-synuclein pH 3.5, D. α-synuclein pH 6.8 post-96 hour aggregation. N.b. the x-axis scale is enlarged for C to reflect the lower charge states displayed. The y-axis scale is altered for D to enable comparison. | ||
This data suggests that the low order oligomeric species identified (dimer to tetramer) vary in their presentation to the gas phase. Such fluctuations in spectra are typical for this protein following nano-ESI-MS analysis under similar experimental conditions.
This shift mirrors a change identified by Frimpong et al. where solution pH was dropped to pH 2.5.41 Our findings suggest that the protein has adopted a more compact structure, thereby decreasing solvent accessibility of protonatable sites and leading to lower charge states prevailing. This correlates with data from multiple physical techniques including IR, SAXs72 and IM-MS,40 which demonstrate an induction of structure72 and a partial collapse of the protein on lowering pH,40 both changes which decrease solvent accessibility and would decrease the average protonation state as shown here (Fig. 1C).
The induction of structure by pH may contribute to the increased detection of higher order species. The increase is clearly seen via comparison of the tetramer species present, z = 29 (Fig. 1A) and 15 ≤ z ≤ 17 (Fig. 1C). The large increase in baseline noise is attributable to an increase in higher ordered aggregates.73 The low sample pH combined with the low pI of α-synuclein (4.67), results in less protein–protein repulsion and is therefore an environment conducive to oligomer formation.34 This is supported by the depletion of the overall signal intensity. A similar effect is also seen under aggregating conditions (ESI† Fig. S1).
The aggregation of α-synuclein is presumed to be a nucleation dependent process and it has been suggested that the dimer is the aggregation nucleus.65,74 As mentioned previously the dimer CSD remains similar to the t0 spectrum (Fig. 1A) suggesting that the soluble population which must act as a feeder stock for the aggregates is comprised of forms of the protein that are conformationally dynamic, even when significant aggregates are present. This is in contrast to earlier work on aggregation of the peptide TTR (105–115)26 where at a mid-point in the aggregation time course a significant increase in the intensity of all observable higher order aggregates was reported followed by a decrease in the late stages and the persistence of dimer and tetramer species. However, it should be noted that the data in all panels in Fig. 1 have been normalised to the base peak, the absolute intensity of the species in Fig. 1D are depleted by almost 50%, compared to the species at t = 0. We speculate that this depletion is the result of low order species being sequestered into much larger oligomers, the presence of which is confirmed by TEM (ESI† Fig. S2) suggesting that this data is taken at a late point in the aggregation time course. Such large fibrils are not detectable by MS in this configuration. This depletion correlates with an increase in baseline noise, also indicative of aggregation (as shown in Fig. 1C) and as previously reported.26
By plotting of the ratio of the sum of the absolute intensity of monomer and dimer peaks of the spectra at four time points along the aggregation course, we observe a shift in ion intensity to dimers with respect to time (Fig. 2). The intensity of even charged dimer peaks will be affected by the mass coincident monomer peaks, although this will not significantly affect this qualitative analysis. The increase in monomer intensity at 96 hours, is similar to the rise in monomer intensity after 4 hours of aggregation of TTR(105–115), which we theorised to be the result of the destabilisation of higher order aggregates in the solution or upon desolvation.26
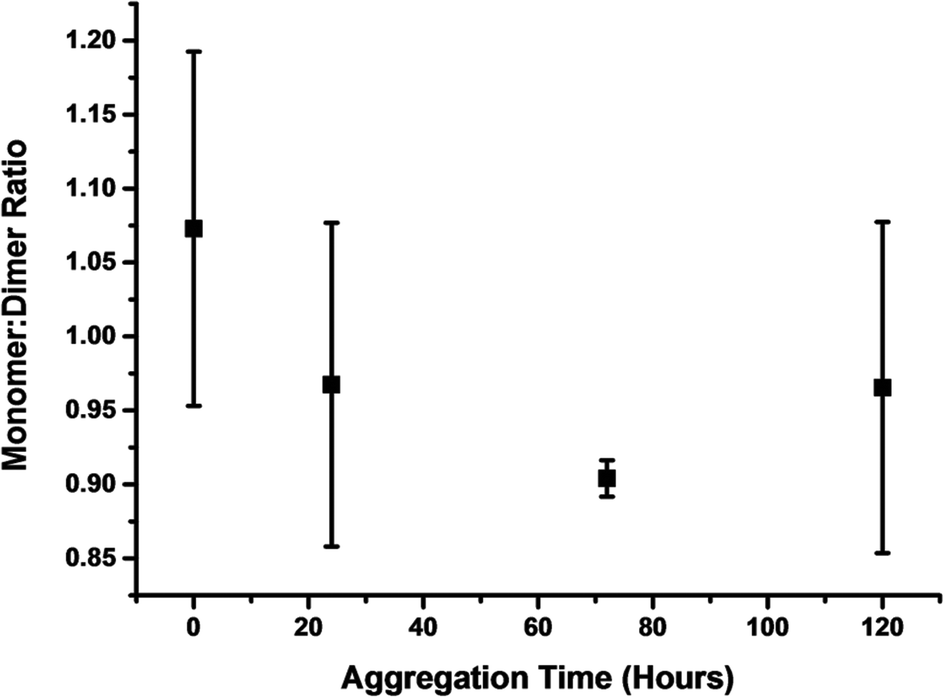 | ||
| Fig. 2 A plot of the monomer: dimer ratio derived from the average sum of the intensity of all monomer and dimer peaks of three independent experiments following an aggregation time course. | ||
IM-MS – following conformations through an aggregation time course
The in vitro aggregation of α-synuclein was conducted as described previously for up to 120 hours, and followed by IM-MS. Collision Cross Section Distributions (CCSD) derived from arrival time distributions for selected monomer and dimer species are shown in Fig. 3 (See ESI† Fig. S3 for MS spectra). At t = 0 for [aSyn + 6H]6+ a narrow CCSD centred on ∼1400 Å2 is observed with a small shoulder attributed to the mass to charge coincident species [(aSyn)2 + 12H]12+ (Fig. 3Ai). The large error bars indicate these species are highly variable and they are no longer present in subsequent time point CCSDs for [aSyn + 6H]6+. At subsequent time points the CCSD remains unchanged with the exception of the loss of the shoulder, perhaps a result of sequestration to higher order oligomeric species, the CCSD remains centred on ∼1400 Å2 (Fig. 3Aiii). For the higher charged monomeric species, [aSyn + 10H]10+ the CCSD is much broader and centred on ∼1800 Å2 (Fig. 3Bi). At subsequent time points for the [aSyn + 10H]10+ CCSD, the majority of species intensity remains centred on ∼1800 Å2. Additionally, a shoulder is consistently present across all time points centred on ∼2300 Å2 (Fig. 3Bi–iii). | ||
| Fig. 3 CCS distributions for the A: [aSyn + 6H]6+, B: [aSyn + 10H]13+, C: [aSyn + 13H]13+ monomer species and for D: [(aSyn)2 + 13H]13+, and E: [(aSyn)2 + 17H]17+, dimer species. The data presented was recorded with 35 V across the drift cell. | ||
In comparison to the data of Bernstein et al.40 despite the fact that a different ionisation mode has been used (negative instead of positive as used here), the CCS values derived are in good agreement for each charge state, although the Bernstein data indicates the presence of an additional slightly larger conformer. However, the CCS values derived for the [aSyn ± 10H]10± species show a discrepancy. The Bernstein CCS value is ∼2500 Å2 (Fig. 4, ref. 40) whereas the major peak in our CCSD plot is centred on ∼1800 Å2 (Fig. 3B). The lower intensity shoulder centred at ∼2300 Å2, represents a more extended conformer. This suggests that we have a cooler source, or we observe a difference resulting from charge location. For [aSyn + 13H]13+, the CCSD is broad and centred on ∼3000 Å2 (Fig. 3Ci). This CCSD also features a shoulder, attributed a compact conformer. Again the large error bars are consistent with a lowly populated species not present in subsequent time points where the intensity remains centred on ∼3000 Å2 (Fig. 3Cii–iii).
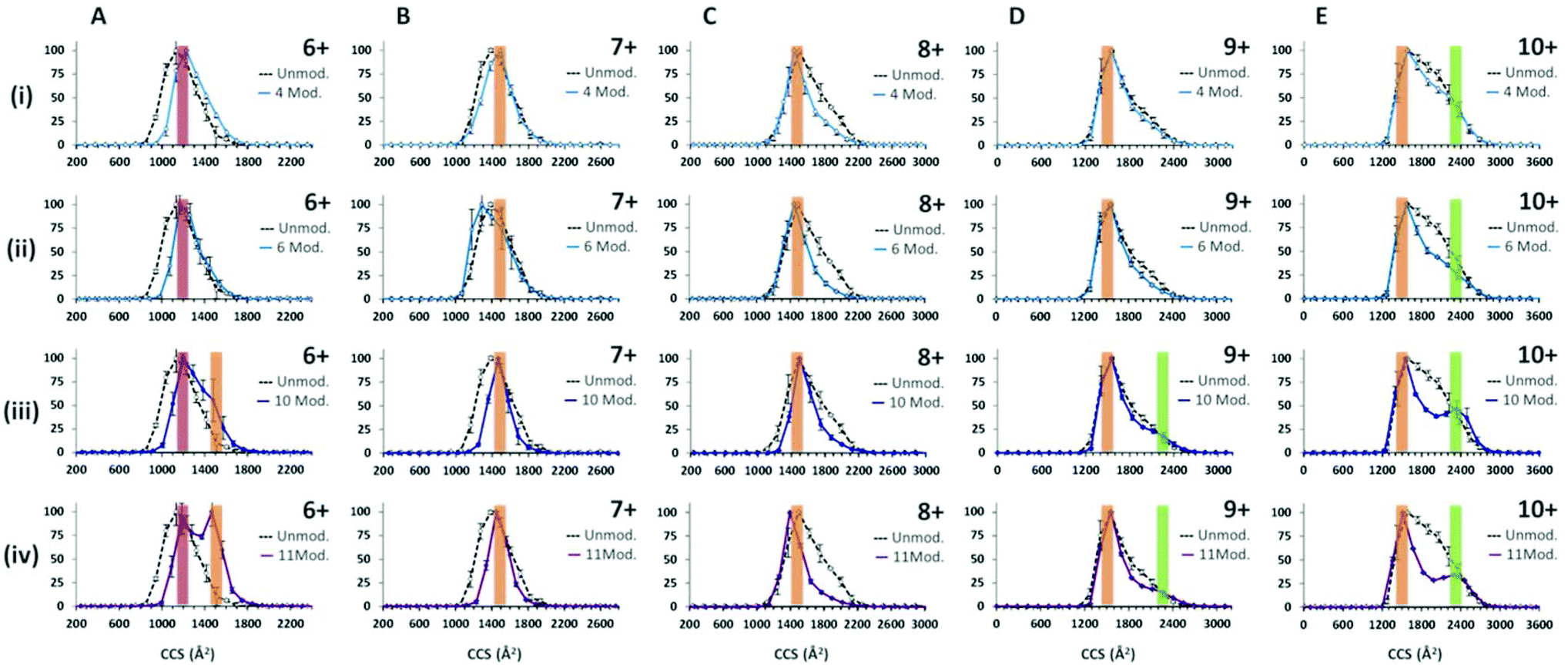 | ||
| Fig. 4 CCS distributions of α-synuclein with and without crosslinking. The dashed (Black) line on each CCSD corresponds to the CCSD derived for each charge state from an un-modified α-synuclein sample run on the instrument intended for comparison. The solid line on each CCSD corresponds to the CCSD derived for each charge state from a cross linked α-synuclein sample, ordered by increasing number of crosslink modifications, (i) 4 modifications, (ii) 6 modifications, (iii) 10 modifications and (iv) 11 modifications. The number of modifications relate to the number of crosslinks present which are a mixture of complete crosslinks and dead end crosslinks. These plots demonstrate the effect of crosslinking and charge state on the conformational constriction of α-synuclein. All data was taken with 30 V across the drift cell. | ||
The CCSD at t = 0 for the dimer, [(aSyn)2 + 13H]13+ is broad, similar to those exhibited for highly charged monomeric species. The ATD provides a CCS of ∼3000 Å2 (Fig. 3Di). There is a small shoulder (∼3600 Å2), which becomes more intense at Day 3 and is present at Day 5 (Fig. 3Dii–iii). The shoulder is obscured on the [(aSyn)2 + 13H]13+ Day 5 CCSD (Fig. 3Diii) by the broadening of the initial peak (∼3000 Å2). For the higher charged dimer [(aSyn)2 + 17H]17+ at t = 0, the CCSD observed is again broad and centred on ∼3900 Å2 with a shoulder centred on ∼3200 Å2 (Fig. 3Ei). This CCSD highlights the variability in the populations in the sample, the large error bars which are present across the CCSD suggesting several lowly populated conformationally dynamic species.
These species were chosen as they represent the breadth of the CSD, remain present throughout the time course, and represent the multimodal distribution present (ESI† Fig. S3). They are representative of the CCSD of all observed species from 7 < z < 22 (data not shown). The large spread in CCS over the entire CSD, ΔCCS ∼ 1600 Å2 (See Fig. 3Ai and Ci) and the large spread in CCS for each charge state (Fig. 3A–E) suggests the protein is flexible in solution and can adopt multiple conformations in solution and gas phase. The average CCS for both monomers and dimers increases with charge state, which we attribute to extended, more solvent exposed and therefore higher protonated forms of the protein in solution an effect which will be exacerbated by Coulombic repulsion in the gas phase as reported previously.75 The TEM data of samples subjected to the same solution and environmental conditions, show no fibrils at t = 0 (ESI† Fig. S2A).
There is a notable lack of change in CCS across the incubation timecourse (Fig. 3) despite the appearance of fibrils in the solution over the same time (ESI† Fig. S2B). The narrow CCSD centred on ∼1400 Å2 at t = 0 for [aSyn + 6H]6+, remains centred on ∼1400 Å2, at t = 3 days and t = 5 days (Fig. 3Ai–iii). The pattern is repeated for higher charged species, at t = 0 for [aSyn + 13H]13+ (Fig. 3Ci), the CCSDs is centred on ∼3000 Å2, and remains so at t = 3 days and t = 5 days (Fig. 3Cii and ii, respectively). There is also no evidence for narrowing of the CCSD which would indicate the adoption of a specific conformer. This pattern is also repeated for the dimeric species, see Fig. 3Ei–iii; the most intense peak remains centred on ∼3900 Å2 and the large error bars of the dimer CCSDs indicate that α-synuclein remains flexible. This suggests that even following long incubation times, the solution contains conformationally dynamic components, which perhaps exist in rapid equilibrium between oligomeric species, prior to their sequestration into larger oligomers or even fibrils. Such species are likely to reorganise during the nESI and even the IM experimental timescales, although with time there is less variability, as evidenced by smaller error bars (Fig. 3ci and iii). A comparison of the [aSyn + 6H]6+ monomer and the [(aSyn)2 + 13H]13+ dimer (Fig. 3) shows no significant decrease in CCS with dimer formation. The similar number of charges, with respect to mass, limits any difference that could be attributed to Coulombic repulsion. This lack of any compaction suggests that the α-synuclein dimer has a small binding interaction site and remains a largely unstructured dimer, or the superposition of many forms. This conclusion concurs with the force spectroscopy studies of α-synuclein dimers of Neupane et al.76 who describe a lack of discrete cooperative unfolding events for most dimers, indicative of lack of structure. In a small number of cases ∼15%, they note multiple unfolding transitions, which is supportive of the number of broad conformational families as well as the day to day variation shown by our MS and IM-MS experiments.76 The observable monomer and dimer species, which dominate the mass spectrum possess a large conformational spread which does not alter significantly during aggregation. This is despite the evidence from TEM (ESI† Fig. S1B) that these solutions contain fibrils, suggesting that the conformations of the soluble feeder stock of the aggregated solution is not altered by it (or at least not that can be observed by this method), or that the fibrils reversibly disassemble during desolvation. This second explanation is less likely since the solutions become harder to spray along the aggregation time course, indicating the presence of aggregates that block the spray tips.
Crosslinking IM-MS to trap solution phase conformers
Following the use of the cross linking reagent, extensive cross linking was seen for monomeric α-synuclein as shown in ESI† Fig. S4. Data for the dimers was not of sufficient intensity and was at too poor resolution to obtain collision cross-section data due to intensity stealing by the different masses of each cross linked form. CCSDs are derived from the arrival time distributions for selected charge states following exposure to the crosslinking reagent are shown in Fig. 4. Analysis of the CCSDs in Fig. 4 shows three conformational families. The first, designated C1, is centred at ∼1200 Å2 and is most clearly visible in the [aSyn + 6H]6+ CCSD (Fig. 4Ai). This compact conformer is present and remains populous even following the addition of eleven modifications (Fig. 4Aiv). The second family of a more extended conformer, E1 is centred at ∼1500 Å2 and is clearly visible in the [aSyn + 6H]6+ CCSD (Fig. 4Aiii–iv) and remains present throughout the intervening charge states ([aSyn + 7H]7+ to [aSyn + 9H]9+) to the [aSyn + 10H]10+ (Fig. 4E). This extended conformer is stabilised by the addition of more crosslink modifications (See Fig. 4Aiii–iv, [aSyn + 6H]6+ CCSD) and remains the dominant conformer. This conformational family matches the Bernstein et al. CCS value for the [aSyn − 8H]8−. The third family, U1 centred at ∼2350 Å2 is most clearly visible in the [aSyn + 10H]10+ CCSD. This unfolded conformer is again stabilised by the addition of crosslink modifications, shown by the gradual defining of the peak in the [aSyn + 10H]10+ CCSD (Fig. 4Ei–iv) and to a smaller extent in the 10 and 11 modification CCSDs of the [aSyn + 9H]9+ charge state (Fig. 4Diii–iv). The small CCS difference between [aSyn + 6H]6+ and [aSyn + 10H]10+ is likely to result from the additional coulombic repulsion suffered by the [aSyn + 10H]10+ species.The ability of the crosslinker modification to restrict the flexibility of the protein can be seen from the narrowing of the charge state distribution (ESI† Fig. S4). This effect can also be seen clearly in the CCSD of single charge state for example that of [aSyn + 7H]7+ (Fig. 4Bi) where narrowing of the CCSD occurs with the addition of as few as four crosslinking modifications and more dramatically with eleven (Fig. 4Biv). This narrowing suggests the crosslinks are preventing the protein from collapsing, preventing the adoption of compact CCS values as displayed in the unmodified sample (See Fig. 4Biv) in addition to decreasing the population of conformers sampled. The same effect is also evident in the CCSDs for other higher charge states including [aSyn + 8H]8+, however highly charged species such as [aSyn + 10H]10+, both unmodified and modified (eleven crosslink modifications, Fig. 4Eiv) exhibit the same most collapsed state (∼1200 Å2), suggesting coulombic repulsion is preventing the complete collapse of the protein which we have previously predicted to be 806 Å2.77 Interestingly, at higher charge states, the crosslinked protein transfers both compact and extended species (Fig. 4Eiv).
ECD – top down sequencing of an IDP
Here we examine the effect of pH on α-synuclein using ECD-FT-ICR MS. Although, ECD is a well-established tool for top-down sequencing of proteins and determination of the position of post translational modifications, information on higher order structure can also be obtained.78 ECD has been previously applied to α-synuclein to investigate its interaction with the naturally occurring polyamine, spermine.63,79 Xie et al.79 focussed on [aSyn + 14H]14+, since the fragmentation efficiency is far higher for higher charge states. The FT-ICR MS mass spectrum is shown in ESI† Fig. S5. Here we examine the fragments from select low charge state monomers, [aSyn + 6H]6+ to [aSyn + 10H]10+ at pH 6.8 and [aSyn + 7H]7+ to [aSyn + 9H]9+ at pH 3.5 (Select annotated fragmentation spectra, ESI† Fig. S6–S9). The fragments from select non-mass coincident dimer species are also examined, with charge states spanning from [(aSyn)2 + 13H]13+ to [(aSyn)2 + 17H]17+ (Select annotated fragmentation spectra, ESI† Fig. S10–S12).Decreasing the pH from 6.8 to 3.5 led to an increase in fragmentation efficiency. There is an increase in the intensity of fragments at pH 3.5 for both monomer and dimer species, which suggests the protein has adopted a less compact form with fewer stabilising non-covalent interactions, more susceptible to ECD. This is at variance with reported IM-MS data which describes the partial collapse of the protein at pH 2.5, a result of the acidic C-terminus being liable to collapse upon protonation40 and far-UV CD, FTIR, ANS fluorescence and SAXS data which demonstrate an increase in structure with a decrease in pH.72 Although the fragmentation pattern appears similar, the majority of fragments resolved for the pH 6.8 sample are from the [aSyn + 10H]10+ charge state. This charge state has been shown via IM-MS to exhibit an extended and therefore fragmentation-susceptible conformation. The comparison of [aSyn + 7H]7+ to [aSyn + 9H]9+ charge states (Fig. 5 and Table 1) for both pHs, highlight the difference in fragmentation with pH. The increase in fragments seen under low solution pH conditions could be a result of the extension of the N-terminus, which has been shown to adopt a more extended form at low pH, in contrast to the collapse experienced by the C-terminal region80
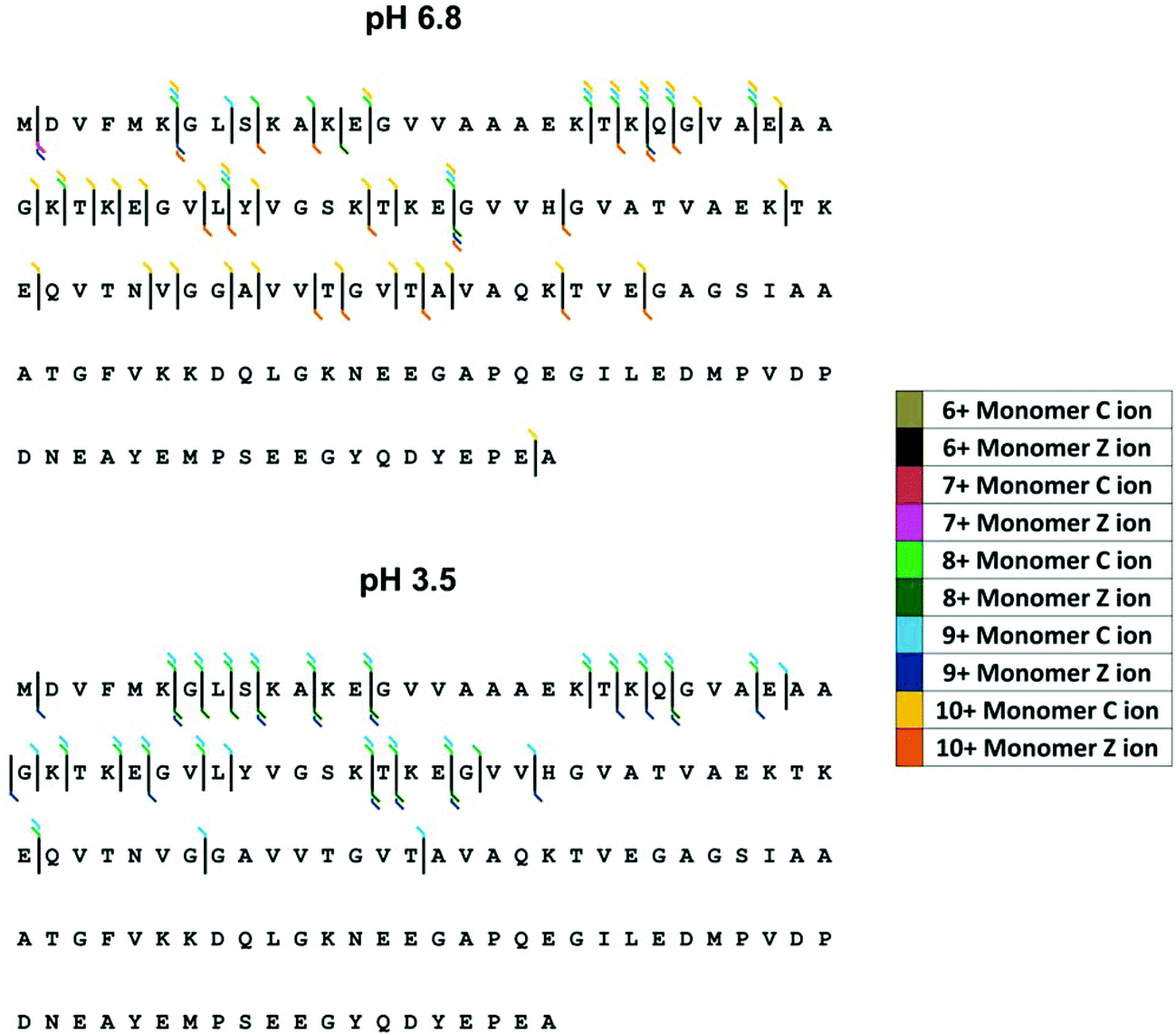 | ||
| Fig. 5 ECD fragmentation map for α-synuclein monomers at pH 6.8 (top) and pH 3.5 (bottom). Fragmentation efficiency is increased at low pH and increases with charge state. | ||
| pH 6.8 | pH 3.5 | ||
|---|---|---|---|
| [13+]/(15+)/17+ | [13+]/(15+)/17+ | ||
| C ion | Z ion | C ion | Z ion |
| (C115) | C279 | (Z274) | |
| (C57) | [C149] | ||
| C35 | C98 | ||
| (C6) | C75 | ||
| C50 | |||
| C47 | |||
| (C46) | |||
| C39 | |||
| (C38) | |||
| C37 | |||
| C35 | |||
| C34 | |||
| (C27) | |||
| (C24) | |||
| (C23) | |||
| (C22) | |||
| (C21) | |||
| C11 | |||
| (C9) | |||
| (C6) | |||
For both monomeric and dimeric parent ions, the fragmentation is limited to the N-terminus of the protein, both at pH 6.8 and 3.5. C-terminal fragments are only observed for the [aSyn + 10H]10+ monomer at pH 6.8 (Fig. 5A). If the α-synuclein monomers [aSyn + 7H]7+ to [aSyn + 9H]9+ at pH 6.8 and 3.5 are considered, no fragmentation is found from Gly47 to Ala140 at pH 6.8 (Fig. 5A) and between Ala76 to Ala140 at pH 3.5 (Fig. 5B). Similarly, for the α-synuclein dimers, limited C-terminal fragmentation occurs (Table 1). This data suggests that under these conditions there is a stable protected core of the protein involving the C-terminus, this correlates with paramagnetic relaxation enhancement and NMR dipolar couplings which demonstrate the presence of long range interactions between the C-terminus and central region of α-synuclein.81,82 In the case of the dimer, this suggests this may be the region of the dimer interface, which is supported by findings from single molecule AFM force spectroscopy measurements.83 However, our results do not correlate with other published data from CD, FTIR and in-cell NMR35 nor with data from HDX MS studies,56,58 which suggest independently a C-termini which is either, protected or unprotected from HDX exchange due to the presence of secondary or higher structure, highlighting the highly dynamic nature of α-synuclein, in particular the C-terminus.
Discussion and outlook
α-synuclein is known to be an extraordinarily conformational plastic protein, characteristic of its intrinsically disordered nature. Our MS data expands on these previous studies, highlighting the presence of multiple conformational families and emphasising the susceptibility of α-synuclein to environmental change, such as pH, as well as highlighting the day to day variability of our subject. The aggregation of α-synuclein is strongly linked to the aetiology of Parkinson's disease; we have demonstrated the ability of MS to follow in vitro aggregation of α-synuclein, through the depletion of the solution phase constituents. This method with little modification will allow the high throughput prototyping of potential aggregation inhibitors, due to the non-discriminatory nature of the technique, a prerequisite for studying the complex aggregation pathways of α-synuclein, as previously highlighted by HDX-MS.59 IM-MS shows a lack of a CCS change with aggregation duration, suggesting that the species in the solution phase remain disordered, adopting aggregation related conformational modifications at the time of sequestration into oligomeric or fibrillar species. This agrees with the intrinsically disordered nature of α-synuclein.The use of gas phase techniques has prompted understandable concern as to the biological relevance of these measurements in comparison with solution assays. Here, we have shown that chemical crosslinking can be used to trap solution phase species prior to gas phase analysis. Following chemical crosslinking we have identified three stable conformational families, compact (∼1200 Å2), extended (∼1500 Å2) and unfolded (∼2350 Å2) which are also present in the uncrosslinked protein albeit at differing abundances. When we extrapolate these values to anticipated Radius of Gyration (Rg) values from solution (ESI† Fig. S14 and Table S1, we find that these correlate to 16.8, 26.5, and 54.1 Å, these values compare well with Rg values calculated for fully globular and random coil 140 amino acid polypeptide chains, of 15.1 Å and 52.1 Å, respectively and the Rg range established encompasses reported experimental Rg values.72 This agreement supports the use of gas phase techniques on intact proteins as a probe of solution conformers.
ECD has been established elsewhere as a technique to probe the location of structure within gas phase protein ions. The large day to day and sample to sample variability of α-synuclein even under tight control could explain the increase in fragmentation efficiency that occurs with dropping the solution pH, a result that contradicts trends observed by multiple structural techniques. Alternatively, this increased fragmentation exhibited at low pH may be a result of the remodelling of α-synuclein. At neutral pH, α-synuclein has been shown to exhibit compact, intermediate and extended conformers by our MS, IM-MS and crosslinking IM-MS data and other studies including Frimpong et al.41 Bernstein et al. identify conformer collapse at pH 2.5,40 which concurs with measurements from other biophysical techniques. In line with the work of Frimpong et al.41 and Ferreon et al.84,85 scanning through a solution condition such as pH or SDS concentration enables access to intermediate conformations of the folding landscape, in our study the pH was lowered to pH 3.5,40 enabling access to intermediate conformations such as those identified in ref. 41. The conformational remodelling of α-synuclein has previously been shown to be substantial, even following small changes; for example, see the unfolding event between the −8 and −9 charge states in ref. 40. We therefore theorise this increase in fragmentation is not the result of the low pH disrupting long range interactions which lock α-synuclein in a semi-flexible state,81,82,86 but rather the induction of large scale conformational remodelling induced by modifying the solution environment. This is a similar effect to that seen by Frimpong et al. via deconvolution of α-synuclein CSDs at increasing pH,41 prior to protein collapse at lower pH (Bernstein et al.40), this intermediate ensemble stage facilitates the greater fragmentation seen at low pH.
The conformational fluidity of α-synuclein, will and does slow the progress of our understanding of this system with all assays. Despite this, MS based techniques can be usefully applied both to obtain structural information and also to follow the complex heterogeneous early stages of aggregation of this amyloidogenic protein.
Acknowledgements
The authors thank the support staff of the University of Manchester and the University of Edinburgh, whose efforts makes ours possible. We acknowledge Sophie R. Harvey, Rebecca Beveridge, C. Logan Mackay, Martin Wear and the EPPF for assistance with experiment design. RASOR Interdisciplinary Research Centre is thanked for the award of an EPSRC funded studentship to A.S.P. We also thank the British Mass Spectrometry Society for the provision of a small grant that has allowed us to pull nano-electrospray tips for 12 years and the Brazilian National Council for Scientific and Technological Development (CNPq), for the visiting student fellowship (246218/2012-6) for A.F.G as part of the Science without Borders (SWB) programme.References
- R. Beveridge, Q. Chappuis, C. Macphee and P. Barran, Analyst, 2013, 138, 32–42 RSC.
- E. Jurneczko, F. Cruickshank, M. Porrini, P. Nikolova, I. D. G. Campuzano, M. Morris and P. E. Barran, Biochem. Soc. Trans., 2012, 40, 1021–U1372 CrossRef CAS PubMed.
- A. B. Kanu, P. Dwivedi, M. Tam, L. Matz and H. H. Hill, J. Mass Spectrom., 2008, 43, 1–22 CrossRef CAS PubMed.
- J. Lim and R. W. Vachet, Anal. Chem., 2004, 76, 3498–3504 CrossRef CAS PubMed.
- A. M. Smith, T. R. Jahn, A. E. Ashcroft and S. E. Radford, J. Mol. Biol., 2006, 364, 9–19 CrossRef CAS PubMed.
- L. A. Woods, G. W. Platt, A. L. Hellewell, E. W. Hewitt, S. W. Homans, A. E. Ashcroft and S. E. Radford, Nat. Chem. Biol., 2011, 7, 730–739 CrossRef CAS PubMed.
- L. A. Woods, S. E. Radford and A. E. Ashcroft, Biochim. Biophys. Acta, Proteins Proteomics, 2013, 1834, 1257–1268 CrossRef CAS PubMed.
- D. P. Smith, S. E. Radford and A. E. Ashcroft, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6794–6798 CrossRef CAS PubMed.
- D. P. Smith, L. A. Woods, S. E. Radford and A. E. Ashcroft, Biophys. J., 2011, 101, 1238–1247 CrossRef CAS PubMed.
- A. C. Leney, C. L. Pashley, C. A. Scarff, S. E. Radford and A. E. Ashcroft, Mol. BioSyst., 2014, 10, 412–420 RSC.
- S. L. Bernstein, T. Wyttenbach, A. Baumketner, J. E. Shea, G. Bitan, D. B. Teplow and M. T. Bowers, J. Am. Chem. Soc., 2005, 127, 2075–2084 CrossRef CAS PubMed.
- M. M. Murray, S. L. Bernstein, V. Nyugen, M. M. Condron, D. B. Teplow and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 6316–6317 CrossRef CAS PubMed.
- S. L. Bernstein, N. F. Dupuis, N. D. Lazo, T. Wyttenbach, M. M. Condron, G. Bitan, D. B. Teplow, J. E. Shea, B. T. Ruotolo, C. V. Robinson and M. T. Bowers, Nat. Chem., 2009, 1, 326–331 CrossRef CAS PubMed.
- M. M. Gessel, C. Wu, H. Y. Li, G. Bitan, J. E. Shea and M. T. Bowers, Biochemistry, 2012, 51, 108–117 CrossRef CAS PubMed.
- X. Y. Zheng, M. M. Gessel, M. L. Wisniewski, K. Viswanathan, D. L. Wright, B. A. Bahr and M. T. Bowers, J. Biol. Chem., 2012, 287, 6084–6088 CrossRef CAS PubMed.
- M. M. Gessel, S. Bernstein, M. Kemper, D. B. Teplow and M. T. Bowers, ACS Chem. Neurosci., 2012, 3, 909–918 CrossRef CAS PubMed.
- L. Sanchez, S. Madurga, T. Pukala, M. Vilaseca, C. Lopez-Iglesias, C. V. Robinson, E. Giralt and N. Carulla, J. Am. Chem. Soc., 2011, 133, 6505–6508 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2009, 131, 18283–18292 CrossRef CAS PubMed.
- N. F. Dupuis, C. Wu, J. E. Shea and M. T. Bowers, J. Am. Chem. Soc., 2011, 133, 7240–7243 CrossRef CAS PubMed.
- L. C. Palmieri, B. Melo-Ferreira, C. A. Braga, G. N. Fontes, L. J. Mattos and L. Lima, Biophys. Chem., 2013, 180, 135–144 CrossRef PubMed.
- M. Landreh, G. Alvelius, J. Johansson and H. Jornvall, Rapid Commun. Mass Spectrom., 2014, 28, 178–184 CrossRef CAS PubMed.
- L. M. Young, P. Cao, D. P. Raleigh, A. E. Ashcroft and S. E. Radford, J. Am. Chem. Soc., 2014, 136, 660–670 CrossRef CAS PubMed.
- L. Young, H. Ndlovu, T. W. Knapman, S. A. Harris, S. E. Radford and A. E. Ashcroft, Int. J. Ion Mobility Spectrom., 2013, 16, 29–39 CrossRef CAS.
- N. Carullaa, M. Zhou, M. Arimon, M. Gairi, E. Giralt, C. V. Robinson and C. M. Dobson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 7828–7833 CrossRef PubMed.
- L. Larini, M. M. Gessel, N. E. LaPointe, T. D. Do, M. T. Bowers, S. C. Feinstein and J. E. Shea, Phys. Chem. Chem. Phys., 2013, 15, 8916–8928 RSC.
- H. L. Cole, J. M. D. Kalapothakis, G. Bennett, P. E. Barran and C. E. MacPhee, Angew. Chem., Int. Ed., 2010, 49, 9448–9451 CrossRef CAS PubMed.
- F. Chiti and C. M. Dobson, in Annual Review of Biochemistry, Annual Reviews, Palo Alto, 2006, vol. 75, pp. 333–366 Search PubMed.
- M. G. Spillantini, R. A. Crowther, R. Jakes, M. Hasegawa and M. Goedert, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6469–6473 CrossRef CAS.
- S. George, N. L. Rey, N. Reichenbach, J. A. Steiner and P. Brundin, Brain Pathol., 2013, 23, 350–357 CrossRef CAS PubMed.
- L. Breydo, J. W. Wu and V. N. Uversky, Biochim. Biophys. Acta, Mol. Basis Dis., 2012, 1822, 261–285 CrossRef CAS PubMed.
- H. A. Lashuel, C. R. Overk, A. Oueslati and E. Masliah, Nat. Rev. Neurosci., 2013, 14, 38–48 CrossRef CAS PubMed.
- J. Burre, S. Vivona, J. J. Diao, M. Sharma, A. T. Brunger and T. C. Sudhof, Nature, 2013, 498, E4–E6 CrossRef CAS PubMed.
- B. Fauvet, M. K. Mbefo, M. B. Fares, C. Desobry, S. Michael, M. T. Ardah, E. Tsika, P. Coune, M. Prudent, N. Lion, D. Eliezer, D. J. Moore, B. Schneider, P. Aebischer, O. M. El-Agnaf, E. Masliah and H. A. Lashuel, J. Biol. Chem., 2012, 287, 15345–15364 CrossRef CAS PubMed.
- E. Coelho-Cerqueira, P. Carmo-Goncalves, A. S. Pinheiro, J. Cortines and C. Follmer, FEBS J., 2013, 280, 4915–4927 CrossRef CAS PubMed.
- A. Binolfi, F. X. Theillet and P. Selenko, Biochem. Soc. Trans., 2012, 40, 950–U292 CrossRef CAS PubMed.
- K. A. Conway, S. J. Lee, J. C. Rochet, T. T. Ding, R. E. Williamson and P. T. Lansbury, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 571–576 CrossRef CAS.
- H. A. Lashuel, B. M. Petre, J. Wall, M. Simon, R. J. Nowak, T. Walz and P. T. Lansbury, J. Mol. Biol., 2002, 322, 1089–1102 CrossRef CAS.
- B. A. Silva, L. Breydo and V. N. Uversky, Mol. Neurobiol., 2013, 47, 446–459 CrossRef CAS PubMed.
- N. Cremades, S. I. A. Cohen, E. Deas, A. Y. Abramov, A. Y. Chen, A. Orte, M. Sandal, R. W. Clarke, P. Dunne, F. A. Aprile, C. W. Bertoncini, N. W. Wood, T. P. J. Knowles, C. M. Dobson and D. Klenerman, Cell, 2012, 149, 1048–1059 CrossRef CAS PubMed.
- S. L. Bernstein, D. F. Liu, T. Wyttenbach, M. T. Bowers, J. C. Lee, H. B. Gray and J. R. Winkler, J. Am. Soc. Mass Spectrom., 2004, 15, 1435–1443 CrossRef CAS PubMed.
- A. K. Frimpong, R. R. Abzatimov, V. N. Uversky and I. A. Kaltashov, Proteins: Struct., Funct., Bioinf., 2010, 78, 714–722 CAS.
- A. Natalello, F. Benetti, S. M. Doglia, G. Legname and R. Grandori, Proteins: Struct., Funct., Bioinf., 2011, 79, 611–621 CrossRef CAS PubMed.
- C. Vlad, K. Lindner, C. Karreman, S. Schildknecht, M. Leist, N. Tomczyk, J. Rontree, J. Langridge, K. Danzer, T. Ciossek, A. Petre, M. L. Gross, B. Hengerer and M. Przybylski, ChemBioChem, 2011, 12, 2740–2744 CrossRef CAS PubMed.
- C. Vlad, M. I. Iurascu, S. Slamnoiu, B. Hengerer and M. Przybylski, Methods Mol. Biol., 2012, 896, 399–412 CAS.
- Y. Peng, C. S. Wang, H. H. Xu, Y. N. Liu and F. M. Zhou, J. Inorg. Biochem., 2010, 104, 365–370 CrossRef CAS PubMed.
- A. Binolfi, G. R. Lamberto, R. Duran, L. Quintanar, C. W. Bertoncini, J. M. Souza, C. Cervenansky, M. Zweckstetter, C. Griesinger and C. O. Fernandez, J. Am. Chem. Soc., 2008, 130, 11801–11812 CrossRef CAS PubMed.
- L. Hong and J. D. Simon, J. Phys. Chem. B, 2009, 113, 9551–9561 CrossRef CAS PubMed.
- Y. Wang, M. Shi, K. A. Chung, C. P. Zabetian, J. B. Leverenz, D. Berg, K. Srulijes, J. Q. Trojanowski, V. M. Y. Lee, A. D. Siderowf, H. Hurtig, I. Litvan, M. C. Schiess, E. R. Peskind, M. Masuda, M. Hasegawa, X. M. Lin, C. Pan, D. Galasko, D. S. Goldstein, P. H. Jensen, H. Yang, K. C. Cain and J. Zhang, Sci. Transl. Med., 2012, 4(121), 121ra20 Search PubMed.
- Y. Lu, M. Prudent, B. Fauvet, H. A. Lashuel and H. H. Girault, ACS Chem. Neurosci., 2011, 2, 667–675 CrossRef CAS PubMed.
- L. L. Liu and K. J. Franz, J. Biol. Inorg. Chem., 2007, 12, 234–247 CrossRef CAS PubMed.
- S. Krishnan, E. Y. Chi, S. J. Wood, B. S. Kendrick, C. Li, W. Garzon-Rodriguez, J. Wypych, T. W. Randolph, L. O. Narhi, A. L. Biere, M. Citron and J. F. Carpenter, Biochemistry, 2003, 42, 829–837 CrossRef CAS PubMed.
- T. Konno, T. Morii, A. Hirata, S. I. Sato, S. Oiki and K. Ikura, Biochemistry, 2005, 44, 2072–2079 CrossRef CAS PubMed.
- C. D. Borsarelli, L. J. Falomir-Lockhart, V. Ostatna, J. A. Fauerbach, H. H. Hsiao, H. Urlaub, E. Palecek, E. A. Jares-Erijman and T. M. Jovin, Free Radicals Biol. Med., 2012, 53, 1004–1015 CrossRef CAS PubMed.
- H. Wei, J. Ahn, Y. Q. Yu, A. Tymiak, J. R. Engen and G. D. Chen, J. Am. Soc. Mass Spectrom., 2012, 23, 498–504 CrossRef CAS PubMed.
- D. Houde and S. A. Berkowitz, J. Pharm. Sci., 2012, 101, 1688–1700 CrossRef CAS PubMed.
- C. Del Mar, E. A. Greenbaum, L. Mayne, S. W. Englander and V. L. Woods, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15477–15482 CrossRef CAS PubMed.
- S. J. C. Lee, J. W. Lee, T. S. Choi, K. S. Jin, S. Lee, C. Ban and H. I. Kim, Anal. Chem., 2014, 86, 1909–1916 CrossRef CAS PubMed.
- S. Mysling, C. Betzer, P. H. Jensen and T. J. D. Jorgensen, Biochemistry, 2013, 52, 9097–9103 CrossRef CAS PubMed.
- W. Paslawski, S. Mysling, K. Thomsen, T. J. D. Jorgensen and D. E. Otzen, Angew. Chem., Int. Ed., 2014, 53, 7560–7563 CrossRef CAS PubMed.
- J. N. Rao, C. C. Jao, B. G. Hegde, R. Langen and T. S. Ulmer, J. Am. Chem. Soc., 2010, 132, 8657–8668 CrossRef CAS PubMed.
- M. Zhu, S. Rajamani, J. Kaylor, S. Han, F. M. Zhou and A. L. Fink, J. Biol. Chem., 2004, 279, 26846–26857 CrossRef CAS PubMed.
- X. Y. Meng, L. A. Munishkina, A. L. Fink and V. N. Uversky, Biochemistry, 2009, 48, 8206–8224 CrossRef CAS PubMed.
- M. Grabenauer, S. L. Bernstein, J. C. Lee, T. Wyttenbach, N. F. Dupuis, H. B. Gray, J. R. Winkler and M. T. Bowers, J. Phys. Chem. B, 2008, 112, 11147–11154 CrossRef CAS PubMed.
- E. Illes-Toth, C. F. Dalton and D. P. Smith, J. Am. Soc. Mass Spectrom., 2013, 24, 1346–1354 CrossRef CAS PubMed.
- S. J. Wood, J. Wypych, S. Steavenson, J. C. Louis, M. Citron and A. L. Biere, J. Biol. Chem., 1999, 274, 19509–19512 CrossRef CAS PubMed.
- M. Hashimoto, L. J. Hsu, A. Sisk, Y. Xia, A. Takeda, M. Sundsmo and E. Masliah, Brain Res., 1998, 799, 301–306 CrossRef CAS.
- A. L. Fink, Acc. Chem. Res., 2006, 39, 628–634 CrossRef CAS PubMed.
- Z. A. Chen, A. Jawhari, L. Fischer, C. Buchen, S. Tahir, T. Kamenski, M. Rasmussen, L. Lariviere, J. C. Bukowski-Wills, M. Nilges, P. Cramer and J. Rappsilber, EMBO J., 2010, 29, 717–726 CrossRef CAS PubMed.
- A. H. Iglesias, L. F. A. Santos and F. C. Gozzo, Anal. Chem., 2010, 82, 909–916 CrossRef CAS PubMed.
- B. J. McCullough, J. Kalapothakis, H. Eastwood, P. Kemper, D. MacMillan, K. Taylor, J. Dorin and P. E. Barran, Anal. Chem., 2008, 80, 6336–6344 CrossRef CAS PubMed.
- R. D. LeDuc, G. K. Taylor, Y. B. Kim, T. E. Januszyk, L. H. Bynum, J. V. Sola, J. S. Garavelli and N. L. Kelleher, Nucleic Acids Res., 2004, 32, W340–W345 CrossRef CAS PubMed.
- V. N. Uversky, J. Li and A. L. Fink, J. Biol. Chem., 2001, 276, 10737–10744 CrossRef CAS PubMed.
- A. E. Counterman, A. E. Hilderbrand, C. A. S. Barnes and D. E. Clemmer, J. Am. Soc. Mass Spectrom., 2001, 12, 1020–1035 CrossRef CAS.
- A. Roostaee, S. Beaudoin, A. Staskevicius and X. Roucou, Mol. Neurodegener., 2013, 8, 5 CrossRef CAS PubMed.
- K. B. Shelimov, D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1997, 119, 2240–2248 CrossRef CAS.
- K. Neupane, A. Solanki, I. Sosova, M. Belov and M. T. Woodside, PLoS One, 2014, 9(1), e86495 Search PubMed.
- R. Beveridge, S. Covill, K. J. Pacholarz, J. M. Kalapothakis, C. E. MacPhee and P. E. Barran, Anal. Chem., 2014, 86, 10979–10991 CrossRef CAS PubMed.
- H. Zhang, W. D. Cui, J. Z. Wen, R. E. Blankenship and M. L. Gross, J. Am. Soc. Mass Spectrom., 2010, 21, 1966–1968 CrossRef CAS PubMed.
- Y. M. Xie, J. Zhang, S. Yin and J. A. Loo, J. Am. Chem. Soc., 2006, 128, 14432–14433 CrossRef CAS PubMed.
- K. P. Wu, D. S. Weinstock, C. Narayanan, R. M. Levy and J. Baum, J. Mol. Biol., 2009, 391, 784–796 CrossRef CAS PubMed.
- C. W. Bertoncini, Y. S. Jung, C. O. Fernandez, W. Hoyer, C. Griesinger, T. M. Jovin and M. Zweckstetter, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 1430–1435 CrossRef CAS PubMed.
- W. B. Zhou, C. M. Long, S. H. Reaney, D. A. Di Monte, A. L. Fink and V. N. Uversky, Biochim. Biophys. Acta, Mol. Basis Dis., 2010, 1802, 322–330 CrossRef CAS PubMed.
- A. V. Krasnoslobodtsev, I. L. Volkov, J. M. Asiago, J. Hindupur, J. C. Rochet and Y. L. Lyubchenko, Biochemistry, 2013, 52, 7377–7386 CrossRef CAS PubMed.
- A. C. M. Ferreon and A. A. Deniz, Biochemistry, 2007, 46, 4499–4509 CrossRef CAS PubMed.
- A. C. M. Ferreon, C. R. Moran, J. C. Ferreon and A. A. Deniz, Angew. Chem., Int. Ed., 2010, 49, 3469–3472 CrossRef CAS PubMed.
- S. McClendon, C. C. Rospigliosi and D. Eliezer, Protein Sci., 2009, 18, 1531–1540 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an02306d |
| This journal is © The Royal Society of Chemistry 2015 |