
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Antibody mimetic receptor proteins for label-free biosensors†
M.
Raina
a,
R.
Sharma
a,
S. E.
Deacon
b,
C.
Tiede
b,
D.
Tomlinson
bc,
A. G.
Davies
a,
M. J.
McPherson
bc and
C.
Wälti
*a
aBioelectronics, School of Electronic and Electrical Engineering, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK. E-mail: c.walti@leeds.ac.uk
bSchool of Molecular and Cellular Biology, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
cAstbury Centre for Structural Molecular Biology, University of Leeds, Woodhouse Lane, Leeds, LS2 9JT, UK
First published on 18th November 2014
Abstract
The development of high sensitivity biosensors, for example for clinical diagnostics, requires the identification of suitable receptor molecules which offer high stability, specificity and affinity, even when embedded into solid-state biosensor transducers. Here, we present an electrochemical biosensor employing small synthetic receptor proteins (Mw < 15 kDa) which emulate antibodies but with improved stability, sensitivity and molecular recognition properties, in particular when immobilized on a solid sensor surface. The synthetic receptor protein is a non-antibody-based protein scaffold with variable peptide regions inserted to provide the specific binding, and was designed to bind anti-myc tag antibody (Mw ∼ 150 kDa), as a proof-of-principle exemplar. Both the scaffold and the selected receptor protein were found to have high thermostability with melting temperatures of 101 °C and 85 °C, respectively. Furthermore, the secondary structures of the receptor protein were found to be very similar to that of the original native scaffold, despite the insertion of variable peptide loops that create the binding sites. A label-free electrochemical sensor was fabricated by functionalising a microfabricated gold electrode with the receptor protein. A change in the phase of the electrochemical impedance was observed when the biosensor was subjected to anti-myc tag antibodies at concentrations between 6.7 pM and 6.7 nM. These findings demonstrate that these non-antibody receptor proteins are excellent candidates for recognition molecules in label-free biosensors.
Introduction
The ability to detect specific biomarkers present in a patient's blood or other biological samples, even at very low concentrations, make electrochemical biosensors a viable technology for diagnosis and monitoring. A typical biosensor comprises a series of receptor molecules immobilized either directly on a conductive substrate,1,2 or indirectly via a self-assembled molecular monolayer.3 When the sensor is exposed to a solution containing the specific biomarker, selective binding of the biomarker to the immobilized receptors occurs, leading to a measurable real-time change in an electrical signal within the sensor.4 The degree of change in the signal provides a measure of the biomarker concentration and, therefore, an indication of disease presence and progression.Among the various receptor molecules used for such biosensors, native antibodies are the most commonly employed as in principle, they are sensitive and selective towards the target biomarker proteins.5 However, despite their widespread use, there can be significant challenges. Antibodies are large, multi-domain proteins with a number of disulphide bonds and can therefore be relatively unstable. They can suffer from loss of affinity and specificity upon immobilization on a solid surface,5,6 non-specific binding to the other regions of the antibody or the surface,7 steric hindrance issues, and instability during regeneration of the functionalized sensor surface.8–10 The bulky multi-meric nature of the antibodies also makes the interaction with the target protein unpredictable, in particular on solid surfaces.11 In addition, antibodies are often difficult to produce recombinantly in a bacterial system, and hence their production can be a time-consuming and expensive process.12
To address these challenges, smaller recombinant antibody fragments have been proposed as alternative receptors.12–14 Their small size was found to reduce steric hindrance, increase surface coverage during immobilization,15 and allow binding to target epitopes that were generally not accessible to full size native antibodies.16 However, antibody fragments are also prone to experience a partial loss of their biological activity owing to change in conformation and/or orientation when immobilized on solid surfaces, and therefore can show reduced affinity to the target molecule.8 In addition, short unconstraint peptides17,18 as well as nucleic acid aptamers19 have been demonstrated as biosensor transducers.
As a further alternative to antibodies, small and easy-to-express non-antibody protein scaffolds, designed to constrain and display variable peptide inserts for target protein recognition, have also been proposed.20–24 The ambition is to develop a robust scaffold that can be used to develop receptor proteins that mimic antibodies, but with improved stability and the surface immobilization properties that are required for biosensor applications.
Here, we report the use of a small receptor protein (Mw ∼ 15 kDa) based on a non-antibody scaffold protein as the target-specific receptor molecule in a label-free electrochemical biosensor. Receptor proteins were isolated that bind anti-myc tag antibody (Mw ∼ 150 kDa) as the model target for this proof-of-principle study. The receptor proteins were characterized in detail and show high thermostability and reproducible refolding. Furthermore, we show that the secondary structure of the scaffold portion of the receptor protein remains intact upon insertion of the variable peptides loops.
Results and discussion
The employed antibody mimetic scaffold provides a robust and flexible alternative to antibodies for developing receptor proteins with highly specific molecular recognition properties. It is based on a consensus protein sequence and provides a structurally highly robust protein able to present one to three ‘variable’ peptides as interaction sites without compromising the structural integrity of the protein. Design of the variable peptide regions allowed the creation of a library from which receptor proteins suitable for highly specific binding to chosen target molecules can be selected. Here, we demonstrate the integration of selected receptor proteins into solid state devices as the molecular recognition element in an electrochemical biosensor. The model target chosen to demonstrate the concept was anti-myc tag antibody, because the interaction of this antibody with the c-myc epitope sequence (EQKLISEEDL) is well characterized and is widely used to detect over-expressed recombinant proteins carrying the myc epitope tag.25Generating receptor proteins
Receptor proteins that mimic the c-myc tag were generated via screening of a phage display library of diversity about 109 generated from library DNA sourced from Sloning BioTechnology GmbH (MorphoSys) in which the three variable peptide regions on the scaffold were replaced with randomised peptides of 6, 9 and 9 amino acids excluding cysteine, respectively. The library was screened against biotinylated anti-myc tag antibody. Phage particles displaying receptor proteins with affinity for the target were eluted after three rounds of panning by exposure to higher concentrations of anti-myc tag antibody.Forty-eight phage clones were arbitrarily selected from the eluted population. Following amplification, the affinity of the receptor proteins displayed on these phage particles for binding to anti-myc tag antibody was determined using a phage ELISA. About 70% of the selected phage clones showed significant binding to anti-myc tag antibody, with almost no binding observed in the control wells. These data confirm the specificity of selected receptor proteins expressed on the surface of the phage particles (Fig. S1 in ESI†).
The twenty receptor proteins with the highest apparent affinity for anti-myc tag antibody were selected and sequenced. For all selected receptor proteins, the amino acid sequence of one of the variable regions was found to resemble the LISE part of the c-myc epitope sequence (Fig. 1). This region is known to be the key sequence in facilitating c-myc epitope binding to anti-myc tag antibody.26 Ten clones with the highest sequence similarity to the c-myc tag were chosen for further analysis. The position of relevant sequences in different loops of the receptor proteins, and their similarity to the c-myc epitope sequence EQKLISEEDL, is also shown in Fig. 1.
 | ||
| Fig. 1 (a) Sequences of the twenty receptor proteins (1–20) selected from the phage ELISA listed in decreasing order of affinity to anti-myc tag antibody estimated from the corresponding phage ELISA signals. NTVR (N-terminal variable region), loop-1 and loop-2 represent the three variable regions where the random amino acid sequences are inserted, with the remaining sequence conserved. (b) Variable regions of the ten selected receptor proteins showing maximum consensus with the c-myc epitope EQKLISEEDL. The amino acid sequence most closely resembling LISE is shown in bold. | ||
The nucleic acid sequences of the ten selected receptor proteins plus 8-histidine tag were inserted at the multiple-cloning region of the pET-11(a) bacterial expression vector (see Fig. S2†). Following transformation into Escherichia coli XL10 Gold cells, colonies were screened for the correct insert by colony PCR and DNA sequencing.
The protein expression conditions were established for a receptor protein recognising glutathione S-transferase (GST). Four different expression hosts, BL21 Star (DE3), C41 (DE3), C43 (DE3), and BL21 Gold cells (DE3), were tested in different auto-induction media. Dot blot analysis revealed that BL21 Star (DE3) cells grown in terrific broth (TB) and yeast extract/tryptone (2YT) showed maximal expression levels (Fig. S3†).
The solubility of the expressed receptor proteins was investigated by analysing aliquots of supernatant and total cell lysate on a 15% SDS (sodium-dodecyl-sulfate) polyacrylamide gel followed by western blot analysis with HRP-conjugated anti-histidine tag antibody. As expected, a band was detected at 12–15 kDa for both supernatant and total cell lysate (Fig. S4†), demonstrating that the receptor proteins were expressed correctly under the optimized conditions.
To identify the receptor protein with the highest affinity for anti-myc tag antibody an ELISA assay was performed. The purified receptor proteins at 10 μg ml−1 together with an anti-GST receptor and the native scaffold as controls, were immobilized on an ELISA plate which was then exposed to 1 μg ml−1 anti-myc tag antibody. Successful binding of the target to the receptor protein was detected with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4000 dilution of anti-mouse antibody conjugated to HRP followed by colorimetric detection (Fig. 2). The specificity and cross-reactivity of the receptor–target binding interaction was investigated using another mouse antibody (anti-β2 microglobulin antibody) and BSA (bovine serum albumin) as controls. Identical ELISA plates to the ones used above were incubated separately with 1 μg ml−1 of each control.
:4000 dilution of anti-mouse antibody conjugated to HRP followed by colorimetric detection (Fig. 2). The specificity and cross-reactivity of the receptor–target binding interaction was investigated using another mouse antibody (anti-β2 microglobulin antibody) and BSA (bovine serum albumin) as controls. Identical ELISA plates to the ones used above were incubated separately with 1 μg ml−1 of each control.
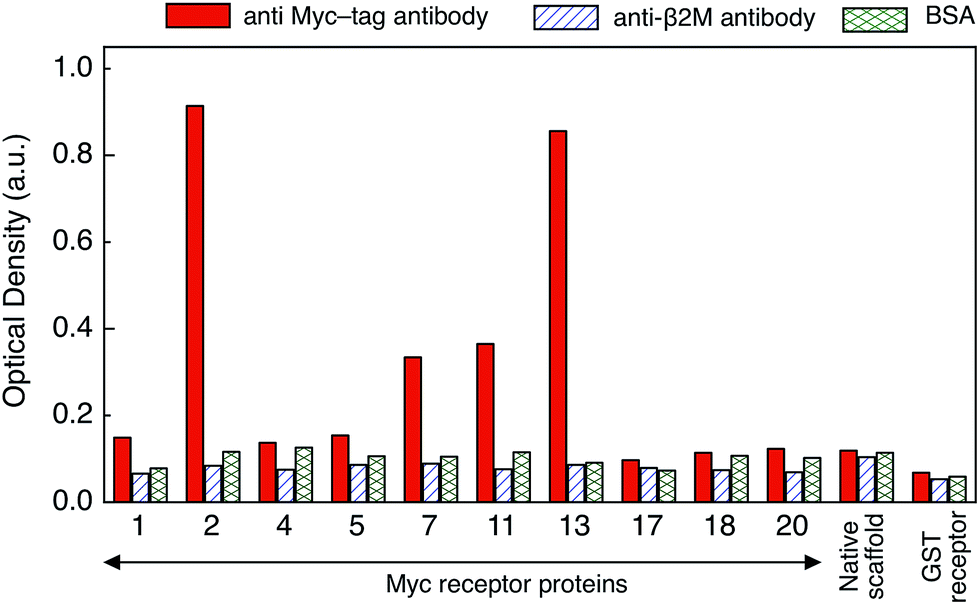 | ||
| Fig. 2 Absorbance measured at 650 nm showing the ELISA response of the ten selected receptor proteins after subjecting them to monoclonal anti-myc tag antibody, anti-β2 microglobulin antibody and BSA. ELISA intensities >0.2 a.u. are considered to represent a positive response. The native scaffold and the GST receptor protein were used as controls, and only minimal response was observed. | ||
Receptor protein-2 and -13 showed highest binding signal for anti-myc tag antibody, followed by receptor protein-11 and -7. The responses of the other receptor proteins were of the same order of magnitude as that of the native scaffold (<0.2 a.u.). A similarly low (<0.2 a.u.) binding response was observed for all receptor proteins when exposed to the controls (BSA and anti-β2 microglobulin antibody), demonstrating specific binding of receptor protein-2, -13, -11 and -7 to the anti-myc tag antibody. However, receptor protein-13 was found to aggregate and precipitate readily, which may be attributed to the presence of the hydrophobic phenylalanine and proline residues in one of the variable loops and which makes the receptor protein-13 prone to aggregation.27 The response of receptor proteins-7 and -11 is only about twice that of the negative controls, and therefore receptor protein-2 was chosen as the receptor protein for label-free biosensing and was characterised in detail.
Characterization
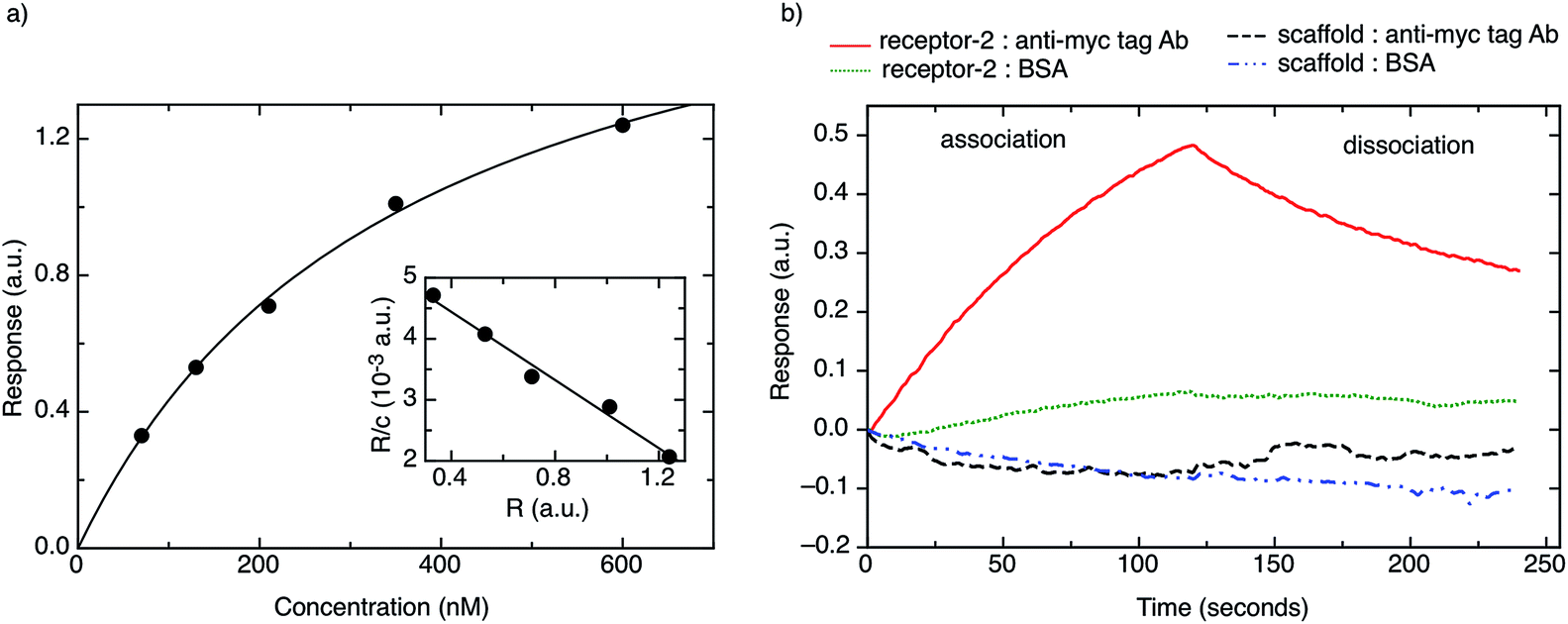 | ||
| Fig. 3 (a) Binding response of monoclonal anti-myc tag antibody to receptor protein-2. The solid line shows the fit to the linearized form of the data. Inset: linearized form of binding data where the y-axis label R/c corresponds to the sensor response (R) divided by anti-myc tag antibody concentration (c), and the x-axis label to the sensor response (R), respectively. (b) Typical sensogram showing the binding response of monoclonal anti-myc tag antibody to receptor protein-2, as well as negative controls (response of: BSA to receptor protein-2, monoclonal anti-myc tag antibody to native scaffold, BSA to native scaffold). | ||
Fig. 3b shows a typical BLI association/dissociation sensogram for a receptor protein-2-functionalised surface when challenged with an anti-myc tag antibody concentration of 130 nM. To demonstrate the specificity of receptor protein-2, analogous binding curves were obtained for the binding of BSA to receptor protein-2 and the binding of anti-myc tag antibody to the native scaffold (Fig. 3b). Both show no detectable binding.
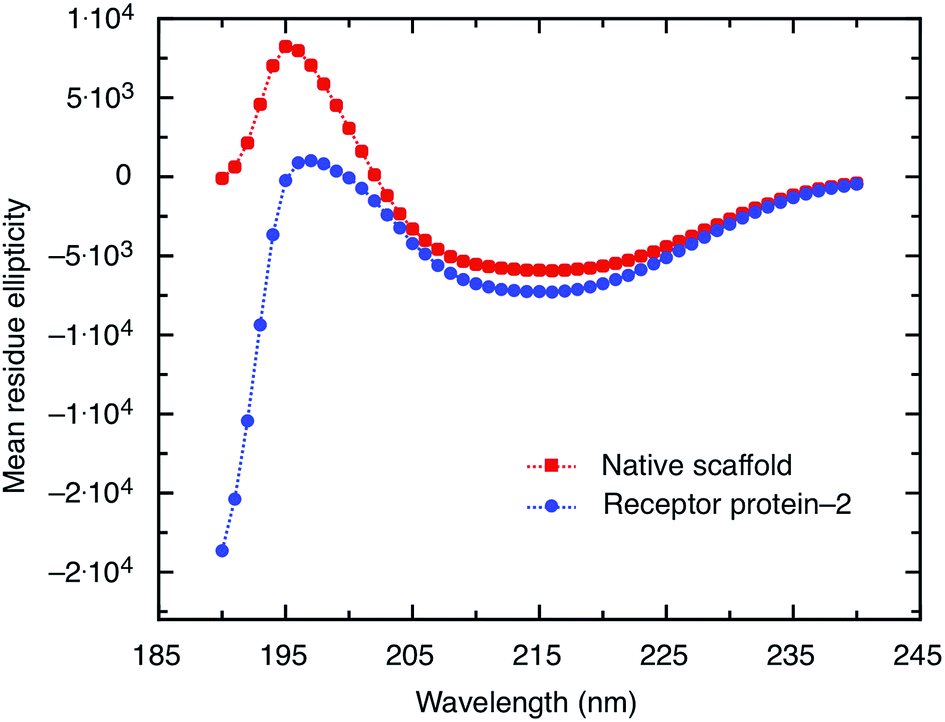 | ||
| Fig. 4 CD spectra of the scaffold and receptor protein-2, both at a concentration of 0.5 mg ml−1. The two spectra show the same characteristic features indicating the presence of predominantly anti-parallel β-pleated sheets. | ||
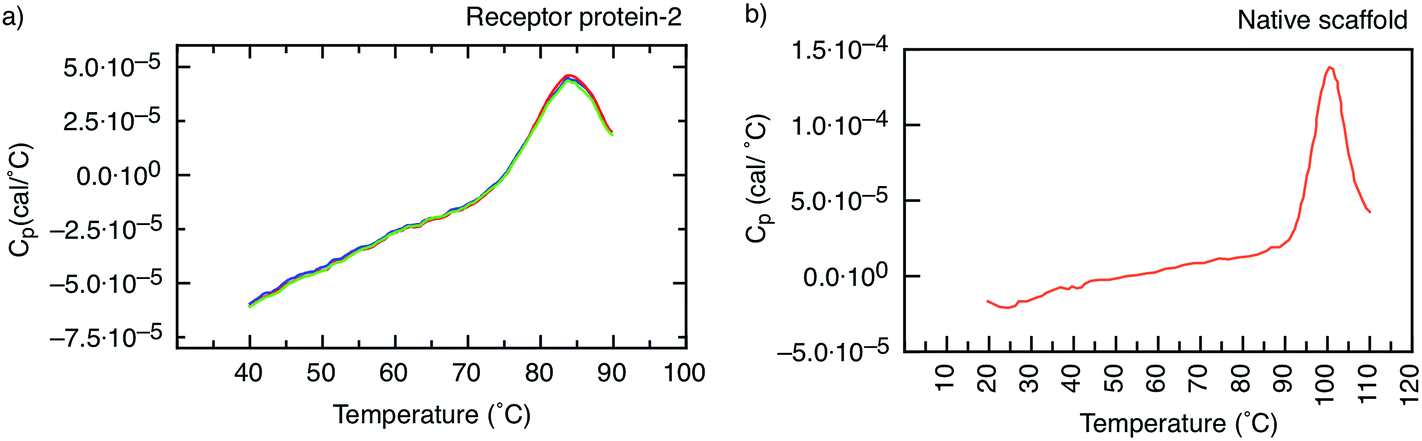 | ||
| Fig. 5 Heat capacity as a function of temperature. (a) Receptor protein-2, showing a melting temperature Tm = 85 °C. The different colours correspond to individual sweeps and show the high reversibility of unfolding. Tm remains the same for at least 30 sweeps. (b) Native scaffold, showing Tm = 101 °C. | ||
In addition, the unfolding of receptor protein-2 was found to be reversible during repeated thermal scans with no degradation observed even after 30 cycles with an identical Tm = 85 °C. However, for the native scaffold, the thermal cycling was not reversible, most likely because the temperatures of >100 °C may have caused aggregation and inhibited the re-folding of the scaffold.
Label-free electrochemical biosensor
The highly stable receptor protein-2 was integrated into a non-faradaic electrochemical impedance biosensor to demonstrate the label-free detection of anti-myc tag antibodies. Following assembly of a monothiol-alkane-PEG-acid (SH–(CH2)11–EG6–COOH) self-assembled monolayer (SAM) onto a gold substrate, the receptor protein-2 was immobilized through EDC/NHS amine coupling chemistry. The COOH was activated by exposure to NHS/EDC for 15 min, and subsequently 500 μl of 10 μg ml−1 receptor protein-2 was deposited onto the surface. This functionalised gold surface formed the working electrode of the three-electrode electrochemical sensor, with an Ag/AgCl saturated KCl electrode as the reference electrode, and a platinum wire as the counter electrode. The electrochemical impedance was measured over a frequency range from 50 mHz to 100 kHz.We have shown previously that the shift in the phase of the electrochemical impedance can be used to monitor binding at the sensor surface,3 and an increase in phase is expected with increasing target concentration. Before challenging the sensor surface with anti-myc tag antibody, the stability of the sensor was ascertained by measuring the phase response of the sensor in 100 mM PB pH 7 after 5 consecutive buffer injections in intervals of 15 minutes. Fig. 6a shows the last three measurements of the phase of the electrochemical impedance at 100 mHz before the application of different concentrations of anti-myc tag antibody, and only minimal noise (<±0.1°) was observed (blue squares in Fig. 6a). The final measurement was then taken as the baseline of the sensor.
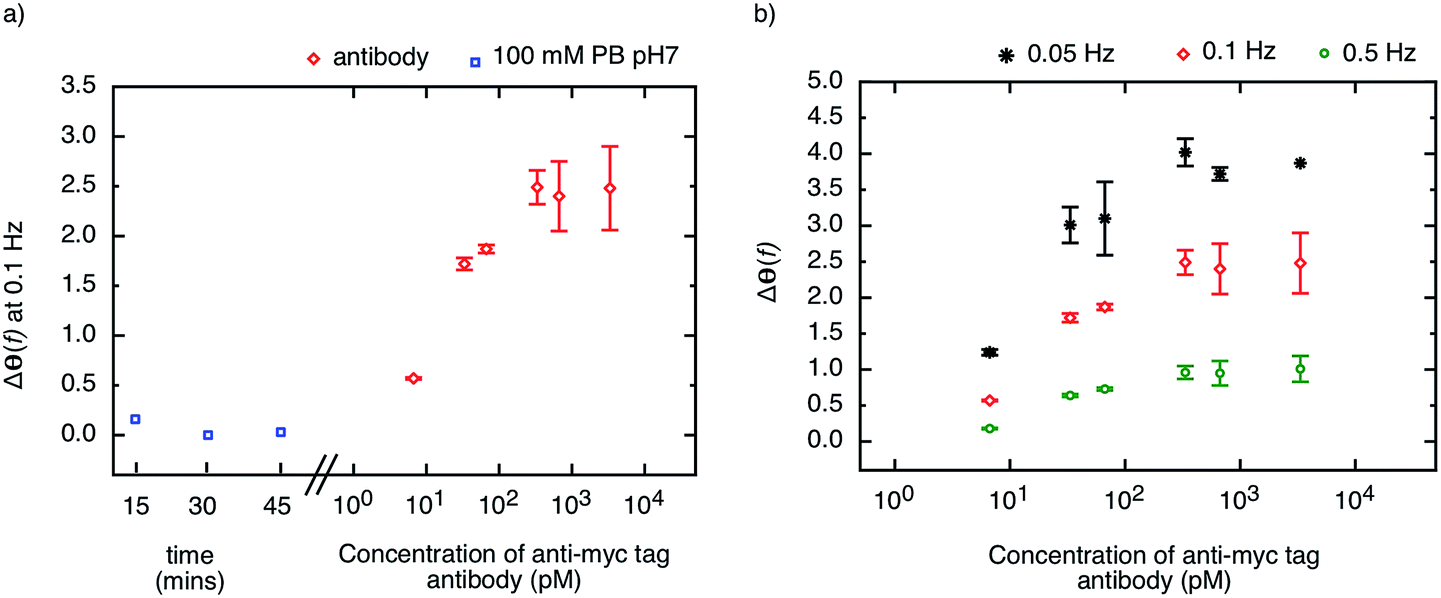 | ||
| Fig. 6 (a) Phase-shift of electrochemical impedance, Δθ(f), at 0.1 Hz of the biosensor, during initial stabilization checks (blue squares) and when exposed to anti-myc tag antibodies at concentrations between 6.7 pM and 3.3 nM in 100 mM PB buffer pH 7. The sensor was incubated for 15 minutes prior to measurements. (b) Sensor response to different concentrations of anti-myc tag antibodies measured at 0.5 Hz, 0.1 Hz and 0.05 Hz. | ||
The sensor was then subjected to different concentrations of the monoclonal anti-myc tag antibody ranging from 6.7 pM to 6.7 nM in 100 mM PB pH 7. The phase of the impedance of the sensor was measured after 15 minutes incubation with the sample. The response of the sensor to different concentrations of anti-myc tag antibody measured at 100 mHz is shown in Fig. 6a (red circles). A sharp increase in the phase of the impedance was observed for the lowest concentrations reported, with a slope of approximately 1.2° per decade. The sensor response saturated above a concentration of 330 pM, yielding a dynamic range of around two orders of magnitude.
We note that the change in phase is larger at smaller frequencies, and for the same concentration of target, the sensor response is most pronounced at the lowest frequency (50 mHz). The phase shift as a result of target binding is shown for different frequencies (50 mHz, 100 mHz, and 500 mHz) in Fig. 6b. However, while the phase shift is largest for 50 mHz, the instrument error at this frequency is significantly larger than at higher frequencies and hence the measurements are less reliable. The signal-to-noise ratio was found to be optimal at around 100 mHz and hence this frequency is used as the sensor response to quantify the amount of binding on the sensor surface.
Conclusion
A non-antibody receptor protein that mimics the c-myc epitope was developed as the capture molecule for a label-free electrochemical biosensor. The native scaffold and selected receptor proteins were characterized. The scaffold and receptor proteins were found to be very stable with melting temperatures of 101 °C and 85 °C for the native scaffold and the receptor protein-2, respectively, and the latter was found to refold after thermal denaturation. The secondary structure of the scaffold portion of receptor protein was found to be maintained despite the insertion of variable peptide sequences and predominantly β-sheet structures were observed. High melting temperature, reliable refolding, and robust secondary structures are important prerequisites for applications where capture molecules are employed far from physiological conditions, such as in biosensors. From BLI experiments, the dissociation constant KD of the receptor protein–monoclonal anti-myc tag antibody complex was found to be approximately 360 nM. However, when the receptor protein was used as the capture molecule in an electrochemical impedimetric biosensor, where it was immobilised on the sensor surface to measure the concentrations of anti-myc tag antibodies, concentrations in the range of 6.7 pM to 330 pM could be measured. These finding demonstrate that such non-antibody receptor proteins are suited as capture molecules for label-free detection of biomarkers in solid-state biosensors.Materials and methods
Receptor protein selection
A phage display library of approximately 1 × 109 clones was generated using a synthetic library construct, containing three variable peptide regions without cysteines, from Sloning BioTechnology GmbH (now Morphosys). The library construct was amplified by Phusion High Fidelity Polymerase (NEB) at 98 °C for 5 min then 10 cycles of 98 °C, 10 seconds; 56 °C, 15 seconds; 72 °C, 15 seconds followed by 72 °C for 5 minutes using the forward primer 5′-TCTGGCGTTTTCTGCGTC-3′ and reverse primer 5′-CTGCGGAACTCCTGCAGTTC-3′. The amplified product was digested with NheI and PstI, gel-extracted and cloned into the similarly digested pBSTG1-Ad phagemid. Ligated products were used for electroporation as previously described.30 Phage display was performed on Maxisorb plates, which were coated with 50 μl of 1 μg ml−1 anti-myc tag antibody overnight at 4 °C and blocked in 2× PBS blocking buffer (Sigma Aldrich, UK) for a minimum of 4 hours at 37 °C. Approximately 1012 cfu pre-panned phage were used for a 30 min panning, wells were washed 30 times and eluted with 50 mM glycine–HCl (pH 2.2) for 10 min, and neutralised with 1 M Tris-HCl (pH 9.1). Eluted phage were used to infect ER2738 cells and new phage were propagated as reported elsewhere.30 A 0.5 μl aliquot of phage suspension was used for the second round of selection using 2 μl of Protein A magnetic beads slurry (Invitrogen). Anti-myc tag antibody immobilized beads were washed and incubated with pre-panned phage for 30 min then washed five times and eluted as before. After two panning rounds 48 randomly chosen clones were tested for specificity by phage ELISA on anti myc tag antibody coated Maxisorb plates. In addition, ELISA was performed as previously described.30We identified 34 positive clones (about 70%) by phage ELISA and determined the sequence of 20 clones of different signal intensities. 19 clones were unique and confirmed the high complexity of the library. From this selection, the ten clones with the highest sequence similarity to the c-myc tag were chosen for further analysis.
The nucleic acid sequences for the selected proteins from the phage ELISA were then cloned into a modified pET-11(a) vector via NheI and NotI restriction sites (see Fig. S2†). The ligated vector was then transformed into competent XL-10 Gold E. coli cells by a heat shock treatment.31 DNA was isolated from a number of colonies and the insert sequences were determined by sequencing (GATC Biotech and Beckmann Coulter Genomics).
Optimization of protein expression
To determine the optimal conditions for recombinant protein expression, four different expression hosts were tested: BL21 Star (DE3); C41 (DE3); C43 (DE3); and, BL21 Gold (DE3). Cells were grown in different autoinduction media: LB (Luria Bertani); TB (Terrific broth); SB (Super broth); and, 2YT (Yeast extract/tryptone) (all sourced from Formedium). The media contained 30 μg ml−1 chloramphenicol and 50 μg ml−1 carbenicilin antibiotics. The pET-11(a) expression plasmid carrying the receptor protein sequence was transformed into each E. coli strain. A colony was picked and 2 ml of ZYP-0.8 G containing carbenicilin and chloramphenicol was inoculated in a well of a 24-well plate. The cells were grown at 37 °C for 6 hours at 1300 rpm in a plate incubator.32 200 μl samples were then harvested into a 96-well plate at time intervals of 16, 24, 40, 48, 64, and 72 hours. The collected samples were lysed using lysis buffer (50 mM HEPES, 25% w/v sucrose, 5 mM MgCl2, 1% triton vol/vol, 10 U/ml omnicleave, 0.1 mg ml−1 lysozyme, pH 8) and an immuno-dot blot assay was performed.33 Lysed cells were dissolved in dot blot solubilisation buffer (50 mM Tris, NaCl 300 mM, 8 M guanidium chloride, pH 8). The blocking buffer consisted of 3% BSA and 1× TBST (Tris buffered saline tween-Tris 50 mM, NaCl 150 mM, 0.1% Tween, pH 7.4).The control comprised various concentrations of histidine-TEVp (His-tagged tobacco etch virus protease). The positive samples identified from the dot blot were further investigated by western blot analysis. The samples were transferred from a 15% SDS PAGE gel (200 V, 1 h) to nitrocellulose membrane using transfer buffer (Tris 0.1 M, glycine 0.192 M, methanol 20%) by applying a constant current of 0.8 mA cm−2 for 1 hour. The immunodetection of the proteins was performed using 1:5000 dilution of anti-histidine antibody conjugated to horseradish peroxidise (HRP) (R&D Systems) and the chemiluminescent detection was performed using SuperSignal West Pico chemiluminescent substrate (Thermo Scientific).
Expression and purification of receptor proteins
The pET-11(a) vector containing anti-myc tag antibody receptor protein sequences was transformed into BL21 Star (DE3) cells. 2 ml of ZYP-0.8G media was inoculated with a single transformed bacterial colony and incubated for 6 hours at 37 °C. A 400 μl aliquot was used to inoculate 400 ml of TB auto-induction medium containing 30 μg ml−1 chloramphenicol and 50 μg ml−1 carbenicilin. The culture was incubated for 48 hours (37 °C, 250 rpm) and the cells were then harvested by centrifugation at 3,000×g for 30 min and frozen at 20 °C.
The cell pellet was re-suspended in 50 ml binding buffer (50 mM HEPES, 20 mM NaCl, 5 mM imidazole, pH 7.5), and sonicated for five 60 s cycles using a Branson sonicator at 50% duty cycle. During sonication, the cell-pellet was kept on ice to avoid over-heating. The sample was then centrifuged in a Sorvall centrifuge at 16,000 rpm for 20 minutes at 4 °C. 40 ml of the supernatant was collected and applied with a syringe onto the 1 ml Hitrap column (GE healthcare) pre-equilibrated with 10 column volumes (CVs) of binding buffer. The column was then washed with 10 CVs of binding buffer followed by 10 CVs of wash buffer (50 mM HEPES pH 7.5, 200 mM NaCl and 40 mM imidazole). The proteins were eluted with 10 CVs of elution buffer (50 mM HEPES pH 7.5, 200 mM NaCl and 500 mM imidazole) and 1 ml fractions were collected in a 96-well plate. The eluted fractions containing proteins were pooled together and dialysed against 1× PBS (Phosphate buffer saline, 136.8 mM NaCl, 2.7 mM KCl, 10.1 mM Na2HPO4, 1.7 mM KH2PO4) using a 3.5 kDa molecular weight cut off dialysis tubing. The purified proteins were then analysed on a 15% SDS (sodium dodecyl sulphate) PAGE gel (Fig. S6†). The SDS PAGE was run at a constant voltage of 120 V with an initial current of approximately 60 mA for 60 minutes, and stained with Coomassie brilliant blue.34 The purified receptor proteins migrated at a position corresponding to 12–15 kDa (Fig. S6†), which is in good agreement with their calculated theoretical molecular weight of 13–14 kDa. As expected, the native scaffold migrated slightly faster owing to the lower molecular weight (Fig. S6b†).
ELISA
A 96-well plate (Nunc MaxiSorp) was coated with 10 μg ml−1 of receptor protein prepared in 100 mM PBS buffer pH 7.4. The control wells were coated with 10 μg ml−1 of scaffold or GST receptor protein. The plate was incubated overnight at 4 °C. The samples were decanted and 250 μl of 2× casein blocking buffer (Sigma Aldrich, UK) was added in each well and incubated for 2 hours at room temperature. The wells were washed 3 times with 1× PBST (0.1% Tween in 1× PBS). Solutions containing 1 μg ml−1 monoclonal anti-myc tag antibody (Abcam UK) aliquots were added to the wells and incubated for 1 hour at room temperature before washing a further three times in 1× PBST. Wells containing immobilised receptor proteins, scaffold, and GST receptor protein were incubated in 1 μg ml−1 BSA (bovine serum albumin) as control. An anti-mouse-antibody conjugated with HRP (1:4000) was added as per manufacturer's protocol (Promega) and incubated for 1 hour at room temperature. After washing ten times with 1× PBST, 1 ml of TMB substrate solution was added and the absorbance at 650 nm was measured after 5 minutes using a Multiskan ascent plate reader (Thermo Fisher Scientific).
Circular dichroism (CD)
CD measurements of the purified receptor proteins and native scaffold were carried out on an Applied Photophysics Chirascan CD spectropolarimeter. 1 mg ml−1 of proteins prepared in 1× PBS was used for analysis. The spectra were collected from 190 nm to 260 nm over two scans. The data were analysed using Dichroweb analysis server35 based on the CDSSTR algorithm calculated using the SP175 reference data sheet.36,37Differential scanning calorimetry (DSC)
DSC measurements were carried out on a VP-DSC (Microcal) instrument. Samples of the binders and 1× PBS buffer were degassed twice under vacuum for 10 minutes. The scanning was performed between 20 °C and 110 °C at a scan rate of 90°C per hour with a 15 minutes pre-scan equilibration. To establish the reversibility of protein folding, repeated heating and cooling was performed on the proteins. The purified binders and the native scaffold were prepared at a concentration of 0.5 mg ml−1 in 1× PBS at pH 7.4.Bio-layer interferometry (BLI)
The affinity and binding kinetics of receptor protein-2–anti-myc tag antibody interaction was measured using bio-layer interferometry (BLItz, Pall Fortebio). Measurements were carried out using the dip and read Ni-NTA biosensors. A 30 seconds response in PBS buffer was first taken to establish the baseline of the sensor. Receptor-proteins were then immobilized on the Ni-NTA sensor via the histidine tags of the receptor proteins by exposing the sensor to a solution of 10 μg ml−1 receptor-proteins in PBS buffer for 120 seconds. After washing the sensor with PBS buffer for 30 seconds, the sensor was then subjected to different concentrations of anti-myc tag antibody or BSA in PBS buffer for 600 seconds to record the association phase. PBS buffer was then reapplied to measure the dissociation. The recorded association and dissociation data were corrected by the baseline response in PBS buffer. A straight line was fitted to the linearised binding curve using ProFit software (Quansoft, Switzerland).Electrochemical impedance spectroscopy (EIS)
EIS measurements were performed using a VSP-3 electrochemical workstation (BioLogic science instruments, France) and a conventional three-electrode cell that comprised a gold working electrode, a Ag/AgCl saturated KCl reference electrode, and a platinum wire as a counter electrode. The measurements were taken in the frequency range 50 mHz to 100 kHz at a DC potential of 80 mV vs. reference (Ag/AgCl) by applying a 30 mV AC potential. The working electrodes were fabricated by evaporation of 20 nm titanium, followed by 80 nm of gold using an electron-beam evaporator (BAK 501, Evatec AG, Switzerland).Prior to use, the gold surface was sonicated in acetone for 5 minutes and then rinsed in ethanol. The surface was incubated for 48 hours in a solution of 1 mM SH–(CH2)11–EG6–COOH (PEG) solution prepared in 200% proof ethanol, thoroughly rinsed in ethanol, and then dried in nitrogen and mounted in the cell. The PEG surface was incubated in 100 mM PB pH 7 for 10 minutes and then washed twice with 100 mM MES buffer, pH 5.5. The carboxyl groups of the PEG monolayer were then activated by incubating the surface in EDC (0.4 M)/NHS (0.1 M) for 15 minutes. EDC-NHS solution was prepared in 100 mM MES buffer, pH 5.5. The surface was incubated in 10 μg ml−1 receptor protein-2 (prepared in 10 mM acetate buffer pH 5.5) for 1 hour, and the remaining active EDC/NHS sites were blocked by incubating the sensor with 1 M ethanolamine at pH 8.5 for 30 minutes. The surface was washed twice with 100 mM PB at pH 7.
Acknowledgements
This work was funded through WELMEC, a Centre of Excellence in Medical Engineering funded by the Wellcome Trust and EPSRC, under grant number WT088908/Z/09/Z. We acknowledge support of the Royal Society and Wolfson Foundation, the University of Leeds through the Biomedical Health Research Centre, and Evatec AG, Switzerland.References
- K. Hernandez and R. Fernandez-Lafuente, Enzyme Microb. Technol., 2011, 48, 107–122 CrossRef CAS PubMed.
- R. A. Williams and H. W. Blanch, Biosens. Bioelectron., 1994, 9, 159–167 CrossRef CAS.
- D. Evans, S. Johnson, S. Laurenson, A. G. Davies, P. Ko Ferrigno and C. Walti, J. Biol., 2008, 7, 3 CrossRef PubMed.
- C. Berggren, B. Bjarnason and G. Johansson, Electroanalysis, 2001, 13, 173–180 CrossRef CAS.
- P. J. Conroy, S. Hearty, P. Leonard and R. J. O'Kennedy, Semin. Cell Dev. Biol., 2009, 20, 10–26 CrossRef CAS PubMed.
- J. E. Butler, L. Ni, W. R. Brown, K. S. Joshi, J. Chang, B. Rosenberg and E. W. Voss, Jr, Mol. Immunol., 1993, 30, 1165–1175 CrossRef.
- Z. Chen and A. Sadana, Biophys. Chem., 1996, 57, 177–187 CrossRef CAS.
- D. Saerens, L. Huang, K. Bonroy and S. Muyldermans, Sensors, 2008, 8, 4669–4686 CrossRef CAS PubMed.
- R. Ohara, A. Knappik, K. Shimada, C. Frisch, F. Ylera and H. Koga, Proteomics, 2006, 6, 2638–2646 CrossRef CAS PubMed.
- F. F. Bier, R. Jockers and R. D. Schmid, Analyst, 1994, 119, 437–441 RSC.
- E. Briand, M. Salmain, C. Compère and C.-M. Pradier, Colloids Surf., B, 2006, 53, 215–224 CrossRef CAS PubMed.
- X. Zeng, Z. Shen and R. Mernaugh, Anal. Bioanal. Chem., 2012, 402, 3027–3038 CrossRef CAS PubMed.
- S. Goodchild, T. Love, N. Hopkins and C. Mayers, in Advances in applied microbiology, Adv. Appl. Microbiol., 2005, vol. 58, pp. 185–226 Search PubMed.
- H. R. Hoogenboom and P. Chames, Immunol. Today, 2000, 21, 371–378 CrossRef CAS.
- L. Huang, G. Reekmans, D. Saerens, J.-M. Friedt, F. Frederix, L. Francis, S. Muyldermans, A. Campitelli and C. V. Hoof, Biosens. Bioelectron., 2005, 21, 483–490 CrossRef CAS PubMed.
- M. Minunni, S. Tombelli, A. Gullotto, E. Luzi and M. Mascini, Biosens. Bioelectron., 2004, 20, 1149–1156 CrossRef CAS PubMed.
- S. Pavan and F. Berti, Anal. Bioanal. Chem., 2012, 402, 3055–3070 CrossRef CAS PubMed.
- J. Yang, S.-E. Kim, M. Cho, I.-K. Yoo, W.-S. Choe and Y. Lee, Biosens. Bioelectron., 2014, 61, 38–44 CrossRef CAS PubMed.
- W. Zhou, P.-J. J. Huang, J. Ding and J. Liu, Analyst, 2014, 139, 2627–2640 RSC.
- P. Colas, J. Biol., 2008, 7, 2 CrossRef PubMed.
- A. Skerra, FEBS J., 2008, 275, 2677–2683 CrossRef CAS PubMed.
- A. E. Nixon and C. R. Wood, Curr. Opin. Drug Discovery Dev., 2006, 9, 261–268 CAS.
- S. Johnson, D. Evans, S. Laurenson, D. Paul, A. G. Davies, P. Ko Ferrigno and C. Wälti, Anal. Chem., 2008, 80, 978–983 CrossRef CAS PubMed.
- E. Wahlberg, C. Lendel, M. Helgstrand, P. Allard, V. Dincbas-Renqvist, A. Hedqvist, H. Berglund, P.-Å. Nygren and T. Härd, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3185–3190 CrossRef CAS PubMed.
- G. I. Evan, G. K. Lewis, G. Ramsay and J. M. Bishop, Mol. Cell. Biol., 1985, 5, 3610–3616 CAS.
- K. Hilpert, G. Hansen, H. Wessner, G. Kuttner, K. Welfle, M. Seifert and W. Hohne, Protein Eng., Des. Sel., 2001, 14, 803–806 CrossRef CAS PubMed.
- C. M. Dobson, Semin. Cell Dev. Biol., 2004, 15, 3–16 CrossRef CAS PubMed.
- W. Schiweck, B. Buxbaum, C. Schätzlein, H. G. Neiss and A. Skerra, FEBS Lett., 1997, 414, 33–38 CrossRef CAS.
- A. Johnson, Q. Song, P. Ko Ferrigno, P. R. Bueno and J. J. Davis, Anal. Chem., 2012, 84, 6553–6560 CrossRef CAS PubMed.
- C. Tiede, A. A. S. Tang, S. E. Deacon, U. Mandal, J. E. Nettleship, R. L. Owen, S. E. George, D. J. Harrison, R. J. Owens, D. C. Tomlinson and M. J. McPherson, Protein Eng., Des. Sel., 2014, 27, 145–155 CrossRef CAS PubMed.
- D. Hanahan, J. Mol. Biol., 1983, 166, 557–580 CrossRef CAS.
- F. W. Studier, Protein Expression Purif., 2005, 41, 207–234 CrossRef CAS PubMed.
- F. C. Bennett and L. C. Yeoman, J. Immunol. Methods, 1983, 61, 201–207 CrossRef CAS.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- L. Whitmore and B. A. Wallace, Nucleic Acids Res., 2004, 32, W668–W673 CrossRef CAS PubMed.
- J. G. Lees, A. J. Miles, F. Wien and B. A. Wallace, Bioinformatics, 2006, 22, 1955–1962 CrossRef CAS PubMed.
- N. Sreerama and R. W. Woody, Anal. Biochem., 2000, 287, 252–260 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an01418a |
| This journal is © The Royal Society of Chemistry 2015 |