
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct deposition of proton exchange membranes enabling high performance hydrogen fuel cells
Matthias
Klingele†
*a,
Matthias
Breitwieser†
*a,
Roland
Zengerle
ab and
Simon
Thiele
ac
aLaboratory for MEMS Applications, IMTEK Department of Microsystems Engineering, University of Freiburg, Georges-Koehler-Allee 103, 79110 Freiburg, Germany. E-mail: matthias.klingele@imtek.uni-freiburg.de; matthias.breitwieser@imtek.uni-freiburg.de; Tel: +49 76120373209 Tel: +49 76120373209
bBIOSS-Centre for Biological Signalling Studies, Universityof Freiburg, Stuebeweg 51, 79104 Freiburg, Germany. E-mail: zengerle@imtek.uni-freiburg.de; Tel: +49 76120373213
cFIT, University of Freiburg, Stefan-Meier-Straße 21, 79104 Freiburg, Germany. E-mail: simon.thiele@imtek.uni-freiburg.de; Tel: +49 76120373247
First published on 2nd April 2015
Abstract
We apply drop-on-demand inkjet printing to fabricate proton exchange membranes for polymer electrolyte fuel cells. This completely substitutes the commonly used membrane foil. A Nafion® dispersion is deposited directly onto the catalyst layers of anode and cathode gas diffusion electrodes, and the two electrodes are pressed together with the membrane layers facing each other. Fuel cells constructed utilizing this method reveal a thin overall membrane thickness of 8–25 μm and a good adhesion of membrane and catalyst layers. This results in a membrane ionic resistance of only 12.7 mΩ cm2 without compromising hydrogen crossover, which was determined to be less than 2 mA cm−2. We achieve a cell power density exceeding 4 W cm−2 with pure oxygen as cathode fuel, which, to our knowledge, is the highest reported power density with a Nafion® membrane hydrogen fuel cell. The membrane shows a stable performance over the entire range of reactant gas humidification from 0 to 100% relative humidity. Power densities exceeding 1.0 W cm−2 are achieved under dry operation with air as cathode fuel. A 576 hour combined mechanical and chemical accelerated stress test reveals no significant degradation in terms of hydrogen crossover, indicating a promising lifetime of the membrane.
Introduction
Current challenges in hydrogen fuel cell production include minimizing production costs and maximizing reliable, long-term power density.1 Besides the reduction of catalyst costs and catalyst degradation, both goals are mainly linked to further development and improvement of the polymer electrolyte membrane (PEM).2 Typically, Nafion® membranes are employed for this task in commercial low temperature polymer electrolyte membrane (PEM) fuel cells, offering sufficient chemical and mechanical stability, reasonable ion conduction, and low reactant gas permeability.3–5 However, when considering PEM fuel cell commercialization, high material and production costs for Nafion® membranes are a considerable obstacle.4,6 The desired functional properties of high ion conductivity, low gas permeability, and long-term stability still need to be further improved.In this work we present a novel membrane electrode assembly (MEA) fabrication technique, where the PEM is directly deposited onto the catalyst layers (CLs) of commercial anode and cathode gas diffusion electrodes (GDEs) via inkjet printing. These directly deposited membrane layers negate the need for a membrane foil, which is the commonly used manufacturing technique. To build a working fuel cell, the membrane coated anode and cathode GDEs are assembled with the membrane layers facing each other, and the layer stack is sealed with a thin PTFE subgasket, preventing fuel crossover at the end faces of the GDEs (Fig. 1a).
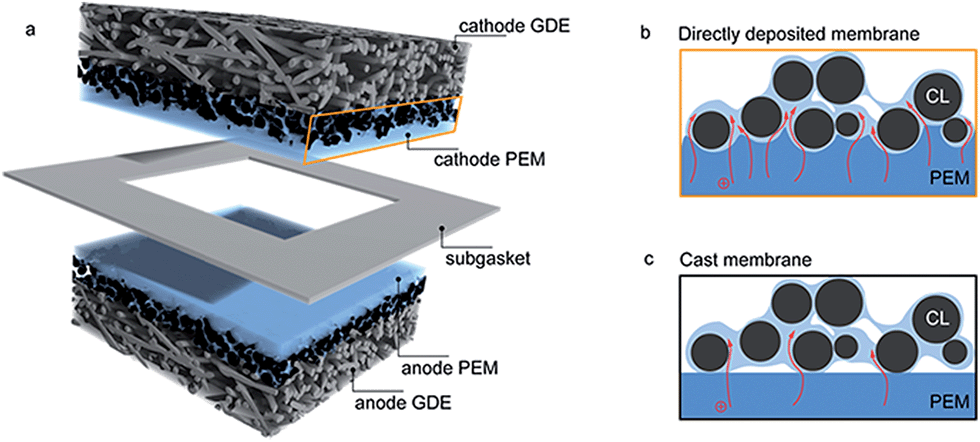 | ||
| Fig. 1 Scheme of the MEA fabricated in this work. A thin PEM layer is inkjet-printed directly on both anode and cathode gas diffusion electrodes. A thin subgasket prevents hydrogen and current crossover through the end faces of the active area (a). The dispersed polymer electrolyte (dark blue) can easily adapt to the catalyst layer surface (b) which leads to relatively thin membranes and an increased electrolyte contact area of membrane and the ionomer phase of the catalyst layer (light blue). This promotes a higher proton conductivity (indicated by red arrows) without compromising fuel crossover. This effect is not as pronounced in conventional MEAs utilizing cast membranes (c). | ||
Our results show that the direct deposition of proton exchange membranes significantly outperforms the classical assembly methods and is able to meet the above-mentioned desired requirements of the Nafion® membrane layer although using the exact same electrolyte material.
The conventional approaches for MEA production include depositing anode and cathode catalyst layers onto a previously cast membrane foil to form catalyst coated membranes (CCMs) or stacking of a membrane foil in between of anode and cathode GDEs. In order to obtain reasonable ion conduction and adhesion of the different functional layers, both of these approaches require several processing steps, such as acidic boiling of the membrane foil for purity issues or hot-pressing the MEA. Furthermore, in both of these techniques, the membrane layer needs to be of supporting character (at least self-supporting) and comprehensively impermeable for reactant gases, which demands a minimum foil thickness. Up to now MEA fabrication via CCMs resulted in much better performance characteristics than fabrication by using GDEs.7 Therefore only scarce efforts have been made in the past to process membranes in between two GDEs such as by die coating8 or improving GDE performance by Nafion® impregnation.9 However, unlike shown in this publication, none of these approaches showed performance characteristics as good or better as MEAs consisting of CCMs.
In our new approach, the direct deposition of the membrane layer on both, anode and cathode GDEs leads to several advantages. The main advantage is that the membrane layer does not need to be self-supporting, since the supporting role is given by the GDE. This allows to arbitrarily reduce membrane thickness, decreasing its intrinsic ionic resistance without compromising mechanical stability of the assembly. Additionally, hot-pressing or similar processing steps are not necessary to obtain a good adhesion of the membrane layer and the catalyst layer. We show that the ionomer layer, in its dispersed form, is able to perfectly mold to the anode and cathode catalyst surface, resulting in a good mechanical adhesion. Moreover, we believe that the dispersed membrane layer partially infiltrates the surface-near pores of the catalyst layers. This concedes almost no gas permeability of the catalyst layer in this region, as well as a strongly increased contact surface area of the membrane layer and the ionomer content of the catalyst layer (Fig. 1b). The latter is believed to be the origin of a drastically decreased ionic contact resistance of the membrane layer.
MEA imaging
Fig. 2a shows a cross-sectional SEM image of the operated fuel cell discussed in this work. | ||
| Fig. 2 SEM image of a MEA cross-section with directly deposited membrane (a) and an edge of a single GDE with the inkjet-printed membrane layer on the top (b) from an approximately 30° angle of view. | ||
In the top and bottom part of the image, the well-cut and partially resin infiltrated gas diffusion layer (GDL) fibers are visible, followed by the micro porous layer (MPL, homogeneous grey layer), the CL (bright layer), and the membrane layer in the center of the stack. From this and other SEM images screening different areas of the MEA, we find a varying membrane thickness between 8 and 25 μm which is partially thinner than commercially available thin Nafion® membranes (such as Nafion® HP, 20 μm). We further find that the membrane layer perfectly adapts to the surface shape of the catalyst layer. Both the thin layer thickness and the good adhesion of the membrane to the catalyst layer are believed to be the origin of the high cell performance, since they lead to low intrinsic and contact resistances of the ion conduction through the membrane layer. In Fig. 2b, roundish features on the surface of the delaminated membrane are revealed, likely originating from evaporation behavior of the dispersion solvent.
Cell polarization and power
Concerning performance, a high power density up to 4.07 W cm−2 and a current density of 5 A cm−2 at 0.6 V are measured for the fuel cell under power optimized conditions (Fig. 3a).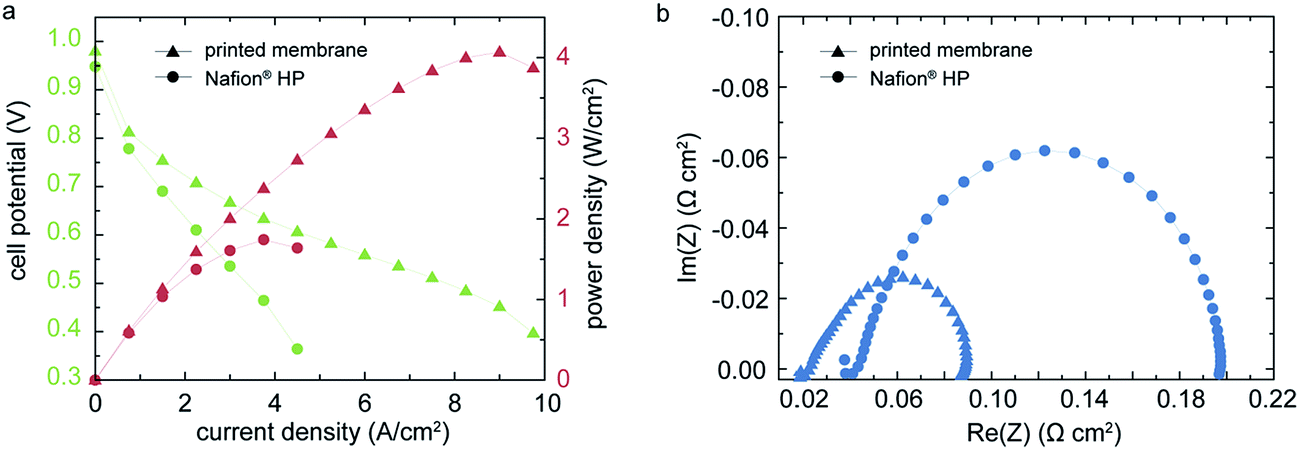 | ||
| Fig. 3 The polarization curves (a) and electrochemical impedance spectroscopy (EIS)-data (b) for the direct deposited membrane MEA and a commercial Nafion® HP based MEA (as reference) under optimized operation conditions (H2/O2; 0.5/0.5 L min−1; 70 °C, 100% RH, 300/300 kPa). The loading of the catalyst layers of anode and cathode is 0.5 mg cm−2 at 70% Pt/C. The peak power density for the printed sample is 4.07 W cm−2 at a current density of 9.0 A cm−2. This is 134% higher than the peak power density of the reference MEA (1.74 W cm−2). The EIS data reveals the significantly lower high frequency resistance and reduced charge transfer resistance of the directly deposited membrane. Both EIS measurements were conducted at a cell voltage of 0.75 V. | ||
To our knowledge, these results exceed any published power densities for Nafion® based membranes by far.4 For reference, we characterized a commercial MEA comprised of a Nafion® HP membrane and the same GDE as used for the printed sample. The Nafion® HP membrane has a nominal thickness of 20 μm, which is in the range of our printed membrane layer. Therefore it is a suitable reference sample, representing the conventional MEA fabrication techniques. Under identical operation conditions, the Nafion® HP based MEA showed a peak power density of 1.74 W cm−2, which is only 42.8% of the power density we measured for the MEA with our direct deposited membrane approach. To compare the high frequency resistance (HFR) and charge transfer resistance Rct of the inkjet-printed and the Nafion® HP fuel cell, the EIS-measurement shown in Fig. 3b are conducted at an identical potential of 0.75 V for both samples. The frequency was swept from 10 kHz to 0.5 Hz. As shown in Fig. 3b, the high frequency resistance and the charge transfer resistance of the directly deposited membrane are significantly lower than the reference MEA with Nafion® HP membrane. We find an Rct of 66.3 mΩ cm2 for the directly deposited membrane whereas the Nafion HP fuel cell reveals an Rct of 153.8 mΩ cm2. This indicates a superior interfacial contact resistance of the membrane and the catalyst layers in our inkjet-printed fuel cells. For the HFR we find the same tendency: We obtain 22.4 mΩ cm2 for the DMD-fuel cell and 43.3 mΩ cm2 for the fuel cell with Nafion® HP membrane.
However, these power optimized operation conditions diverge from normal cell operation parameters in regular application, where low flow rates, low humidification and air as cathode gas are typically desired.
Fig. 4 shows the polarization curves of the same cell under normal operation conditions with different states of humidification. The highest power density of 1.27 W cm−2 and a current density of 1.75 A cm−2 is reached at 90% relative humidity and a potential of 0.6 V. Further, no significant drop in power density for operation with 20% or 35% relative humidity (RH) is observed, in contrast to the widely reported characteristics of Nafion® membranes.4,10,11
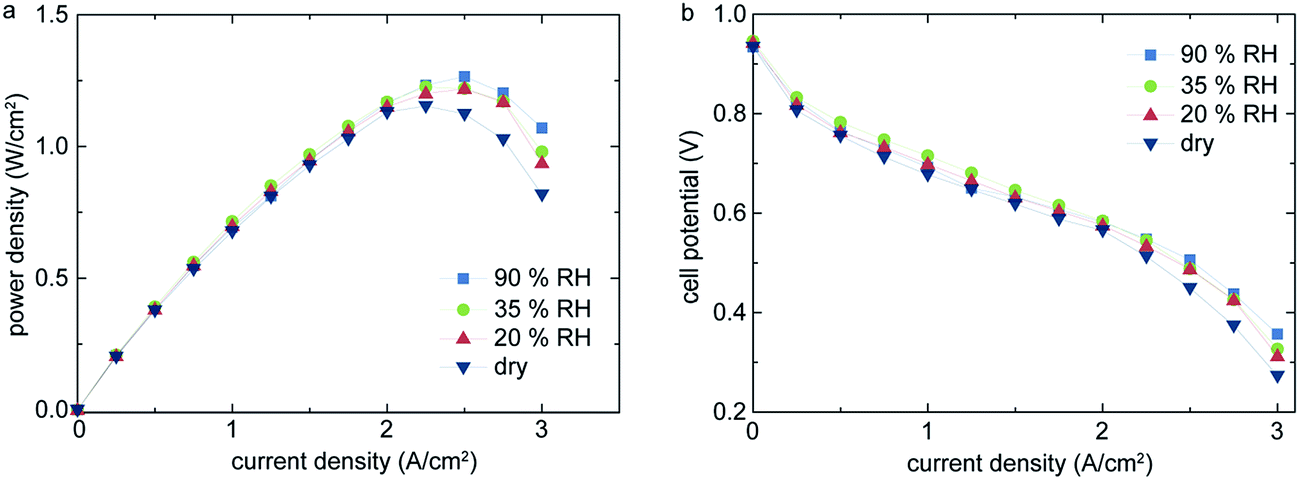 | ||
| Fig. 4 Comparison of different states of humidification for the operation under normal conditions (H2/air; 1.2/2.0 stoichiometric ratio; 70 °C; 300/300 kPa). Shown are the power density (a) and the cell polarization (b) over current density. | ||
Under entirely dry conditions, we still obtain 1.15 W cm−2 peak power density, which is a performance drop of only 120 mW cm−2 when compared to humidified operation. Even with very low, or no, humidification, the membrane is sufficiently humidified only by back-diffusion of water generated by the cathode reaction. The effect of back-diffusion is favored by the low thickness of the single membrane layers and the steep water concentration gradient from cathode to anode side. This effect has been previously recognized in literature.10,12,13 However, to our knowledge, all previously published measurements of PEM fuel cells without external humidification showed only little cell performance. Typically, 20–40% lower power densities for dry operation than under externally humidified conditions are reported.10,12,13 For comparison, in this work we observe a drop of power density of <9.45% compared to fully humidified conditions.
Membrane resistance
As shown in Fig. 3, the high power density of fuel cells with directly deposited membranes is primarily due to very low losses in the ohmic region of the cell polarization, which typically is linked to the ionic resistance of the membrane.11 As published by Pivovar et al., it is important to take the electrical resistance of the gas diffusion electrodes into account to reveal the effective membrane resistance in high frequency resistance (HFR) measurements.14Fig. 5 shows the evolution of the calculated membrane resistance (Rmembrane = HFR − Relectrode) over the current density for different levels of humidification. As expected, the membrane resistance increases with lower humidification, but the overall membrane resistance ranges well below typical values for state-of-the-art Nafion® membranes of low thickness (such as NR-211) even for the dry operation conditions.4 Under dry operation conditions, we do not observe the increase of the HFR at high current densities, which may be related to the enhanced water generation on the cathode side under high load conditions. The steeper water gradient from the cathode side to the anode side in this state of operation enables increased water back-diffusion and thus better humidification and nearly constant protonic conductivity of the membrane in the range of 1.5–3.0 A cm−2.
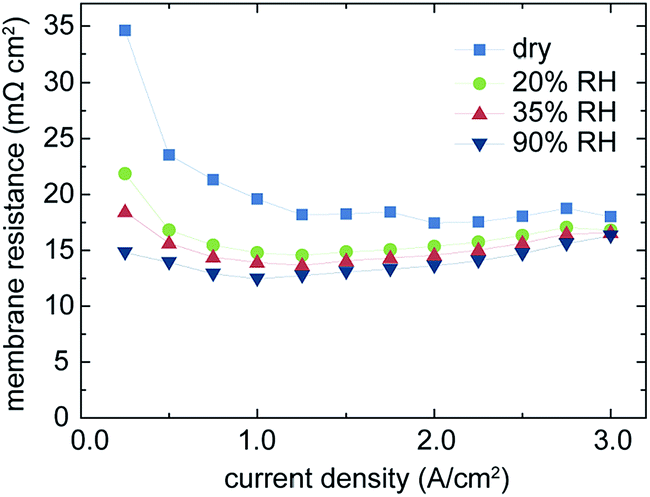 | ||
| Fig. 5 The evolution of the membrane resistance over the current density is compared for different states of humidification. The resistance values are extracted by the evaluation of the high frequency intercept of in situ EIS. The fuel cell is operated at 70 °C, 300 kPa, 1.2/2.0 H2/air. | ||
R electrode was determined by assembling the fuel cell with the two GDEs, face to face as proposed by Jiang et al., without the two inkjet-printed membrane layers and the subgasket.15 The cell resistance was measured by applying an increasing voltage to the cell and measuring the current increase. The slope of the resulting linear dependence corresponds to the ohmic resistance of the 5 cm2 GDEs, including all resistances linked to the flow fields and cables. By linear fitting we obtain 11.3 ± 0.3 mΩ cm2 as electrode resistance, which is in accordance to the values published elsewhere.15 The uncertainty emerges from the standard deviation of repeated measurements. By subtracting this electrode resistance from the HFR-value, the effective ionic membrane resistance can be extracted.
HFR-values are evaluated by means of impedance spectroscopy and extracting the high-frequency intersect of the spectrum.16 From this HFR-value, the previously measured electrode resistance of 11.3 mΩ cm2 is subtracted to obtain the membrane resistance. As lowest value we obtain 12.7 mΩ cm2 (Fig. 5). This resistance is dramatically below the resistances of commercial Nafion® or other commercial membranes, typically ranging between 50 and several hundred mΩ cm2 depending on thickness, morphology and level of humidification.4,11,14,15,17 The obtained membrane resistance is only slightly higher than the electrical resistance of the support material, emphasizing the extreme improvement originating from our novel direct membrane deposition approach for MEA production. Thus, in terms of series resistance, the membrane and the electrodes are contributing nearly equally to the overall cell polarization in the ohmic region.
The very low membrane resistance can be explained by the advantages of directly depositing the membrane on the GDE. First, as published by Wang et al., printing Nafion® directly onto the catalyst layer leads to an reduced contact resistance between the catalyst layer and the membrane, resulting in a lower overall protonic resistance.18 Second, as the SEM-cross-section in Fig. 2a reveals, we obtain a membrane thickness of 8–25 μm. The locally thin regions of the membrane are believed to be of very low ion resistance and therefore contribute dominantly to the overall low membrane resistance.
Long-term stability
A primary goal of membrane development is to improve the long-term stability. For automotive fuel cell applications, a lifetime of 5000 hours is typically targeted.19 Accelerated stress tests (AST) are widely used to simulate the degradation behavior within a more reasonable timeframe.20 Despite the promising power densities and the robust operation under dry conditions, as shown in the upper sections, a degradation study of the directly deposited membrane is crucial to evaluate the quality of these membranes in a long-term perspective. In order to degrade the membrane layer chemically and mechanically, we use a combined stress cycle recommended by US fuel cell council (USFCC) and developed by DuPont.21 The cycle consists in periodic sequences of 24 hours of RH-cycling (30 min dry operation/30 min 100% RH operation, N2/N2, 80 °C) and 24 hours of load cycles (7 min operation at 10 mA cm−2, 3 min operation at 800 mA cm−2, H2/O2, 80 °C, 50% RH). The break-down criterion is a hydrogen crossover current density of >10 mA cm−2. Compared to separate degradation protocols, a combined cycle as used in this work leads to faster degradation of the membrane due to multiple degradation influences.22 The crossover current density is measured every 48 h (after each cycle) via linear sweep voltammetry (LSV). The LSV measurements are conducted with 0.2 L min−1 fixed hydrogen flow and 0.05 L min−1 fixed nitrogen flow at 70 °C cell temperature, 100% RH and atmospheric pressure. The cell potential is swept from open circuit voltage (OCV) to 0.6 V with a potentiostat. For the pristine MEA, we obtain an initial crossover current density <2 mA cm−2 (Fig. 6a). This value is in the range of published crossover current densities for thin commercial Nafion® membranes, such as Nafion® NR211 with 25.4 μm thickness.4,23 The LSV measurement also reveals a soft internal electrical short through the membrane layer.24 By linear fitting to the raising current density against the cell voltage, we extract an electrical resistance of 112.5 Ω cm2, which is below typical insulating values of >1000 Ω cm2.16 These electrical losses likely originate from electrode material pin-holing the membrane and should be addressed in the future by improving the surface quality of the membrane and the catalyst layer.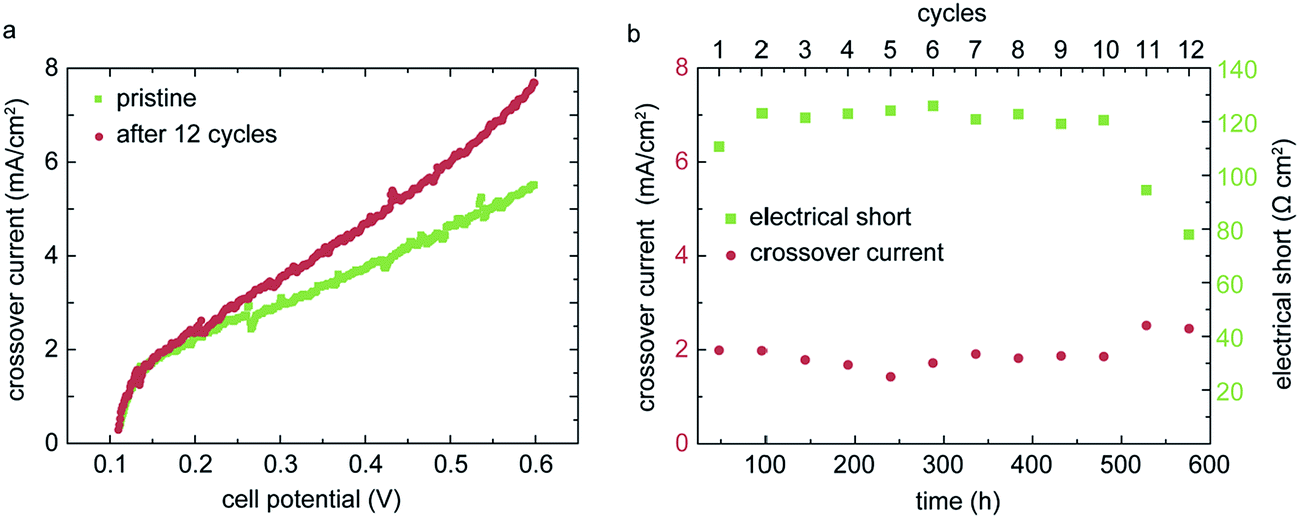 | ||
| Fig. 6 LSV measurements of the pristine MEA and the same MEA after 12 cycles of combined AST (a) and the time evolution of the crossover current and electrical short over time (b). The crossover current was extracted from LSV measurements at the point of maximum curvature. The electrical short resistance is extracted from the linear slope of the LSV measurement in the range of 0.3 V to 0.6 V cell potential. | ||
Fig. 6b shows the evolution of the hydrogen crossover and electrical resistance of the MEA over time. For the first 10 cycles, the hydrogen crossover current density remains more or less constant at about 2 mA cm−2. The crossover slightly increases within the 11th and 12th cycle, showing first signs of degradation. After 12 cycles, the AST was ended. The electrical resistance also remains constant at about 120 Ω cm2 up to the 10th cycle. During the 11th and 12th cycle, the resistance drops to about 80 Ω cm2, revealing an increased electrical short. Both these degradation phenomena can be explained by the membrane thinning during fuel cell operation, as described in Lim et al.22 The thinning of the membrane promotes slightly higher gas permeability, as well as the exposure of initially coated electrode features pin-holing the membrane. However, the gas impermeability remains good even after degradation and thinning of the membrane. This can be explained by a clogging of the surface-near pores of the CL. The impregnation (or clogging) of the pores leads to a high gas impermeability of the CL itself, which can only be achieved when the membrane is deposited in dispersed form. This indicates that our MEA production favors low hydrogen cross-over in the long-term perspective. In comparison, Wu et al. obtained a crossover of 20 mA cm−2 for a PTFE reinforced Nafion® membrane after only 6 cycles, with an equivalent AST-protocol.25
These results confirm a promising long-term stability for the inkjet-printed membrane and give a favorable outlook for possible future applications of this new MEA fabrication approach. As already mentioned previously, the soft electrical short should be addressed in further process developments. But as the AST revealed, this electrical short was stable over 576 hours of accelerated degradation, thus having only minor impact on the long-term fuel cell performance.
Experimental
The conventionally used membrane foil,11 which is typically cast from a polymer electrolyte dispersion, is completely substituted by a directly deposited membrane and a subgasket. A layer of polymer electrolyte dispersion is deposited on top of the catalyst layers (CLs) of both anode and cathode gas diffusion electrodes (GDE), via drop-on-demand inkjet printing. Both GDEs consist of a gas diffusion layer (GDL), a micro porous layer (MPL), and a catalyst layer (CL). The membrane electrode assembly (MEA) for a working fuel cell is formed by assembling the membrane-coated anode and cathode GDEs with the membrane layers facing each other. A 50 μm thick PTFE-subgasket, positioned between the GDEs, prevents hydrogen and parasitic current crossover through the end faces of the active area (see Fig. 1). Two main gaskets are surrounding the GDEs (not shown in Fig. 1).For the deposition of the polymer electrolyte dispersion onto the catalyst layer, a BioFluidix BioSpot® printer with a PipeJet™ Printhead (dispensing pipe with 500 μm inner diameter and 13 mm length) has been employed. The advantages of using this printer for the purpose of printing polymer dispersions include high dispensing accuracy and rather large nozzles, which avoids the problem of clogging with large particles. This large nozzle size allows the dispensing of polymer electrolyte dispersion with polymer content as high as 3.3 vol%, which, in our experiments, could not be achieved by printers with smaller nozzles. In general, other techniques to produce MEAs, like screen-printing or spray coating, could be equally suitable for direct membrane deposition as well (this especially accounts for possible large scale production). However, for research purposes, inkjet printing is favorable because it enables quick variation of process parameters, such as droplet size, pitch, and polymer ink composition.
For the MEAs presented in this work, the printed polymer electrolyte dispersion consisted of 1 part Nafion® D2020 Dispersion (21 wt% Nafion®, 34 wt% water, 44 wt% 1-propanol, 1 wt% ethanol and mixed ethers; Ion Power GmbH) and 2 parts 2-propanol. This mixture resulted in a Nafion® content of 0.07 g mL−1 of the printed dispersion. One layer of this electrolyte dispersion was printed on top of each catalyst layer of the anode and cathode GDEs (Paxitech SAS, 5 cm2, 0.5 mg cm−2, 70% Pt/C) and subsequently tempered for 2 hours at 85 °C on a hotplate in order to evaporate residual solvents. The dispensing process was optimized for a reliable and reproducible droplet dispensing, rather than process speed. Coating two 5 cm2 GDEs, to form one MEA, takes about 18 minutes at a dispense frequency of 4 Hz. It was found that a 25 μm stroke length (s) of the inkjet piezo actuator, a downstroke velocity (vd) of 125 μm ms−1, and an upstroke velocity (vu) of 5 μm ms−1 (Fig. 7a) are suitable parameters for the printer with this dispersion. This resulted in a droplet volume of 30 nL, as determined from stroboscopic droplet observation. In order to obtain a homogeneous membrane layer, the droplet pitch was found to be optimal at a distance of 0.5 mm between droplets on the catalyst layer substrate (Fig. 7b). The print head velocity was 2 mm s−1. By assembling the fuel cell as schematically shown in Fig. 1, the resulting active fuel cell area is reduced to 4 cm2, due to the dimensions of the subgasket. No further pre- or post-treatment steps were performed.
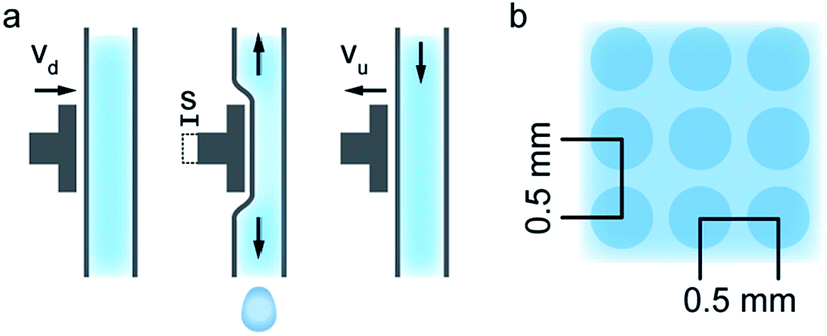 | ||
| Fig. 7 The inkjet dispense cycle. A voltage pulse controls the stroke of a piezo-actuator, which displaces the polymer electrolyte dispersion and dispenses it out of the dispensing pipe (a). A dispense pattern with a droplet pitch of 0.5 mm results in the droplets merging together on the substrate, and to a homogeneous membrane layer (b). | ||
As suitable reference sample we used a MEA with a 20 μm thin Nafion® HP membrane and the identical catalyst layers. The MEA was produced by hot-pressing the same gas diffusion layers as used in the inkjet-printed GDEs on the catalyst coated Nafion® HP membrane.
To operate the assembled fuel cell, we used a Scribner 850e fuel cell testing system with an integrated frequency response analyzer (FRA). Polarization curves, impedance spectroscopy (IS), and linear sweep voltammetry (LSV) measurements were recorded in order to characterize the cell. For performance optimized conditions, the fuel cell was operated with a high fixed flow of 0.5 L min−1 H2 and 0.5 L min−1 O2 as reactant gases, with 100% relative humidity (RH), 70 °C cell temperature, and 300 kPa pressure on both anode and cathode side. These conditions were set to minimize mass transport effects, and their corresponding performance limitations. To simulate realistic operation, the fuel cell was operated with a stoichiometric ratio of 1.2 H2 and 2.0 air, with a cell temperature of 70 °C and pressure of 300 kPa. To condition the fuel cell assembly properly, the fuel cell was initially operated for 30 min at open circuit voltage (OCV). Subsequently, 30 polarization curves were performed until stable fuel cell performance was established. The level of gas humidification was varied between 0% to 90% RH. In order to ensure that the data obtained at dry operation (0% RH) is not falsified by remaining water from precedent operation under humidified conditions, the whole MEA, including all gaskets, was dried for 14 hours on a hot plate at 35 °C prior to the measurement. Under humidification, we operated the cell for at least one hour at each state of humidity, until stable conditions were fulfilled. Furthermore, we increasingly varied the states of humidity from 20 to 90% RH, in order to avoid artefacts from prior measurements. After this conditioning procedure, polarization curves, LSV, high frequency resistance (HFR) measurements, and the accelerated stress test were performed. In order to record the polarization data, each current density was held steady for 5 min prior to measurement, and the I–V-characteristic was measured from high to low voltages. All polarization measurements were repeated on four separately produced sample MEAs to check their reproducibility. It was found that all samples yielded reproducible results.
A scanning electron microscope (Phenom Pro) was used for optical investigation of embedded cross-sections of the MEA. A 1 cm2 section of the active area of the operated MEA was embedded in epoxy resin. To obtain a proper cross-section of the MEA, the embedded assembly was cut with an ultra-microtome (PowerTome, RMC products) and subsequently investigated in the scanning electron microscope. Another section of the MEA was delaminated and investigated in the SEM without epoxy embedding, in order to reveal the surface properties of the membrane after operation. The images were recorded with an electron acceleration voltage of 5 kV.
Conclusion
In this work, the MEA is fabricated by directly depositing a Nafion® dispersion, as membrane layer on anode and cathode GDEs. This results in a high cell power density of 4.07 W cm−2 under pure hydrogen/oxygen operation, which is primarily due to a very low membrane resistance of about 12.7 mΩ cm2 and a reduced charge transfer resistance.The main reasons for the lower resistance values are believed to be the relatively thinner membrane, and a strongly decreased contact resistance of membrane and the proton conducting phase of the catalyst layer.
Additionally, we show stable power densities above 1 W cm−2 under completely dry conditions with air as cathode gas. This desirable but very untypical behavior for a Nafion® membrane is attributed to internal humidification of the thin membrane via back-diffusion of the produced water from the cathode to the anode side. This provides sufficient membrane humidification and a stable ion conduction even at low RH. Furthermore, we demonstrated that the directly deposited membrane shows promising degradation stability in terms of hydrogen crossover, when compared to other Nafion® membranes of considerably higher membrane resistances.
Our novel approach of MEA fabrication by direct deposition has a large potential for cost saving: The consumption of cost intensive Nafion® polymer is significantly reduced by printing thin Nafion® membranes and using a PTFE subgasket. Further, enabled by the much higher power density, the cost per Watt ratio of the MEAs is largely improved. Finally, as operation under dry conditions is possible with the direct deposition approach, costly humidifiers may not be necessary for fuel cell operation anymore. As cost is a main factor for the introduction of fuel cell applications, our results may contribute to significantly accelerate the introduction of fuel cells into mass market.
Future research for the direct membrane deposition approach must be dedicated to develop the further understanding of the significantly decreased membrane resistance. Also the processes leading to the encountered electrical short within the degradation experiment have to be investigated and diminished. Generally our approach is well suited for upscaling to large scale high efficiency and cost effective MEA production, for both automotive and stationary applications. For this scale, well established coating techniques like screen-printing or spray-coating may be used in order to enable a larger throughput.
Author contributions
M. Klingele and M. Breitwieser contributed equally to all scientific developments and ideas of this work and to writing the paper. M. Klingele conducted the sample preparation. M. Breitwieser conducted the sample characterization. R. Zengerle and S. Thiele supervised the scientific work.Acknowledgements
This work was funded by the German Federal Ministry of Education BMBF within the project GECKO (grant number 03SF0454C). We gratefully thank Kevin Cooper (Scribner Associates Inc.), Dietmar Gerteisen (Fraunhofer ISE) and Steven Holdcroft (Simon Fraser University) for valuable discussions. We acknowledge Lukas Zielke for support in the graphical artwork.References
- Y. Wang, K. S. Chen, J. Mishler, S. C. Cho and X. C. Adroher, Appl. Energy, 2011, 88(4), 981 CrossRef CAS PubMed.
- C. Houchins, G. Kleen, J. Spendelow, J. Kopasz, D. Peterson, N. Garland, D. Ho, J. Marcinkoski, K. Martin, R. Tyler and D. Papageorgopoulos, Membranes, 2012, 2(4), 855 CrossRef CAS PubMed.
- A. Gruger, A. Régis, T. Schmatko and P. Colomban, Vib. Spectrosc., 2001, 26, 215 CrossRef CAS.
- J. Peron, A. Mani, X. Zhao, D. Edwards, M. Adachi, T. Soboleva, Z. Shi, Z. Xie, T. Navessin and S. Holdcroft, J. Membr. Sci., 2010, 356(1–2), 44 CrossRef CAS PubMed.
- S. Kundu, L. C. Simon, M. Fowler and S. Grot, Polymer, 2005, 46(25), 11707 CrossRef CAS PubMed.
- K. D. Kreuer, J. Membr. Sci., 2001, 185(1), 29 CrossRef CAS.
- C.-Y. Liu and C.-C. Sung, J. Power Sources, 2012, 220, 348 CrossRef CAS PubMed.
- X. Ding, S. Didari, T. F. Fuller and T. A. L. Harris, J. Electrochem. Soc., 2012, 159(6), 746 CrossRef PubMed.
- C.-C. Sung, C.-Y. Liu and C. C. Cheng, Int. J. Hydrogen Energy, 2014, 39(22), 11700 CrossRef CAS PubMed.
- J. Zhang, Y. Tang, C. Song, X. Cheng, J. Zhang and H. Wang, Electrochim. Acta, 2007, 52(15), 5095 CrossRef CAS PubMed.
- F. Barbir, PEM Fuel Cells: Theory and Practice, Elsevier Science, San Diego, 2012 Search PubMed.
- F. N. Büchi and S. Srinivasan, J. Electrochem. Soc., 1997, 144(8), 2767 CrossRef PubMed.
- M. V. Williams, H. Kunz and J. M. Fenton, J. Power Sources, 2004, 135(1–2), 122 CrossRef CAS PubMed.
- B. S. Pivovar and Y. S. Kim, J. Electrochem. Soc., 2007, 154(8), B739 CrossRef CAS PubMed.
- R. Jiang, C. K. Mittelsteadt and C. S. Gittleman, J. Electrochem. Soc., 2009, 156(12), B1440 CrossRef CAS PubMed.
- H. H. Wang, X.-Z. Yuan and H. Li, PEM fuel cell diagnostic tools, CRC Press/Taylor & Francis, Boca Raton, FL, 2012 Search PubMed.
- F. N. Buchi and G. G. Scherer, J. Electrochem. Soc., 2001, 148(3), A183–A188 CrossRef CAS PubMed.
- Z. Wang and Y. Nagao, Electrochim. Acta, 2014, 129, 343 CrossRef CAS PubMed.
- J. Wu, X. Z. Yuan, J. J. Martin, H. Wang, J. Zhang, J. Shen, S. Wu and W. Merida, J. Power Sources, 2008, 184(1), 104 CrossRef CAS PubMed.
- W. Liu, K. Ruth and G. Rusch, J. New Mater. Electrochem. Syst., 2001, 4, 227 CAS.
- S. Zhang, X. Yuan, H. Wang, W. Merida, H. Zhu, J. Shen, S. Wu and J. Zhang, Int. J. Hydrogen Energy, 2009, 34(1), 388 CrossRef CAS PubMed.
- C. Lim, L. Ghassemzadeh, F. van Hove, M. Lauritzen, J. Kolodziej, G. G. Wang, S. Holdcroft and E. Kjeang, J. Power Sources, 2014, 257, 102 CrossRef CAS PubMed.
- X.-Z. Yuan, S. Zhang, H. Wang, J. Wu, J. C. Sun, R. Hiesgen, K. A. Friedrich, M. Schulze and A. Haug, J. Power Sources, 2010, 195(22), 7594 CrossRef CAS PubMed.
- M. M. Mench, E. C. Kumbur and T. N. Veziroğlu, Polymer electrolyte fuel cell degradation, Academic press, Amsterdam, Boston, 2012, p. 2011 Search PubMed.
- B. Wu, M. Zhao, W. Shi, W. Liu, J. Liu, D. Xing, Y. Yao, Z. Hou, P. Ming, J. Gu and Z. Zou, Int. J. Hydrogen Energy, 2014, 39(26), 14381 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |