
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mixed-ligand complexes of paddlewheel dinuclear molybdenum as hydrodehalogenation catalysts for polyhaloalkanes†
Hayato
Tsurugi
*,
Akio
Hayakawa
,
Shun
Kando
,
Yoshitaka
Sugino
and
Kazushi
Mashima
*
Department of Chemistry, Graduate School of Engineering Science, Osaka University, CREST, Toyonaka, Osaka 560-8531, Japan. E-mail: tsurugi@chem.es.osaka-u.ac.jp; mashima@chem.es.osaka-u.ac.jp
First published on 31st March 2015
Abstract
We developed a hydrodehalogenation reaction of polyhaloalkanes catalyzed by paddlewheel dimolybdenum complexes in combination with 1-methyl-3,6-bis(trimethylsilyl)-1,4-cyclohexadiene (MBTCD) as a non-toxic H-atom source as well as a salt-free reductant. A mixed-ligated dimolybdenum complex Mo2(OAc)2[CH(NAr)2]2 (3a, Ar = 4-MeOC6H4) having two acetates and two amidinates exhibited high catalytic activity in the presence of nBu4NCl, in which [nBu4N]2[Mo2{CH(NAr)2}2Cl4] (9a), derived by treating 3a with ClSiMe3 and nBu4NCl, was generated as a catalytically-active species in the hydrodehalogenation. All reaction processes, oxidation and reduction of the dimolybdenum complex, were clarified by control experiments, and the oxidized product, [nBu4N][Mo2{CH(NAr)2}2Cl4] (10a), was characterized by EPR and X-ray diffraction studies. Kinetic analysis of the hydrodehalogenation reaction as well as a deuterium-labelling experiment using MBTCD-d8 suggested that the H-abstraction was the rate-determining step for the catalytic reaction.
Introduction
The M2L4-type paddlewheel dinuclear complexes having monoanionic bridging ligands comprise the simplest metal cluster motif, and intensive investigation has been focused on their structures, redox behaviors, and spectroscopic properties, as well as their catalytic applications.1–3 Dinuclear paddlewheel complexes of the late transition metals such as rhodium and ruthenium act as useful and versatile catalysts for cyclopropanation of olefins and for functionalizing C–H, O–H, N–H, and Si–H bonds via the generation of metal-carbene, [M2L4(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CR1R2)], metal-alkoxide, and -nitride species, [M2L4(Z)] (Z = OR, N).3c–3e,4–6 In all cases, because four supporting ligands tightly coordinate to the adjacent two metal centers while maintaining the dinuclear paddlewheel skeleton and the metal–metal bond, the architecture of the four supporting ligands was tunable to control redox behavior and catalytic performance of the paddlewheel complexes. In sharp contrast, few studies have examined the catalytic application of paddlewheel complexes of the early transition metals. As an example, Mo2(OAc)4 was used for an aza-Diels–Alder reaction of acyl hydrazones and dienes;7 however, the original paddlewheel structure was not maintained during the reaction. We and others have continued to investigate the stoichiometric and catalytic application of quadruply bonded M2 complexes of group 6 metals for organic radical generation by designing bridging ligands,8–10 and have achieved catalytic radical addition and polymerization reactions.9 During these transformations, the metal–metal bond responds to the one-electron redox processes without decomposition of the dinuclear structure. The structural stability of the dinuclear motif owes to both the surrounding four ligands and the metal–metal multiple bonds. In a further catalytic application of the M2 complexes, we used a cyclohexadiene derivative instead of α-olefins as a substrate for the organic radicals generated from polyhaloalkanes, leading to the formation of hydrodehalogenated products. Although hydrodehalogenation is one of the key reactions for decomposing environmentally unfriendly polyhaloalkanes, precious metal catalysts are often used.11 Herein, we report that paddlewheel Mo2 complexes, as shown in Fig. 1, act as catalysts for hydrodehalogenation reactions of polyhaloalkanes upon combination with 1-methyl-3,6-bis(trimethylsilyl)-1,4-cyclohexadiene (MBTCD)12 as an H-atom donor, and reveal the mechanism and actual active species in this catalytic process.
CR1R2)], metal-alkoxide, and -nitride species, [M2L4(Z)] (Z = OR, N).3c–3e,4–6 In all cases, because four supporting ligands tightly coordinate to the adjacent two metal centers while maintaining the dinuclear paddlewheel skeleton and the metal–metal bond, the architecture of the four supporting ligands was tunable to control redox behavior and catalytic performance of the paddlewheel complexes. In sharp contrast, few studies have examined the catalytic application of paddlewheel complexes of the early transition metals. As an example, Mo2(OAc)4 was used for an aza-Diels–Alder reaction of acyl hydrazones and dienes;7 however, the original paddlewheel structure was not maintained during the reaction. We and others have continued to investigate the stoichiometric and catalytic application of quadruply bonded M2 complexes of group 6 metals for organic radical generation by designing bridging ligands,8–10 and have achieved catalytic radical addition and polymerization reactions.9 During these transformations, the metal–metal bond responds to the one-electron redox processes without decomposition of the dinuclear structure. The structural stability of the dinuclear motif owes to both the surrounding four ligands and the metal–metal multiple bonds. In a further catalytic application of the M2 complexes, we used a cyclohexadiene derivative instead of α-olefins as a substrate for the organic radicals generated from polyhaloalkanes, leading to the formation of hydrodehalogenated products. Although hydrodehalogenation is one of the key reactions for decomposing environmentally unfriendly polyhaloalkanes, precious metal catalysts are often used.11 Herein, we report that paddlewheel Mo2 complexes, as shown in Fig. 1, act as catalysts for hydrodehalogenation reactions of polyhaloalkanes upon combination with 1-methyl-3,6-bis(trimethylsilyl)-1,4-cyclohexadiene (MBTCD)12 as an H-atom donor, and reveal the mechanism and actual active species in this catalytic process.
 | ||
| Fig. 1 Paddlewheel dimolybdenum complexes 1–5. | ||
Results and discussion
We began by searching for the best paddlewheel Mo2 catalyst among the homoleptic and mixed-ligand complexes listed in Fig. 1 for the hydrodehalogenation reaction of 1,1,1,3-tetrachloropropane, as a model substrate, in CD3CN in the presence of 1.2 equiv. of MBTCD to give 1,1,3-trichloropropane (Table 1).13 Simple homoleptic complexes 1 and 5 exhibited very low activities (runs 1 and 7), while mixed-ligand complexes of dimolybdenum bearing both acetate and amidinate ligands exhibited moderate catalytic activities (runs 2, 3, and 6). The stereochemistry of the mixed-ligand complexes was a key factor: trans-Mo2(OAc)2[CH(NAr)2]2 (Ar = 4-MeOC6H4) (3a) afforded the hydrodehalogenated product in 64% yield, whereas cis-Mo2(OAc)2[CH(NAr)2]2 (3b) afforded only a very low yield of the dehalogenated product (run 4). In addition, a trans-arranged mixed-ligated complex 3c having bulky 2,6-dimethylphenyl groups at the nitrogen atoms of the amidinate ligand exhibited almost no activity, probably because the substrate approach was sterically prevented (run 5). Accordingly, we examined the solvent and additive effects using 3a as the catalyst. In THF-d8 and C6D6, the yield of 1,1,3-trichloropropane decreased (runs 8 and 9). Positive additive effects of nBu4NCl were observed: addition of nBu4NCl (10 mol%) to the reaction mixture in acetonitrile led to the formation of 1,1,3-trichloropropane in 84% yield (run 10). In the blank reaction without MBTCD, no reaction proceeded since MBTCD acts as the H-donor and reductant for the catalytic reaction. When we used HSiEt3 as a commercially available H-donor instead of MBTCD, the yield decreased (11%), probably due to the inefficient reducing ability of the in situ generated intermediate [Mo2]5+ species (vide infra).|
|
||||
|---|---|---|---|---|
| Run | Cat. | Solvent | Additive | Yielda (%) |
| a Determined using 1H NMR measurements. Yield in parentheses was the isolated yield. b n Bu4NCl (10 mol%) was added to the reaction mixture. | ||||
| 1 | 1 | CD3CN | — | 4 |
| 2 | 2 | CD3CN | — | 47 |
| 3 | 3a | CD3CN | — | 64 |
| 4 | 3b | CD3CN | — | 9 |
| 5 | 3c | CD3CN | — | 1 |
| 6 | 4 | CD3CN | — | 31 |
| 7 | 5 | CD3CN | — | <1 |
| 8 | 3a | THF-d8 | — | 59 |
| 9 | 3a | C6D6 | — | 42 |
| 10b | 3a | CD3CN | n Bu4NCl | 84 (73) |
| 11 | 7a | CD3CN | — | 71 |
| 12b | 7a | CD3CN | n Bu4NCl | 85 |
| 13b | 8a | CD3CN | n Bu4NCl | 32 |
| 14 | 9a | CD3CN | — | 85 |
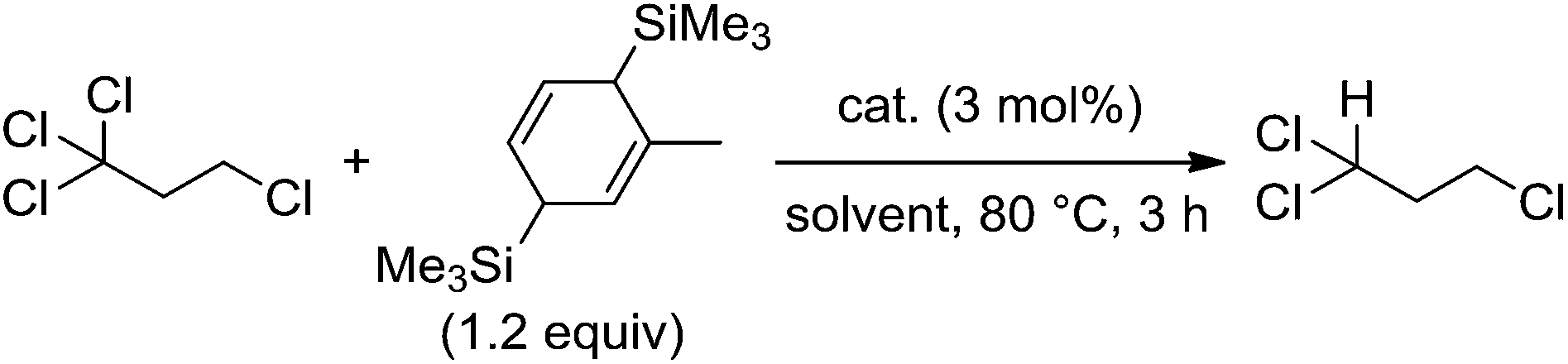
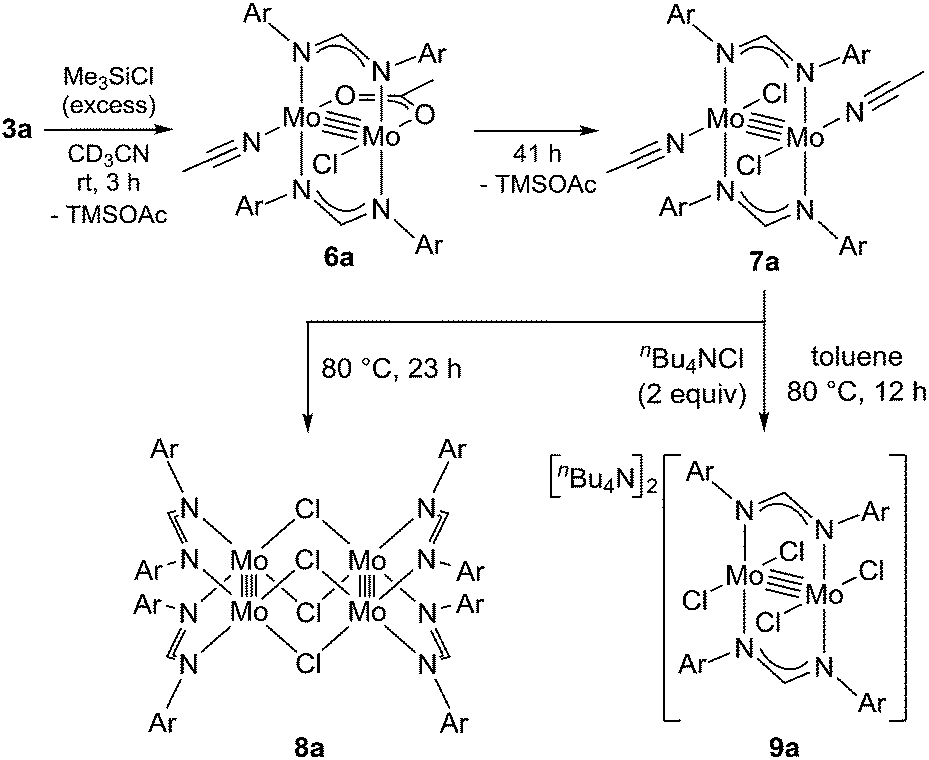
To reveal the additive effects of nBu4NCl to 3a, we conducted control experiments. We observed the formation of trimethylsilyltoluene and ClSiMe3 as the reaction byproducts derived from MBTCD, along with Me3SiOAc, which was the reaction product of the acetate ligand and ClSiMe3. Because the ligand replacement reaction of an acetate ligand by ClSiMe3 was reported by Cotton et al.,14 we examined the reaction of 3a with excess ClSiMe3 in CD3CN (Scheme 1). In the 1H NMR spectrum, new resonances assignable to the Mo2 complex 6a appeared within 30 minutes at room temperature, and, after 41 h, all of 3a was converted to 7a, which was isolated as a purple powder.15 Subsequent heating of a solution of 7a at 80 °C resulted in the formation of a dimeric molybdenum cluster, [Mo2{CH(NAr)2}2(μ-Cl)2]2 (8a),16 which was previously isolated and structurally characterized. The dimeric complex 8a exhibited low catalytic activity even in the presence of nBu4NCl, probably due to the low solubility of 8a in the reaction media (run 13). In contrast, treatment of complex 3a with ClSiMe3 in the presence of nBu4NCl in toluene at 80 °C afforded a dianionic Mo2 species, [nBu4N]2[Mo2{CH(NAr)2}2Cl4] (9a), in which two amidinates and four chloride ligands coordinated to the Mo2 core, based on single crystal X-ray diffraction analysis (Fig. 2(a)).15,17 Notably, no conversion of 9a to the Cl-bridged dimer 8a was observed, even upon prolonged heating of 9a in CH3CN. The catalytic activity of 9a was equal to that of 3a/nBu4NCl (runs 10 and 14). By monitoring the reaction progress using 1H NMR spectroscopy, we found the induction period for the 3a/nBu4NCl catalyst. Complexes 3a or 7a were slowly catalyzing the hydrodehalogenation reaction. Interestingly, complex 9a initiated the catalytic hydrodehalogenation reaction without any induction period (Fig. 3), indicating that the dianionic complex 9a was the identity of the catalytically active species.
 | ||
| Scheme 1 Ligand substitution and dimerization reactions of 3a by the addition of Me3SiCl. | ||
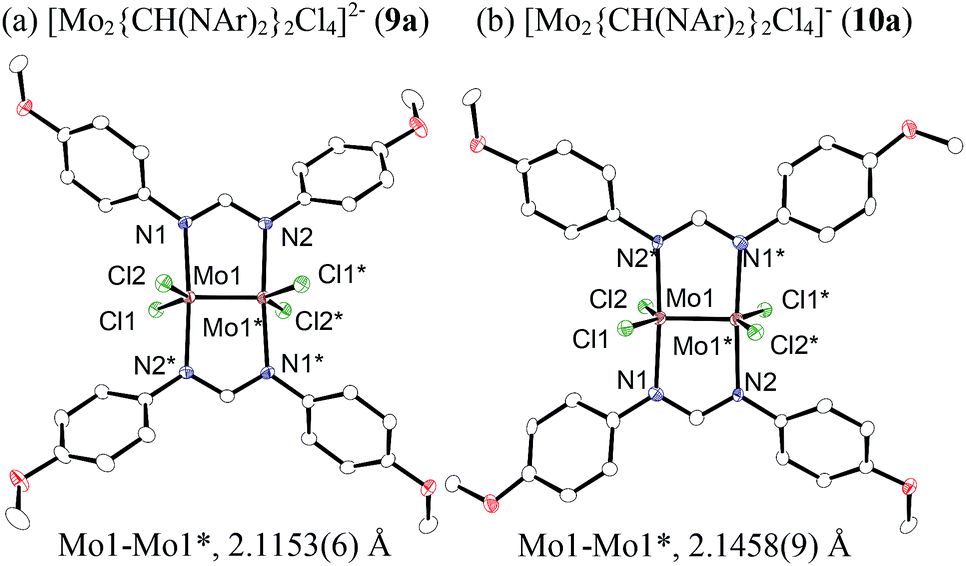 | ||
| Fig. 2 ORTEP drawing of the molecular structure for dimolybdenum complexes (a) 9a and (b) 10a. Hydrogen atoms and the cationic part are omitted for clarity. | ||
 | ||
| Fig. 3 Reaction profile for the hydrodehalogenation catalyzed by Mo2 complexes (3 mol%) with MBTCD (1.2 equiv.) at 60 °C. | ||
Because of the inactivity of MBTCD toward [Mo2]4+ complexes, the catalytic reaction of 9a (3a/nBu4NCl) was assumed to be initiated by the first reductive cleavage of a carbon–halogen bond, which produced a carbon radical and an [Mo2]5+ species. Thus, we next performed an oxidation reaction of 9a with CCl4 at room temperature, and the oxidized [Mo2]5+ species, [nBu4N][Mo2{CH(NAr)2}2Cl4] (10a), was isolated in 88% yield (Scheme 2, path a). During the reaction, nBu4NCl was eliminated, and the ligand system of the Mo2 core remained intact. A single crystal X-ray diffraction study of 10a confirmed the elongation of the Mo–Mo bond by ∼0.04 Å from 9a, and the distance of the Mo–Mo bond is typical for [Mo2]5+ species (Fig. 2(b)).8a,15,18 The formation of [Mo2]5+ was further confirmed using EPR spectroscopic analysis, in which resonances typical of [Mo2]5+ species were detected (g = 1.955). The high catalytic activity of 9a was ascribed to the relatively negative Eox([Mo2]4+/5+) value: the Eox ([Mo2]4+/5+) value of 9a was −0.29 V, which shifted to a more negative value compared to that of 7a (−0.14 V) and 8a (−0.08 V). The other aspect of the high catalytic activity of 9a is the stability of the Mo2(L)2Cl4 structure and solubility during the redox processes. In fact, complex 7a reacted with CCl4 to form a [Mo2]6+ species, [Mo2{CH(NAr)2}2Cl4(CH3CN)2] (11a), as poorly soluble dark-red microcrystals that precipitated from the reaction mixture (eqn (1)).15
 | (1) |
 | ||
| Scheme 2 Redox reactions of 9a and 10a. | ||
When the oxidized species 10a was treated with MBTCD in the absence of nBu4NCl or without MBTCD in the presence of nBu4NCl to reduce 10a, no reaction was observed, even after heating. On the other hand, in the presence of MBTCD, nBu4NCl, and AIBN as a carbon radical source, we observed the formation of 9a together with ClSiMe3, trimethylsilyltoluene, and isobutyronitrile, after heating at 80 °C for 1.5 h (Scheme 2, path b). This indicated that the AIBN-derived carbon radical abstracted one hydrogen atom from MBTCD to generate isobutyronitrile and a radical derivative of MBTCD, which subsequently reduced 10a to give 9a, along with ClSiMe3 and trimethylsilyltoluene.
By using the complex 9a as a catalyst for the hydrodehalogenation of 1,1,1,3-tetrachloropropane, we checked the initial reaction rate dependence for the catalyst, 1,1,1,3-tetrachloropropane, and MBTCD. A first-order rate dependence on the catalyst and MBTCD concentration was observed, whereas the reaction was not dependent on the concentration of 1,1,1,3-tetrachloropropane (Fig. 4(a)–(c)). During the catalytic reaction as described in Table 1, we did not find any byproducts such as radical homo-coupling and disproportionated products, suggesting that the reaction of 9a and 1,1,1,3-tetrachloropropane is in fast equilibrium with 10a before reacting with MBTCD. Zero-order in 1,1,1,3-tetrachloropropane might indicate the saturation of the reactive intermediate composed of 10a and the carbon radical in the coordination sphere, which is consistent with the large negative entropy value (vide infra). In addition, MBTCD-d8 was applied to the catalytic reaction: the KIE value was 1.71, suggesting that the H-abstraction from MBTCD by the organic radical was involved in the rate-determining step. Furthermore, the rate of the catalytic reaction over a temperature ranging from 50 to 65 °C was monitored by 1H NMR spectroscopy. Eyring kinetic analyses of the reaction profile afforded the activation parameters of ΔH‡ = 25.7 ± 0.9 kJ mol−1, ΔS‡ = −56.5 ± 0.7 e.u., and ΔG‡(298 K) = 96.2 ± 1.8 kJ mol−1 (Fig. 4(d)). A large negative ΔS‡ value indicated an ordered transition state for the H-abstraction step: we presume that the organic radical derived from 1,1,1,3-tetrachloropropane stays in the coordination sphere of 10a after C–Cl reductive cleavage, while the radical abstracts the H-atom from MBTCD.
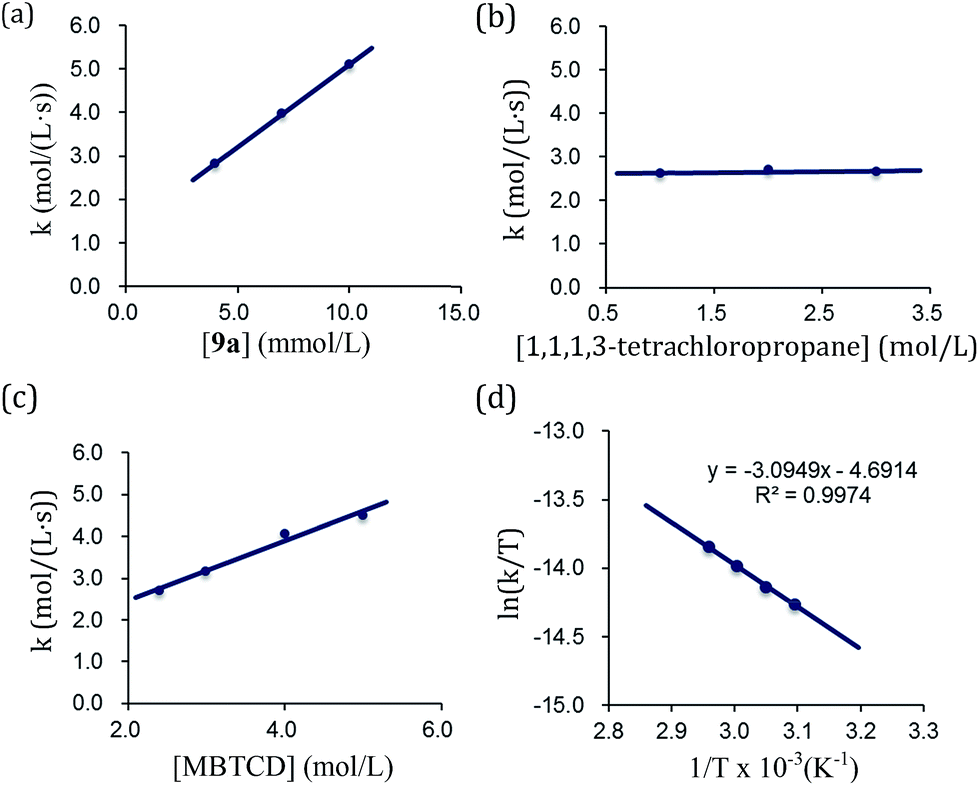 | ||
| Fig. 4 Kinetic analyses of the catalytic hydrodehalogenation reaction catalyzed by 9a. (a) Rate-dependence on [9a]. (b) Rate-dependence on [1,1,1,3-tetrachloropropane]. (c) Rate-dependence on [MBTCD]. (d) Eyring plot in the range of 50–65 °C. | ||
Based on the above observations for the kinetic study and redox reactions of the Mo2 complexes, we propose a plausible catalytic cycle as shown in Scheme 3. In the initial stage, the dinuclear metal cluster 9a is in equilibrium with [Mo2]5+ species 10a in the presence of alkyl halides, which is often observed for the carbon radical generation by low-valent metal species with alkyl halides.19 Because of the zero-order dependence on the substrate concentration and no observation of the radical homo-coupling and disproportionated compounds, this equilibrium is very fast, and the reactive intermediate, 10a and the carbon radical in the coordination sphere, is generated. Next, the carbon radical abstracts a hydrogen atom from MBTCD, which is a rate-determining step in this catalytic cycle, to form hydrodehalogenated products and a radical derivative of MBTCD. Finally, the [Mo2]5+ cluster is reduced by the MBTCD-derived radical to regenerate the [Mo2]4+ species 9a together with ClSiMe3 and trimethylsilyltoluene.
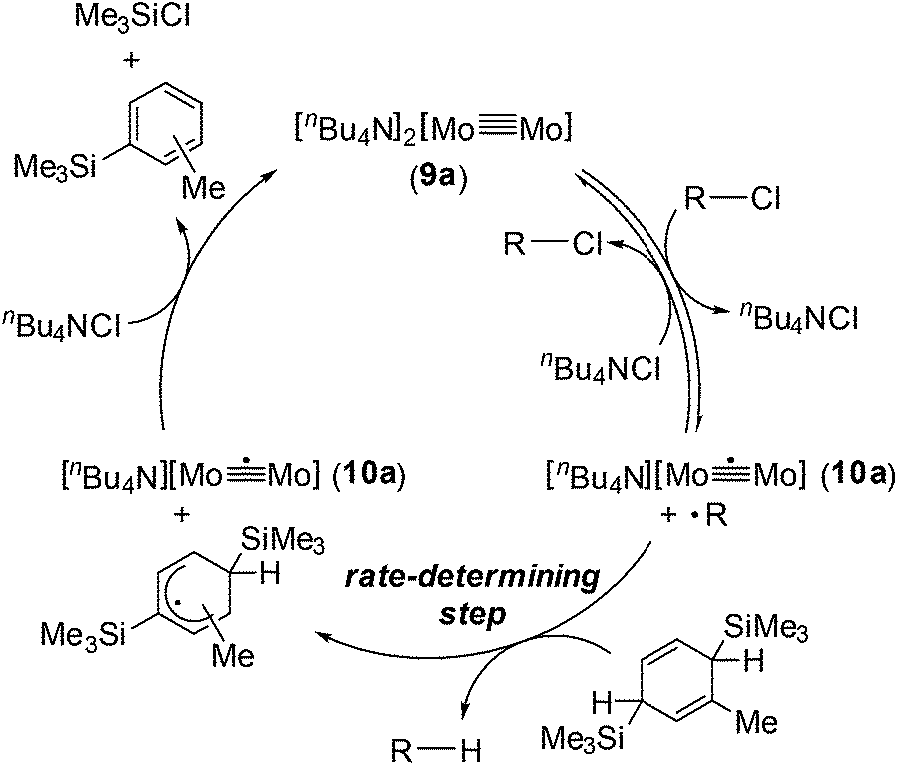 | ||
| Scheme 3 Plausible mechanism for hydrodehalogenation reactions of alkyl halides catalyzed by the Mo2 complex 9a. | ||
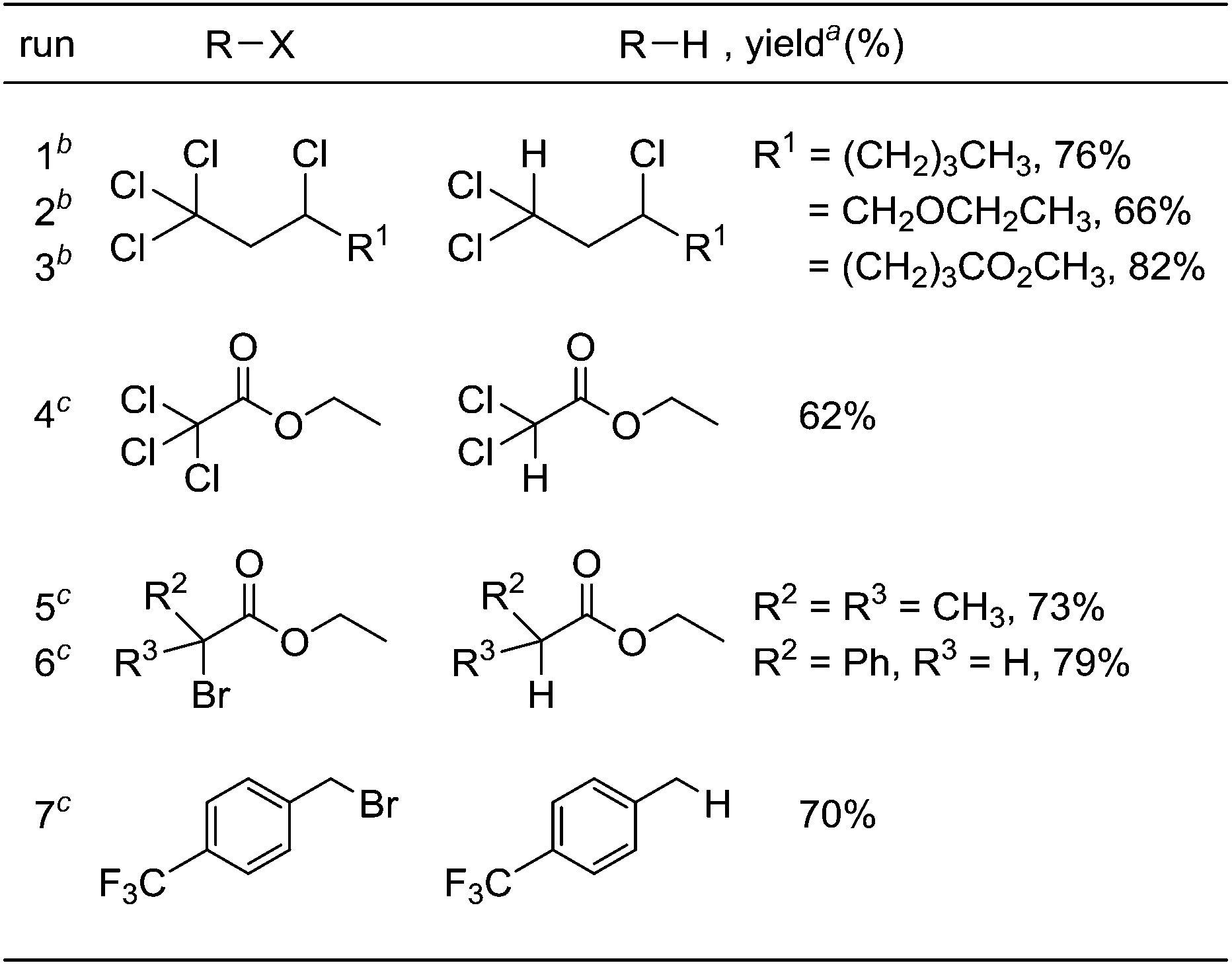
Under optimized conditions for the catalytic hydrodehalogenation reaction using 3a/nBu4NCl in CH3CN at 80 °C as described in Table 1, the substrate scope was surveyed for haloalkanes having a trichloromethyl or a bromomethyl group, and the results are summarized in Table 2. Tetrachloroalkanes having a longer alkyl chain, ether, and ester groups were effectively dehalogenated to give the corresponding trichloroalkanes in 66%–82% yields (runs 1–3). In addition, α-halocarbonyl ester derivatives and benzyl bromide derivatives were applicable for the hydrodehalogenation reaction in the presence of 2 equiv. of MBTCD to afford the corresponding products in good yields (runs 4–7).
Conclusions
We developed catalytic hydrodehalogenation reactions using paddlewheel Mo2 complexes in the presence of MBTCD as an H-atom donor as well as a reductant. Trans-Mo2(OAc)2[CH(NAr)2]2 (3a, Ar = 4-MeOC6H4) exhibited high catalytic activity for the catalytic reaction in the presence of nBu4NCl. Control experiments revealed that the ligand exchange of two acetate ligands of 3a by ClSiMe3 and nBu4NCl to form an ionic [nBu4N]2[Mo2{CH(NAr)2}2Cl4] (9a) were key reaction events to generate the catalytically active species. In addition, kinetic analysis of the catalytic reaction profile, a deuterium-labeling experiment using MBTCD-d8, and isolation of the oxidized species 10a clarified the catalytic cycle and the rate-determining step in the catalytic reaction. Further studies of the applications of one electron redox processes by paddlewheel Mo2 complexes toward various organic transformations are ongoing in our laboratory.Acknowledgements
H.T. acknowledges financial support by a Grant-in-Aid for Scientific Research on Innovative Areas “New Polymeric Materials Based on Element-Blocks (no. 2401)” and a Grant-in-Aid for Young Scientists (A) of The Ministry of Education, Culture, Sports, Science, and Technology, Japan. This work was financially supported by the Core Research for Evolutional Science and Technology (CREST), the program of the Japan Science and Technology Agency (JST).Notes and references
- F. A. Cotton, C. A. Murillo and R. A. Walton, Multiple Bonds between Metal Atoms, Springer, New York, 2005 Search PubMed.
- (a) M. H. Chisholm, Angew. Chem., Int. Ed., 1986, 25, 21 CrossRef PubMed; (b) F. A. Cotton, C. Lin and C. A. Murillo, Acc. Chem. Res., 2001, 34, 759 CrossRef CAS PubMed.
- (a) T. Ren, Coord. Chem. Rev., 1998, 175, 43 CrossRef CAS; (b) M. Mikuriya, D. Yoshioka and M. Handa, Coord. Chem. Rev., 2006, 250, 2194 CrossRef CAS PubMed; (c) H. M. L. Davies and J. R. Manning, Nature, 2008, 451, 417 CrossRef CAS PubMed; (d) J. Hansen and H. M. L. Davies, Coord. Chem. Rev., 2008, 252, 545 CrossRef CAS PubMed; (e) A. F. Trindade, J. A. S. Coelho, C. A. M. Afonso, L. F. Veiros and P. M. P. Gois, ACS Catal., 2012, 2, 370 CrossRef CAS.
- (a) M. P. Doyle, M. McKervey and T. Ye, Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanation to Ylides, Wiley, New York, 1998 Search PubMed; (b) T. Ye and A. McKervey, Chem. Rev., 1994, 94, 1091 CrossRef CAS; (c) M. P. Doyle and D. C. Forbes, Chem. Rev., 1998, 98, 911 CrossRef CAS PubMed; (d) H. M. L. Davies and R. E. J. Beckwith, Chem. Rev., 2003, 103, 2861 CrossRef CAS PubMed; (e) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed.
- (a) S. Uemura and S. R. Patil, Chem. Lett., 1982, 1743 CrossRef CAS; (b) C. J. Moody and F. N. Palmer, Tetrahedron Lett., 2002, 43, 139 CrossRef CAS; (c) A. J. Catino, R. E. Forslund and M. P. Doyle, J. Am. Chem. Soc., 2004, 126, 13622 CrossRef CAS PubMed; (d) A. J. Catino, J. M. Nichols, H. Choi, S. Gottipamula and M. P. Doyle, Org. Lett., 2005, 7, 5167 CrossRef CAS PubMed; (e) A. J. Catino, J. M. Nichols, B. J. Nettles and M. P. Doyle, J. Am. Chem. Soc., 2006, 128, 5648 CrossRef CAS PubMed; (f) H. Choi and M. P. Doyle, Org. Lett., 2007, 9, 5349 CrossRef CAS PubMed; (g) S. J. Na, B. Y. Lee, N.-N. Bui, S.-I. Mho and H.-Y. Jang, J. Organomet. Chem., 2007, 692, 5523 CrossRef CAS PubMed; (h) E. C. McLaughlin and M. P. Doyle, J. Org. Chem., 2008, 73, 4317 CrossRef CAS PubMed; (i) E. C. McLaughlin, H. Choi, K. Wang, G. Chiou and M. P. Doyle, J. Org. Chem., 2009, 74, 730 CrossRef CAS PubMed; (j) G. J. Chuang, W. Wang, E. Lee and T. Ritter, J. Am. Chem. Soc., 2011, 133, 1760 CrossRef CAS PubMed.
- (a) J. S. Pap, S. D. George and J. F. Berry, Angew. Chem., Int. Ed., 2008, 47, 10102 CrossRef CAS PubMed; (b) J. S. Pap, J. L. Snyder, P. M. B. Piccoli and J. F. Berry, Inorg. Chem., 2009, 48, 9846 CrossRef CAS PubMed; (c) A. K. M. Long, R. P. Yu, G. H. Timmer and J. F. Berry, J. Am. Chem. Soc., 2010, 132, 12228 CrossRef PubMed; (d) A. K. M. Long, G. H. Timmer, J. S. Pap, J. L. Snyder, R. P. Yu and J. F. Berry, J. Am. Chem. Soc., 2011, 133, 13138 CrossRef CAS PubMed; (e) G. H. Timmer and J. F. Berry, Chem. Sci., 2012, 3, 3038 RSC.
- Y. Yamashita, M. M. Salter, K. Aoyama and S. Kobayashi, Angew. Chem., Int. Ed., 2006, 45, 3816 CrossRef CAS PubMed.
- (a) F. A. Cotton, L. M. Daniels, C. A. Murillo and D. J. Timmons, Chem. Commun., 1997, 1449 RSC; (b) J. A. M. Canich, F. A. Cotton, K. R. Dunbar and L. R. Falvello, Inorg. Chem., 1988, 27, 804 CrossRef CAS; (c) F. A. Cotton, N. E. Gruhn, J. Gu, P. Huang, D. L. Lichtenberger, C. A. Murillo, L. O. van Dorn and C. C. Wilkinson, Science, 2002, 298, 1971 CrossRef CAS PubMed; (d) F. A. Cotton, P. Huang, C. A. Murillo and X. Wang, Inorg. Chem. Commun., 2003, 6, 121 CrossRef; (e) F. A. Cotton, J. P. Donahue, D. L. Lichtenberger, C. A. Murillo and D. Villagrán, J. Am. Chem. Soc., 2005, 127, 10808 CrossRef CAS PubMed; (f) F. A. Cotton, C. A. Murillo, X. Wang and C. C. Wilkinson, Dalton Trans., 2006, 4623 RSC; (g) F. A. Cotton, C. A. Murillo, X. Wang and C. C. Wilkinson, Dalton Trans., 2007, 3943 RSC.
- H. Tsurugi, K. Yamada, M. Moumita, Y. Sugino, A. Hayakawa and K. Mashima, Dalton Trans., 2011, 9358 RSC.
- Reductive cleavage of carbon–halogen bonds were reported previously, see: (a) F. A. Cotton, L. M. Daniels, C. A. Murillo, D. J. Timmons and C. C. Wilkinson, J. Am. Chem. Soc., 2002, 124, 9249 CrossRef CAS PubMed; (b) F. A. Cotton, J. P. Donahue, N. E. Gruhn, D. L. Lichtenberger, C. A. Murillo, D. J. Timmons, L. O. van Dorn, D. Villagrán and X. Wang, Inorg. Chem., 2006, 45, 201 CrossRef CAS PubMed; (c) F. A. Cotton, C. A. Murillo, X. Wang and C. C. Wilkinson, Inorg. Chem., 2006, 45, 5493 CrossRef CAS PubMed.
- (a) V. V. Lunin and E. S. Lokteva, Russ. Chem. Bull., 1996, 45, 7 Search PubMed; (b) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2002, 102, 4009 CrossRef CAS PubMed; (c) J. D. Nguyen, E. M. D'Amato, J. M. R. Narayanam and C. R. J. Stephenson, Nat. Chem., 2012, 4, 854 CrossRef CAS PubMed; (d) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756 CrossRef CAS PubMed.
- R. A. Müller, H. Tsurugi, T. Saito, M. Yanagawa, S. Oda and K. Mashima, J. Am. Chem. Soc., 2009, 131, 5370 CrossRef PubMed.
- Bis(trimethylsilyl)cyclohexadiene derivatives were converted to aromatic compounds in the presence of dioxygen or peroxides with release of H atom, see: (a) J. Dunogues, D. N'Gabe, M. Laguerre, N. Duffaut and R. Calas, Organometallics, 1982, 1, 1525 CrossRef CAS; (b) H. Sakurai, M. Kira and H. Sugiyama, Chem. Lett., 1983, 599 CrossRef CAS; (c) K. A. Walker, L. J. Markoski and J. S. Moore, Synthesis, 1992, 1265 CrossRef CAS.
- (a) P. Agaskar and F. A. Cotton, Inorg. Chim. Acta, 1984, 83, 22 CrossRef; (b) M. L. H. Green, G. Parkin, J. Bashkin, J. Fail and K. Prout, J. Chem. Soc., Dalton Trans., 1982, 2519 RSC; (c) J. D. Arenivar, V. V. Mainz, H. Ruben, R. A. Andersen and A. Zalkin, Inorg. Chem., 1982, 21, 2649 CrossRef; (d) F. A. Cotton and G. L. Powell, Polyhedron, 1985, 4, 1669 CrossRef CAS.
- See ESI.†.
- F. A. Cotton, C. Y. Liu, C. A. Murillo and X. Wang, Chem. Commun., 2003, 2190 RSC.
- Dianionic [Mo2(L)2(Cl)4] complexes, see: (a) C. D. Garner, S. Parkes, I. B. Walton and W. Clegg, Inorg. Chim. Acta, 1978, 31, 451 CrossRef; (b) W. Clegg, C. D. Garner, S. Parkes and I. B. Walton, Inorg. Chem., 1979, 18, 2250 CrossRef CAS.
- (a) F. A. Cotton, X. Feng and M. Matusz, Inorg. Chem., 1989, 28, 594 CrossRef CAS; (b) P. J. Bailey, S. F. Bone, L. A. Mitchell, S. Parsons, K. J. Taylor and L. J. Yellowlees, Inorg. Chem., 1997, 36, 867 CrossRef CAS.
- Importance for the equilibrium between the low-valent metals/alkyl halides and their oxidized metal species/carbon radicals is discussed in atom-transfer radical addition/polymerization mechanisms, see: (a) K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921 CrossRef CAS PubMed; (b) M. Kamigaito, T. Ando and M. Sawamoto, Chem. Rev., 2001, 101, 3689 CrossRef CAS PubMed; (c) T. Pintauer and K. Matyjaszewski, Chem. Soc. Rev., 2008, 37, 1087 RSC; (d) W. Tang, N. V. Tsarevsky and K. Matyjaszewski, J. Am. Chem. Soc., 2006, 128, 1598 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis and characterization of Mo2 complexes, kinetic analysis of the reactions, 1H NMR spectra of the catalytic reactions, identification of the products, CV of selected Mo2 complexes, and crystal data for 7a (CCDC 1046579), 9a (CCDC 1046580), 10a (CCDC 1046581), and 11a (CCDC 1046582). CCDC 1046579–1046582. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00721f |
| This journal is © The Royal Society of Chemistry 2015 |