
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C–H arylation of triphenylene, naphthalene and related arenes using Pd/C†
Karl D.
Collins
,
Roman
Honeker‡
,
Suhelen
Vásquez-Céspedes‡
,
Dan-Tam D.
Tang
and
Frank
Glorius
*
Westfälische Wilhelms-Universität Münster, Organisch-Chemisches Institut, Corrensstraße 40, 48149 Münster, Germany. E-mail: glorius@uni-muenster.de
First published on 22nd December 2014
Abstract
A highly selective arylation of a number of polyaromatic hydrocarbons (PAHs) with aryliodonium salts and Pd/C as the only reagent is reported. The first C–H functionalization of triphenylene is explored, and proceeds at the most sterically hindered position. This non-chelate assisted C–H functionalization extends the reactivity profile of Pd/C and provides controlled access to π-extended PAHs, an important aspect of work towards the preparation of nanographenes. Mechanistic studies suggest in situ formation of catalytically active insoluble nanoparticles, and that the reaction likely proceeds via a Pd(0)/Pd(II) type reaction manifold.
Introduction
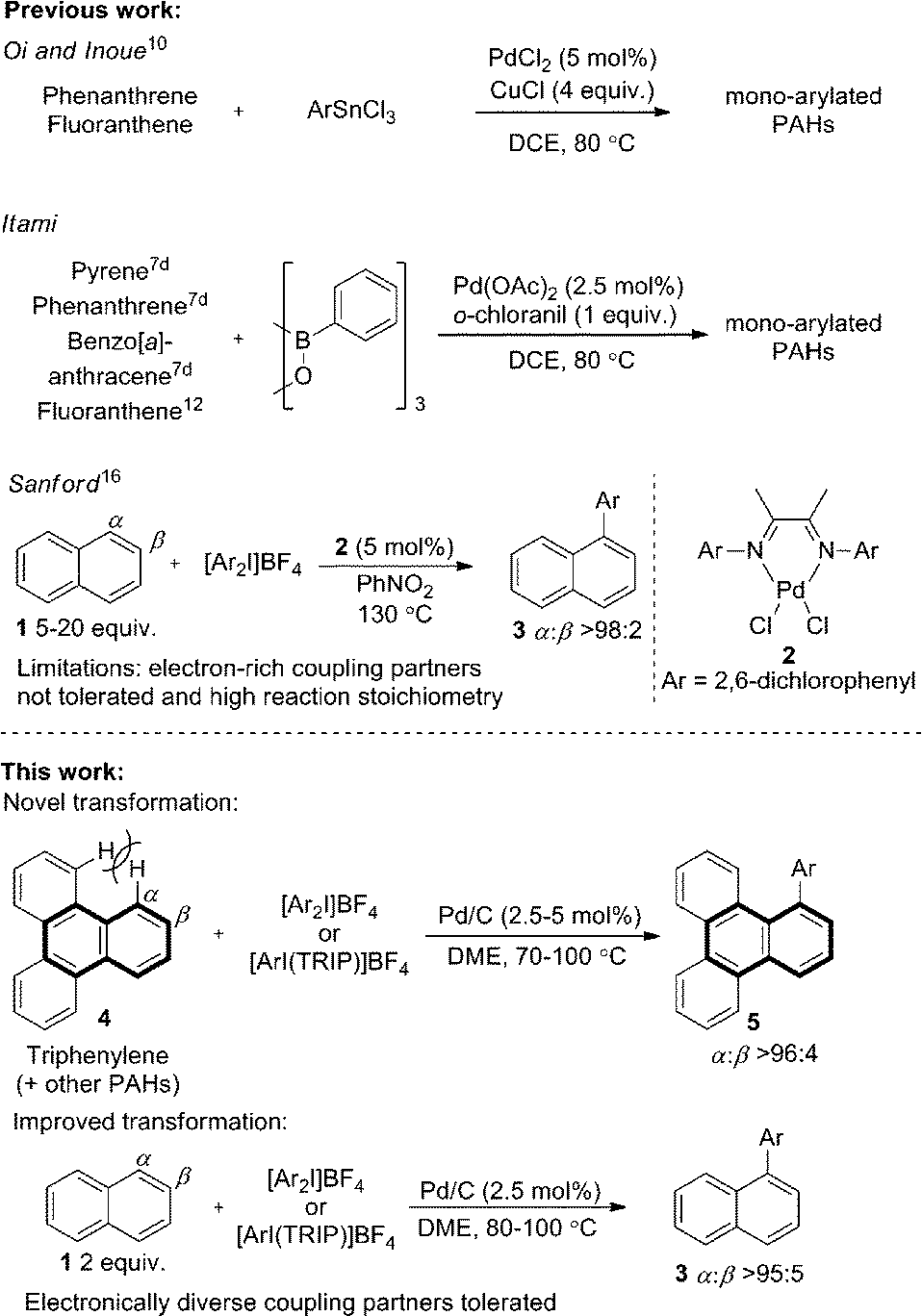
Pd/C has long been established as an efficient catalyst in hydrogenation and cross-coupling reactions.1 Although supported catalysts are formally heterogeneous in nature, studies of Pd/C suggest it typically acts as a reservoir of homogeneous active catalytic species, following leaching of palladium from the support into the solution.1c,1e,2 Our recent work identified that Pd/C can mediate highly selective direct C–H functionalizations of heteroaromatic systems, representing novel reactivity beyond its traditional applications.3 A surprising facet of these works was that studies suggested a heterogeneous active catalytic species despite the high temperature or strongly oxidizing nature of the reaction conditions.4,5 Furthermore, Pd/C was clearly superior in terms of regioselectivity and yield to all homogeneous palladium sources screened, and no ligands were employed. Following these unexpected observations, we were encouraged to explore the more challenging selective C–H functionalization of non-heteroatom containing arenes,6 with a view to unveiling further surprising selectivity or reactions exploiting heterogeneous catalytic species.The direct arylation of PAHs is an important and unmet challenge for the ‘bottom-up’ preparation of π-extended PAHs.7 Controlled access to such defined graphene like two-dimensional sheets of sp2-hybridized carbon atoms could have significant implications for the preparation of larger graphene structures,8 which are of immense interest to numerous scientific fields.9 Despite the potential applications, only limited precedent for the direct arylation of larger PAHs is reported. Seminal work by Oi and Inoue reported an effective arylation of phenanthrene and fluoranthene with aryltin trichlorides.10 One example of phenanthrene arylation has also been reported by Shi.11 Important studies by the group of Itami demonstrated a more general solution (Scheme 1) using prepared boroxines as coupling partners.7d
 | ||
| Scheme 1 C–H functionalization of naphthalene and polyaromatic hydrocarbons (PAHs). TRIP is 2,4,6-triisopropylphenyl. | ||
Using Pd(OAc)2 and o-chloroanil as oxidant, they have described arylations of pyrene, phenanthrene and benzo[a]anthracene at the more reactive olefin like K-region7d of these PAHs. Notably, the more challenging C–H arylation of triphenylene (4), which does not contain a K-region is not reported. Selective coupling of fluoranthene12 and the polyarylation of corannulenes13 and perylenes14 have also been demonstrated. C–H arylation of naphthalene (1), has been explored more extensively, though typically proceeds with moderate yields and poor α![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :β selectivity.15 A study from the Sanford group employing a bulky Pd(II) diimine catalyst (2) in nitrobenzene at 130 °C represents the only highly selective α-arylation of naphthalene (1), and scope is limited to a small number of electron-deficient or electron-neutral aryliodonium salts as coupling partners (Scheme 1).16,17
:β selectivity.15 A study from the Sanford group employing a bulky Pd(II) diimine catalyst (2) in nitrobenzene at 130 °C represents the only highly selective α-arylation of naphthalene (1), and scope is limited to a small number of electron-deficient or electron-neutral aryliodonium salts as coupling partners (Scheme 1).16,17
In this work we first present a Pd/C (Heraeus: 5 wt% Pd/C type K-0219, <3% H2O) catalyzed arylation of triphenylene (4) with aryliodonium salts.18 To the best of our knowledge this represents both the first C–H functionalization of triphenylene, and a rare example of C–H functionalization of non-heteroaromatic systems using a simple heterogeneous catalyst (Scheme 1).19 Notably, the reaction occurs with complete selectivity at the most hindered position. The arylation of several other PAHs is also demonstrated, including the hitherto unknown direct C–H functionalization of anthracene at C1–H.20 This simple catalytic system also enabled a highly selective arylation (α:β > 95:5) of naphthalene (1) with electronically diverse diaryliodonium salts. Empirical data suggests the in situ generation of catalytically active insoluble nanoparticles from a Pd/C precatalyst, and further mechanistic studies support a Pd(0)/Pd(II) reaction manifold.
Results and discussion
Initially exploring the arylation of simple arenes using Pd/C, we found that subjecting naphthalene (1) to our previously reported conditions3b identified traces of an arylated product. Following an initial solvent screen, we then explored the reactivity of larger PAHs and were very excited to observe a highly selective arylation of triphenylene (4). Optimization established that 5 mol% of Pd/C in DME at 100 °C gave 5a in 65% yield with excellent α:β selectivity (>97:3), with no efforts made to exclude air or moisture. The suppression of polyarylation in direct C–H functionalization of PAHs is still an unmet challenge, and accounts in part for the remaining material. X-ray studies confirmed that the arylation occurs at the significantly more hindered position. The scope of the reaction was explored and we were pleased to find that this challenging transformation proceeds effectively with a range of functionalized diaryliodonium salts (Scheme 2). The diversity of functionalities tolerated in the reaction provides multiple options for further derivatization of these novel structures towards more complex π-extended systems. The arylation of related π-extended PAHs was also demonstrated: we observed an unprecedented selectivity for the arylation of anthracene at the C1–H position, as confirmed by X-ray crystallographic analysis. The K-region7d of pyrene and phenanthrene is preferentially arylated to give 7 and 8 respectively. The high yields observed for the mono-arylation of pyrene when compared to the literature precedent are particularly noteworthy considering the value of these structures.7d Selective arylation of the zigzag edge of both acenaphthene and fluoranthene to give 9 and 10 respectively, was also achieved. For the arylation of naphthalene (1) a reduced catalyst loading of 2.5 mol% was employed, with no additional reagents or ligands required to achieve consistently excellent α:β selectivities of >94:6 (Scheme 3). We observed synthetically useful yields with only 2 equivalents of naphthalene, providing practical access to arylated products; previous studies15–17 have typically employed 5–100 equivalents of naphthalene to achieve acceptable yields, which significantly impedes purification. If required, the yield of the reaction can be further increased by using a greater excess of naphthalene. Notably, this simple catalytic system enabled the reaction of electronically diverse iodonium salts, with o-substitution and unsymmetrical iodonium salts containing a cheap, non-transferable arene (2,4,6-triisopropylphenyl) also well tolerated.
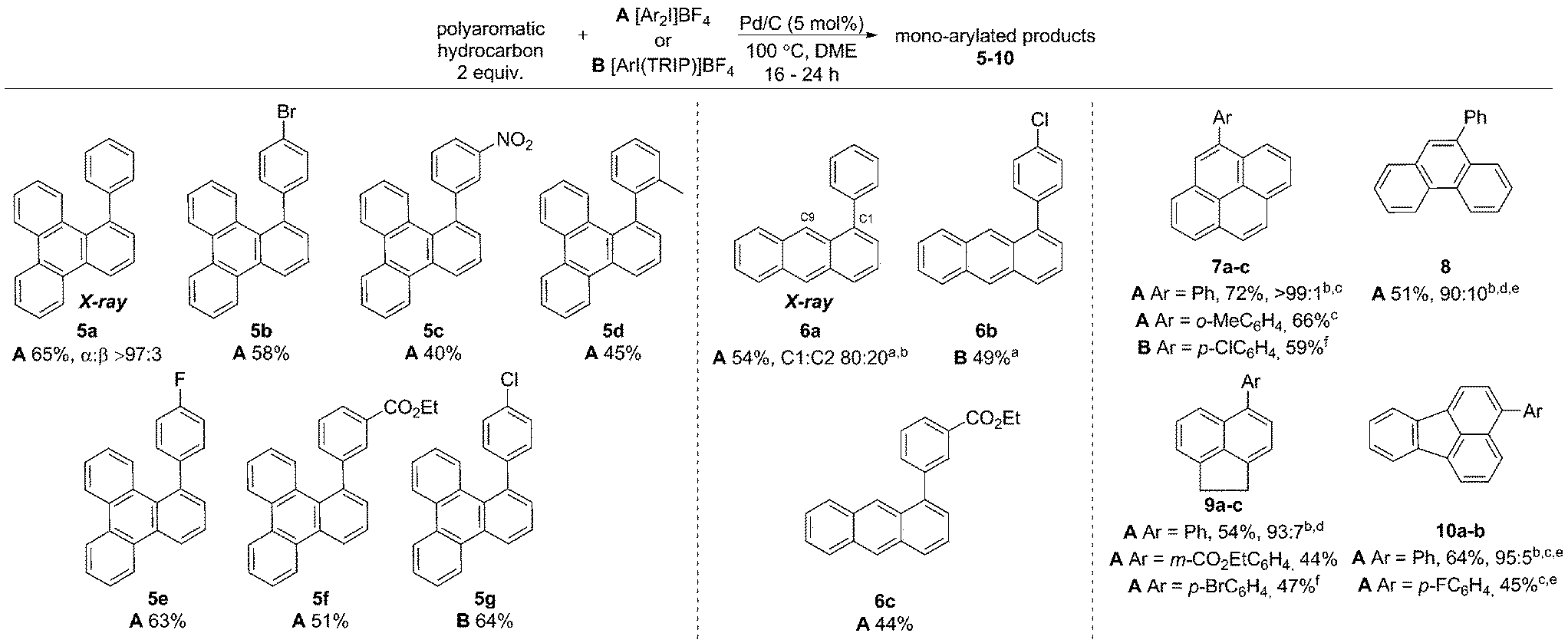 | ||
| Scheme 2 Arylation of larger polyaromatic hydrocarbons. Yields given are isolated yields for the major regioisomer unless noted otherwise. aConditions: anthracene (5 equiv.), Pd/C (2.5 mol%), A at 80 °C and B at 100 °C. bRatio of major regioisomer to any other regioisomers. cConditions: 80 °C, Pd/C (2.5 mol%). dConditions: 70 °C, Pd/C (2.5 mol%). eYield is for major isomer only, though isolated as a mixture. fConditions: 100 °C, Pd/C (2.5 mol%). TRIP is 2,4,6-triisopropylphenyl. | ||
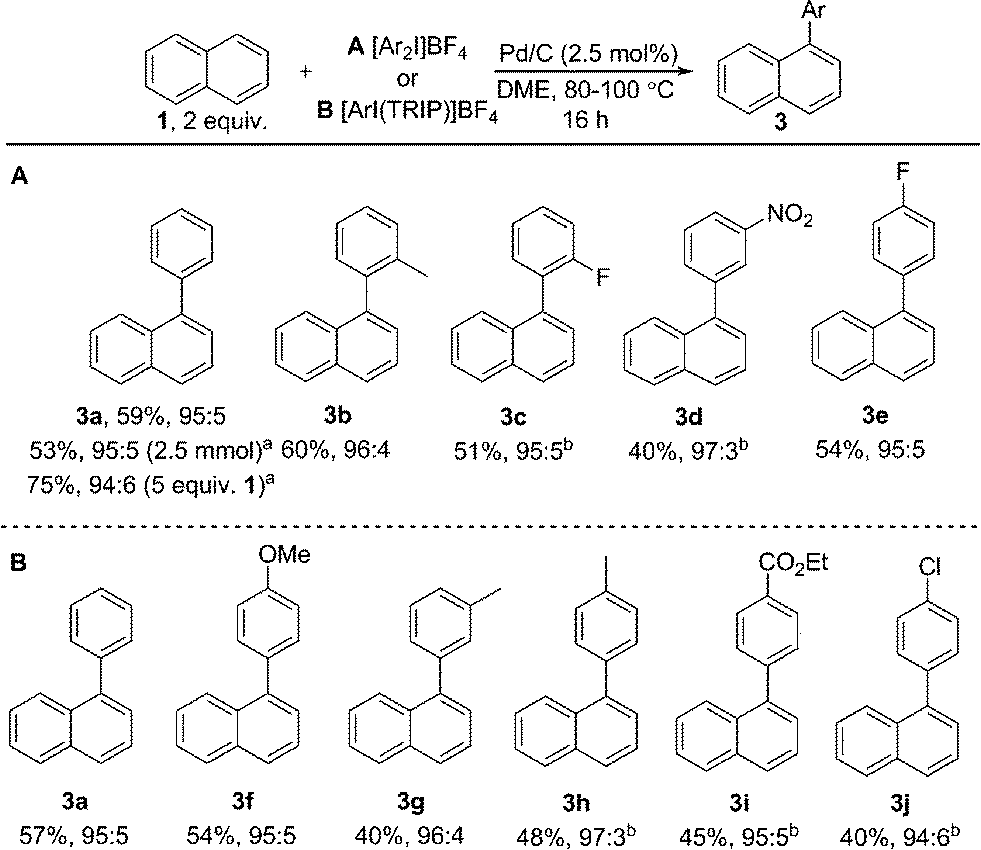 | ||
| Scheme 3 Scope of naphthalene arylation. Yields given are isolated yields for the major regioisomer. All ratios refer to α:β. aGC yield. bReaction temperature 100 °C. TRIP is 2,4,6-triisopropylphenyl. | ||
We have also investigated the reaction of a large number of substituted naphthalenes, though unfortunately we observed either no reactivity or poor selectivity (see ESI† for all examples). Electronically distinct arylated naphthalenes 3a, 3f and 3i were subjected to the reaction conditions a second time to identify any electronic influence on the selectivity of a second arylation, though all substrates yielded a near identical distribution (7:3) of bis-arylated products. Despite significant effort, we were unable to isolate bis-arylated triphenylene or anthracene structures: mono-arylated compounds were subjected to the reaction conditions for a second time resulting in conversion of the starting materials, though gave intractable mixtures of compounds.
Heterogeneity studies
Our preliminary experiments in this study were indicative of a reaction mechanism distinct to that of our previously reported C–H functionalization of heteroaromatic systems using Pd/C and aryliodonium salts.3b Consequently, investigation of the hetero- or homogeneous nature of the active catalytic species and the reaction mechanism were undertaken to gain further insight. Studying the arylation of triphenylene (4) and naphthalene (1) with [Ph2I]BF4 we investigated the nature of the active catalytic species. A heterogeneous catalytic species was indicated by several established methods:1d,4,21(1) A standard Hg-poisoning test resulted in inhibition of the reaction, suggesting the active catalytic species is likely to be heterogeneous in nature.
(2) A hot filtration test indicated that all active catalytic species are removed by the filtration process, which is consistent with a catalytically active heterogeneous species.
(3) Several three-phase tests using polymer-bound naphthalene were undertaken, and in no instance was any arylated product detected on the polymer support.22 Should Pd/C act simply as a reservoir for homogeneous palladium, reaction of the supported naphthalene would be expected. Control experiments indicated that the support does not inhibit reaction.
While these methods for evaluating the nature of the active catalyst have individual limitations, taken as a whole these results are strongly indicative of a catalytically active heterogeneous component. Considering the high temperature and oxidative nature of our reaction conditions it is probable that palladium does leach from the support into solution,1c,1e,2 though does not generate catalytically active homogeneous palladium species. These experiments provide no indication of whether the heterogeneous species is supported on carbon or not.
Preliminary studies of the active catalytic species
For the reaction of naphthalene, kinetic studies indicated a significant induction period and a sigmoidal reaction profile,23 which is consistent with the formation of the active catalytic species in situ (Fig. 1). Leaching of palladium into solution and formation of homogeneous active catalytic species could account for this observation, though is contrary to our studies. Consequently, we investigated the potential origins of this induction period.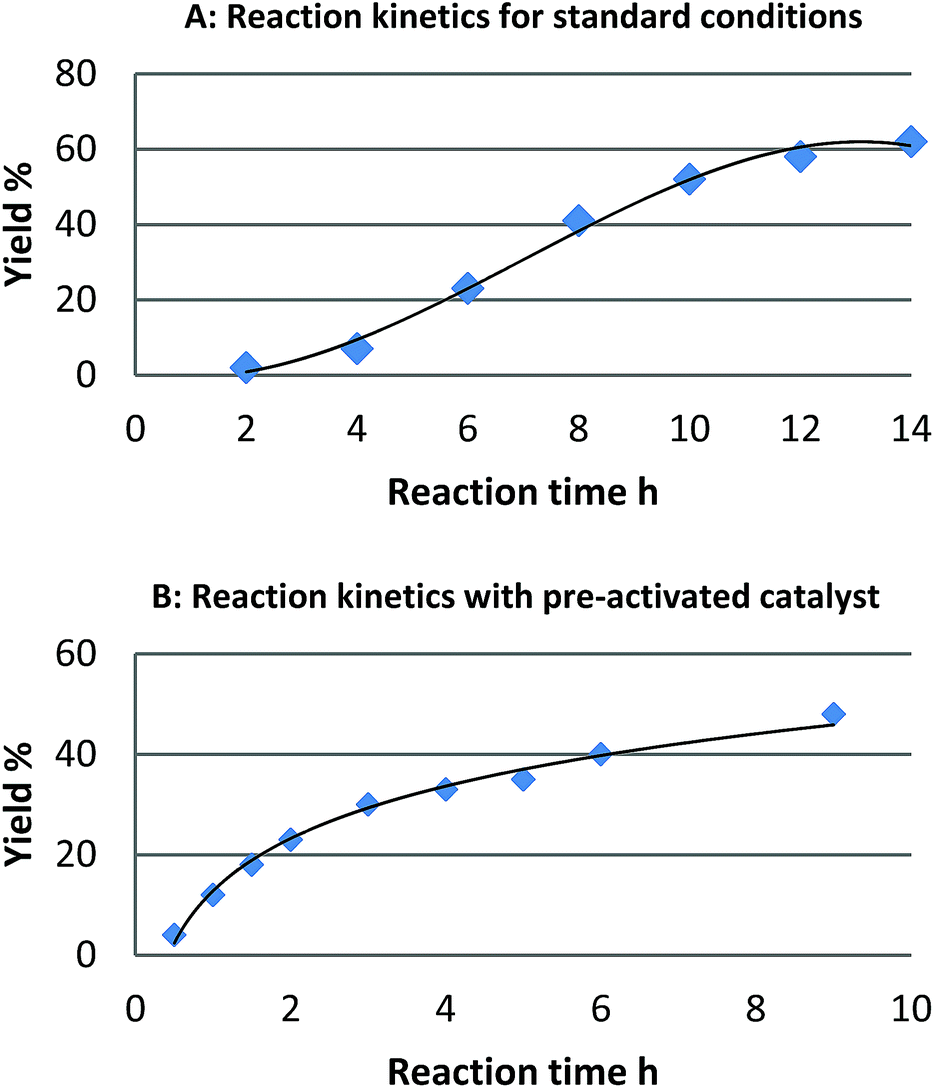 | ||
| Fig. 1 Reaction profile for the reaction of naphthalene (1) and [Ph2I]BF4: (A) standard reaction conditions and (B) after pre-activation of the catalyst with (diacetoxy)iodobenzene. | ||
We found that pretreatment of the catalyst under the standard reaction conditions for 2 h with diaryliodonium salts or (diacetoxy)iodobenzene prior to the introduction of naphthalene, resulted in a significant reduction of the induction period. We hypothesized that pretreatment of the catalyst with these strong oxidants may result in oxidation of Pd(0) to Pd(II) and initiation of a Pd(II)/Pd(IV) catalytic cycle as proposed by Sanford.16 Alternatively, a physical modification of Pd/C resulting in the formation of the active catalyst may also account for the induction period. To investigate the likelihood of a Pd(II)/Pd(IV) catalytic cycle we investigated the reactivity of six homogeneous or heterogeneous Pd(II) sources for the arylation of naphthalene (Table 1).
|
|
||
|---|---|---|
| Catalyst | Yieldb % |
α:βb selectivity |
| a Standard conditions: naphthalene (0.2 mmol), [Ph2I]BF4 (0.5 equiv.), catalyst 5.0 mol%, DME (1 mL), 80 °C, 16 h. b Determined by GC-FID. c 2.5 mol% catalyst. nd is not determined. | ||
| K2PdCl4 | 8 | 95:5 |
| PdCl2(dppe) | 3 | 82:18 |
| PdCl2(PPh3)3 | 9 | 96:4 |
| Pd(OH)2/Cc | 2 | nd |
| Pd(OH)2/Fe3O4c | 12 | 96:4 |
| Pd(OAc)2 | 52 | 93:7 |
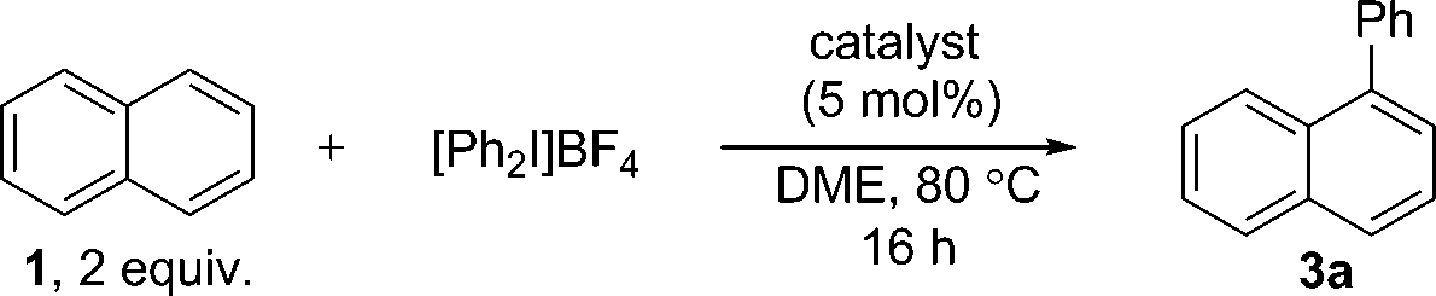
With the exception of Pd(OAc)2 only negligible reactivity was identified, which does not support that the catalytic cycle is initiated by a Pd(II) species. The reactivity observed with Pd(OAc)2 can readily be explained by the well-established thermal reduction of Pd(OAc)2 to form catalytically active heterogeneous Pd(0) nanoparticles.24 Kinetic studies employing Pd(OAc)2 as the catalyst demonstrated an induction period consistent with the generation of nanoparticles, and the formation of heterogeneous material could be observed visually. Further comparative studies (vide infra) support this hypothesis. A preliminary analysis of the results presented so far indicates that the activation of the catalyst with (diacetoxy)iodobenzene or an iodonium salt is unlikely to be due to an oxidation of Pd(0) to Pd(II)/Pd(IV). The formation of a new heterogeneous Pd(0) species following leaching of Pd into solution25 possibly via an Ostwald ripening mechanism,26 or catalyst modification without leaching of the metal could account for the observations reported. The reaction is shown to be first order in palladium, and a reduced induction period with increased catalyst loading is also observed. These results are consistent with the formation of the active catalyst being rate-limiting.
A more extensive catalyst screening was undertaken, importantly highlighting significant differences in the reactivity of several commercial Pd/C sources. Interestingly, we found that in addition to Pd/C and Pd(OAc)2, Pd/Al2O3 proved a proficient catalyst for arylation, and we subsequently undertook a short comparison of all three systems (Table 2).
|
|
|||
|---|---|---|---|
| Pd/C | Pd(OAc)2c | Pd/Al2O3 | |
| a Standard conditions: naphthalene (1) (2 equiv.), iodonium salt (1 equiv.), catalyst (2.5 mol%), DME (0.1 M), 80 °C, 16 h. Yields given are GC yields for the major regioisomer unless noted otherwise. het indicates that the results suggest a heterogeneous active catalytic species. b Isolated yield.. c 5 mol% of catalyst used. nd is not determined. | |||
| Hg poisoning | het | het | het |
| Hot filtration test | het | het | het |
| Standard reaction | 63% | 52% | 64% |
| 5 equiv. 1 | 75% | 50% | 79% |
| Reaction with [p-MeOPhI(TRIP)]BF4 | 54%b | 30% | Trace |
| Reaction with [o-MePh2I]BF4 | 60%b | nd | Trace |
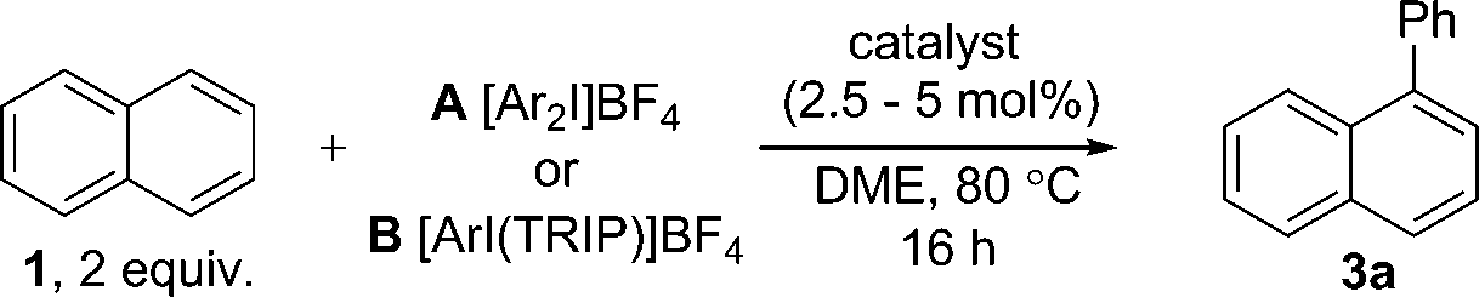
For all catalysts Hg poisoning and hot filtration studies indicate a heterogeneous active catalytic species, supporting the earlier hypothesis that Pd(OAc)2 is reduced to Pd(0) nanoparticles under the reaction conditions. When 5 equivalents of naphthalene are used, both supported systems give a higher yield than in the standard reaction, but with Pd(OAc)2 this is not the case. This indicates that the presence of a support is not crucial for reactivity but it appears to increase the turnover number of the catalyst. Finally, although Pd/Al2O3 proved highly proficient for the standard reaction, only trace reactivity was observed with electron-rich or ortho-substituted arenes. It would appear that while several palladium sources can mediate the basic transformation, Pd/C is typically superior.
Reaction mechanism
While postulating traditional ‘homogeneous’ type reaction mechanisms for processes involving nanoparticles is unlikely to be reflective of the true processes, we sought we investigate the feasibility of a number of reaction pathways:(1) Undirected C–H activation of naphthalene/triphenylene.6y
(2) Oxidation of Pd(0) to Pd(II) and initiation of Pd(II)/Pd(IV) reaction manifold.11
(3) Radical reaction pathways.16
(4) Insertion of an ArPd(II)X species.15a
(5) Electrophilic attack of naphthalene/triphenylene (SEAr/electrophilic palladation).7d,27
We immediately discounted a mechanism that proceeds initially via an undirected C–H activation. Firstly, an unusual inverse kinetic isotope effect (KIE) of 0.54 was determined by parallel initial rate studies of naphthalene and d8-naphthalene, ruling out a rate-limiting C–H activation (Scheme 4).28 Secondly, the reaction is shown to be zero-order in naphthalene precluding its involvement in the rate-determining step. Finally, deuteration experiments employing pre-activated catalyst indicated no deuterium incorporation into naphthalene or triphenylene, suggesting that a Pd(0) catalyzed C–H activation is unlikely.
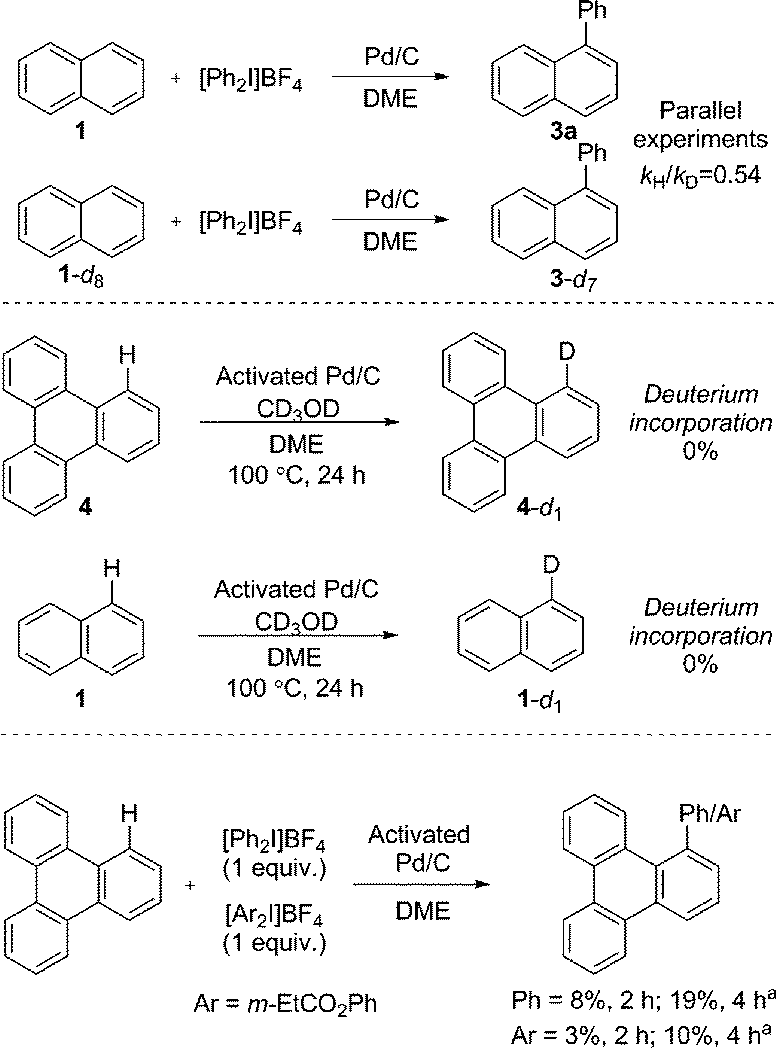 | ||
| Scheme 4 Determination of KIE, deuteration and competition experiments. a GC yield. | ||
 | ||
| Fig. 2 Pd(II)/Pd(IV) reaction manifold proposed by Sanford. | ||
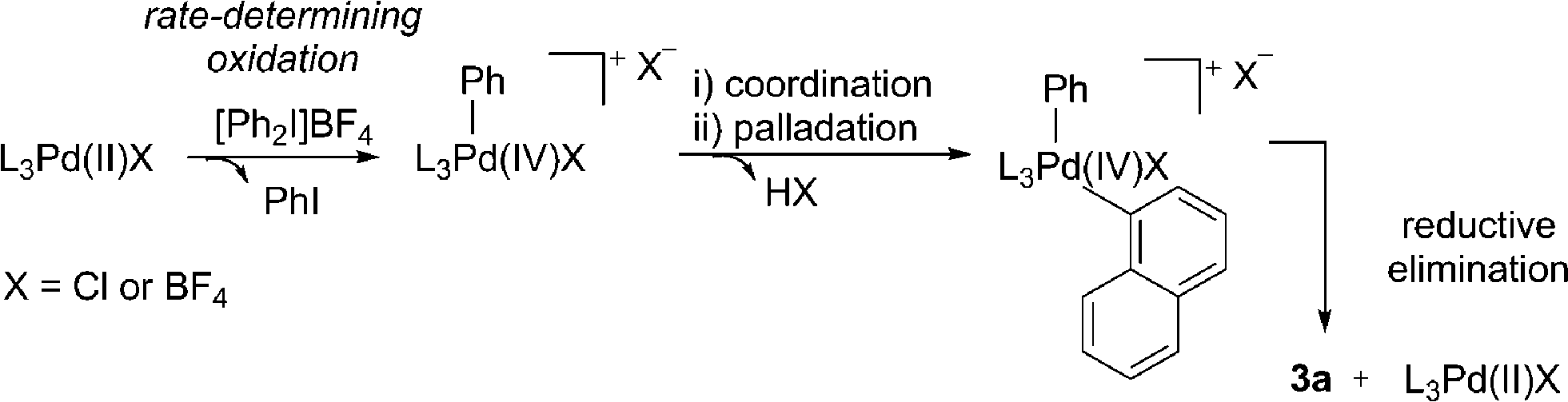
We next considered a Pd(II)/Pd(IV) manifold for the arylation of naphthalene (1) with diaryliodonium salts as proposed by Sanford16 (Fig. 2). Sanford proposed a rate-determining oxidation of Pd(II) to Pd(IV) accounts for the failed reaction of electron-rich iodonium salts. In contrast to her report, our studies show facile reaction of electron-rich iodonium salts, with electron-deficient coupling partners often requiring higher temperatures. A competition experiment also demonstrated more electron-rich iodonium salts react more rapidly, indicating a rate-limiting oxidation of palladium is unlikely (Scheme 4). Furthermore, our described catalyst activation studies suggested entry into a Pd(II)/Pd(IV) catalytic cycle was unlikely. An alternative rate-limiting reductive elimination form Pd(IV) is also improbable.
Radical coupling pathways have been proposed by Rodríguez for related reactions (Fig. 3).15a In our studies we have found that the reaction is inhibited by TEMPO, BHT and BQ which is consistent with a radical component within the reaction mechanism (Fig. 3). Identification of a Ph–BHT adduct further supports this proposition, and suggests that an aryl radical is derived from the iodonium salt. We feel that direct addition of aryl radicals to naphthalene (Fig. 3, A) is unlikely in this instance, as this addition is well precedented to occur with regioselectivities dependent on the electronic nature of the aryl radical, something we do not observe.15a,29 The formation of radical species via hydrogen atom abstraction (Fig. 3, B) is also unlikely when bond dissociation energies (BDE) and/or energetics of the radical formation are considered.30 Firstly, the C–H BDE of benzene is highly comparable to that of naphthalene and therefore would be likely to react under these reaction conditions.30a,b Under our conditions several monoarenes gave only trace reactivity. Secondly, as selectivity of the hydrogen atom abstraction will be governed by the BDE/stability of the formed radical, arylation of anthracene would be expected at the bis-benzylic C9–H in contrast to the C1–H selectivity observed in our study (Scheme 2).30a,c,d The relative BDE for the α and β C–H bonds in naphthalene are still debated, though are very similar and selectivity on this basis seems unlikely.30 Consequently we propose should a radical process be in operation, palladium is nevertheless involved in the bond forming step.
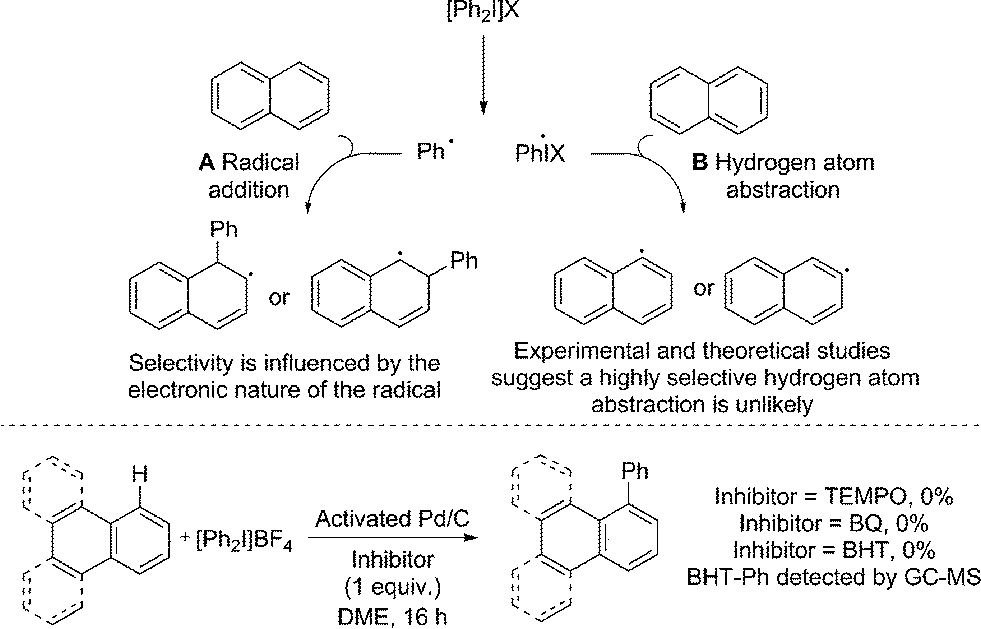 | ||
| Fig. 3 Previously proposed radical pathways for the arylation of naphthalene; radical inhibitor studies. | ||
Based on additional experiments and the data presented so far, a speculative mechanism using naphthalene as a representative substrate is proposed (Fig. 4). As previously noted, it is important to recognize the limitations in attributing homogeneous type reaction mechanisms to heterogeneous processes. We hypothesize that Pd/C acts as a metal reservoir, and following leaching of Pd from the support, catalytically proficient insoluble nanoparticles A are formed. While we cannot rule out modification of the catalyst without leaching of palladium, the product formation when employing Pd(OAc)2 indicates that the support is not essential for reactivity. Favorable interactions between extended π-systems and nanoparticles suggest saturation of the surface to give B is likely, particularly in the presence of a large excess of naphthalene (1). Experimental evidence supports this hypothesis: under the standard reaction conditions PhI is generated throughout the duration of the reaction in proportion to product formation. In contrast, when [Ph2I]BF4 is treated with catalytic Pd/C in the absence of the arene, 80% yield of PhI is identified after only 2 h (Fig. 4). This result suggests that naphthalene inhibits the direct reaction of the diaryliodonium salt and Pd. That the reaction is shown to be zero-order in naphthalene is also consistent with saturation type kinetics associated with heterogeneous catalysis in the presence of a large excess of reagent (pseudo first-order conditions).31
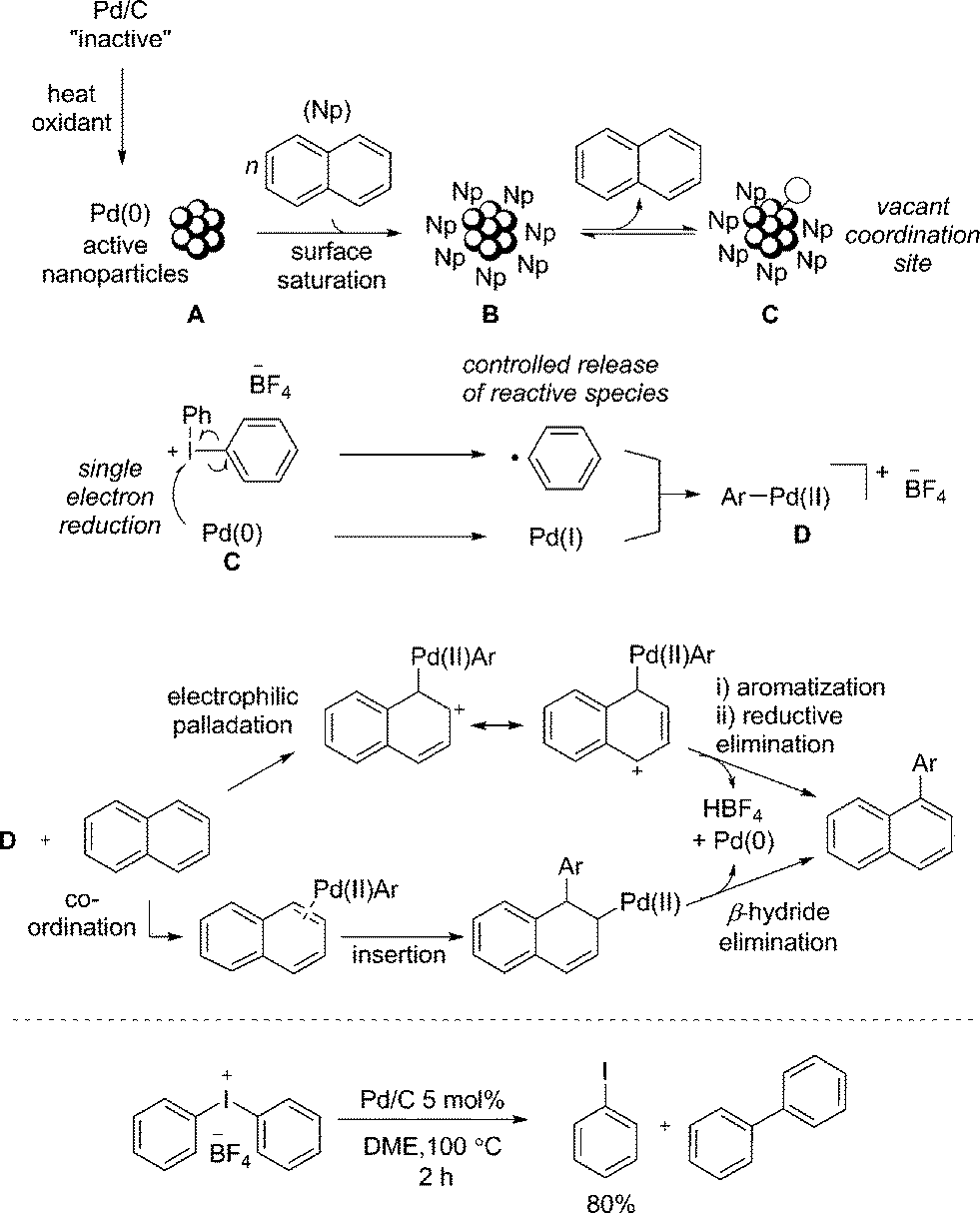 | ||
| Fig. 4 Proposed reaction mechanisms for the arylation of PAHs. Naphthalene is used as a representative substrate. Counterions omitted for clarity when required; treatment of [Ph2I]BF4 with Pd/C in the absence of a coupling partner. | ||
The creation of a vacant coordination via reversible dissociation of naphthalene to give species C, would then enable reaction with the iodonium salt. Single electron reduction of the iodonium salt results in a controlled release of a phenyl radical, and recombination with Pd(I) would generate a formal Pd(II) cationic species D. Single electron processes involving hypervalent iodine(III) reagents are well established32 and the ability of Pd to partake in single electron processes is widely discussed.33 Furthermore, our mechanistic studies are consistent with the formation of an aryl radical, though no evidence for a free radical in solution has been established. A 2-electron oxidative addition process to give D directly from Pd(0) and the iodonium salt cannot be ruled out. As discussed, the presence of polyaromatic species appears to inhibit the reaction of diaryliodonium salt and Pd; an important consequence of this appears to be the controlled release of a reactive phenyl species, helping to minimize the formation of biphenyl. Unlike in the reaction of only [Ph2I]BF4 and Pd/C (Fig. 4), trace amounts of biphenyl are formed under the standard reaction conditions, suggesting a low effective concentration of the reactive phenyl component. Following formation of an Ar–Pd(II) species D, a facile electrophilic palladation followed by re-aromatization and reductive elimination, or an insertion and subsequent β-hydride elimination would provide access to the product. Similar mechanisms have been proposed by Itami for the arylation of pyrene.7d
The selective transfer of the least sterically hindered arene when employing unsymmetrical diaryliodonium salts is consistent with an electrophilic arylation using transition metal catalysis,27 though does not rule out an insertion pathway. Alternatively, the reactivity of naphthalene in contrast to simple mono-arenes could be accounted for when considering an insertion pathway. It is reported that naphthalene has a greater propensity than simple benzenes to form η2-complexes with metals, a prerequisite for activation of the arene towards insertion.34 Finally, as the reaction is shown to be zero-order in naphthalene, neither palladation process would be rate-limiting, though the unusual inverse KIE is yet to be accounted for. Although the fundamental origin is unclear, Perrin has demonstrated decreasing acidity of benzoic acids and phenols for deuterated species relative to the corresponding proteated compounds.35 This would suggest an effect equivalent to increasing the electron-density at the oxygen atom. Consequently, a slightly faster reductive elimination or β-hydride elimination when employing 1-d8 due to the cumulative effect of several deuterium atoms increasing the electron-density at palladium could account for the unusual KIE, should this be the rate-limiting step of the catalytic cycle. Inverse KIEs for reductive eliminations are reported.36
Conclusions
We have reported the first general C–H arylation of triphenylene. This reaction proved applicable to several other PAHs and naphthalene, and without recourse to ligands or other additives provides a practical and highly selective arylation using electronically and sterically diverse aryliodonium salts. Mechanistic studies are consistent with the formation of insoluble nanoparticles following leaching of Pd into solution from Pd/C. The reaction has been demonstrated as likely to proceed via a Pd(0)/Pd(II) manifold rather than the commonly proposed Pd(II)/Pd(IV) often cited for similar transformations.Acknowledgements
Generous financial support by the Deutsche Forschungsgemeinschaft (Leibniz award; SFB 858) and the Fonds der Chemischen Industrie (R.H.) is gratefully acknowledged. We thank Steffen Greßies for preliminary experiments and Theresa Olyschläger for experimental support. We also greatly thank Constantin G. Daniliuc for X-ray crystallographic analysis.Notes and references
- (a) F.-X. Felpin, T. Ayad and S. Mitra, Eur. J. Org. Chem., 2006, 2679 CrossRef CAS; (b) M. Pal, Synlett, 2009, 2896 CrossRef CAS PubMed; (c) M. Seki, Synthesis, 2006, 2975 CrossRef CAS PubMed; (d) L. Yin and J. Liebscher, Chem. Rev., 2007, 107, 133 CrossRef CAS PubMed; (e) F.-X. Felpin, Synlett, 2014, 25, 1055 CrossRef PubMed.
- (a) H.-U. Blaser, A. Indolese, A. Schnyder, H. Steiner and M. Studer, J. Mol. Catal. A: Chem., 2001, 173, 3 CrossRef CAS; (b) M. Genelot, V. Dufaud and L. Djakovitch, Adv. Synth. Catal., 2013, 355, 2604 CrossRef CAS; (c) B. H. Lipshutz, S. Tasler, W. Chrisman, B. Spliethoff and B. Tesche, J. Org. Chem., 2003, 68, 1177 CrossRef CAS PubMed; (d) I. W. Davies, L. Matty, D. L. Hughes and P. J. Reider, J. Am. Chem. Soc., 2001, 123, 10139 CrossRef CAS.
- (a) D.-T. D. Tang, K. D. Collins and F. Glorius, J. Am. Chem. Soc., 2013, 135, 7450 CrossRef CAS PubMed; (b) D.-T. D. Tang, K. D. Collins, J. B. Ernst and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 1809 CrossRef CAS PubMed.
- L. Huang, P. K. Wong, J. Tan, T. P. Ang and Z. Wang, J. Phys. Chem. C, 2009, 113, 10120 CAS.
- M. Lamblin, L. Nassar-Hardy, J.-C. Hierso, E. Fouquet and F.-X. Felpin, Adv. Synth. Catal., 2010, 352, 33 CrossRef CAS.
- Recent reviews on C–H activation: (a) R. Jazzar, J. Hitce, A. Renaudat, A. J. Sofack-Kreutzer and O. Baudoin, Chem.–Eur. J., 2010, 16, 2654 CrossRef CAS PubMed; (b) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (c) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (d) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (e) S. H. Cho, J. Y. Kim, J. Kwak and S. Chang, Chem. Soc. Rev., 2011, 40, 5068 RSC; (f) J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (g) O. Baudoin, Chem. Soc. Rev., 2011, 40, 4902 RSC; (h) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (i) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (j) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (k) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (l) B.-J. Li and Z.-J. Shi, Chem. Soc. Rev., 2012, 41, 5588 RSC; (m) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (n) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Chem. Rev., 2012, 112, 5879 CrossRef CAS PubMed; (o) Z. Shi, C. Zhang, C. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (p) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (q) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (r) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (s) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (t) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (u) K. M. Engle and J.-Q. Yu, J. Org. Chem., 2013, 78, 8927 CrossRef CAS PubMed; (v) R. Rossi, F. Bellina, M. Lessi and C. Manzini, Adv. Synth. Catal., 2014, 356, 17 CrossRef CAS; (w) S. De Sarkar, W. Liu, S. I. Kozhushkov and L. Ackermann, Adv. Synth. Catal., 2014, 356, 1461 CrossRef CAS; (x) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS ; for a specific review on non-chelate assisted C–H activation, see ; (y) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed.
- (a) K. Itami, Pure Appl. Chem., 2012, 84, 907 CrossRef CAS PubMed; (b) T. N. Hoheisel, S. Schrettl, R. Szilluweit and H. Frauenrath, Angew. Chem., Int. Ed., 2010, 49, 6496 CrossRef CAS PubMed; (c) D. Wei and Y. Liu, Adv. Mater., 2010, 22, 3225 CrossRef CAS PubMed; (d) K. Mochida, K. Kawasumi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2011, 133, 10716 CrossRef CAS PubMed . The K-region is defined by Itami as an exposed outer π-bond of PAHs. Although aromatic, these outer π-bonds possess partial olefinic character and are less stabilized, and therefore more reactive, than the other π-bonds within the PAH.
- (a) J. Cai, P. Ruffieux, R. Jaafar, M. Bieri, T. Braun, S. Blankenburg, M. Muoth, A. P. Seitsonen, M. Saleh, X. Feng, K. Müllen and R. Fasel, Nature, 2010, 466, 470 CrossRef CAS PubMed; (b) X. Feng, W. Pisula and K. Müllen, Pure Appl. Chem., 2009, 81, 2203 CrossRef CAS; (c) E. H. Fort and L. T. Scott, Angew. Chem., Int. Ed., 2010, 49, 6626 CrossRef CAS PubMed; (d) R. E. Smalley, Y. Li, V. C. Moore, B. K. Price, R. Colorado, H. K. Schmidt, R. H. Hauge, A. R. Barron and J. M. Tour, J. Am. Chem. Soc., 2006, 128, 15824 CrossRef CAS PubMed.
- (a) M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed; (b) A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed; (c) J. Wu, W. Pisula and K. Müllen, Chem. Rev., 2007, 107, 718 CrossRef CAS PubMed.
- H. Kawai, Y. Kobayashi, S. Oi and Y. Inoue, Chem. Commun., 2008, 1464 RSC.
- S.-D. Yang, C.-L. Sun, Z. Fang, B.-J. Li, Y.-Z. Li and Z.-J. Shi, Angew. Chem., Int. Ed., 2008, 47, 1473 CrossRef CAS PubMed.
- K. Kawasumi, K. Mochida, T. Kajino, Y. Segawa and K. Itami, Org. Lett., 2012, 14, 418 CrossRef CAS PubMed.
- Q. Zhang, K. Kawasumi, Y. Segawa, K. Itami and L. T. Scott, J. Am. Chem. Soc., 2012, 134, 15664 CrossRef CAS PubMed.
- K. Kawasumi, K. Mochida, Y. Segawa and K. Itami, Tetrahedron, 2013, 69, 4371 CrossRef CAS PubMed.
- (a) S. Castro, J. J. Fernández, R. Vicente, F. J. Fañanás and F. Rodríguez, Chem. Commun., 2012, 48, 9089 RSC; (b) K. Funaki, H. Kawai, T. Sato and S. Oi, Chem. Lett., 2011, 40, 1050 CrossRef CAS; (c) O. Kobayashi, D. Uraguchi and T. Yamakawa, Org. Lett., 2009, 11, 2679 CrossRef CAS PubMed; (d) R. Li, L. Jiang and W. Lu, Organometallics, 2006, 25, 5973 CrossRef CAS; (e) W. Liu, H. Cao and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 2004 CrossRef CAS PubMed; (f) T.-H. Park, A. J. Hickman, K. Koh, S. Martin, A. G. Wong-Foy, M. S. Sanford and A. J. Matzger, J. Am. Chem. Soc., 2011, 133, 20138 CrossRef CAS PubMed; (g) C. Qin and W. Lu, J. Org. Chem., 2008, 73, 7424 CrossRef CAS PubMed; (h) T. E. Storr and M. F. Greaney, Org. Lett., 2013, 15, 1410 CrossRef CAS PubMed; (i) N. Yamaoka, K. Sumida, I. Itani, H. Kubo, Y. Ohnishi, S. Sekiguchi, T. Dohi and Y. Kita, Chem.–Eur. J., 2013, 19, 15004 CrossRef CAS PubMed.
- A. J. Hickman and M. S. Sanford, ACS Catal., 2011, 1, 170 CrossRef CAS . For an impressive study on the selective β-arylation of naphthalene with Na2PtCl4 see: A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710 CrossRef PubMed.
- Limited examples of high selectivity are also reported by Lu, ref. 15d, though yields are poor (16–32%).
- Iodonium salts were prepared according to the methods developed by Olofsson, Gaunt and Reismann: (a) M. Bielawski, D. Aili and B. Olofsson, J. Org. Chem., 2008, 73, 4602 CrossRef CAS PubMed; (b) M. Bielawski, J. Malmgren, L. M. Pardo, Y. Wikmark and B. Olofsson, ChemistryOpen, 2014, 3, 19 CrossRef CAS PubMed; (c) M. Bielawski and B. Olofsson, Chem. Commun., 2007, 2521 RSC; (d) M. Bielawski, M. Zhu and B. Olofsson, Adv. Synth. Catal., 2007, 349, 2610 CrossRef CAS; (e) M. E. Kieffer, K. V. Chuang and S. E. Reisman, Chem. Sci., 2012, 3, 3170 RSC; (f) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed.
- For a directed C–H functionalization of arenes using Pd–polyoxometalate catalysis see: (a) L. L. Chng, J. Zhang, J. Yang, M. Amoura and J. Y. Ying, Adv. Synth. Catal., 2011, 353, 2988 CrossRef CAS ; arene functionalization employing Pd/C was recently reported, though studies indicated a homogeneous active catalytic species ; (b) L. L. Chng, J. Yang, Y. Wei and J. Y. Ying, Chem. Commun., 2014, 50, 9049 RSC . For heterogenous Pd-catalyzed C–H functionalization of heteroarenes see ref. 3 and ; (c) S. Mukhopadhyay, G. Rothenberg, D. Gitis, M. Baidossi, D. E. Ponde and Y. Sasson, J. Chem. Soc., Perkin Trans. 2, 2000, 1809 RSC; (d) L. M. Neal and H. E. Hagelin-Weaver, J. Mol. Catal. A: Chem., 2008, 284, 141 CrossRef CAS PubMed; (e) L. M. Neal, S. D. Jones, M. L. Everett, G. B. Hoflund and H. E. Hagelin-Weaver, J. Mol. Catal. A: Chem., 2010, 325, 25 CrossRef CAS PubMed; (f) D. Saha, L. Adak and B. C. Ranu, Tetrahedron Lett., 2010, 51, 5624 CrossRef CAS PubMed; (g) Y. M. A. Yamada, Y. Yuyama, T. Sato, S. Fujikawa and Y. Uozumi, Angew. Chem., Int. Ed., 2014, 53, 127 CrossRef CAS PubMed; (h) V. A. Zinovyeva, M. A. Vorotyntsev, I. Bezverkhyy, D. Chaumont and J.-C. Hierso, Adv. Funct. Mater., 2011, 21, 1064 CrossRef CAS ; for heterogeneous Pd catalyzed C–H functionalization of carbonyl C–H bonds see ; (i) Q.-L. Zhu, N. Tsumori and Q. Xu, Chem. Sci., 2014, 5, 195 RSC; (j) L. Adak, S. Bhadra and B. C. Ranu, Tetrahedron Lett., 2010, 51, 3811 CrossRef CAS PubMed ; for heterogeneous Ni-catalyzed C–H functionalization of heteroarenes see ; (k) N. T. S. Phan, C. K. Nguyen, T. T. Nguyen and T. Truong, Catal. Sci. Technol., 2014, 4, 369 RSC ; for a single example of arene C–H functionalization with Raney®-Ni (no investigation of heterogeneity) see; (l) R. Zhang, C.-X. Miao, S. Wang, C. Xia and W. Sun, ChemCatChem, 2012, 4, 192 CrossRef CAS ; for selected reviews on the application of nanocatalysts see ; (m) V. Olshettiwar, R. Luque, A. Fihri, H. Zhu, M. Bouhrara and J.-M. Basset, Chem. Rev., 2011, 111, 3036 CrossRef PubMed; (n) K. V. S. Ranganath and F. Glorius, Catal. Sci. Technol., 2011, 1, 13 RSC; (o) A. Schätz, O. Reiser and W. J. Stark, Chem.–Eur. J., 2010, 16, 8950 CrossRef PubMed; (p) S. Shylesh, V. Schünemann and W. R. Thiel, Angew. Chem., Int. Ed., 2010, 49, 3428 CrossRef CAS PubMed; (q) A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed; (r) D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed ; for a recent example see ; (s) E. Gross, J. H.-C. Liu, F. D. Toste and G. A. Somorjai, Nat. Chem., 2012, 4, 947 CrossRef CAS PubMed.
- A single report for functionalization of the more reactive C9–H bond is reported by Kita: see ref. 15i.
- (a) R. H. Crabtree, Chem. Rev., 2012, 112, 1536 CrossRef CAS PubMed; (b) M. Pagliaro, V. Pandarus, R. Ciriminna, F. Béland and P. D. Carà, ChemCatChem, 2012, 4, 432 CrossRef CAS; (c) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS.
- Polymer-supported triphenylene is not readily synthetically accessible.
- An induction period of <15 min in the case of triphenylene is attributed to the more rapid formation of the active catalyst at higher temperature/concentration. The induction period for the reaction of naphthalene was significantly reduced under identical conditions.
- A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed . Ligand stabilization of other Pd catalysts minimizes nanoparticle formation.
- For an excellent recent study, and related works, on the formation of soluble Pd nanoparticles from a homogeneous palladium precatalyst see: (a) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS PubMed; (b) T. Diao, D. Pun and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8205 CrossRef CAS PubMed; (c) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed; (d) Y. Izawa, D. Pun and S. S. Stahl, Science, 2011, 333, 209 CrossRef CAS PubMed.
- (a) W. Ostwald, Z. Phys. Chem., 1900, 34, 495 Search PubMed; (b) J. Alemán, A. V. Chadwick, J. He, M. Hess, K. Horie, R. G. Jones, P. Kratochvil, I. Meisel, I. Mita, G. Moad, S. Penczek, R. F. T. Stepto and R. G. Jones, Pure Appl. Chem., 2007, 79, 1801 CrossRef.
- (a) M. Ochiai, T. Shu, T. Nagaoka and Y. Kitagawa, J. Org. Chem., 1997, 62, 2130 CrossRef CAS PubMed; (b) B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS PubMed; (c) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458 CrossRef CAS PubMed; (d) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260 CrossRef CAS PubMed; (e) T. B. Petersen, R. Khan and B. Olofsson, Org. Lett., 2011, 13, 3462 CrossRef CAS PubMed; (f) S. R. Neufeldt and M. S. Sanford, Adv. Synth. Catal., 2012, 354, 3517 CrossRef CAS PubMed.
- E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed.
- B. A. Marshall and W. A. Waters, J. Chem. Soc., 1959, 381 RSC.
- (a) K. Hemelsoet, V. Van Speybroeck, D. Moran, G. Marin, L. Radom and M. Waroquier, J. Phys. Chem. A, 2006, 110, 13624 CrossRef CAS PubMed; (b) D. R. Reed and S. R. Kass, J. Mass Spectrom., 2000, 35, 534 CrossRef CAS; (c) R. H. Chen, S. A. Kafafi and S. E. Stein, J. Am. Chem. Soc., 1989, 111, 1418 CrossRef CAS; (d) K. Hemelsoet, V. Van Speybroeck and M. Waroquier, J. Phys. Chem. A, 2008, 112, 13566 CrossRef CAS PubMed.
- R. L. Augustine, Heterogeneous catalysis for the synthetic chemist, Marcel Dekker, New York, 1996 Search PubMed.
- (a) P. Stang and V. V. Zhdankin, Chem. Rev., 1996, 96, 1123 CrossRef CAS PubMed; (b) T. Wirth, M. Ochiai, A. Varvgolis, V. V. Zhdankin, G. F. Koser, H. Tohma and Y. Kita, Topics in Current Chemistry: Hypervalent Iodine Chemistry. Modern Developments in Organic Synthesis, ed. T. Wirth, Springer-Verlag, Berlin, 2002 Search PubMed.
- (a) D. C. Powers and T. Ritter, Top. Organomet. Chem., 2011, 503, 129 CrossRef . For a recently proposed single electron transfer from Pd(0) see ; (b) J.-W. Gu, W.-H. Guo and X. Zhang, Org. Chem. Front., 2015, 2, 38 RSC . For a very relevant proposed single electron reduction of aryl chlorides with Pd/C see ; (c) S. Mukhopadhyay, G. Rothenberg, H. Wiener and Y. Sasson, Tetrahedron, 1999, 55, 14763 CrossRef CAS.
- S. T. Belt, L. Dong, S. B. Duckett, W. D. Jones, M. G. Partridge and R. N. Perutz, J. Chem. Soc., Chem. Commun., 1991, 266 RSC.
- (a) C. L. Perrin and A. Flach, Angew. Chem., Int. Ed., 2011, 50, 7674 CrossRef CAS PubMed; (b) C. L. Perrin and Y. Dong, J. Am. Chem. Soc., 2007, 129, 4490 CrossRef CAS PubMed.
- (a) D. G. Churchill, K. E. Janak, J. S. Wittenberg and G. Parkin, J. Am. Chem. Soc., 2003, 125, 1403 CrossRef CAS PubMed; (b) W. D. Jones, Acc. Chem. Res., 2003, 36, 140 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1013416 and 1013417. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03051f |
| ‡ Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |