
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comparative study of the coordination behavior of cyclo-P5 and cyclo-As5 ligand complexes towards the trinuclear Lewis acid complex (perfluoro-ortho-phenylene)mercury†
Martin
Fleischmann
a,
James S.
Jones
b,
François P.
Gabbaï
b and
Manfred
Scheer
*a
aInstitut für Anorganische Chemie, Universität Regensburg, 93040 Regensburg, Germany. E-mail: manfred.scheer@chemie.uni-regensburg.de; Fax: +49-941-943-4439
bDepartment of Chemistry, Texas A&M University, College Station, Texas 77843-3255, USA
First published on 1st October 2014
Abstract
Reactions of the cyclo-E5 sandwich complexes [Cp*Fe(η5-P5)] (1) and [Cp*Fe(η5-As5)] (2) with the planar Lewis acid trimeric (perfluoro-ortho-phenylene)mercury [(o-C6F4Hg)3] (3) afford compounds that show distinctly different assemblies in the solid state. The phosphorus containing ligand 1 forms dimeric coordination units with two molecules of 3, with one P atom of each cyclo-P5 ligand positioned in close proximity to the center of a molecule of 3. In contrast to the coordination behavior of 1, the arsenic analog 2 shows simultaneous interaction of three As atoms with the Hg atoms of 3. A DFT study and subsequent AIM analyses of the products suggest that electrostatic forces are prevalent over donor–acceptor interactions in these adducts, and may play a role in the differences in the observed coordination behavior. Subsequently, a series of [CpRFe(η5-P5)] (CpR = C5H5−ntBun, n = 1–3, 6a–c) sandwich complexes was prepared and also reacted with [(o-C6F4Hg)3]. In the solid state the obtained products 7a–c with increasing steric demand of the CpR ligands show no significant change in their assembly compared to the Cp* analog 4. All of the products were characterized by single crystal X-ray structure analysis, mass spectrometry and elemental analysis as well as NMR spectroscopy and IR spectrometry.
Introduction
The research area of substituent-free group 15 element ligands in the coordination sphere of transition metal complexes has shown to be a prosperous field in chemistry.1–5 Some of these complexes possess planar E3, E4, E5 and E6 rings. From their appealing symmetry, to the lively discussion of their possible aromaticity,6–10 these main group ligands induce a fascination to chemists on their own. Among these, the ferrocene analogous sandwich complexes [Cp*Fe(η5-E5)] (E = P (1), E = As (2))11,12 bearing a planar E5 ring as an end-deck are of special interest as ligands in supramolecular coordination chemistry, since the cyclo-E5 moieties show a large variety of coordination modes depending on the nature of the Lewis acid used. While reactions of the cyclo-P5 complex 1 with strongly coordinating CuI halides lead to an abundance of coordination polymers,13,14 and also spherical aggregates,15–19 the As analogue 2 has so far lead only to the isolation of coordination polymers.20 In these products the P atoms are mainly σ coordinating the Cu centers via their lone pairs while the As5 ring mainly shows π coordination via As–As bonds. The reaction of 1 with Ag+ ions under weakly coordinating conditions affords a soluble one-dimensional coordination polymer.21 Recently, we were able to show that both E5 complexes 1 and 2 reveal a similar η5 coordination of the E5 end deck to the group 13 cations Tl+ and In+.22,23 Since investigations of the reactivity of cyclo-P5 and cyclo-As5 complexes including a direct comparison are rare, it seems worthwhile to analyze their coordination chemistry towards the unusual Lewis acid trimeric (perfluoro-ortho-phenylene)mercury [(o-C6F4Hg)3] (3).24The latter is a planar, electron deficient molecule containing three sterically available Hg atoms in close proximity. Compound 3 forms weak Lewis acid/base adducts with a large variety of O, N and S donor Lewis bases as well as some anions (Scheme 1 a).25,26 Additionally, it readily builds up alternating binary stacks with different electron rich aromatic hydrocarbons27–32 and forms double sandwich complexes with the metallocenes [Cp2Ni] and [Cp2Fe] (Scheme 1b).33 Accordingly, we reported the reaction of 3 with the triple decker complex [(CpMo)2(μ,η6:η6 P6)] bearing two Cp rings and a cyclo P6 middle deck.34 In this case, the obtained products show a one dimensional polymeric structure which is based on weak P–Hg interactions and no Hg–Cp interactions are observed.
 | ||
| Scheme 1 (a) The planar Lewis acid 3 can simultaneously interact through all three Hg atoms with Lewis bases; (b) double-sandwich complexes built from 3 and the simple metallocenes [Cp2Fe] and [Cp2Ni]. | ||
The presented results raise the question of how the ferrocene analog cyclo E5 complexes 1 and 2 will interact with the planar Lewis acid 3. Will they form Lewis acid/base adducts via the lone pairs of the group 15 elements or will they show a π interaction of the aromatic E5 ligands, comparable to pure ferrocene? To address this question we reacted the cyclo-E5 complexes 1 and 2 with [(o-C6F4Hg)3] (3) in CH2Cl2 and subsequently determined the solid state structure of the products. To gain further insight into the Hg–E interactions, the electrostatic potential surfaces of the complexes 1–3 were obtained from DFT calculations. Additionally an atoms in molecules (AIM) analysis was performed on the experimentally determined geometries. To investigate the impact of sterical demand on these compounds, we prepared a series of cyclo-P5 sandwich complexes [CpRFe(η5-P5)] (CpR = C5H5−ntBun, n = 1–3, 6a–c) with increasing sizes of the Cp ligands and subsequently reacted them with compound 3.‡ The resulting adducts were analysed by X-ray crystallography and a Hirshfeld surface analysis was performed to better compare the involved intermolecular contacts in the solid state.
Results and discussion
Synthesis of the sandwich complexes [CpRFe(η5-P5)] (CpR = C5H5−ntBun, n = 1–3, 6a–c)
For the present work all three complexes were prepared by the thermolysis of [CpRFe(CO)2]2 with P4 in decalin. Chromatographic workup afforded the pure compounds as dark green solids.Synthesis of the compounds 4, 5, 7a–c
Since [(o-C6F4Hg)3] (3) forms Lewis acid/base adducts with donor solvents like THF or MeCN, the syntheses were all conducted in CH2Cl2 to prevent any competition between the En ligand complexes and the solvent molecules. Nevertheless, in some of the reactions we could isolate two solvates of [(o-C6F4Hg)3] containing only CH2Cl2 (see ESI†). For the current study, the En ligand complexes were combined with a stoichiometric (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) amount of [(o-C6F4Hg)3] and the mixture was dissolved in pure CH2Cl2. After filtration, the solvent was evaporated to the limit of solubility. The supersaturated solution was stored at +4 °C or −30 °C which afforded crystals of the compounds 4, 5 and 7a–c in a matter of several hours to some days.
:1) amount of [(o-C6F4Hg)3] and the mixture was dissolved in pure CH2Cl2. After filtration, the solvent was evaporated to the limit of solubility. The supersaturated solution was stored at +4 °C or −30 °C which afforded crystals of the compounds 4, 5 and 7a–c in a matter of several hours to some days.
General considerations
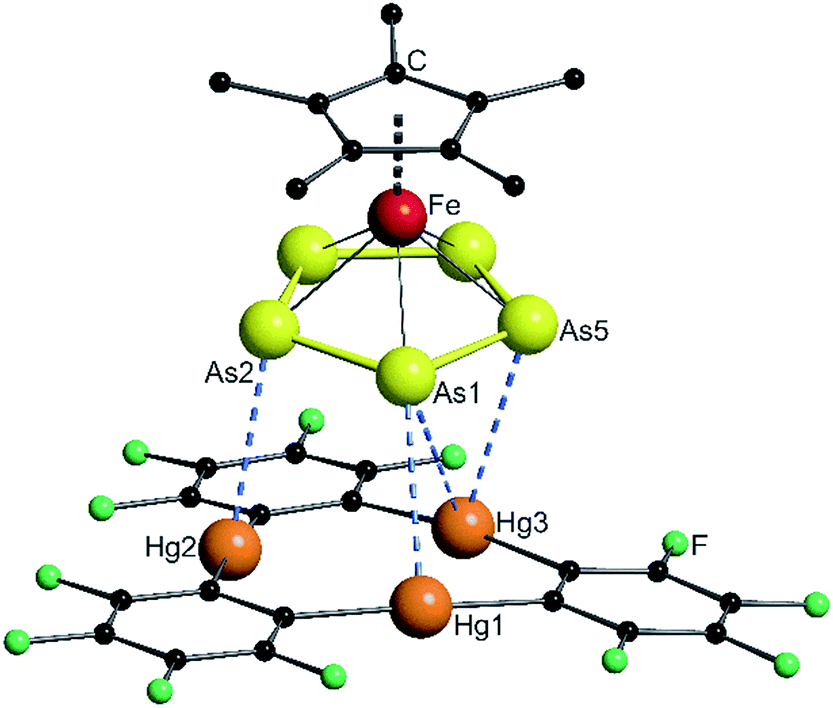
The solid state structures of the formed assemblies are based on weak interactions of the Hg atoms of [(o-C6F4Hg)3] and the phosphorus or arsenic atoms of the sandwich complexes [Cp*Fe(η5-E5)]. The van der Waals (vdW) radius of Hg in different compounds is discussed in the literature with reported values ranging from 1.7 Å up to 2.2 Å.35–38 In the following discussion the shortest value of 1.7 Å was taken as a reference. Therefore, Hg–E distances that are within the sum of the vdW radii39 of 3.6 Å for E = P or 3.7 Å for E = As are highlighted by fragmented blue lines in the following Fig. 1, 2 and 6. | ||
| Fig. 1 Solid state structure of 4; selected bond lengths [Å] and angles [°]: Hg1–P1 3.2878(9), Hg2–P1 3.3592(9), Hg3–P3 3.5281(11), Hg1′–P3 3.5265(10), angle P5-plane – Hg3-plane 62.29(2). | ||
 | ||
| Fig. 2 Solid state structure of 5; selected bond lengths [Å] and angles [°]: Hg1–As1 3.4059(4), Hg2–As2 3.3014(4), Hg3–As1 3.6325(5), Hg3–As5 3.4201(5), angle As5-plane – Hg3-plane 10.68(2). | ||
When the starting compounds 1 and 3 are dissolved in CH2Cl2, the solution shows the dark green color of the pure complex 1. The crystals which were obtained by storing a concentrated solution at −30 °C are pleochromic showing a green to brown color. Compound 4 crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The solid state structure is depicted in Fig. 1.
. The solid state structure is depicted in Fig. 1.
In 4 the P5 ring of 1 is approaching the center of the three Hg atoms of 3 with the phosphorus atom P1 and on the other side, the atom P3 is coordinating to the second Hg3 moiety. The P–P bond lengths are very uniform with an average value of 2.111(4) Å, which is the same as in the starting compound 1 (2.120(5) Å).21 The angle enclosed by the cyclo-P5 plane and the Hg3 plane constitutes 62.29(2)°. The observed assembly resembles the weak Lewis acid/base adducts that are formed from 3 with several Lewis bases and significantly differs from a cofacial arrangement that was found for the double-sandwich complexes formed by [Cp2Fe] and 3.33 The closest Hg–P distance Hg1–P1 of 3.2878(9) Å is a bit longer than the closest Hg–P contact (3.195(3) Å) found in the polymeric chains of [(o-C6F4Hg)·{(CpMo)2(μ,η6:η6-P6)}]n but is comparable with other observed Hg–P distances of these compounds.34 The shortest intermolecular P⋯P distance is 3.9443(13) Å and all of the others lie above 4 Å. In summary, the best description of the solid state structure of 4 is the enclosure of two cyclo-P5 sandwich complexes by two planar molecules of 3 held together by weak Hg⋯P interactions.
In CD2Cl2 solution at room temperature 4 shows a singlet in both the 1H NMR spectrum and the 31P{1H} NMR spectrum. The signal is only shifted 0.04 ppm upfield in the case of the methyl protons and 2.5 ppm downfield in the case of the phosphorus atoms compared to the free complex 1. When cooled to 193 K, these shifts increase to 0.13 ppm upfield for the 1H and 7.6 ppm downfield for the 31P nuclei. In all of the experiments we could not resolve any coupling to the NMR active 199Hg (I = 1/2, 16.84% natural abundance) or 201Hg (I = 3/2, 13.22% natural abundance) nuclei. The 19F NMR spectrum shows two multiplets that correspond to the fluorine atoms of 3 in the ortho and para positions to the Hg atoms.40 The mass spectrum (FD or ESI†) of 4 shows no adducts in the gas phase. Only the starting materials 1 and 3 can be detected. Thus, the small differences of the chemical shifts and the absence of any coupling in the NMR spectra as well as the absence of any product peaks in the mass spectrum are in good agreement with the expected weak Hg⋯P interactions.
During the further investigation we also added the cyclo-As5 complex 2 to the Lewis acid 3. The brown solution of both compounds in CH2Cl2 could easily be distinguished from the olive green color of the pure sandwich complex 2. The obtained crystals of compound 5 were a medium brown color. The solid state structure is shown in Fig. 2.
Compound 5 crystallizes in the triclinic space group P. The As–As bond lengths are very uniform with an average value of 2.326(6) Å which is the same as found in the starting compound 2 (2.327(6) Å).23 The assembly of the As5 ring significantly differs from the P5 ring in 4. The angle enclosed by the As5 plane and the Hg3 plane of 10.68(2)° shows an almost parallel arrangement. The center of the Hg3 triangle is not situated directly below the center of the As5 ring, but rather below the arsenic atom As1. The resulting Hg–As distances show four contacts below the sum of the vdW radii with the closest one between Hg2 and As2 of 3.3014(4) Å. The assembly can best be described as the coordination of three As atoms to the Hg3 platform. The observation of different assemblies for 1 and 2 with the weak Lewis acid 3 was surprising, since we observed a similar η5-coordination mode of the E5 end-decks of 1 and 2 to the weak Lewis acids Tl+ and In+ previously.23 There is no second molecule of 3 stacked directly on top of the sandwich complex 2 to form a double-sandwich structure, as was observed for ferrocene. Nevertheless, there is a close contact (3.383(2) Å) between a carbon atom of the Cp* ring to a carbon atom of a fluorinated phenyl ring of the next molecule of 3 which may indicate possible stabilizing π–π-interactions between the electron rich Cp* ring and the electron deficient molecules of 3 or even F–H interaction to the methyl groups.
In order to better understand the difference in the nature of the Hg–E interactions in 4 and 5, their constituent compounds 1, 2, and 3 were first subjected to optimization by DFT methods.§ The computed magnitudes of the respective HOMO–LUMO gaps between 1 and 3 and 2 and 3 of 3.70 and 3.36 eV suggest that efficient mixing of the HOMOs of 1 and 2 with the LUMO of 3 is not likely to be prevalent in 4 and 5. Instead, we envisage that electrostatic and dispersion forces may play a large role in the stabilization of these adducts. To investigate the role played by electrostatic forces in 4 and 5, we decided to inspect the electrostatic potential surfaces of the individual components, as shown in Fig. 3. For 1 and 2, a distinct accumulation of negative character is observed at the center of the E5 ring. This feature is reminiscent of that observed for simple aromatic units such as benzene or the cyclopentadienide ligands of metallocenes.41 A closer inspection of the surfaces shows a greater accumulation of negative character at the phosphorus atoms in 1. This accumulation of negative character appears to be directly aligned with the phosphorus lone pairs that point outward from the center of the P5 ring. Such areas of negative electrostatic potential concentration are much less developed on the surface of the As5 ring in 2, a difference that we assign to the more electropositive character of arsenic and the more diffuse nature of its orbitals. Bearing in mind that the electrostatic potential surface at the center of the 3 is positive,26,42 the formation of the adducts 4 and 5 is driven, at least in part, by electrostatic forces as shown by the complementarity of the surfaces that come into contact in the adducts.
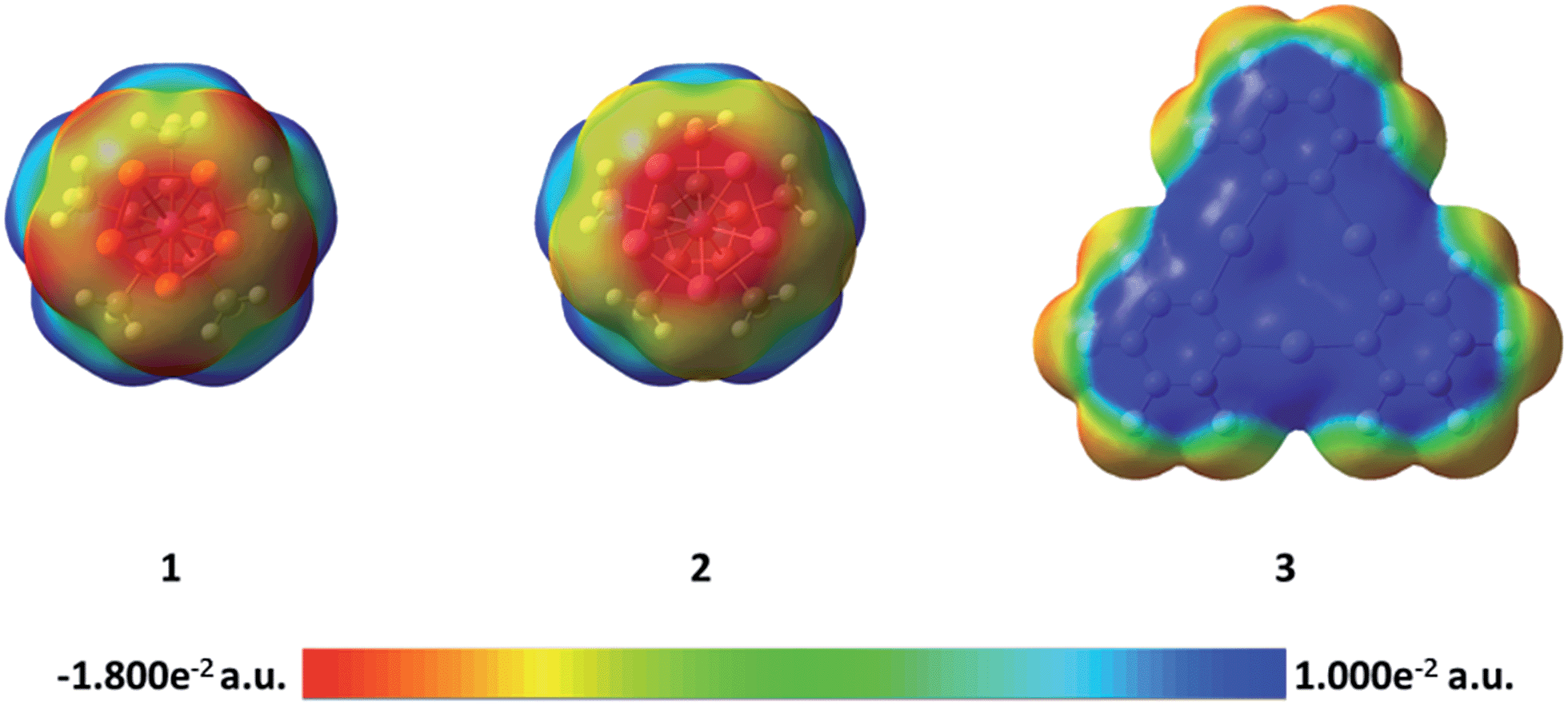 | ||
| Fig. 3 Electrostatic potential surfaces of compounds 1, 2, and 3. | ||
The side-on coordination of the phosphorus complex 1 to the center of 3 in adduct 4 can be correlated to the concentration of negative charges on each of the phosphorus atoms. Similarly, the more co-planar arrangement of the As5 ring and Hg3 plane in 5 is proposed to result from the complementarity of the negative and positive electrostatic potential concentrations at the centers of the As5 and Hg3 units, respectively.
In an effort to further investigate the nature of the Hg–E interactions in 4 and 5, atoms in molecules (AIM)43 analyses were carried out at the experimentally determined geometries. XYZ plots featuring selected bond critical points between the cyclo-E5 units and 3 are shown in Fig. 4. Relevant features of the calculated electron density distributions for selected Hg–E bond critical points (BCP) found in 4 and 5 are shown in Tables 1 and 2, respectively. Tables of the electron density distribution features at all bond critical points found between units of 1 and 3 and 2 and 3 are provided in the ESI.† In 4, four bond critical points were found between the cyclo-P5 moiety of 1 and the two molecules of 3, as shown in Fig. 4. P1, which is positioned above the center of a unit of 3, shares a BCP with each of the proximal Hg atoms, with the electron densities at the critical points ranging from 0.072 to 0.105 e Å−3. A critical point with a similar electron density (0.072 e Å−3) was also found between P3 and Hg1′ of the second unit of 3. In 5, the AIM analysis found three BCPs between the cyclo-As5 moiety of 2 and 3. The three As atoms closest to 3 (As1, As2, and As5) each share a single critical point with a proximal Hg atom, with the electron densities at these critical points ranging from 0.091 to 0.109 e Å−3.
 | ||
| Fig. 4 Sections of the solid state structure of 4 (left) and 5 (right), including selected bond critical points located via AIM analysis. Bond critical points are shown in blue. | ||
| BCP no. (A–B) | d(A-BCP) (Å) | d(B-BCP) (Å) | ρ(rBCP) (e Å−3) | ∇2ρ(rBCP) (e Å−5) | H(rBCP)/ρ(rBCP) (Eh e−1) | ε |
|---|---|---|---|---|---|---|
| 1 (Hg1–P1) | 1.657 | 1.630 | 0.105 | 0.876 | 0.012 | 0.048 |
| 2 (Hg2–P1) | 1.664 | 1.696 | 0.101 | 0.835 | 0.020 | 0.046 |
| 3 (Hg3–P1) | 1.754 | 1.776 | 0.072 | 0.620 | 0.055 | 0.046 |
| 4 (Hg1′–P3) | 1.737 | 1.789 | 0.072 | 0.622 | 0.065 | 0.051 |
| BCP no. (A–B) | d(A-BCP) (Å) | d(B-BCP) (Å) | ρ(rBCP) (e Å−3) | ∇2ρ(rBCP) (e Å−5) | H(rBCP)/ρ(rBCP) (Eh e−1) | ε |
|---|---|---|---|---|---|---|
| 1 (Hg1–As1) | 1.698 | 1.707 | 0.091 | 0.757 | 0.044 | 0.031 |
| 2 (Hg2–As2) | 1.654 | 1.648 | 0.109 | 0.885 | 0.018 | 0.052 |
| 3 (Hg3–As5) | 1.695 | 1.731 | 0.096 | 0.762 | 0.027 | 0.344 |
The values of the electron density, ρ(r), found at the Hg–E BCPs in both 4 and 5 are relatively small, being similar in magnitude to those found for weak hydrogen bonds.44 The positive values of the Laplacian of the electron density at the Hg–E BCPs, ∇2ρ(rBCP), are also suggestive of closed shell interactions. The relatively small magnitude of the ρ(r) and ∇2ρ(rBCP) values found at the bond critical points are not conclusive evidence of the weakness of the Hg–E interactions, as ρ(r) values tend to become smaller with increasing diffuseness of the electrons involved.45 However, the positive values of H(rBCP)/ρ(rBCP), the total energy density at the BCP relative to ρ(r),43 found at the Hg–E BCPs suggest that any donor–acceptor46,47 contribution to the Hg–E bonding is weak.48 Instead, we note that positive H(rBCP)/ρ(rBCP) values are usually encountered in systems stabilized by electrostatic and/or van der Waals interactions.45 Hence, while donor–acceptor bonding cannot be entirely neglected in 4 and 5, electrostatic forces as supported by the preceding potential map analysis must play a prevalent role in the formation of these adducts. Dispersion forces, which are inherently more difficult to visualize, may also play an important role.
The ellipticity values (ε), which provide information on the anisotropy of the electron density perpendicular to the bond path, at the Hg–P BCPs in 4 are small and uniform, indicating that there is no preferential plane of electron density accumulation. This is a characteristic of σ interactions, in agreement with the orientation of the phosphorus lone pairs toward the mercury atoms. In contrast to those found in 4, the ellipticities at the Hg–As BCPs in 5 are not uniform. The ellipticity value of 0.344 found at the BCP between Hg3 and As5 is substantially larger than the values obtained for the two other Hg–As BCPs. Considering the relative uniformity of the ρ(r) values found for all three Hg–As BCPs, the large ellipticity value found for the Hg3–As5 BCP suggests the involvement of an As–As π-bond in the interaction with Hg3.
Whether the different assembly of the cyclo-P5 and the cyclo-As5 complexes towards the planar Lewis acid 3 might be caused by packing effects due to the longer As–As bonds (≈2.33 Å) compared to the P–P bonds (≈2.12 Å) is hard to answer. Considering all the presented experimental data we can assume the Hg⋯P interactions found in 4 to be weak. Both E5 complexes 1 and 2 exhibit two degenerate orbitals as their HOMO which are localized on the E5 rings.20 Consequently, we rationalized that it might be possible to direct the P5 complex to also show an almost cofacial arrangement to the molecular plane of Lewis acid 3. Therefore, we followed a synthetic approach by increasing the steric bulk of the CpR ligand on the cyclo-P5 sandwich complex to induce a change of its orientation towards the Hg3 plane of 3 in the solid state. For this reason we decided to compare complexes with mono-, di- and trisubstituted tert-butyl-cyclopentadienyl ligands [Cp′Fe(η5-P5)] (6a), [Cp′′Fe(η5-P5)] (6b), [Cp′′′Fe(η5-P5)] (6c). The compounds are obtained by reacting the suitable CpR substituted dimeric iron–dicarbonyl complexes [CpRFe(CO)2]2 with white phosphorus at elevated temperature.
The determined solid state structures of 6a–c are shown in Fig. 5. All three of the analyzed complexes 6a–c show the expected sandwich structure with two parallel five-membered rings. Table 3 summarizes some geometric data for a better comparison. The distances between the Fe atom and the center of both rings increase very little when the size of the Cp ligand increases. When looking at the top row in Fig. 5 it can be seen that the P5 rings are in an almost eclipsed position with the Cp rings in all cases. This could be explained by steric effects when looking closer at the bottom row, since two methyl groups of each tert-butyl group are pointing between two P atoms of the P5 rings. The volume of the complexes was determined by dividing the unit cell volume by the number of molecules within the cell. Here it can be seen, that each additional tert-butyl group adds about 100 Å3 to the size of the complexes (Table 3).
 | ||
| Fig. 5 Solid state structures of the complexes [Cp'Fe(η5-P5)] 6a (left), [Cp′′Fe(η5-P5)] 6b (middle), [Cp′′′Fe(η5-P5)] 6c (right); (top) viewing direction perpendicular to the P5 plane revealing a nearly eclipsed arrangement of the Cp rings to the P5 rings for all three complexes. (Bottom) side view of the complexes 6a–c. | ||
| d(P5-Fe) | d(Cp-Fe) | Volume/Å3 | |
|---|---|---|---|
| 6a | 1.5396(2) | 1.7026(2) | 338.6 |
| 6b | 1.5514(13) | 1.7122(13) | 436.5 |
| 6c | 1.5615(2) | 1.7174(2) | 530.2 |
With these cyclo-P5 complexes 6a–c in hand, we prepared and fully characterized the compounds 7a–c formed by the reaction of the cyclo-P5 complex with [(o-C6F4Hg)3] in a 1:1 stoichiometry. The solid state structures of 7a–c are shown in Fig. 6. The obtained compounds each exhibit a similar assembly to that found in 4, with two cyclo-P5 complexes enclosed by two molecules of 3 held together by weak Hg⋯P interactions.
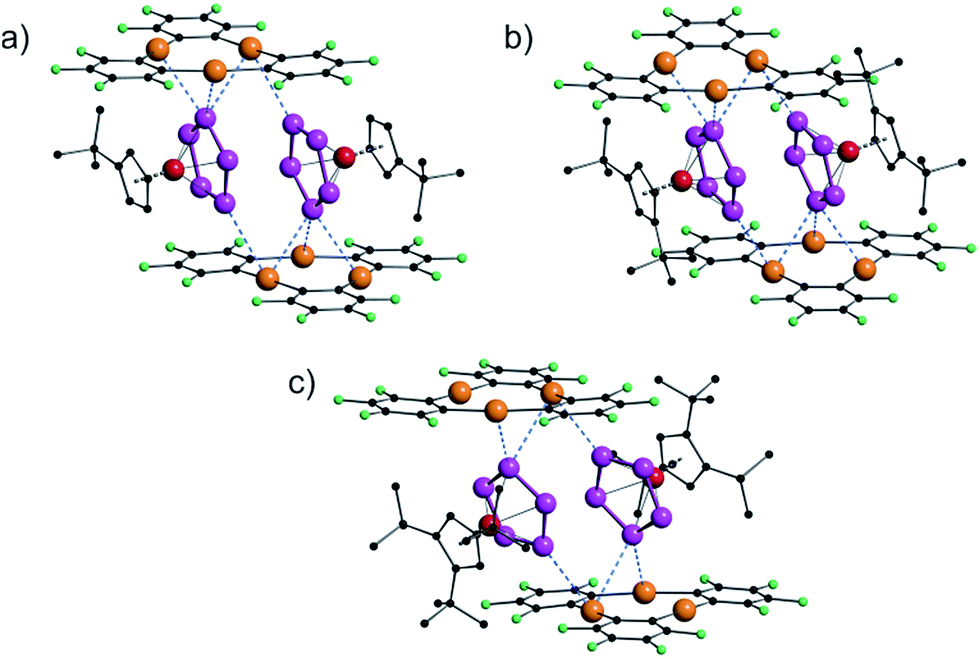 | ||
| Fig. 6 Solid state structures of [{Cp′Fe(η5-P5)}·{(o-C6F4Hg)3}] (7a) (a), [{Cp′′Fe(η5-P5)}·{(o-C6F4Hg)3}] (7b) (b), and [{Cp′′′Fe(η5-P5)}·{(o-C6F4Hg)3}] (7c) (c). | ||
There are small differences in the assemblies caused by the steric demand of the Cp ligands, but the general arrangement of the cyclo-P5 ring towards the molecular plane of 3 did not change dramatically, although the central phosphorus atom in 7c (Fig. 6c) shows only two contacts below the sum of the vdW radii to the Hg atoms of 3.
In order to better visualize the different interactions of the cyclo-P5 and the cyclo-As5 ligand towards the planar Lewis acid 3 we performed a Hirshfeld surface analysis49–52 of all of the described compounds.53Fig. 7 shows a representation of the Hirshfeld surfaces (HS) of the planar Lewis acid 3 which is facing the cyclo-E5 ligands 1 or 2 derived from the solid state structures of 4 (a + c) and 5 (b + d), respectively. While the first row shows dnorm values which are used to identify close intermolecular contacts mapped onto the HS, the second row displays the corresponding shape index. The yellow ellipses highlight the contact regions to the pnictogen atoms of the cyclo-E5 ligands. Fig. 7a and c exhibit a pronounced indentation of the HS in the center of the molecule for 4. Fig. 7a additionally shows three close contacts as white to red dots in this region on the HS which arise from interaction of the three Hg atoms of 3 with one P atom of the P5 ring.54 In Fig. 7b we can identify a contact area, highlighted in yellow, which shows five small indentations for 5 instead. These can be seen even better in the representation of the respective shape index in Fig. 7d, which resembles a face to face arrangement of the As5 plane to the Hg3 plane.
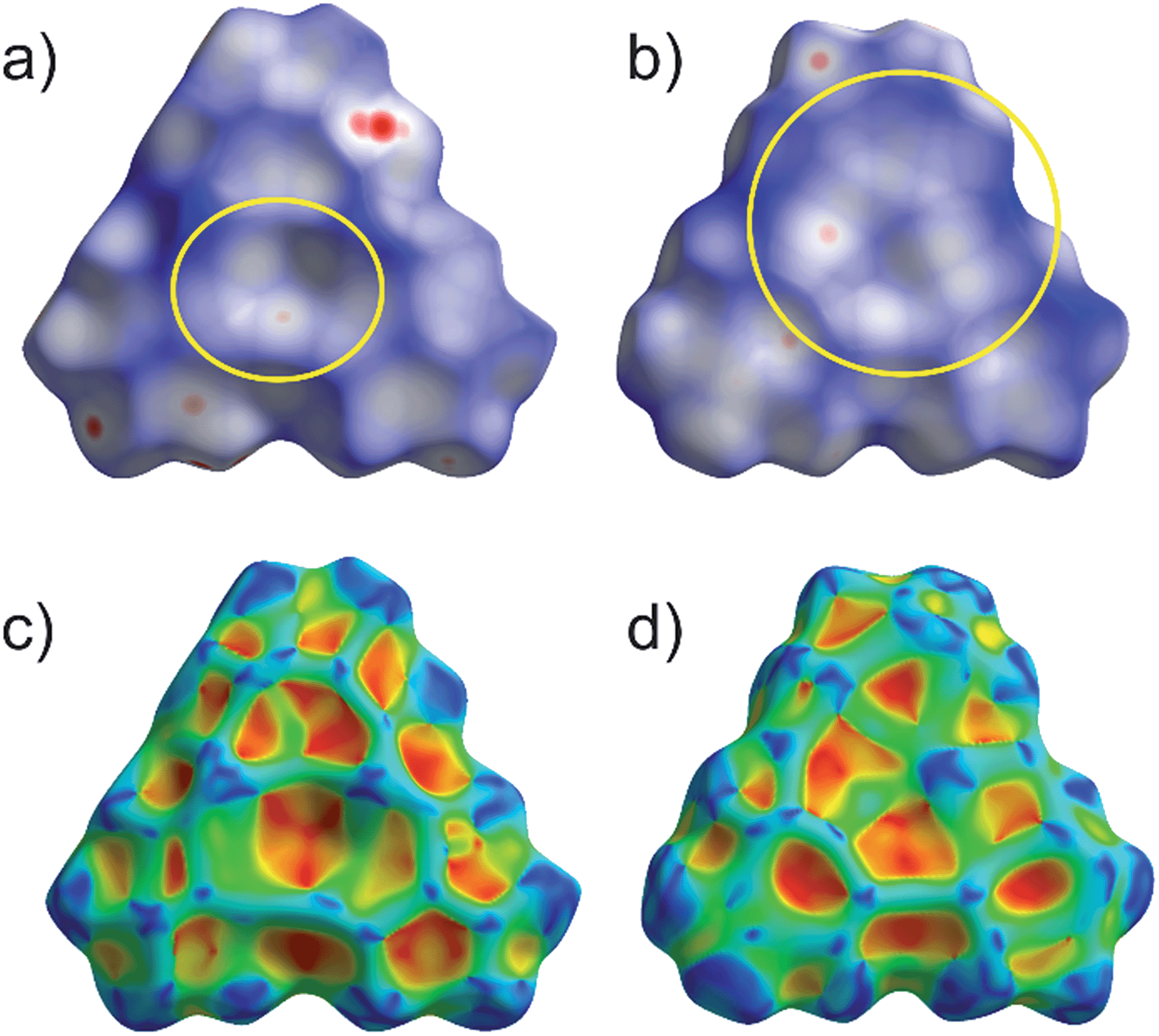 | ||
| Fig. 7 Representation of the Hirshfeld surfaces (HS) of the planar Lewis acid 3 in compounds 4 (a + c) and 5 (b + d). The first row shows dnorm values mapped onto the HS while the second row shows the shape index. Highlighted in yellow are the contact areas of the planar molecule 3 with the cyclo-E5 ligands of [Cp*Fe(η5-P)5] in (a) and [Cp*Fe(η5-As)5] in (b), respectively. | ||
A detailed HS analysis including decomposed fingerprint plots of all of the described compounds enabled us to further analyze and compare important intermolecular distances. Fig. 8 shows the fingerprint plots of the planar Lewis acid 3 in 4 and 5 with highlighted regions of contact atom pairs.53
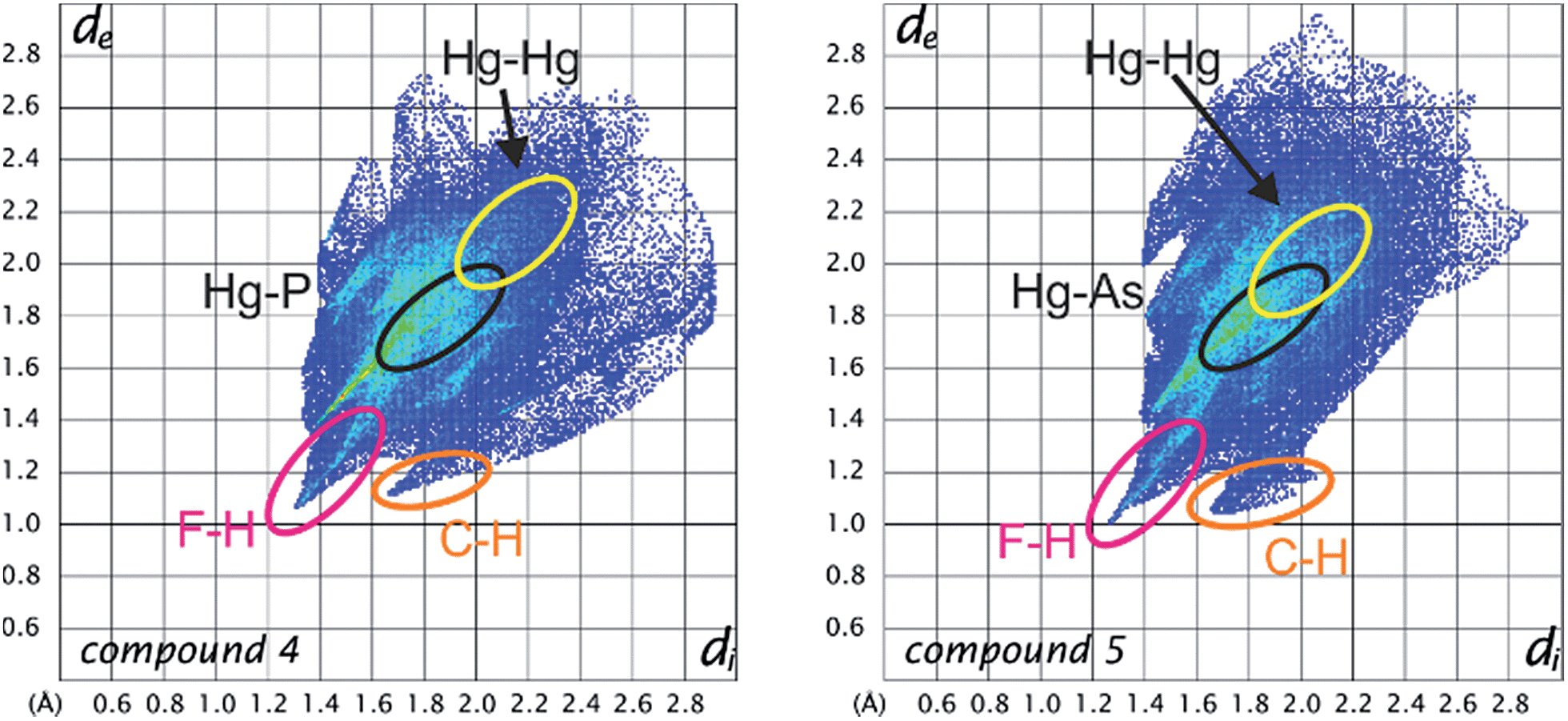 | ||
| Fig. 8 Fingerprint plots of the Hirshfeld surfaces of the planar Lewis acid 3 in the compounds 4 (left) and 5 (right). Regions of the shortest intermolecular distances depending on particular atom pairs are highlighted. | ||
The fingerprint plots of the Lewis acid 3 show some similar features for all of the compounds. While the F–H and C–H distances naturally represent the shortest intermolecular contacts, the Hg–Hg contacts are already at the edge of Hg–Hg interactions and only contribute less than 2% to the Hirshfeld surface. In 7c there are no Hg–Hg contacts at all. However, the Hg–P and Hg–As distances represent short intermolecular contacts for their respective atom types below the sum of the vdW radii (see general considerations). The Hg–P contact area generally contributes about 4–5% to the Hirshfeld surface in all of the cyclo-P5 compounds (4, 7a–c) and does not change upon Cp ligand exchange of the P5 complexes. In contrast, the F–H and F–F contacts for example are significantly influenced by the respective cyclo-P5 complex (the rising H content of the Cp ligand results in a rising F–H contact area).53 Therefore, in accordance with the single-crystal X-ray structure analyses, it can be assumed that the observed arrangement in the solid state of two cyclo-P5 sandwich complexes enclosed by two planar Lewis acidic molecules (3) is relatively stable and can resist a considerable increase in size of the adjacent ligands on the cyclo-P5 sandwich complexes.
Conclusion
A systematic comparison of the coordination behavior of the cyclo-E5 complexes [Cp*Fe(η5-P5)] (1) and [Cp*Fe(η5-As5)] (2) towards the planar trinuclear Lewis acid [(o-C6F4Hg)3] (3) is presented. While one phosphorus atom of the P5 ring of 1 interacts simultaneously with all three Hg atoms of 3 resembling a weak Lewis acid/base adduct, the analogous cyclo-As5 complex 2 interacts with the Hg atoms of 3via only three As atoms instead showing an almost cofacial arrangement of the As5 plane to the Hg3 plane of 3 in the solid state. The assemblies are supported by weak Hg–E interactions which are in agreement with the small shifts in the NMR spectra as well as the absence of any adduct signals in the mass spectra of 4 and 5.Large energy gaps between the HOMOs of 1 and 2 and the LUMO of 3, along with the complementarity of their respective electrostatic potential surfaces, suggests that electrostatic forces play a prominent role in the stabilization and coordination behavior of 4 and 5. AIM analyses of 4 and 5 corroborate the observed weakness of the Hg–E interactions, and suggest the involvement of an As–As π bond in the Hg–As interactions present in compound 5.
Subsequently, the cyclo-P5 sandwich complexes 6a–c as well as their adducts with the Lewis acid 3 (7a–c) were prepared and fully characterized. By determining the solid state structure and performing a detailed Hirshfeld surface analysis for all of the compounds we could demonstrate that the general arrangement that was found for 4 is relatively stable and can resist a considerable increase of steric demand of the cyclo-P5 complexes. A comparison of the HS of 4 and 5 shows quite different contact areas, as expected.
In conclusion the presented results show that although the characterized compounds are only supported by weak interactions instead of strong covalent dative bonds a preference of σ-interaction with the cyclo-P5 complex 1 and π-interaction with the cyclo-As5 complex 2 is observed.
Acknowledgements
This work was financially supported by the Deutsche Forschungsgemeinschaft (Sche 384/26-2), the National Science Foundation (CHE-1300371) and the Welch Foundation (A-1423). Christian Marquardt is gratefully acknowledged for recording the crystal data for [3·(CH2Cl2)] (9) (see ESI†). Bianca Attenberger is acknowledged for recording the analytical data for [Cp′FeP5] 6a.Notes and references
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed.
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- B. P. Johnson, G. Balázs and M. Scheer, Coord. Chem. Rev., 2006, 250, 1178–1195 CrossRef CAS.
- O. J. Scherer, Angew. Chem., Int. Ed., 1990, 29, 1104–1122 CrossRef.
- O. J. Scherer, Acc. Chem. Res., 1999, 32, 751–762 CrossRef CAS.
- D. Y. Zubarev and A. I. Boldyrev, in Encyclopedia of Inorganic and Bioinorganic Chemistry, John Wiley & Sons, Ltd, 2011 Search PubMed.
- N. H. Martin and J. D. Robinson, J. Mol. Graphics Modell., 2012, 38, 26–30 CrossRef CAS PubMed.
- Z. Li, C. Zhao and L. Chen, J. Mol. Struct., 2007, 810, 1–6 CrossRef CAS.
- F. De Proft, P. W. Fowler, R. W. A. Havenith, P. V. R. Schleyer, G. Van Lier and P. Geerlings, Chem.–Eur. J., 2004, 10, 940–950 CrossRef CAS PubMed.
- E. J. P. Malar, J. Org. Chem., 1992, 57, 3694–3698 CrossRef CAS.
- O. J. Scherer and T. Brück, Angew. Chem., Int. Ed., 1987, 26, 59 Search PubMed.
- O. J. Scherer, C. Blath and G. Wolmershäuser, J. Organomet. Chem., 1990, 387, C21–C24 CrossRef CAS.
- J. Bai, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2002, 41, 1737–1740 CrossRef CAS.
- F. Dielmann, A. Schindler, S. Scheuermayer, J. Bai, R. Merkle, M. Zabel, A. V. Virovets, E. V. Peresypkina, G. Brunklaus, H. Eckert and M. Scheer, Chem.–Eur. J., 2012, 18, 1168–1179 CrossRef CAS PubMed.
- J. Bai, A. V. Virovets and M. Scheer, Science, 2003, 300, 781–783 CrossRef CAS PubMed.
- M. Scheer, A. Schindler, C. Gröger, A. V. Virovets and E. V. Peresypkina, Angew. Chem., Int. Ed., 2009, 48, 5046–5049 CrossRef CAS PubMed.
- M. Scheer, A. Schindler, J. Bai, B. P. Johnson, R. Merkle, R. Winter, A. V. Virovets, E. V. Peresypkina, V. A. Blatov, M. Sierka and H. Eckert, Chem.–Eur. J., 2010, 16, 2092–2107 CrossRef CAS PubMed.
- S. Welsch, C. Gröger, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2011, 50, 1435–1438 CrossRef CAS PubMed.
- A. Schindler, C. Heindl, G. Balázs, C. Gröger, A. V. Virovets, E. V. Peresypkina and M. Scheer, Chem.–Eur. J., 2012, 18, 829–835 CrossRef CAS PubMed.
- H. Krauss, G. Balazs, M. Bodensteiner and M. Scheer, Chem. Sci., 2010, 1, 337–342 RSC.
- M. Scheer, L. J. Gregoriades, A. V. Virovets, W. Kunz, R. Neueder and I. Krossing, Angew. Chem., Int. Ed., 2006, 45, 5689–5693 CrossRef CAS PubMed.
- S. Welsch, L. J. Gregoriades, M. Sierka, M. Zabel, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2007, 46, 9323–9326 CrossRef CAS PubMed.
- M. Fleischmann, S. Welsch, H. Krauss, M. Schmidt, M. Bodensteiner, E. V. Peresypkina, M. Sierka, C. Gröger and M. Scheer, Chem.–Eur. J., 2014, 20, 3759–3768 CrossRef CAS PubMed.
- P. Sartori and A. Golloch, Chem. Ber., 1968, 101, 2004–2009 CrossRef CAS.
- M. R. Haneline, R. E. Taylor and F. P. Gabbaï, Chem.–Eur. J., 2003, 9, 5188–5193 CrossRef PubMed.
- T. J. Taylor, C. N. Burress and F. P. Gabbaï, Organometallics, 2007, 26, 5252–5263 CrossRef CAS.
- T. J. Taylor, C. N. Burress, L. Pandey and F. P. Gabbaï, Dalton Trans., 2006, 4654–4656 RSC.
- C. N. Burress, M. I. Bodine, O. Elbjeirami, J. H. Reibenspies, M. A. Omary and F. P. Gabbaï, Inorg. Chem., 2007, 46, 1388–1395 CrossRef CAS PubMed.
- A. S. Filatov, E. A. Jackson, L. T. Scott and M. A. Petrukhina, Angew. Chem., Int. Ed., 2009, 48, 8473–8476 CrossRef CAS PubMed.
- M. Tsunoda and F. P. Gabbaï, J. Am. Chem. Soc., 2000, 122, 8335–8336 CrossRef CAS.
- M. R. Haneline, M. Tsunoda and F. P. Gabbaï, J. Am. Chem. Soc., 2002, 124, 3737–3742 CrossRef CAS PubMed.
- M. R. Haneline, J. B. King and F. P. Gabbaï, Dalton Trans., 2003, 2686–2690 RSC.
- M. R. Haneline and F. P. Gabbaï, Angew. Chem., Int. Ed., 2004, 43, 5471–5474 CrossRef CAS PubMed.
- M. Fleischmann, C. Heindl, M. Seidl, G. Balázs, A. V. Virovets, E. V. Peresypkina, M. Tsunoda, F. P. Gabbaï and M. Scheer, Angew. Chem., Int. Ed., 2012, 51, 9918–9921 CrossRef CAS PubMed.
- A. J. Canty and G. B. Deacon, Inorg. Chim. Acta, 1980, 45, L225–L227 CrossRef CAS.
- S. S. Batsanov, J. Chem. Soc., Dalton Trans., 1998, 1541–1546 RSC.
- P. Pyykko and M. Straka, Phys. Chem. Chem. Phys., 2000, 2, 2489–2493 RSC.
- K. R. Flower, V. J. Howard, S. Naguthney, R. G. Pritchard, J. E. Warren and A. T. McGown, Inorg. Chem., 2002, 41, 1907–1912 CrossRef CAS PubMed.
- A. F. Holleman, E. Wiberg and N. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, 2007 Search PubMed.
- R. E. Taylor and F. P. Gabbaï, J. Mol. Struct., 2007, 839, 28–32 CrossRef CAS.
- J. Rodríguez-Otero, E. M. Cabaleiro-Lago, Á. Peña-Gallego and M. Merced Montero-Campillo, Tetrahedron, 2009, 65, 2368–2371 CrossRef.
- A. Burini, J. P. Fackler, R. Galassi, T. A. Grant, M. A. Omary, M. A. Rawashdeh-Omary, B. R. Pietroni and R. J. Staples, J. Am. Chem. Soc., 2000, 122, 11264–11265 CrossRef CAS.
- R. W. F. Bader, Atoms in Molecules: A Quantum Theory, Cambridge University Press, Oxford, UK, 1991 Search PubMed.
- R. Parthasarathi, V. Subramanian and N. Sathyamurthy, J. Phys. Chem. A, 2006, 110, 3349–3351 CrossRef CAS PubMed.
- P. Macchi and A. Sironi, Coord. Chem. Rev., 2003, 238–239, 383–412 CrossRef CAS.
- G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040–6046 CrossRef CAS PubMed.
- D. Himmel, I. Krossing and A. Schnepf, Angew. Chem., Int. Ed., 2014, 53, 370–374 CrossRef CAS PubMed.
- D. Cremer and E. Kraka, Angew. Chem., Int. Ed., 1984, 23, 627–628 CrossRef.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814–3816 RSC.
- J. J. McKinnon, A. S. Mitchell and M. A. Spackman, Chem.–Eur. J., 1998, 4, 2136–2141 CrossRef CAS.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- M. A. Spackman, Phys. Scr., 2013, 87, 48103 CrossRef.
- A detailed discussion of the Hirshfeld surface analyses for the compounds 4, 5 and 7a–c can be found in the ESI including representations of discussed surfaces and selected decomposed fingerprint plots.†.
- Unfortunately, the close contacts are not very pronounced as red contact areas since the crystal explorer software uses shorter vdW radii as a reference. A detailed description of the matter can be found in the ESI.†.
- M. Scheer, G. Friedrich and K. Schuster, Angew. Chem., Int. Ed., 1993, 32, 593–594 CrossRef.
- O. J. Scherer, T. Hilt and G. Wolmershäuser, Organometallics, 1998, 17, 4110–4112 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of the syntheses, X-ray diffraction analyses of all presented and related products and details of the DFT calculations. CCDC 1012615–1012624. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02353f |
| ‡ The complexes for n = 2, 3 were reported before.55,56 During this work the first solid state structure for a complex with n = 2 and a new polymorph for n = 3 were analyzed by X-ray diffraction analysis. |
| § Calculations were performed using the Gaussian program with the B3LYP functional and mixed basis sets: Hg, cc-pVTZ-PP; P/Fe/As, 6-311++G**; F, 6-31G(d′); C/H, 6-31G. |
| This journal is © The Royal Society of Chemistry 2015 |