
Carbazole/fluorene based conjugated small molecules: synthesis and comparative studies on the optical, thermal and electrochemical properties†
Madiha Irfana,
Kevin D. Belfieldbc and
Aamer Saeed*a
aDepartment of Chemistry, Quaid-i-Azam University, Islamabad, 44000 Pakistan. E-mail: aamersaeed@yahoo.com; Fax: +92-51-9064-2241; Tel: +92-51-9064-2128
bDepartment of Chemistry, University of Central Florida (UCF), Orlando 162366, Florida, USA
cCollege of Science and Liberal Arts, New Jersey Institute of Technology, University Heights, Newark, NJ 07102, New Jersey, USA
First published on 15th May 2015
Abstract
A series of new molecular materials with varying carbazole and fluorene contents were prepared employing Suzuki coupling and their properties were compared. A variation of the core nucleus from fluorene to carbazole was a key point to study its effect on the thermal, optical and electrochemical properties. The materials were characterized by FTIR, 1H-, 13C-NMR, APCI-MS and elemental microanalyses. The synthesized materials exhibit slightly bathochromic shifted UV/Vis spectra in dilute solution and emission maxima in the blue region along with good thermal stability. The dyes with a fluorene core are electrochemically and spectrally stable with high lying HOMO levels, in contrast to the carbazole only materials, having potential as light emitting and hole transporting materials in organic light emitting diodes.
Introduction
π-Conjugated small molecules have been extensively explored over two decades for various photonics, electronics, optoelectronic devices owing to their easy purification, high thermal and chemical stabilities, and exceptional electronic and optical properties.1–3 Moreover, the advantages of oligomers includes the tuning of their properties by varying the molecular design by selecting suitable side chain and terminal groups for excellent processing, changing the end-capping groups, or by insertion of certain groups. Thus, by alteration of the conjugated cores charge transporting and emissive properties can be controlled. Consequently, the optoelectronic properties can be optimized by divergent molecular design along with the processing.4–6 Molecular materials can serve as model systems for polymeric systems due to their well-defined structure, monodispersity as well as high purity since the purification of small molecules is much easier, as compared to the polymers.7The excellent hole-transporting capacity, high charge carrier mobility, the formation of stable radical cations coupled with efficient thermal, morphological and photophysical properties8 of carbazole makes possible its proficient use in organic semiconductor devices like OLED,9 solar cells,10 and non-linear optical (NLO) materials11 either as a main core/or as a substituent. Zhu et al. have reported the synthesis of carbazole only oligomers with improved properties for OLEDs.12 A series of phosphine oxide hosts with different hole transporting carbazolyl units were designed for blue phosphorescent OLEDs with improved efficiency.13–16 Recently, aryl amine modified phosphine oxide hosts have also been developed.17
Conversely, on account of the synthetic versatility of the fluorene moiety at aromatic 2,7 and C-9 positions along with thermal and chemical stability18 its use in optoelectronic devices has extensively been investigated. Fluorene derivatives have been used as blue-emitting materials in (OLEDs).19 Solubility and photoluminescent quantum yield of a number of fluorene based compounds are appropriate for use in OLEDs20,21 but the drawback that limits their use in OLEDs is the difficulty to inject charges in these materials. It can be improved by introducing different electron donor and acceptor substituents in their structure in order to adjust the HOMO and LUMO energy levels. For this purpose, carbazole/fluorene based dendrimers were synthesized with improved hole transporting properties for multilayer OLEDs applications.22 Nobile et al. reported the synthesis of bifluorene core based materials with different electron donating and accepting moieties to tailor the properties by the use of suitable functional groups and end-capping materials.23 Furthermore, oligomer like systems having fluorenyl moieties were developed for selective tuning of optical and electrical properties.24 Similarly, hybrids based on fluorene phosphine oxide having deep blue electoluminance with high efficiency have been constructed.25,26 Recently, carbazole/fluorene copolymers were synthesized through Suzuki-coupling with enhanced spectral stability for use as blue OLEDs.27
In this paper, we describe the synthesis of carbazole and fluorene based molecular materials possessing different ratios of carbazole/fluorene using the Suzuki coupling reaction in order to investigate and compare their effect on optical, thermal and electrochemical properties.
Experimental
Materials and measurements
All reagents and chemicals were of commercial grades and used as received unless otherwise stated. Solvents were dried and degassed following standard procedures. Melting points were determined using digital Gallenkamp (Sanyo) model MPD BM 3.5 with digital thermometer and are uncorrected. Infrared spectra were recorded using an FTX 3000 MX spectrophotometer using the ATR method. 1H NMR and 13C NMR spectra were obtained using a Bruker AM (400 MHz, 100 MHz) spectrometer respectively in CDCl3 solution using TMS as an internal reference. APCI-MS were recorded on Agilent 6210 TOF. Elemental analyses were performed on CHNS 932 LECO instrument. Thermogravimetric analysis (TGA) was performed using a PerkinElmer Thermal analysis System 409. Differential Scanning Calorimetry (DSC) measurements were performed using Bruker Reflex II thermosystem. UV-Vis spectra were taken using CECIL-7400 UV/Visible spectrophotometer and fluorescence spectra were recorded using the Hitachi FL solutions 7000 fluorescence spectrophotometer. Cyclic voltammetry was performed on CH-800C potentiostat using 0.1 M TBAP as internal reference in DCM (purged with argon for 10 minutes) on platinum (0.2 mm diameter) as working electrodes versus Ag/AgCl reference electrode and platinum wire as counter electrode at room temperature.General procedure for Pd-catalyzed Suzuki coupling
A 2 M solution of K2CO3 (20 mL) was added to a mixture of boronic acid (1.0 eq.), and the bromo derivative (2.26 eq.) dissolved in toluene (15 mL). The mixture was evacuated and filled with nitrogen thrice in order to remove air. Tetrakis triphenylphospine palladium (0.05 g, 0.04 mmol, 5 mol%) was added in three portions and after each addition, successive degassing was carried out. The mixture was refluxed for 72 h under nitrogen and filtered through a short silica plug to remove the catalyst. The filtered mixture was extracted with dichloromethane and organic phase separated, dried over MgSO4, rotary evaporated and crude products were purified by column chromatography on silica gel using suitable mobile phases and vacuum dried under reduced pressure.9-(7-(6-(7-(9H-Carbazol-9-yl)-9,9-dioctyl-9H-fluoren-2-yl)-9-octyl-9H-carbazol-3-yl)-9,9-dioctyl-9H-fluoren-2-yl)-9H-carbazole (CFT)
The compound was synthesized by same procedure described above using the corresponding boronic acid 5 (0.2 g, 0.54 mmol), 9-(2-bromo-9,9-dioctylfluoren-7-yl)-9H-carbazole 11 (0.78 g, 1.23 mmol), toluene (15 mL), 2 M K2CO3 (20 mL), Pd(PPh3)4 (0.05 g, 0.04 mmol, 5 mol%). Crude product was purified by column chromatography on silica gel (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DCM = 7:1) to obtain as off-white solid (0.45 g, 67%).
:DCM = 7:1) to obtain as off-white solid (0.45 g, 67%).
Yield: 67%; Mp: 88 °C; Rf: 0.27; IR (ν/cm−1, ATR): 3074 (sp2 C–H stretch), 2991, 2865 (sp3 C–H stretch), 1600 (Ar C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1464 (CH2 bend), 1376 (CH3 bend), 1311 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.52 (2H, d, J = 7.56 Hz), 8.20–8.18 (m, 4H, Ar–H), 7.96 (d, J = 8.64 Hz, 2H, Ar–H), 7.90–7.79 (m, 6H, Ar–H), 7.76–7.75 (m, 2H, Ar–H), 7.58–7.51 (m, 6H, Ar–H), 7.48–7.42 (m, 8H, Ar–H), 7.34–7.31 (m, 4H, Ar–H); 4.42 (t, J = 7.23 Hz, 2H, N–CH2), 2.24–2.07 (m, 10H), 1.21–1.19 (m, 10H), 1.14–1.05 (m, 40H), 0.91 (quint, 8H), 0.88 (t, J = 7.11 Hz, 15H, CH3); 13C NMR (100 MHz, CDCl3): δ 152.6, 150.2, 141.3, 140.9, 140.2, 138.5, 136.0, 132.3, 131.5, 129.9, 128.7, 126.0, 125.4, 123.9, 121.8, 121.0, 120.6, 118.8, 116.6, 109.7, 108.9, 55.7, 45.4, 41.2, 31.9, 29.6, 28.2, 27.3, 22.1, 14.1; MS (m/z, APCI): calcd 1385.9 (M+), found 1387.9 (M + 2); anal for C102H119N3 (%): calcd C = 88.32, H = 8.65, N = 3.03, found C = 88.30, H = 8.62, N = 8.64.
C), 1464 (CH2 bend), 1376 (CH3 bend), 1311 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.52 (2H, d, J = 7.56 Hz), 8.20–8.18 (m, 4H, Ar–H), 7.96 (d, J = 8.64 Hz, 2H, Ar–H), 7.90–7.79 (m, 6H, Ar–H), 7.76–7.75 (m, 2H, Ar–H), 7.58–7.51 (m, 6H, Ar–H), 7.48–7.42 (m, 8H, Ar–H), 7.34–7.31 (m, 4H, Ar–H); 4.42 (t, J = 7.23 Hz, 2H, N–CH2), 2.24–2.07 (m, 10H), 1.21–1.19 (m, 10H), 1.14–1.05 (m, 40H), 0.91 (quint, 8H), 0.88 (t, J = 7.11 Hz, 15H, CH3); 13C NMR (100 MHz, CDCl3): δ 152.6, 150.2, 141.3, 140.9, 140.2, 138.5, 136.0, 132.3, 131.5, 129.9, 128.7, 126.0, 125.4, 123.9, 121.8, 121.0, 120.6, 118.8, 116.6, 109.7, 108.9, 55.7, 45.4, 41.2, 31.9, 29.6, 28.2, 27.3, 22.1, 14.1; MS (m/z, APCI): calcd 1385.9 (M+), found 1387.9 (M + 2); anal for C102H119N3 (%): calcd C = 88.32, H = 8.65, N = 3.03, found C = 88.30, H = 8.62, N = 8.64.
9-(9,9-Dioctyl-7-(9-octyl-9H-carbazol-3-yl)-9H-fluoren-2-yl)-9H-carbazole (CCF)
The compound was synthesized by same procedure described above from corresponding boronic acid 6 (1.0 g, 3.1 mmol), 9-(7-bromo-9,9-dioctylfluoren-2-yl)-9H-carbazole 11 (2.16 g, 3.4 mmol) under Suzuki coupling conditions as a white solid (1.60 g, 62%) after column chromatography (hexane:DCM 6:1).
Yield: 62%; Mp: 72 °C; Rf: 0.43; IR (ν/cm−1, ATR): 3087 (sp2 C–H stretch), 2971, 2852 (sp3 C–H stretch), 1596 (Ar CC), 1465 (CH2 bend), 1375 (CH3 bend), 1318 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.21–8.18 (m, 4H, Ar–H), 7.97 (dd, J = 8.01, 3.72 Hz, 2H, Ar–H), 7.73–7.59 (m, 4H, Ar–H), 7.36–7.29 (m, 3H, Ar–H), 7.19–7.07 (m, 8H, Ar–H), 4.37 (t, J = 7.11 Hz, 2H, N–CH2), 2.09–2.01 (m, 4H, C–CH2), 1.94–1.82 (m, 2H, N–CH2–CH2), 1.41–1.22 (m, 30H), 0.97 (t, J = 7.89 Hz, 9H, CH3), 0.81 (quint, 4H); 13C NMR (100 MHz, CDCl3): δ 151.2, 149.4, 144.1, 141.9, 140.3, 139.9, 138.8, 136.0, 132.5, 130.5, 128.7, 126.2, 125.8, 123.3, 122.9, 121.7, 120.6, 120.1, 119.8, 118.9, 109.7, 108.8, 55.5, 43.5, 40.1, 31.7, 30.4, 29.9, 29.2, 22.5, 14.1; MS (m/z, APCI): calcd 832.5 (M+), found 833.5 [M + H]+; anal for C61H72N2 (%): calcd C = 87.93, H = 8.71, N = 3.36, found C = 87.91, H = 8.70, N = 3.38.
9-(7-(7-(7-(9H-Carbazol-9-yl)-9,9-dioctyl-9H-fluoren-2-yl)-9,9-dioctyl-9H-fluoren-2-yl)-9,9-dioctyl-9H-fluoren-2-yl)-9H-carbazole (CFD)
The compound was prepared by same method using corresponding boronic acid 8 (0.17 g, 0.35 mmol), 9-(7-bromo-9,9-dioctylfluoren-2-yl)-9H-carbazole 11 (0.51 g, 0.80 mmol), 2 M K2CO3 (20 mL), toluene (15 mL), Pd(PPh3)4 (0.05 g, 0.043 mmol) and purified by column chromatography over silica gel using (hexanes:DCM 6:1) as a light yellow solid (0.35 g, 66%) yield.
Yield: 66%; Mp: 77 °C; Rf: 0.29; IR (ν/cm−1, ATR): 3077 (sp2 C–H stretch), 2982, 2861 (sp3 C–H stretch), 1599 (Ar CC), 1465 (CH2 bend), 1376 (CH3 bend), 1314 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.21–8.17 (m, 4H, Ar–H), 8.01–7.92 (m, 2H, Ar–H), 7.89–7.75 (m, 2H, Ar–H), 7.70–7.68 (m, 4H, Ar–H), 7.64 (m, 4H, Ar–H), 7.59–7.52 (m, 6H, Ar–H), 7.47–7.41 (9m, 8H, Ar–H), 7.35–7.30 (m, 4H, Ar–H), 2.14–2.03 (m, 12H), 1.22–1.15 (m, 60H), 0.82–0.73 (m, 30H); 13C NMR (100 MHz, CDCl3): δ 150.3, 148.8, 141.6, 140.2, 139.7, 138.4, 136.2, 132.8, 130.9, 129.5, 126.7, 125.2, 123.9, 122.7, 121.8, 120.6, 119.7, 116.4, 114.2, 112.8, 45.7, 42.4, 33.5, 31.2, 30.4, 29.7, 23.8, 14.3; MS (m/z, MALDI): calcd 1496.1 (M+), found 1498 [M+ + 2]; anal for C111H136N2 (%): calcd C = 88.98, H = 9.15, N = 1.87, found, 88.99, H = 9.06, N = 1.84.
9-(9,9′-Dioctyl-7-(9,9′-dioctyl-9H-fluorene-2-yl)-9H-fluorene-2-yl)-9H-carbazole (CFM)
The compound was synthesized by same procedure using corresponding boronic acid 10 (0.18 g, 0.41 mmol), 2 M K2CO3 (15 mL), 9-(7-bromo-9,9-dioctylfluoren-2-yl)-9H-carbazole 11 (0.29 g, 0.45 mmol), toluene (20 mL), Pd(PPh3)4 (0.05 g, 0.04 mmol, 5 mol%) for reaction time of 48 hours, purified by column chromatography upon silica gel (hexane:DCM 5:1) as white solid (0.25 g, 64%).
Yield: 64%; Mp: 69 °C; Rf: 0.48; IR (ν/cm−1, ATR): 3071 (sp2 C–H stretch), 2984, 2873 (sp3 C–H stretch), 1594 (Ar CC), 1465 (CH2 bend), 1375 (CH3 bend), 1314 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.20 (d, J = 7.76 Hz, 2H, Ar–H), 7.97 (d, J = 8.36 Hz, 2H, Ar–H), 7.90–7.87 (m, 2H, Ar–H), 7.76–7.70 (m, 4H, Ar–H), 7.59–7.57 (m, 2H, Ar–H), 7.51–7.43 (m, 5H, Ar–H), 7.34 (ddd, J = 8.04, 7.52, 1.4 Hz, 4H, Ar–H), 2.16–2.07 (m, 8H, C–CH2), 1.14–1.07 (m, 40H), 0.93 (t, J = 7.1 Hz, 12H, CH3), 0.86 (quint, 8H, CH2–CH3); 13C NMR (100 MHz, CDCl3): δ 151.5, 148.2, 147.6, 143.4, 141.6, 140.8, 138.8, 136.9, 132.4, 131.3, 130.1, 129.6, 128.7, 126.5, 125.8, 123.6, 122.5, 121.7, 120.7, 119.8, 117.2, 114.4, 48.2, 42.7, 34.1, 31.3, 29.7, 26.4, 23.5, 14.3; MS (m/z, APCI): calcd 943.7 (M+), found 944.7 [M + H]+; anal for C70H89N (%): calcd C = 89.02, H = 9.50, N = 1.48, found C = 88.98, H = 9.54, N = 1.47.
9-(6-(6-(6-(9H-Carbazol-9-yl)-9-octyl-9H-carbazol-3-yl)-9-octyl-9H-carbazol-3-yl)-9-octyl-9H-carbazol-3-yl)-9H-carbazole (CCP)
Synthesized by method reported above using corresponding boronic acid 5 (0.2 g, 0.54 mmol), 9-(6-bromo-9-octyl-9H-carbazol-3-yl)-9H-carbazole 4 (0.65 g, 1.24 mmol), Pd(PPh3)4 (0.05 g, 0.04 mmol, 5 mol%), toluene (15 mL), 2 M K2CO3 (20 mL) and purified by column chromatography on silica gel (hexane:DCM = 5:1) to afford very light yellow solid (0.37 g, 59%).
Yield: 59%; Mp: 91 °C; Rf: 0.21; IR (ν/cm−1, ATR): 3077 (sp2 C–H stretch), 2972, 2849 (sp3 C–H stretch), 1598 (Ar CC), 1465 (CH2 bend), 1375 (CH3 bend), 1312 (C–N); 1H NMR (CDCl3, 400 MHz): 8.26–8.25 (m, 2H, Ar–H), 8.19 (ddd, J = 6.4, 4.1, 1.4 Hz, 6H, Ar–H), 7.61–7.59 (m, 6H, Ar–H), 7.55–7.49 (m, 6H, Ar–H), 7.42–7.39 (m, 8H, Ar–H), 7.33–7.29 (ddd, J = 6.4, 4.1, 1.4 Hz, 6H, Ar–H), 4.41 (t, J = 7.30 Hz, 6H, N–CH2), 2.09–1.92 (m, 6H, N–CH2–CH2), 1.41–1.24 (m, 30H), 0.91 (t, J = 7.13 Hz, 9H, CH3); 13C NMR (100 MHz, CDCl3): δ 145.9, 141.0, 138.4, 133.5, 131.2, 129.9, 125.7, 124.9, 122.1, 120.6, 119.8, 119.1, 117.2, 114.4, 112.5, 111.3, 109.8, 109.0, 61.1, 38.4, 31.8, 29.4, 27.3, 22.6, 14.0; MS (m/z, APCI): calcd 1163.7 (M+), found 1164.8 [M + H]+; anal for C84H85N5 (%): calcd C = 86.63, H = 7.36, N = 6.01, found C = 86.65, H = 7.35, N = 6.02.
9-(9-Octyl-6-(9-octyl-9H-carbazol-3-yl)-9H-carbazol-3-yl)-9H-carbazole (CCT)
The compound was synthesized by same method from boronic acid 6 (1.0 g, 3.1 mmol), 9-(6-bromo-9-octyl-9H-carbazol-3-yl)-9H-carbazole 4 (1.8 g, 3.4 mmol), 2 M K2CO3 (30 mL), toluene (20 mL), Pd(PPh3)4 (0.05 g, 0.04 mmol, 5 mol%) except the reaction time was 48 hours and purified by column chromatography (hexane:DCM 6:1) as white semi-solid (1.34 g, 61%).
Yield: 61%; Mp: 79 °C; Rf: 0.39; IR (ν/cm−1, ATR): 3082 (sp2 C–H stretch), 2971, 2859 (sp3 C–H stretch), 1595 (Ar CC), 1466 (CH2 bend), 1375 (CH3 bend), 1317 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.26 (d, J = 7.34 Hz, 2H, Ar–H), 8.19–8.02 (m, 2H, Ar–H), 7.87–7.76 (m, 4H, Ar–H), 7.67 (d, J = 8.23 Hz, 2H, Ar–H), 7.58–7.49 (m, 2H, Ar–H), 7.44–7.40 (m, 5H, Ar–H), 7.31 (ddd, J = 6.3, 4.1, 1.5 Hz, 4H, Ar–H), 4.41 (t, J = 7.3 Hz, 4H, N–CH2), 2.04–1.91 (m, 4H, N–CH2–CH2), 1.51–1.25 (m, 20H), 0.91 (t, J = 6.9 Hz, 6H, CH3); 13C NMR (100 MHz, CDCl3): δ 148.2, 143.4, 139.6, 136.5, 133.1, 129.2, 127.3, 125.6, 124.3, 123.6, 122.4, 120.8, 118.2, 116.9, 115.7, 115.1, 114.3, 59.4, 35.6, 32.1, 29.9, 28.1, 23.3, 14.2; MS (m/z, APCI): calcd 721.4 (M+), found 722.4 [M + H]+; anal for C52H55N3 (%): calcd C = 86.50, H = 7.68, N = 5.82, found C = 86.47, H = 7.66, N = 5.83.
2,7(6-(9-Octyl-9H carbazolyl)3-yl)9,9′-dioctyl-9H fluorene (CFC)
The compound was synthesized by same method using boronic acid 8 (0.2 g, 0.41 mmol), 9-(6-bromo-9-octyl-9H-carbazol-3-yl)-9H-carbazole 4 (0.5 g, 0.95 mmol), 2 M K2CO3 (20 mL), toluene (15 mL), Pd(PPh3)4 (0.05 g, 0.04 mmol, 5 mol%) and purified by column chromatography using hexane:ethyl acetate (9:1) to afford a light yellow solid (0.36 g, 68%).
Yield: 68%; Mp: 66 °C; Rf: 0.24; IR (ν/cm−1, ATR): 3069 (sp2 C–H stretch), 2982, 2857 (sp3 C–H stretch), 1598 (Ar CC), 1463 (CH2 bend), 1375 (CH3 bend), 1314 (C–N); 1H NMR (CDCl3, 400 MHz): δ 8.32 (m, 2H, Ar–H), 8.15–8.07 (m, 4H, Ar–H), 7.94 (d, J = 7.76 Hz, 2H, Ar–H), 7.78 (d, J = 7.76 Hz, 2H, Ar–H), 7.66–7.58 (m, 2H, Ar–H), 7.51–7.49 (m, 4H, Ar–H), 7.40–7.34 (m, 6H, Ar–H), 7.31–7.29 (m, 2H, Ar–H), 7.19–7.17 (m, 2H, Ar–H), 7.12–7.07 (m, 8H, Ar–H); 4.10 (t, J = 7.7 Hz, 4H, N–CH2), 2.15.2.12 (m, 4H, N–CH2–CH2), 2.02–1.99 (m, 4H), 1.41–1.33 (m, 20H), 1.18–1.09 (m, 20H), 0.98 (t, J = 7.81 Hz, 12H, CH3), 0.79 (quint, 4H); 13C NMR (100 MHz, CDCl3): δ 151.2, 142.1, 140.6, 139.5, 138.3, 135.4, 133.8, 130.9, 129.8, 128.8, 126.1, 125.6, 124.7, 123.1, 121.5, 120.3, 119.8, 119.3, 118.7, 114.6, 109.9, 108.6, 55.3, 43.7, 40.5, 31.9, 30.1, 29.4, 27.2, 23.9, 22.6, 14.1; MS (m/z, APCI): calcd 1274.8 (M+), found 1275.8 [M + H]+; anal for C93H102N4 (%): calcd C = 87.55, H = 8.06, N = 4.39, found C = 87.52, H = 8.08, N = 4.36.
Results and discussion
Synthetic methodology
In the newly synthesized series of molecular materials a systematic variation in carbazole and fluorene content was carried out in order to evaluate the changes in properties. The synthetic strategy adopted for the preparation of intermediates is outlined in Scheme 1. The alkylation of both carbazole and fluorene at the 9th position by C-8 chain was conducted in order to achieve the better solubility.28,29 Selective bromination of alkylated carbazole was achieved by Br2/CH3COOH and NBS/DMF30,31 to convert the latter to their corresponding boronic acids.32,33 In a similar manner 2-bromo- and 2,7-dibromo alkylated fluorenes were also converted to the corresponding boronic acids.34 3,6-Dibromo-9-octylcarbazole and 2,7-dibromo-9,9′-dioctylfluorene was coupled with carbazole under Ulmann coupling conditions.35,36 The target compounds were finally obtained by Suzuki-cross coupling via Pd(0) catalyzed reaction by the end capping of synthesized bromo-derivatives (4, 11) with the various synthesized boronic acids (Scheme 2).37,38 All of the target compounds were soluble in common organic solvents and their structures were established by FTIR, 1H-, 13C-NMR, APCI-MS spectroscopic techniques and the microelemental analyses. | ||
| Scheme 1 Synthetic route to intermediates. | ||
 | ||
| Scheme 2 Synthesis of targeted compounds under Suzuki coupling. | ||
In FTIR a strong absorption in the range of ν 3087–3069 cm−1 appeared due to sp2 C–H stretchings of the aromatic ring and an sp3 C–H stretchings of alkyl chain in the range of ν 2991–2849 cm−1 indicated the successful coupling. The presence of new intense signals in both the aliphatic and aromatic regions of the 1H- as well as 13C-NMR spectra, further supported the coupling. Further confirmation was achieved by APCI-MS analysis with a [M + H]+ peaks at 1387.9, 833.5, 1498.1, 944.7, 1164.8, 722.4, 1275.8 for CFT, CCF, CFD, CFM, CCP, CCT and CFC respectively proved the assigned structures. Elemental analysis showed the close consistency of the found and calculated composition of atoms.
Thermal properties
The thermal gravimetric analysis was carried out in the temperature range of 0–850 °C under a nitrogen atmosphere at a heating rate of 10 °C min−1. The synthesized compounds showed good thermal stability with the onset of decomposition temperature (Td) in the range of 232–435 °C. The singly end capped materials display low Td as compared to the di end-capped materials. This trend is observed due to the symmetry and rigidity of these materials in contrast to the singly substituted material. The first stage of decomposition was probably due to loss of alkyl groups. The thermal decomposition temperature increased with increase in carbazole moieties at the periphery. Fig. 1 shows a comparison of TGA profiles of CFT, CCF, CFC, and CCT.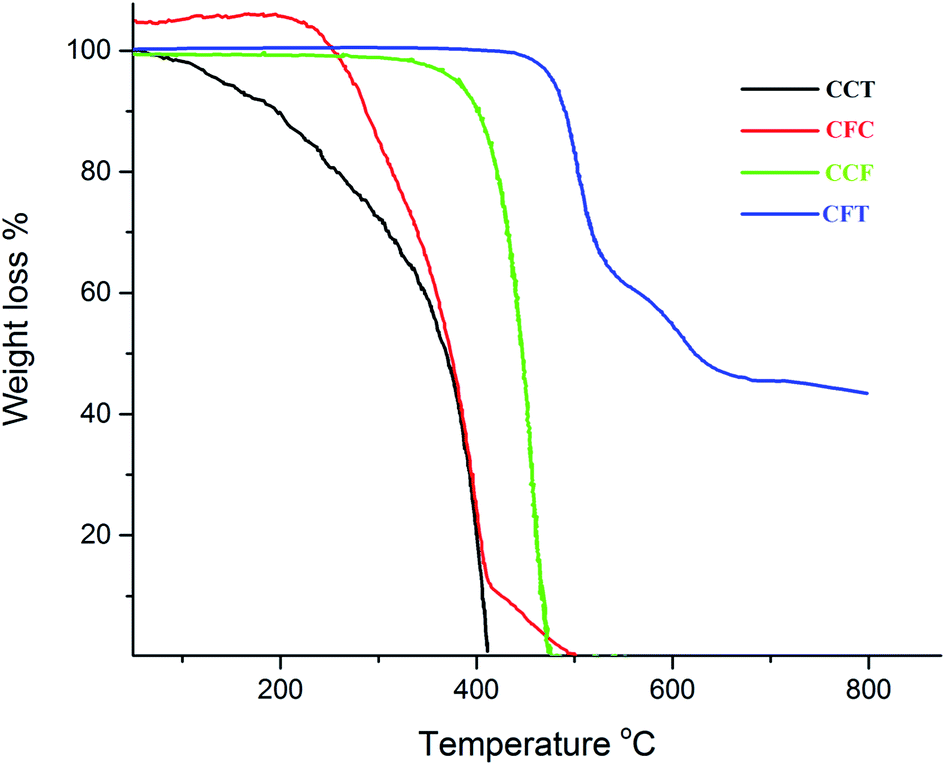 | ||
| Fig. 1 TGA curves of CFT, CCF, CFC, CCT. | ||
The compound CFT is a di-end-capped material with a Td of 435 °C. This compound decomposes in three stages. The second stage comes to an end at 665 °C. It is apparent that the process of disintegration gets slower by increase in temperature. The completion of the degradation process at 812 °C leaves 43% residue.
The compound CCF seems to degrade in a single step having Td 344 °C, whereas Tf (the temperature at which 100% mass loss is observed) is found to be 470 °C. This indicates the resistance offered by the bonds of the compound in the beginning of the pyrolysis. Near the last stage of the decomposition, however, the bond-breaking process cannot withstand the soaring thermal energy. There is no residue left at the end of this run.
The compound CFC begins to degrade at 232 °C and with a mass loss of 88% for the first stage which terminates at 418 °C. The second stage (final stage) comes to an end at 500 °C as the intermediate formed at the completion of first step exhibits no thermal stability. No residue was found. The compound CCT exhibits one-step degradation. The decomposition starts at 80 °C and 100% mass loss occurs at a temperature of 412 °C. The earlier decomposition reflects the ease of bond cleavage probably due to the incorporation of central bis-octylcarbazole and moisture.
Thermal behavior was determined by DSC in the range of 25–400 °C up at a heating rate of 10 °C min−1 under inert atmosphere. All compounds exhibited a clear endothermic melting peak during the first heating scan. Additional minor phase transitions were also detected by DSC. Upon cooling, no crystallization peaks were observed. The DSC thermogram of (CFT) is depicted in the Fig. 2.
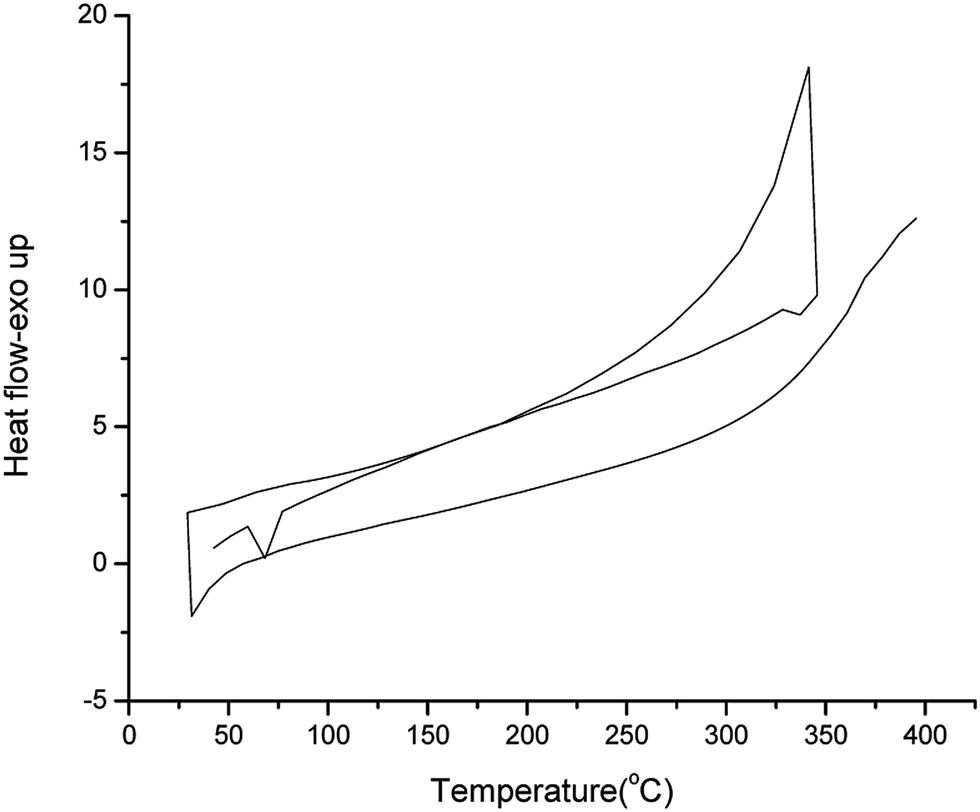 | ||
| Fig. 2 DSC thermogram of CFT. | ||
The first peak (endothermic in nature) at 68 °C is believed to rise due to the melting of the compound. The exothermic peak at 350 °C may be due to the change in phase of the substance. Apparently the compounds are supercooled liquids with semi-crystalline nature and negligible glass transition. XRD was also performed to check the nature and results were supportive to DSC with absence of crystallinity. XRD of CFT provided in Fig. 3. More or less all the dyes show similar DSC results (Table 1). The results of thermal studies were indicative of the stability of dyes that were end capped on both sides and that the dyes incorporating the central fluorene unit are more stable in contrast to the corresponding dyes possessing central carbazole unit.
 | ||
| Fig. 3 XRD of CFT. | ||
| Tda (°C) | Tm/Tcrb (°C) | λmax,absc (nm) | λmax,emd (nm) | Stoke shift (nm) | HOMOe (eV) | LUMOf (eV) | Band gapg (eV) | |
|---|---|---|---|---|---|---|---|---|
| a Thermal decomposition temperature Td, determined through TGA at heating rate of 10 °C min−1 under nitrogen.b Melting temperature Tm determined through DSC at heating rate of 10 °C min−1 under nitrogen.c Determined in dilute CHCl3 as 10−6 M solution.d Determined in dilute CHCl3 as 10−6 M solution.e HOMO was calculated employing ferrocene value of 4.8 eV below the vacuum level.f Estimated from HOMO and band gap by using a relation band gap (Eo–o) = HOMO − LUMO.g Calculated from the absorption threshold of UV-visible absorption spectrum. | ||||||||
| CFT | 435 | 78/— | 349, (292) | 400 | 51 | −5.86 | −2.694 | 3.16 |
| CCF | 344 | 58/— | 334, (293) | 452, 550 | 118 | −5.38 | −2.186 | 3.19 |
| CFD | 433 | — | 350, (292) | 400 | 50 | −5.50 | −2.451 | 3.05 |
| CFM | 402 | 72/— | 340, (291) | 402, 507 | 62 | −5.72 | −2.510 | 3.23 |
| CCP | 419 | 95/— | 343, (297) | 400 | 57 | −6.12 | −2.81 | 3.31 |
| CCT | 133 | 91/— | 337, (290) | 450, 550 | 113 | −5.93 | −2.70 | 3.23 |
| CFC | 232 | 78/— | 346, (295) | 400 | 54 | −5.43 | −2.21 | 3.22 |
Optical properties
Fig. 4a and b represents the absorption and emission spectra of the synthesized compounds recorded in chloroform. Compound CFT shows the maximum absorption at 349 nm due to π–π* transitions of conjugated backbone. The introduction of 3,6-linked carbazole did not significantly affect the decrease in absorption wavelength (blue shift) due to decrease in conjugation associated with the meta linked carbazole. A small band found at 292 nm corresponds to the π–π* transition of carbazole moiety. While its monosubstituted analogue CCF shows a hypsochromic shift at 334 nm due to decreased conjugation in contrast to CFT. The maximum absorption in the case of CCP (carbazole only material) is blue shifted to a value of 343 nm contrary to CFT that incorporates the fluorene moieties also. This blue shift indicates the decrease in conjugation length or no conjugation over long distance due to meta-linked carbazole. For the singly end-capped carbazole i.e. CCT λmax appeared at 337 nm showing a decrease in absorption position contrary to doubly end-capped material CCP. Absorption in the case of CFC appeared at 346 nm. In the case of CFD maximum absorption occurs at 350 nm due to conjugated backbone. The red shift in absorption position was due to greater fluorene content that cause the increase in conjugation length. The compound CFM (mono analogue of CFD) has λmax at 340 nm, blue shifted from CFD by 10 nm due to decreased fluorene content. The presence of small bands for these materials in the range of 267–293 nm was due to transitions from the carbazole moiety. It is evident that the UV/Vis spectra of all doubly end-capped materials were almost similar and also the singly end-capped materials possess the similar spectra. But the spectra of doubly and singly end-capped material differ from one another due to bathochromic shifts resulting from large conjugation in the case of di-substituted material It is interesting to note that extent of this red shift is lesser in case of carbazole only material due to minimization of conjugation whereas by increasing fluorene content the shifts become larger. Furthermore, on switching of the central core from carbazole to fluorene a slight red shift was observed. The presence of alkyl groups have no significant effect on the optical properties. | ||
| Fig. 4 (a) Absorption spectra (b) emission spectra of carbazole/fluorene dyes in CHCl3 10−6 M. | ||
The synthesized dyes exhibited blue emission properties in solution with the maximum emission for all the compounds in the range of 400–450 nm. The di-substituted material possess a sharp emission peak centered at 400 nm while the singly substituted materials have a broad emission peak that is more bathochromically shifted having shifts in the range of 120–110 nm and also a shoulder peak present in these materials above 500 nm (Fig. 4b). The broadened emission peaks and larger Stokes shifts are indicative of spectral instability of the singly end-capped materials. The tail might be due to excimer formation that interrupts its use in devices. While the di-end-capped materials have more localized emission peaks in the blue region. Moreover, it is important to consider that Stokes shift of di-substituted materials are low in the range of 50–57 nm as compared to mono-substituted probably due to deviation from planarity. Also the emission spectra of di-substituted material are alike in the absorption spectra and independent of fluorene content with no pronounced change on the alteration of central carbazole to fluorene nucleus while the mono-substituted materials possess the similar trend comparable to their absorption spectra with the exception of compound CFM having a sharp peak pattern. Results of optical studies indicate the improved properties for di-end-capped materials in contrast to singly end capped material. The results are depicted in Table 1.
Electrochemical properties
The electrochemical properties of dyes were determined through cyclic voltammetry on platinum electrode in the 1 mM solution for each dye containing 0.1 M tetra-n-butyl ammonium perchlorate (TBAP) as a supporting electrolyte at a potential scan rate of 50 mV s−1 (Fig. 5). All the potentials were measured vs. Ag/AgCl as a reference electrode and for each measurement the ferrocene/ferrocenium (Fc) redox system was employed as an internal standard. The HOMO, LUMO, band gap values of all the dyes are listed in the Table 1. | ||
| Fig. 5 Cyclic voltammograms of c/f dyes (1 mM each) recorded at platinum electrode in DCM solution containing 0.1 M tetra-n-butyl ammonium perchlorate (TBAP) as a supporting electrolyte employing potential scan rate of 50 mV s−1. | ||
Almost all the compounds show similar electrochemical behavior with no observed reduction peaks. The values of HOMO level range from 5.38–6.12 eV with the highest value for compound CCF with a hole injection barrier of 0.38 eV from ITO (5.0 eV) employing its use in OLEDs as hole transporting material. Such behavior for compound CCF was observed due to the presence of terminal electron donating alkylated carbazole that favors the oxidation. Whereas, the compound CFT has low lying HOMO level. Similarly the dyes with central fluorene content incorporated in them, possess the high lying HOMO level for CFC, CFD and CFM respectively suggestive of their use as a stable hole carrying material. While unexpectedly the carbazole only material CCP, CCT possess the low lying HOMO and LUMO level with increased band gap consistent with their optical properties. Additionally CCP has a HOMO value lower than CCT due to enhancement in carbazole core which reduces the oxidation potential. Furthermore the oxidation peak for CCP is irreversible suggesting its electrochemical instability. The high HOMO value of CCT was again due to terminal alkyl carbazole. The reason for such behavior of the carbazole only material is that the HOMO value of the nitrogen-containing material depends on the position of nitrogen atom. The LUMO level of the dyes matched with the high work function cathodes used in OLEDs. Results of electrochemical studies reveal that the fluorene containing material is electrochemically stable and an attractive candidate for hole transportation in OLEDs relative to the corresponding carbazole material. In order to investigate the effect of stability, successive scans on both singly and di-end-capped materials were carried out. By comparison disubstituted dyes show negligible change while their mono-analouges changes the position of its potential indicating the more stability of di-end-capped compounds. The comparison of successive electrochemical scans of CFT and CCF was depicted in Fig. 6. The high electrochemical stability of disubstituted materials shows increased lifetime in device operation.
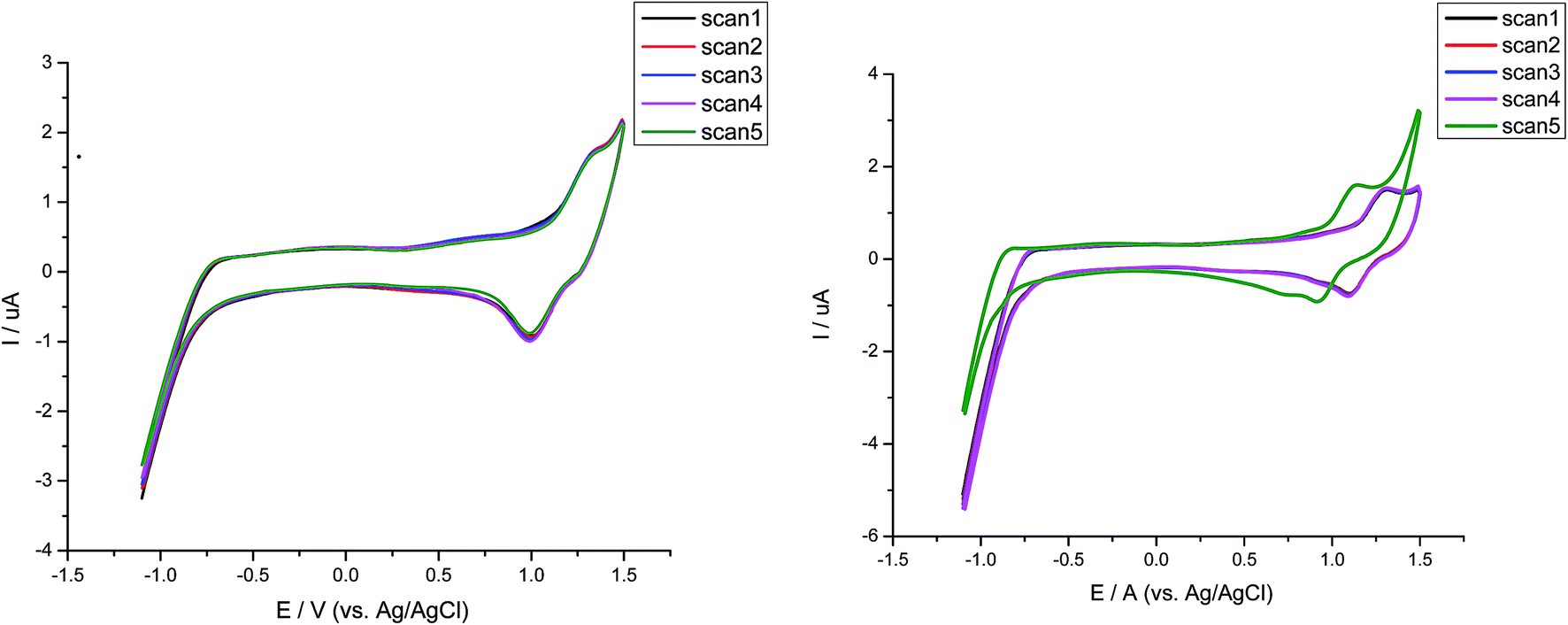 | ||
| Fig. 6 Comparison of successive CV curves of CFT (a) and CCF (b) upon 10 scans. | ||
Conclusions
Synthesis of molecular materials with varying carbazole/fluorene cores were carried out in order to combine the advantages of carbazole and fluorene and compare their effect on different properties. The synthesized dyes are thermally stable and possess good solubility behavior and blue emission. An investigation of properties reveals that the disubstituted analogues show pure blue emission with increased thermal stability in contrast to monosubstituted derivatives. The derivatives containing carbazole/fluorene moieties have much superior properties than carbazole only molecular materials, due to combination of both fluorene and carbazole cores. Moreover, the modification of the central core from carbazole to fluorene indicates the superior properties of fluorene containing dyes due to enhanced conjugation. Electrochemical studies were indicative of electrochemical stability of disubstituted materials which in turn suggest the better device lifetime and also the better alignment of HOMO level of fluorene materials with the PEDOT:PSS employs their use as hole transporting material in OLEDs. Depending upon their properties these blue light emitting fluorescent materials are expected to show good device performance as an HTL/emissive material in OLEDs. This work provides the comparative study of carbazole and fluorene carrying materials that might be helpful for the design of other molecular materials for OLEDs.Acknowledgements
This work was supported by research grant under the Pak-USAID joint project i.e. no. 4-279/PAK-US/HEC/2010/917 and the U.S. National Academy of Sciences (PGA-P210877). M.I. gratefully acknowledges a research scholarship from HEC Islamabad under the HEC Indigenous PhD Scholarship 5000 Scheme. KDB would also like to acknowledge support from the National Science Foundation (CHE-0832622). We acknowledge the mass spectrometry facility at the University of Florida and National Science Foundation for MS instrumentation (CHE-1040016) for providing the facility of mass analysis.References
- J. A. E. H. Van Haare, L. Gronendaal, E. E. Havinga, R. A. J. Janssen and E. W. Meijer, Angew. Chem., Int. Ed., 1996, 35, 638–640 CrossRef CAS.
- J. Roncali, P. Leriche and A. Cravino, Adv. Mater., 2007, 19, 2045–2060 CrossRef CAS.
- K. Brunner, A. Van Dijken, H. Borner, J. J. A. M. Bastiaansen, N. M. M. Kiggen and B. M. W. Langeveld, J. Am. Chem. Soc., 2004, 126, 6035–6042 CrossRef CAS PubMed.
- A. Pogantsch, F. P. Wenzl, E. J. W. List, G. Leising, A. C. Grimsdale and K. Mullen, Adv. Mater., 2002, 14, 1061–1064 CrossRef CAS.
- D. Ma, J. M. Lupton, I. D. W. Samuel, S.-C. Lo and P. L. Burn, Appl. Phys. Lett., 2002, 81, 2285–2287 CrossRef CAS.
- P. Furuta, J. Brooks, M. E. Thompson and J. M. J. Fréchet, J. Am. Chem. Soc., 2003, 125, 13165–13172 CrossRef CAS PubMed.
- (a) Q. Liu, W. Liu, B. Yao, H. Tian, Z. Xie, Y. Geng and F. Wang, Macromolecules, 2007, 40, 1851–1857 CrossRef CAS; (b) C. Chi, C. Im, V. Enkelmann, A. Ziegler, G. Lieser and G. Wegner, Chem.–Eur. J., 2005, 11, 6833–6845 CrossRef CAS PubMed; (c) C.-Z. Zhou, T. Liu, J.-M. Xu and Z.-K. Chen, Macromolecules, 2003, 36, 1457–1464 CrossRef CAS.
- M.-H. Ho, B. Balaganesan, T.-Y. Chu, T.-M. Chen and C. H. Chen, Thin Solid Films, 2008, 517, 943–947 CrossRef CAS.
- P. Kundu, K. R. J. Thomas, J. T. Lin, Y.-T. Tao and C.-H. Chien, Adv. Funct. Mater., 2003, 13, 445–452 CrossRef CAS.
- (a) S. Beaupre, P.-L. T. Boudreault and M. Leclerc, Adv. Mater., 2010, 22, E6–E27 CrossRef CAS PubMed; (b) Z. He, C. Zhang, X. Xu, L. Zhang, L. Huang, J. Chen, H. Wu and Y. Cao, Adv. Mater., 2011, 23, 3086–3089 CrossRef CAS PubMed; (c) N. Blouin, A. Michaud and M. Leclerc, Adv. Mater., 2007, 19, 2295–2300 CrossRef CAS.
- Z. Kotler, J. Segal, M. Sigalov, A. Ben-Asuly and V. Khodorkovsky, Synth. Met., 2000, 115, 269–273 CrossRef CAS.
- W.-L. Gong, F. Zhong, M. P. Aldred, Q. Fu, T. Chen, D.-K. Huang, Y. Shen, X.-F. Qiao, D. Ma and M.-Q. Zhu, RSC Adv., 2012, 2, 10821–10828 RSC.
- C. Han, Z. Zhang, H. Xu, J. Li, G. Xie, R. Chen, Y. Zhao and W. Huang, Angew. Chem., Int. Ed., 2012, 51, 10104–10108 CrossRef CAS PubMed.
- Y. Tao, J. Xiao, C. Zheng, Z. Zhang, M. Yan, R. Chen, X. Zhou, H. Li, Z. An, Z. Wang, H. Zu and W. Huang, Angew. Chem., Int. Ed., 2013, 52, 10491–10495 CrossRef CAS PubMed.
- W. Yang, Z. Zhang, C. Han, Z. Zhang, H. Xu, P. Yan, Y. Zhao and S. Liu, Chem. Commun., 2013, 49, 2822–2824 RSC.
- C. Han, L. Zhu, F. Zhao, Z. Zhang, J. Wang, Z. Deng, H. Xu, J. Li, D. Ma and P. Yan, Chem. Commun., 2014, 50, 2670–2672 RSC.
- C. Han, L. Zhu, J. Li, F. Zhao, H. Xu, D. Ma and P. Yan, Chem.–Eur. J., 2014, 20, 16350–16359 CrossRef CAS PubMed.
- J. U. Wallace and S. H. Chen, Adv. Polym. Sci., 2008, 212, 145–186 CAS.
- S. Tao, Y. Zhou, C.-S. Lee, X. Zhang and S.-T. Lee, Chem. Mater., 2010, 22, 2138–2141 CrossRef CAS.
- L. Feng, C. Zhang, H. Bie and Z. Chen, Dyes Pigm., 2005, 64, 31–34 CrossRef CAS.
- E. Wang, C. Li, Y. Mo, Y. Zhang, G. Ma, W. Shi, J. Peng, W. Yang and Y. Cao, J. Mater. Chem., 2006, 16, 4133–4140 RSC.
- O. Usluer, S. Demic, D. A. M. Egbe, E. Birckner, C. Tozlu, A. Pivrikas, A. M. Ramil and N. S. Sariciftci, Adv. Funct. Mater., 2010, 20, 4152–4161 CrossRef CAS.
- R. Grisorio, A. Dell'Aquila, G. Romanazzi, G. P. Suranna, P. Mastrorilli, P. Cosma, D. Acierno, E. Amendola, G. Ciccarella and C. F. Nobile, Tetrahedron, 2006, 62, 627–634 CrossRef CAS.
- C. Han, L. Zhu, J. Li, F. Zhao, Z. Zhang, H. Xu, Z. Deng, D. Ma and P. Yan, Adv. Mater., 2014, 26, 7070–7077 CrossRef CAS PubMed.
- D. Yu, F. Zhao, Z. Zhang, C. Han, H. Xu, J. Li, D. Ma and P. Yan, Chem. Commun., 2012, 48, 6157–6159 RSC.
- D. Yu, F. Zhao, C. Han, H. Xu, J. Li, Z. Zhang, Z. Deng, D. Ma and P. Yan, Adv. Mater., 2012, 24, 509–514 CrossRef CAS PubMed.
- H.-J. Jiang and J.-L. Zhang, Polym. Adv. Technol., 2014, 25, 644–650 CrossRef CAS.
- Y. He, C. Zhong, A. He, Y. Zhou and H. Zhang, Mater. Chem. Phys., 2009, 114, 261–266 CrossRef CAS.
- K. D. Belfield, K. J. Schafer, W. Mourad and B. A. Reinhardt, J. Org. Chem., 2000, 65, 4475–4481 CrossRef CAS PubMed.
- M. Park, J. R. Buck and C. J. Rizzo, Tetrahedron, 1998, 54, 12707–12714 CrossRef CAS.
- S. H. Kim, I. Cho, M. K. Sim, S. Park and S. Y. Park, J. Mater. Chem., 2011, 21, 9139–9148 RSC.
- X. Xiao, Y. Fu, M. Sun, L. Li and Z. Bo, J. Polym. Sci., Part A: Polym. Chem., 2007, 2410–2424 CrossRef CAS.
- M.-G. Ren, H.-J. Guo, F. Qi and Q.-H. Song, Org. Biomol. Chem., 2011, 9, 6913–6916 CAS.
- M. P. Aldred, C. Li, G.-F. Zhang, W.-L. Gong, A. D. Q. Li, Y. Dai, D. Ma and M.-Q. Zhu, J. Mater. Chem., 2012, 22, 7515–7528 RSC.
- L. Qin-De, L. Jianping, D. Jianfu and T. Ye, Macromol. Chem. Phys., 2008, 209, 1931–1941 CrossRef.
- Y.-H. Sun, X.-H. Zhu, Z. Chen, Y. Zhang and Y. Cao, J. Org. Chem., 2006, 71, 6281–6284 CrossRef CAS PubMed.
- (a) O. Paliulis, J. Ostrauskaite, V. Gaidelis, V. Jankauskas and P. Strohriegl, Macromol. Chem. Phys., 2003, 204, 1706–1712 CrossRef CAS; (b) C. Yang, H.-s. Song and D.-b. Liu, J. Mater. Sci., 2012, 47, 3315–3319 CrossRef CAS; (c) K. Takagi, H. Takao and T. Nakagawa, Polym. J., 2013, 45, 396–400 CrossRef CAS.
- M. P. Aldred, C. Li, G.-F. Zhang, W.-L. Gong, A. D. Q. Li, Y. Dai, D. Ma and M.-Q. Zhu, J. Mater. Chem., 2012, 22, 7515–7528 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and 1H-NMR, APCI-MS. See DOI: 10.1039/c5ra01141h |
| This journal is © The Royal Society of Chemistry 2015 |