
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chelator free gallium-68 radiolabelling of silica coated iron oxide nanorods via surface interactions†
Benjamin P.
Burke
ab,
Neazar
Baghdadi
a,
Alicja E.
Kownacka
a,
Shubhanchi
Nigam
ab,
Gonçalo S.
Clemente
bc,
Mustafa M.
Al-Yassiry
a,
Juozas
Domarkas
ab,
Mark
Lorch
a,
Martin
Pickles
d,
Peter
Gibbs
d,
Raphaël
Tripier
e,
Christopher
Cawthorne
bc and
Stephen J.
Archibald
*ab
aDepartment of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: s.j.archibald@hull.ac.uk
bPositron Emission Tomography Research Centre, University of Hull, Cottingham Road, Hull, HU6 7RX, UK
cSchool of Biological, Biomedical and Environmental Sciences, University of Hull, Cottingham Road, Hull, HU6 7RX, UK
dCentre for Magnetic Resonance Investigations, Hull York Medical School at University of Hull, Anlaby Road, Hull, HU3 2JZ, UK
eUniversité de Brest, UMR-CNRS 6521/SFR148 ScInBioS, UFR Sciences et Techniques, 6 Avenue Victor le Gorgeu, C.S. 93837, 29238 Brest, France
First published on 13th August 2015
Abstract
The commercial availability of combined magnetic resonance imaging (MRI)/positron emission tomography (PET) scanners for clinical use has increased demand for easily prepared agents which offer signal or contrast in both modalities. Herein we describe a new class of silica coated iron–oxide nanorods (NRs) coated with polyethylene glycol (PEG) and/or a tetraazamacrocyclic chelator (DO3A). Studies of the coated NRs validate their composition and confirm their properties as in vivo T2 MRI contrast agents. Radiolabelling studies with the positron emitting radioisotope gallium-68 (t1/2 = 68 min) demonstrate that, in the presence of the silica coating, the macrocyclic chelator was not required for preparation of highly stable radiometal-NR constructs. In vivo PET-CT and MR imaging studies show the expected high liver uptake of gallium-68 radiolabelled nanorods with no significant release of gallium-68 metal ions, validating our innovation to provide a novel simple method for labelling of iron oxide NRs with a radiometal in the absence of a chelating unit that can be used for high sensitivity liver imaging.
1. Introduction
Medical imaging can be defined as the non-invasive visualisation of the intact human body for clinical analysis. Positron Emission Tomography (PET) is a nuclear medicine technique based on the detection of positron emitting radionuclides to give a high sensitivity functional image.1–3 However, as a molecular imaging technique, it does not provide anatomical information and is, therefore, often combined with other techniques, such as computed tomography (CT) or magnetic resonance imaging (MRI). Although the combination of PET with CT is most prevalent in the clinic, recent developments in detector design have led to the creation of combined PET/MR scanners that offer advantages over PET/CT, such as increased soft tissue contrast, lower radiation dose and the potential application of MRI contrast agents.4,5 The introduction of clinical PET/MRI scanners increases the demand for multimodal contrast agents, achieved either by using a cocktail of known imaging agents or by developing new dual modality contrast agents in which both components are linked into one construct.6–9Super-paramagnetic iron oxide nanoparticles (SPIONs) are used in MRI to reduce the time taken for proton nuclei to transfer their spin to neighbouring proton nuclei, resulting in a loss of transverse magnetisation (T2 relaxation), causing a relative darkening in regions of SPION accumulation.10,11 Iron–oxide based nanospheres have been the most commonly used T2 contrast agents clinically,12–14 largely because of their low toxicity and the potential for facile modification of the surface.15–17 Iron–oxide based nanorods (NRs) are less common but may offer advantages, such as improved MRI contrast18–20 and improved properties as drug delivery vehicles.21
68Ga (t1/2 = 68 min) is an attractive radioisotope for PET due to high proportion of positron decay (89%) and its availability from a generator based system as a decay product of a parent isotope with a long half-life (68Ge, t1/2 = 271 days), eliminating the need for an onsite cyclotron.22,23 In aqueous solution and at physiological pH, 68Ga is exclusively in the oxidation state +3, thus most synthetic procedures are carried out in the presence of weakly coordinating ligands such as citrate, acetate or oxalate at low pH in order to avoid the formation of insoluble Ga(OH)3 and soluble Ga(OH)4−, both of which dramatically slow the kinetics of complex formation.24,25 Gallium(III) generally forms six coordinate complexes and is classified as a hard Lewis acid, therefore, it binds to borderline and hard Lewis base donor atoms such as nitrogen and oxygen, found in routinely used chelators for biomedical imaging.26,27 Incorporation of 68Ga into a targeting vector is generally achieved by the use of bifunctional chelating agents (BCAs), molecules containing a chelating moiety responsible for trapping 68Ga in a stable in vivo form, functionalised with a reactive group allowing its conjugation to a bioactive molecule, protein or peptide.28,29 1,4,7,10-Tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) is the most widely employed BCA due to the relative ease of its synthesis (due to its chemical symmetry) and the ability to complex, with adequate stability, various metal ions relevant for a range of biomedical applications.3,22,30,31
Herein we describe the development of silica coated iron oxide NRs radiolabelled with gallium-68 for PET/MR multimodal imaging in which the inclusion of a bifunctional chelating agent (such as DOTA) is not required for stable gallium(III) complexation in vivo.
2 Results & discussion
2.1. Synthesis and surface characterisation
Iron–oxide nanorods, coated with various ratios of a siloxane terminated tetraazamacrocycle (siloxane-DO3A) and a siloxane polyethylene glycol (PEG) derivative, were synthesised using a modified method, similar to that previously reported by our group.32 Briefly, uncoated iron oxide nanorods and the selected ratio of functionalised siloxanes were stirred in a basic 60% ethanol/water solution, as shown in Scheme 1. The preparation includes a rigorous washing procedure followed by resuspension of coated NRs in water and removal of macro aggregates by filtration through a 0.45 μm filter. Macrocycle coated nanoparticles were subsequently subjected to trifluoroacetic acid (TFA) hydrolysis to unmask the protected acetate arms to give nanoparticles coated using 100% macrocycle (1) and 50% macrocycle, 50% PEG (2). The 100% PEG coated NRs (3) do not require a hydrolysis step.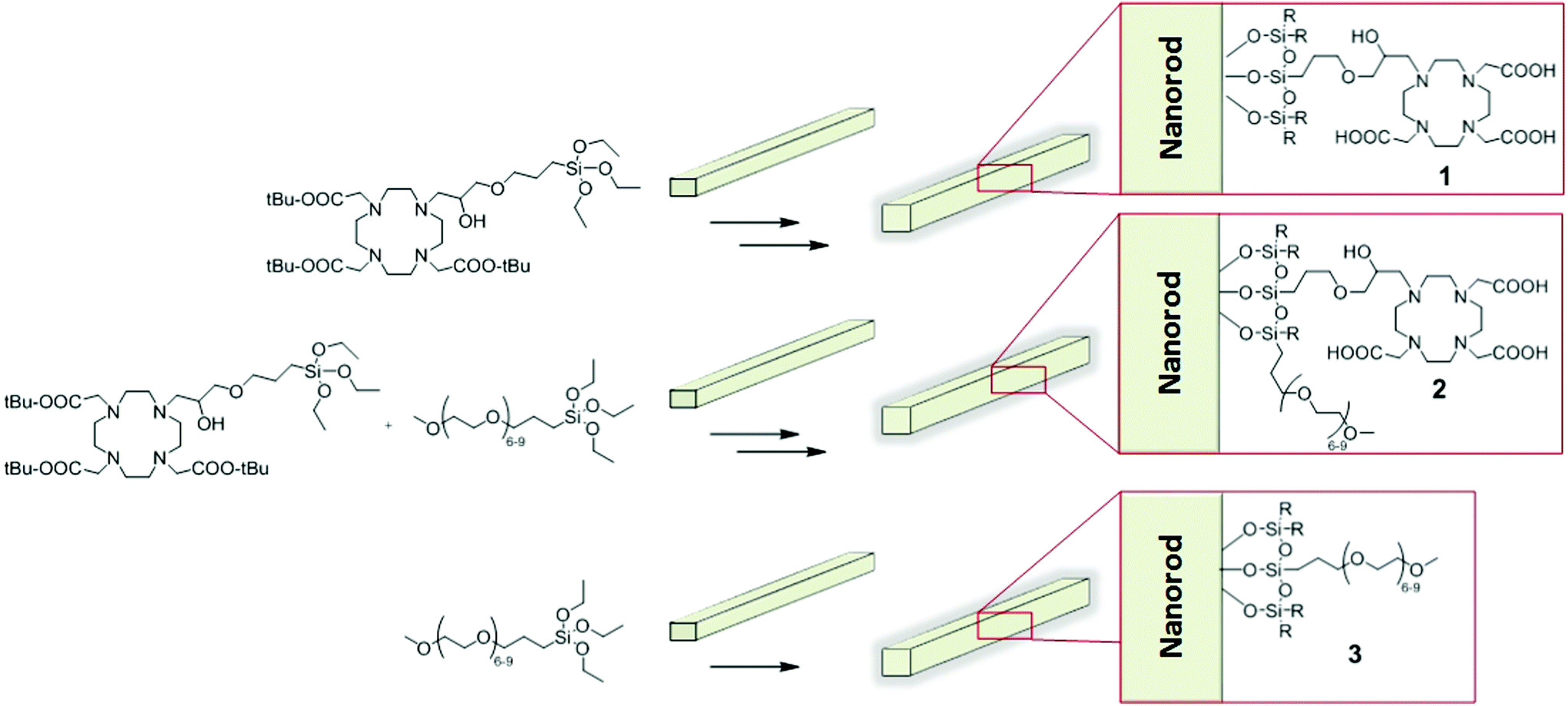 | ||
| Scheme 1 Synthesis of silica coated iron oxide nanorods 1–3. | ||
Transmission electron microscopy (TEM) analysis was carried out on the final constructs (1–3) to ensure that synthetic coating protocols do not affect the core iron–oxide structure, see Fig. 1. All constructs were found to have a similar size distribution, with rods of length 80–130 nm. Fourier transform infra-red (FTIR) spectroscopy was carried out to validate the presence of the surface coating, see Fig. 2. All coating types had the expected stretches of Fe–O, Si–O–C and Si–O–Fe between 600 and 1000 cm−1.33 Surface coatings can be differentiated due to the independent peak characteristics of each type. In case of 1, characteristic macrocycle-derived C–N stretches can be assigned at ca. 1200 cm−1. In case of 3, C–O–C linkage, characteristic to PEG, can be assigned at 1117 cm−1. 2 was designed to contain a mixture of coating component and was produced using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of the two coating components, with the FTIR showing a broad peak in the region overlapping both regions. The chemical composition of surface coating is corroborated by combustion analysis of coated NRs nitrogen content (0.79, 0.29 and 0.00% for 1, 2 and 3, respectively) as the only inclusion of nitrogen is from the tetraazamacrocycle coating, decreasing for 2 relative to 1 as expected, see Fig. S1.†
:1 ratio of the two coating components, with the FTIR showing a broad peak in the region overlapping both regions. The chemical composition of surface coating is corroborated by combustion analysis of coated NRs nitrogen content (0.79, 0.29 and 0.00% for 1, 2 and 3, respectively) as the only inclusion of nitrogen is from the tetraazamacrocycle coating, decreasing for 2 relative to 1 as expected, see Fig. S1.†
 | ||
| Fig. 1 TEM images of iron oxide nanorods 1–3. | ||
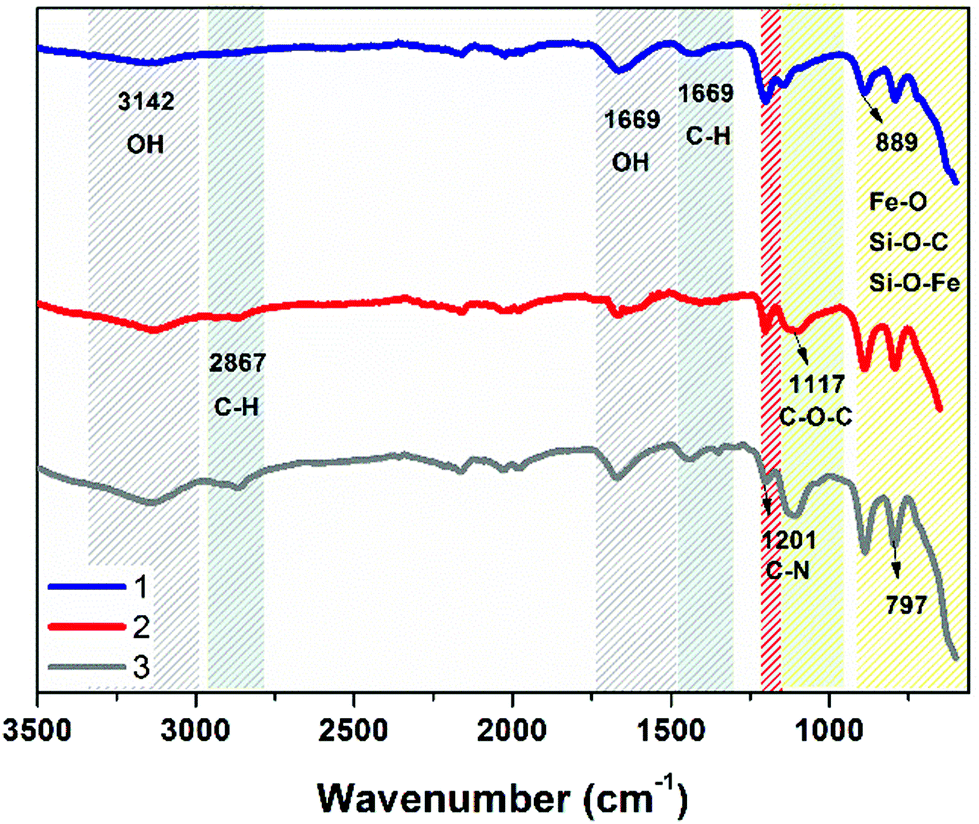 | ||
| Fig. 2 FTIR spectra of nanorods 1–3. | ||
2.2. Nanorod construct analysis
The surface charge of nanoparticles gives an indication of their aqueous suspension stability, an essential requirement for translation to in vivo studies. A large negative or positive value would suggest an increased electrostatic repulsion between individual nanoparticles, decreasing the tendency for aggregation.34–36 The zeta potential of the constructs (1–3) was measured at a constant concentration (7.5 mM Fe) at pH 7 in PBS, see Fig. S2.† All constructs have an overall negative charge between −10 and −20 mV. Analogous PEG methyl ether terminating nanoparticles have a similar surface charge37 and DOTA based systems have deprotonated carboxylic acid groups at pH 7.38,39 All constructs have a relatively similar zeta potential, with 2 having the highest negative potential at −17.9 mV. The high negative surface charge of all constructs translates to high stability in aqueous solution, with no visible precipitation observed for any of the samples after 2 months.The size of nanoparticles is an important characteristic governing their in vivo biodistribution pathways and, therefore, determining their potential for application as MRI contrast agents. Very small (<5 nm) nanoparticles are generally eliminated via the renal system and large (>200 nm) nanoparticles are excreted via the spleen and liver. Either of these elimination pathways results in short blood circulation times.10,40,41 Samples of all three constructs were suspended in water (at a concentration of 0.1 mM Fe) and their hydrodynamic size was measured by nanoparticle tracking analysis (NTA) in which mean and modal sizes were determined, see Fig. S3.† The constructs had mean sizes in the range of 99–155 nm, with an increase in size correlating with the addition of macrocycle to the surface coating, all are an acceptable size for in vivo applications. There are challenges with NTA analysis and the use of rods rather than spheres: (a) how do these numbers relate to the different nanorod axes?; and (b) are the in vivo excretion pathways of the nanorods different to that of the nanospheres and, if so, what is the effect of length?
Prior to radiolabelling and in vivo studies, the magnetic behaviour of the NR constructs was investigated in vitro at 3 T, a magnetic field strength correlating to common clinical MRI scanners. The r1 and r2 relaxation rates were calculated for all three constructs (1–3), see Fig. 3 and 4. All constructs are relatively weak T1 contrast agents. As T2 contrast agents, all constructs showed fast relaxivity with improvement correlated to the increasing PEG content of the NR coating.
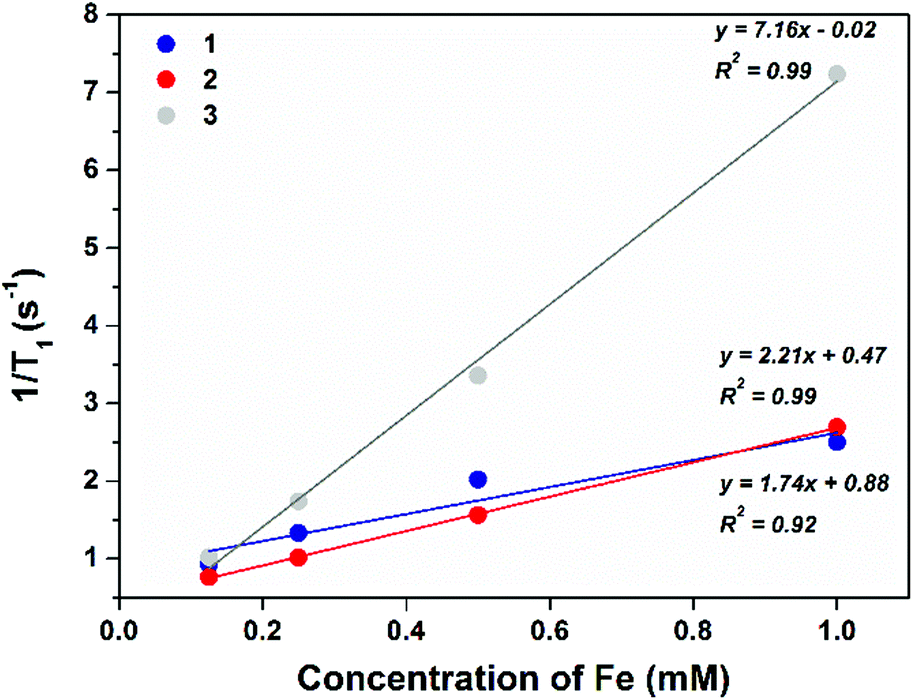 | ||
| Fig. 3 T 1 MRI relaxivity plots of 1–3 at a range of Fe concentrations (0.1–1 mM) at 3 T to determine r1. | ||
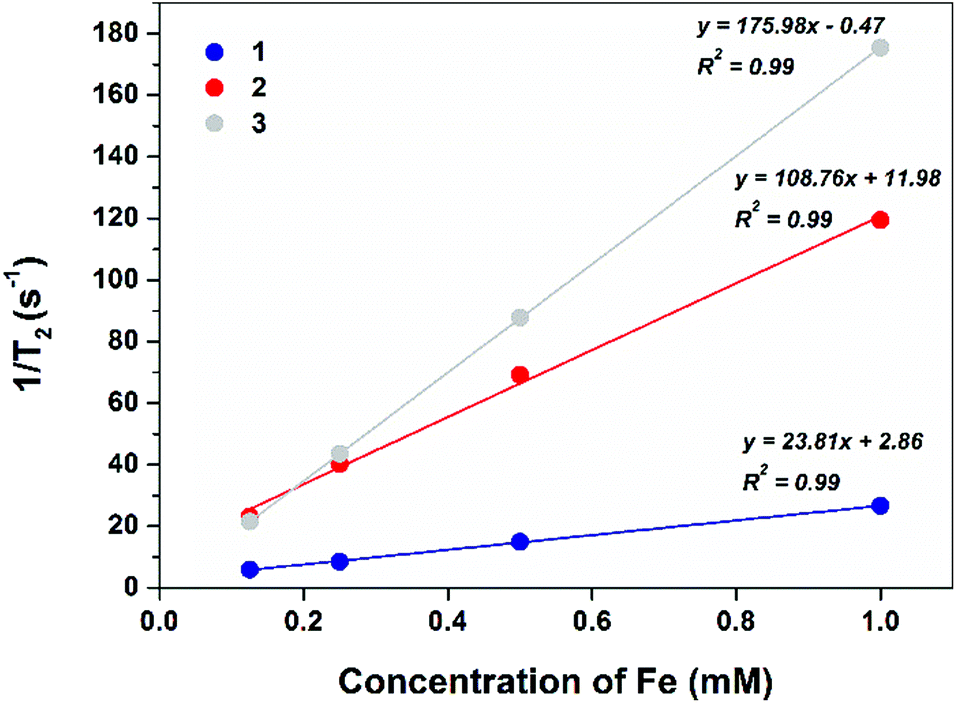 | ||
| Fig. 4 T 2 MRI relaxivity plots of 1–3 at a range of Fe concentrations (0.1–1 mM) at 3 T to determine r2. | ||
The large variability in both r1 and r2 values, as a function of chemical composition of surface coating, could be explained by a combination of factors, such as variation of hydrodynamic size, hydrophilicity and permeability of the different coatings to water molecules.42 The increase in hydrodynamic size inversely correlates with both r1 and r2 values. Although superparamagnetic contrast agents generate a long-range magnetic field for the spin-spin relaxation (T2) process, this effect decreases as distance increases. Similarly, as the mechanism for transverse (T1) relaxation is proximity based, an increase in coating thickness leads to a decrease in r1 values.43 The hydrophilicity and permeability of the surface coating also has an effect on relaxivity rates,44 although more relevant for T1 relaxation, varying the ease of water molecule access to the iron surface. The more easily that water molecules can transfer into close proximity with the iron surface, and exchange rapidly, the faster the relaxivity rates.
2.3. Gallium-68 radiolabelling and stability
In aqueous solutions, gallium is almost exclusively found in the oxidation state +3, and is highly influenced by pH, with the optimal coordination conditions situated between pH 3 and 5, since a more acidic environment may inhibit the complex formation due to protonation of donor atoms, and a neutral or basic environment causes unreactive hydroxides to form. The constructs described in this study were prepared and radiolabelled with 68GaCl3 following a standard procedure.32 Briefly, to a dried aliquot of 68GaCl3 was added a sample of NRs in 0.2 M sodium acetate solution and the reaction mixture was heated to 90 °C for 15 minutes, whilst monitoring by radio-thin layer chromatography (radio-TLC). In accordance with our previous results, the macrocyclic chelator was not required for the radiochemical incorporation of gallium-68 and quantitative radiolabelling was achieved in a 15 minute reaction time.32 In order to simulate conditions of possible gallium(III) ion release in vivo, two assays were performed, see Fig. 5. Firstly, the radiolabelled constructs were incubated in human serum. All three constructs (1–3) showed high stability (>95%) after 3 hours at 37 °C, a reasonable time point considering the short half-life of gallium-68 (68 min). As gallium(III) has a similar atomic radius to iron(II), the primary mechanism for gallium(III) bioprocessing in vivo is via iron ion pathways, such as complexation with apo-trasnferrin in blood plasma.45 In order to probe this property, the radiolabelled constructs (1–3) were incubated with an excess of apo-transferrin (0.5 mg) at 37 °C. Again, after 3 hours >95% of gallium-68 remained attached to the constructs, with no significant difference observed between differently coated NRs in either assay.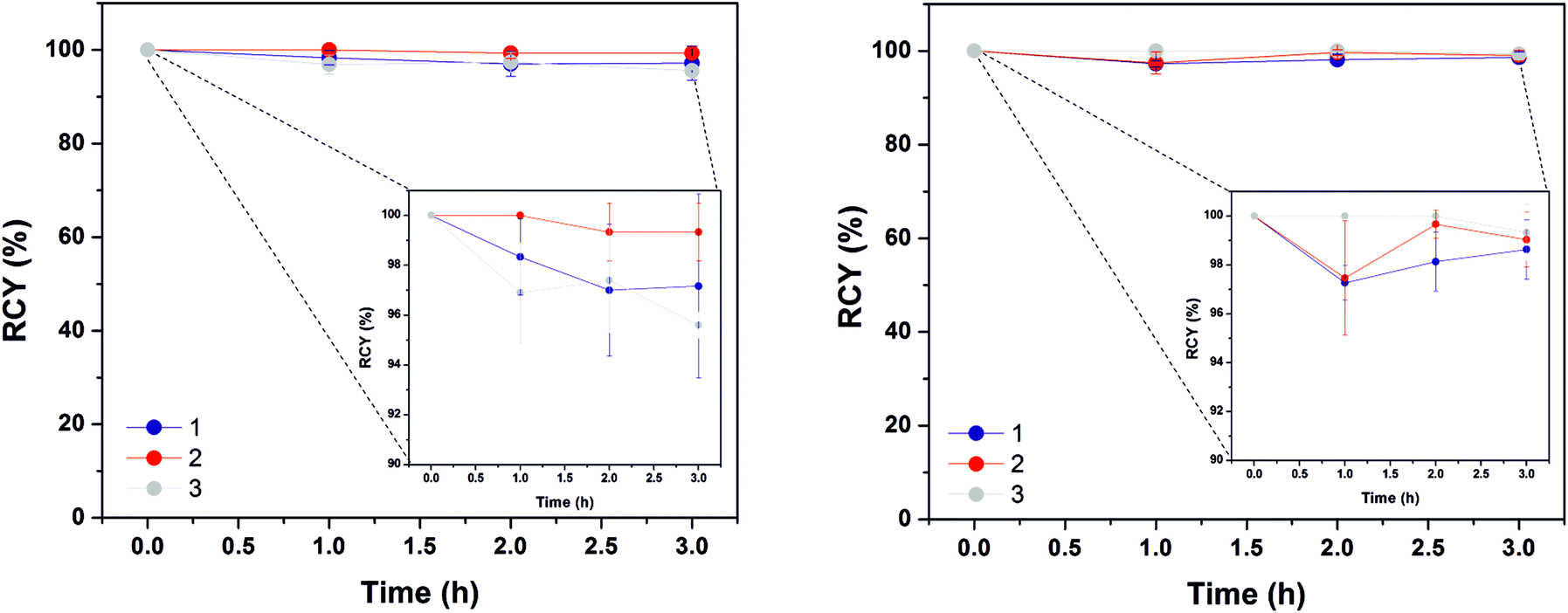 | ||
| Fig. 5 Radiochemical stability of gallium-68 radiolabelled 1–3 in human serum (left) and apo-transferrin (right) over 3 hours, measured via radio-TLC. | ||
Importantly, the 68Ga radiolabelling results indicate that a bifunctional chelating agent is not required for rapid radiolabelling or to stably attach the metal ion in vitro, as demonstrated in chelator competition assays, with 3 showing analogous labelling stability to 1 and 2. Experiments on comparable non-PEG silica coatings using triethoxy(ethyl)silane (data not shown) yielded equivalent radiochemical results, indicating that the effect is not solely a property of the PEG chains. Spectroscopic analysis of non-radioactive 69/71Ga isotope analogues failed to provide data on the existence of Ga–O–Si or Ga–O bonds as the peaks were masked by the intense Fe–O–Si and Fe–O peaks. Due to the polymeric nature of the surface coating and the high density, determined to be 37% w/w or 5.62 ligands per nm2 of iron oxide for 3 (see Fig. S4†),46 we assume that the attachment of gallium(III) to the core is unlikely. In order to assess the hypothesis of the formation of Ga–O–Si bonds, we attempted the 68Ga radiochemistry at a lower pH, which would cause siloxy protonation and hence would not be able to react with the gallium(III). Reactions performed at pH 2 with 3 using the same temperature and reaction time yielded only 4.4% ± 0.9 (n = 3) RCY. Also, by modifying the pH of the same reactions back up to 5 and heating again for 15 minutes, the RCY increased to 96.7% ± 1.5 (n = 3), supporting this hypothesis.
2.4. In vivo imaging biodistribution and stability
All three radiolabelled NRs demonstrate comparable in vitro stability. The most interesting construct is 3 due to its lack of a chelator moiety. Thus, PET-CT and MR imaging studies were carried out in mice using gallium-68 labelled 3 to assess the co-localisation of PET and MRI signals and evaluate the in vivo stability of the construct.Iron oxide nanoparticles of a similar size to ones used in this study are quickly taken up by the reticuloendothelial system (RES) and accumulate in the liver and spleen.12,37,47 PET scanning revealed that the 68Ga-3 construct rapidly (<5 min) accumulated in the liver (Fig. 6). Some limited uptake in the lung was noted in some scans; this has been observed previously with iron oxide nanoparticles and has been attributed to possible embolisation caused by post-injection aggregation.48,49 After the PET-CT study the mouse was transferred to the MR scanner and both T1 and T2 MR images were acquired (see Fig. 6 and S6†). As expected the liver T1 signal was not significantly affected by the 68Ga-3 construct. Whereas T2 weighted images revealed hypointense liver signal intensity, indicating localisation of the iron oxide NRs in the liver.
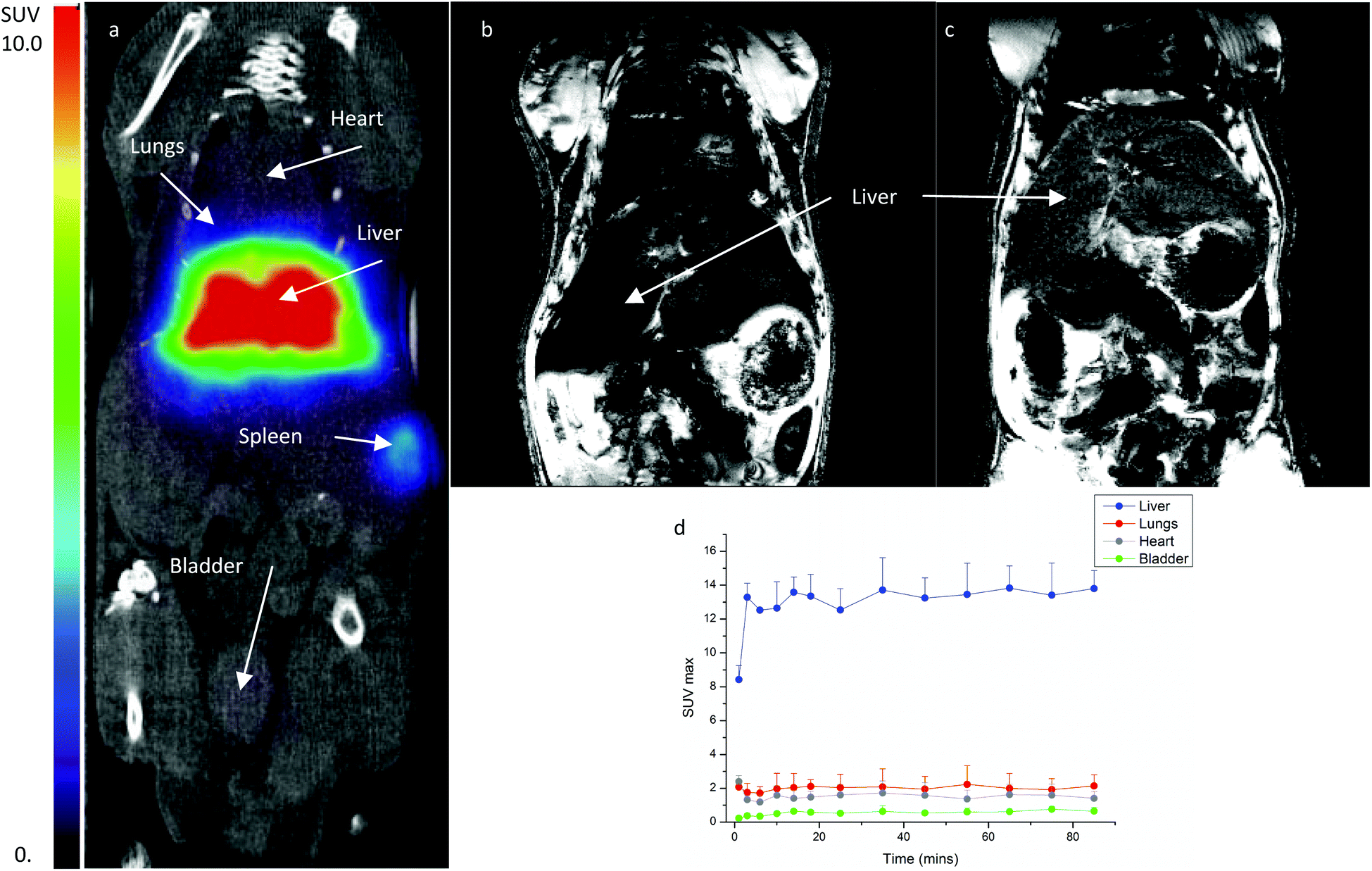 | ||
| Fig. 6 In vivo mouse images of (a) 10 MBq/50 μg Fe of 68Ga-3, fused PET-CT coronal slice image showing areas of main organ uptake at 80–90 minutes post-injection, (b) 10 MBq/50 μg Fe 68Ga-3, T2 weighted MR image at 90 minutes post-injection, (c) control mouse without nanoparticle administration, T2 weighted MR image at 90 minutes post-injection and (d) time-activity-curve for major organs from PET image after administration of 10 MBq/50 μg Fe of 68Ga-3. | ||
As a comparison, 68Ga3+ was administered as gallium(III) citrate and its biodistributon assessed in order to observe the biodistribution that may occur if the 68Ga were released from the nanorod in vivo. PET scans show that the ‘free’ 68Ga3+ is distributed more widely throughout multiple organs and is cleared much more slowly from the blood pool, see Fig. S5.† A significantly lower amount of radioactivity is noted in the liver, which is expected due to 68Ga coordination to transferrin.
The presence of 68Ga on the nanoparticle was also confirmed by ex vivo analysis, see Fig. S7.† After sacrifice (90 min post-injection), the liver was harvested and processed to separate the nanoparticles from the cell pellet, after which radio-TLC was used to determine if the radioactivity was still attached to the nanoparticle. As a comparison, the same experiment was carried out after independent administration of 68Ga-citrate and 68Ga-transferrin. It can be seen that after administration of 68Ga3+ or 68Ga-transferrin, the majority of the radioactivity is attached to transferrin. When 68Ga-3 is administered, >90% of the radioactivity is still attached to the nanorods even after analysis 90 min post-injection, which is a relatively late in vivo time point for gallium-68 imaging given its short half-life.
Debate is ongoing as to the best design parameters and applications for combined PET/MR imaging agents.50 Iron–oxide nanoparticles of this size localise in the liver via phagocytosis into the Kupffer cells and malignant liver legions often lack normal Kupffer cells and remain “bright” in an MRI scan upon SPION administration.51,52 However, the lower MRI sensitivity means that differentiation between regions with and without SPION may be challenging and may limit widespread clinical utility. Clinical utility could be increased by using PET radioisotope labelled SPIONs such as those developed in this study due to the increased sensitivity of PET and subsequent clarity of low uptake regions.
3 Conclusion
In this study, we have demonstrated that the positron emitting radioisotope 68Ga can be used to radiolabel silica coated iron–oxide nanorods via surface interactions in the absence of a bifunctional chelating agent. Studies of 68Ga-3 (NRs coated with 100% PEG) confirm that the construct is stable in vivo and demonstrate its applicability as a PET/MR multimodal imaging agent. This is particularly interesting, because coated NRs can be easily obtained in one step from commercial materials, removing challenging and complex organic synthesis steps. Thus, this study establishes a novel straight-forward method for the preparation of PET/MR multimodal imaging agents which have the potential to combine the high sensitivity of PET with MR contrast for malignant liver lesion imaging and also offer the potential for facile surface labelling methods that can be applied to currently licensed iron oxide MRI contrast agents for clinical use. This would facilitate more rapid progress through patient trials to regulatory approval.4 Experimental
4.1. Materials and methods
Tri-tert-butyl-2,2′,2′′-(10-(2-hydroxy-3-(3-(triethoxysilyl)propoxy)propyl)-1,4,7,10-tetraazacyclododecane-1,4,7-triyl)triacetate (Siloxane-DO3A) and uncoated iron oxide nanorods were synthesised following literature proceedures.32 2-Methoxy(polyethyleneoxy) propyltrimethoxysilane (Siloxane-PEG, MW 1120–1250) was purchased from Gelest Inc. (Germany). All other chemicals were purchased from Sigma Aldrich, Acros or Alfa Aesar. External rare earth magnet (N42, NdFeB, 30 × 30 × 40 mm) was purchased from MagnetExpert Ltd. Transition Emission Microscopy (TEM) was performed on air dried suitable diluted samples deposited on a carbon-coated copper grid. Images were obtained using a Gatan US4000 digital camera (Gatan UK, Abingdon, Oxford) mounted onto a JEOL 2010 transmission electron microscope (Jeol UK) running at 200 kV. FTIR spectra were collected using a Fourier transform infra-red spectrometer manufactured by Perkin Elmer (model Spectrum RXI). Nanoparticle tracking analysis (NTA) was carried out with a LM10-HS microscope from NanoSight® using a 75 mW laser at 532 nm (green). Zeta potential was measured with Zeta Nanosizer (Malvern Instruments, ZEN3600, UK) at 25 °C, the conversion of the electrophoretic mobility (μ) into zeta potential (ζ) was performed using the Smoluchowski equation. Elemental analysis for carbon, hydrogen and nitrogen was carried out using a CHN analyser EA1108 (Carlo Erba). Inductively Coupled Optical Emission Spectroscopy (ICP-OES) analysis was performed using a Perkin Elmer Optima 5300 DV. All the samples were in solid state, digested with aqua regia in glass sample vials on a hotplate. Calibration standards were prepared at 1 and 10 ppm from 1000 ppm concentrates of iron and silicon which were purchased from Romil UK. Gallium-68 was eluted from a 740 MBq 68Ge/68Ga generator (iThemba LABS/IDB Holland) and prepared for synthesis following literature methods.32 Radio-TLC analyses were carried out using either a 1480 Wizard 3′′ Gamma Counter or Lablogic Scan-Ram, equipped with a NaI detector at a speed of 10 mm min−1. Data were recorded using Lablogic Laura (version 4.1.7.70) after developing silica plates (TLC-SG, Merck KGaA) in aqueous 0.1 M citric acid to separate 68Ga-NRs (Rf = 0), 68Ga-transferrin (Rf = 0.5) and 68Ga3+ (Rf = 1).4.2. Synthesis of coated iron–oxide nanorods
General procedure for coating nanorods with siloxanes: freshly prepared nanorods were coated with siloxane-DO3A and 2-methoxy(polyethyleneoxy) propyltrimethoxysilane (Siloxane-PEG) in different ratios. In each reaction, bare NRs (0.3 g, 1.3 mmol) were suspended under nitrogen in 60% ethanol (100 ml) by extensive sonication. Ammonium hydroxide (20 ml, 35%) was added drop-wise and the solution was stirred for 30 min. The designated ratio of siloxane-DO3A and siloxane-PEG (total 0.34 mmol) was dissolved in ethanol (10 ml) and added over 10 min, the reaction was then stirred for 48 hours at room temperature. The solid precipitate formed was attracted to the bottom of the flask using an external rare earth magnet and the clear aqueous supernatant liquid was decanted. The washing and decanting procedure was repeated with 60% ethanol (2 × 100 ml), ethanol (2 × 100 ml), methanol (2 × 100 ml) and diethyl ether (100 ml) and dried. The crude sample was re-suspended in water (20 ml) filtered through a 5 μm filter followed by a 0.45 μm filter (Merck®) and dried under high vacuum to give coated NRs as a black/brown powder. Nanorods functionalised with 100% siloxane-DO3A or with 50% siloxane-DO3A were suspended in DCM (20 ml), TFA (2 ml) was added and the reaction was shaken at room temperature for 24 hours. The solid precipitate was attracted to the bottom of the flask using an external rare earth magnet and the clear aqueous supernatant liquid was decanted. The washing and decanting procedure was repeated with ethanol (2 × 20 ml), methanol (20 ml) and diethyl ether (100 ml) and dried. As previously, the crude sample was re-suspended in water (20 ml) filtered through a 5 μm filter followed by a 0.45 μm filter (Merck®) and dried under high vacuum to give coated NRs as a black/brown powder. (1 = 124 mg, 2 = 154 mg, 3 = 97 mg).4.3. MRI relaxivity studies
Relaxivity measurements were carried out on a clinical 3.0 T Discovery MR750 General Electric scanner. Longitudinal (T1) and transverse (T2) relaxation times were determined at a range of iron concentrations: 1, 0.5, 0.25 and 0.125 mM in water. A linear plot of concentration versus 1/relaxation time gives r1 and r2 as the plot gradient.4.4. PET/CT image acquisition and image analysis
CD1 nude female mice (Charles River, UK) were anaesthetised with 1–2% Isoflurane, the tail vein was catheterised and they were placed in the Minerve Small-Animal Imaging Cell (Minerve, France) and transferred to the SuperArgus 2R preclinical PET/CT scanner (Sedecal, Spain). At the start of the PET acquisition, animals were injected with ∼10 MBq, 50 μg Fe of 68Ga-3 intravenously(IV) via the tail vein and a 90-minute whole-body imaging sequence acquired (2 bed positions of 2 × 60 s, 4 × 120 s and 7 × 300 s). CT images were acquired via 360 projections/8 shots at a tube voltage of 45 kV and current of 140 μA. Respiration and temperature were monitored throughout using the SA Instruments 1025 T monitoring system (SA Instruments, CA, USA). All procedures were approved by the University of Hull Animal Welfare Ethical Review Body (AWERB) and carried out in accordance with the Animals in Scientific Procedures Act 1986 and the UKCCCR Guideline 201053 by approved protocols following institutional guidelines (Home Office Project License number 60/4549 held by Dr. Cawthorne).4.5. MR image acquisition
Magnetic resonance imaging was performed on a wide bore vertical 11.7 T MR scanner (Bruker, Germany). With a 4 cm vertical field of view with slide thickness’ of 1.0 mm and interslice distances of 1.5 mm. T1 images were acquired using a RARE inverse recovery sequence in which TE = 7.5 ms, TR = 3500 ms, flip angle = 180°, SW = 79365.1 Hz. T2 images were acquired using a T2 weighted FLASH sequence in which TE = 6.9 ms, TR = 350 ms, flip angle = 30°, SW = 50505.1 Hz.
4.6. 68Ga-3in vivo liver stability
000 RPM for 10 minutes. The supernatant was separated from the cell pellet. Radio-TLC was carried out on the supernatant to determine gallium attachment.
Acknowledgements
The authors would like to thank Ann Lowry for carrying out TEM experiments, Bob Knight for performing the ICP-OES measurements and Carol Kennedy for carrying out CHN analyses. We gratefully acknowledge the Daisy Appeal Charity for funding (Grant: DAhul0211) and the University of Hull for support of the PET Research Centre. NB would like to thank the Saudi Arabian Cultural Bureau for a Saudi Government Scholarship (S2042). The research leading to these results has received funding (AK) from the People Programme (Marie Curie Actions) of the European Union's Seventh Framework Programme FP7/2007–2013/under REA grant agreement No. 607868 (iTERM). This work was supported by STSM Grants from MPNS EU COST Action TD1007 “Bimodal PET-MRI molecular imaging technologies and applications for in vivo monitoring of disease and biological processes”. MDP and PG express their thanks to Yorkshire Cancer Research for the charity's continuing support. We thank Dr Assem Allam for his generous donation and ongoing support to the PET Research Centre at the University of Hull.Notes and references
- P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998 CrossRef CAS PubMed.
- P. Blower, Dalton Trans., 2006, 1705 RSC.
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260 RSC.
- R. R. Raylman, S. Majewski, S. Lemieux, S. S. Velan, B. Kross, V. Popov, M. F. Smith, A. G. Weisenberger and R. Wojcik, Nucl. Instrum. Methods Phys. Res., Sect. A, 2006, 569, 306 CrossRef CAS.
- F. Gerstl, C. Windischberger, M. Mitterhauser, W. Wadsak, A. Holik, K. Kletter, E. Moser, S. Kasper and R. Lanzenberger, NeuroImage, 2008, 41, 204 CrossRef PubMed.
- X. Q. Yang, H. Hong, J. J. Grailer, I. J. Rowland, A. Javadi, S. A. Hurley, Y. L. Xiao, Y. A. Yang, Y. Zhang, R. Nickles, W. B. Cai, D. A. Steeber and S. Q. Gong, Biomaterials, 2011, 32, 4151 CrossRef CAS PubMed.
- L. E. Jennings and N. J. Long, Chem. Commun., 2009, 3511 RSC.
- J.-s. Choi, J. C. Park, H. Nah, S. Woo, J. Oh, K. M. Kim, G. J. Cheon, Y. Chang, J. Yoo and J. Cheon, Angew. Chem., Int. Ed., 2008, 47, 6259 CrossRef CAS PubMed.
- L. Frullano, C. Catana, T. Benner, A. D. Sherry and P. Caravan, Angew. Chem., Int. Ed., 2010, 49, 2382 CrossRef CAS PubMed.
- M. Colombo, S. Carregal-Romero, M. F. Casula, L. Gutierrez, M. P. Morales, I. B. Bohm, J. T. Heverhagen, D. Prosperi and W. J. Parak, Chem. Soc. Rev., 2012, 41, 4306 RSC.
- E. C. Cho, C. Glaus, J. Y. Chen, M. J. Welch and Y. N. Xia, Trends Mol. Med., 2010, 16, 561 CrossRef CAS PubMed.
- R. Qiao, C. Yang and M. Gao, J. Mater. Chem., 2009, 19, 6274 RSC.
- J. W. M. Bulte and D. L. Kraitchman, NMR Biomed., 2004, 17, 484 CrossRef CAS PubMed.
- C. Sun, J. S. H. Lee and M. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1252 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995 CrossRef CAS PubMed.
- A. K. Gupta, R. R. Naregalkar, V. D. Vaidya and M. Gupta, Nanomed., 2007, 2, 23 CrossRef CAS PubMed.
- Y. X. J. Wang, S. M. Hussain and G. P. Krestin, Eur. Radiol., 2001, 11, 2319 CrossRef CAS PubMed.
- W.-W. Wang and J.-L. Yao, Mater. Lett., 2010, 64, 840 CrossRef CAS.
- A. F. Rebolledo, S. Laurent, M. Calero, A. Villanueva, M. Knobel, J. F. Marco and P. Tartaj, ACS Nano, 2010, 4, 2095 CrossRef CAS PubMed.
- S. Nath, C. Kaittanis, V. Ramachandran, N. S. Dalal and J. M. Perez, Chem. Mater., 2009, 21, 1761 CrossRef CAS PubMed.
- P. Kolhar, A. C. Anselmo, V. Gupta, K. Pant, B. Prabhakarpandian, E. Ruoslahti and S. Mitragotri, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 10753 CrossRef CAS PubMed.
- B. P. Burke, G. S. Clemente and S. J. Archibald, J. Labelled Compd. Radiopharm., 2014, 57, 239 CrossRef CAS PubMed.
- M. D. Bartholomä, Inorg. Chim. Acta, 2012, 389, 36 CrossRef.
- J.-F. Morfin and E. Toth, Inorg. Chem., 2011, 50, 10371 CrossRef CAS PubMed.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858 CrossRef CAS PubMed.
- R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686 CrossRef CAS.
- E. Wong, P. Caravan, S. Liu, S. J. Rettig and C. Orvig, Inorg. Chem., 1996, 35, 715 CrossRef CAS.
- D. J. Berry, Y. Ma, J. R. Ballinger, R. Tavare, A. Koers, K. Sunassee, T. Zhou, S. Nawaz, G. E. D. Mullen, R. C. Hider and P. J. Blower, Chem. Commun., 2011, 47, 7068 RSC.
- S. Liu and D. S. Edwards, Bioconjugate Chem., 2001, 12, 7 CrossRef CAS PubMed.
- B. P. Burke and S. J. Archibald, Annu. Rep. Prog. Chem., Sect. A: Inorg. Chem., 2013, 109, 232 RSC.
- G. J. Stasiuk and N. J. Long, Chem. Commun., 2013, 49, 2732 RSC.
- B. P. Burke, N. Baghdadi, G. S. Clemente, N. Camus, A. Guillou, A. E. Kownacka, J. Domarkas, Z. Halime, R. Tripier and S. J. Archibald, Faraday Discuss., 2014, 175, 59 CAS.
- E. K. U. Larsen, T. Nielsen, T. Wittenborn, H. Birkedal, T. Vorup-Jensen, M. H. Jakobsen, L. Ostergaard, M. R. Horsman, F. Besenbacher, K. A. Howard and J. Kjems, ACS Nano, 2009, 3, 1947 CrossRef CAS PubMed.
- M. Ma, Y. Zhan, Y. Shen, X. Xia, S. Zhang and Z. Liu, J. Nanopart. Res., 2011, 13, 3249 CrossRef CAS.
- M. Mahmoudi, S. Sant, B. Wang, S. Laurent and T. Sen, Adv. Drug Delivery Rev., 2011, 63, 24 CrossRef CAS PubMed.
- C. Nazli, T. I. Ergenc, Y. Yar, H. Y. Acar and S. Kizilel, Int. J. Nanomed., 2012, 7, 1903 CAS.
- A.-C. Faure, S. Dufort, V. Josserand, P. Perriat, J.-L. Coll, S. Roux and O. Tillement, Small, 2009, 5, 2565 CrossRef CAS PubMed.
- J. Moreau, E. Guillon, J. C. Pierrard, J. Rimbault, M. Port and M. Aplincourt, Chem. – Eur. J., 2004, 10, 5218 CrossRef CAS PubMed.
- C. M. Fisher, E. Fuller, B. P. Burke, V. Mogilireddy, S. J. A. Pope, A. E. Sparke, I. Dechamps-Olivier, C. Cadiou, F. Chuburu, S. Faulkner and S. J. Archibald, Dalton Trans., 2014, 43, 9567 RSC.
- H. S. Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. I. Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165 CrossRef CAS PubMed.
- P. Bouziotis, D. Psimadas, T. Tsotakos, D. Stamopoulos and C. Tsoukalas, Curr. Top. Med. Chem., 2012, 12, 2694 CrossRef CAS PubMed.
- F. Ye, S. Laurent, A. Fornara, L. Astolfi, J. Qin, A. Roch, A. Martini, M. S. Toprak, R. N. Muller and M. Muhammed, Contrast Media Mol. Imaging, 2012, 7, 460 CrossRef CAS PubMed.
- L. Josephson, J. Lewis, P. Jacobs, P. F. Hahn and D. D. Stark, Magn. Reson. Imaging, 1988, 6, 647 CrossRef CAS PubMed.
- H. Duan, M. Kuang, X. Wang, Y. A. Wang, H. Mao and S. Nie, J. Phys. Chem. C, 2008, 112, 8127 CAS.
- W. R. Harris and V. L. Pecoraro, Biochemistry, 1983, 22, 292 CrossRef CAS PubMed.
- Y. B. Sun, X. B. Ding, Z. H. Zheng, X. Cheng, X. H. Hu and Y. X. Peng, Chem. Commun., 2006, 42, 2765 RSC.
- F. Alexis, E. Pridgen, L. K. Molnar and O. C. Farokhzad, Mol. Pharm., 2008, 5, 505 CrossRef CAS PubMed.
- E. Locatelli, L. Gil, L. L. Israel, L. Passoni, M. Naddaka, A. Pucci, T. Reese, V. Gomez-Vallejo, P. Milani, M. Matteoli, J. Llop, J. P. Lellouche and M. C. Franchini, Int. J. Nanomed., 2012, 7, 6021 CAS.
- M. Jauregui-Osoro, P. A. Williamson, A. Glaria, K. Sunassee, P. Charoenphun, M. A. Green, G. E. D. Mullen and P. J. Blower, Dalton Trans., 2011, 40, 6226 RSC.
- R. T. M. de Rosales, J. Labelled Compd. Radiopharm., 2014, 57, 298 CrossRef PubMed.
- D.-M. Koh, Cancer Imaging, 2012, 12, 363 CrossRef PubMed.
- S. Maurea, P. P. Mainenti, A. Tambasco, M. Imbriaco, C. Mollica, E. Laccetti, L. Camera, R. Liuzzi and M. Salvatore, Quant. Imaging Med. Surg., 2014, 4, 181 Search PubMed.
- P. Workman, E. O. Aboagye, F. Balkwill, A. Balmain, G. Bruder, D. J. Chaplin, J. A. Double, J. Everitt, D. A. H. Farningham, M. J. Glennie, L. R. Kelland, V. Robinson, I. J. Stratford, G. M. Tozer, S. Watson, S. R. Wedge, S. A. Eccles, V. Navaratnam, S. Ryder and I. Natl Canc Res, Br. J. Cancer, 2010, 102, 1555 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5nr02753e |
| This journal is © The Royal Society of Chemistry 2015 |