DOI:
10.1039/C4MB00710G
(Paper)
Mol. BioSyst., 2015,
11, 1029-1040
Cancer-associated fibroblasts induce trastuzumab resistance in HER2 positive breast cancer cells†
Received
13th December 2014
, Accepted 20th January 2015
First published on 4th February 2015
Abstract
Whether and how cancer-associated fibroblasts induce trastuzumab resistance in HER2+ breast cancer is still elusive. We analyzed the percentage of cancer stem cells and multiple pathway status before and after trastuzumab treatment in HER2 positive breast cancer cells co-cultured with conditional medium (CM) from CAFs. The results suggest that CAFs induce trastuzumab resistance by expanding cancer stem cells and activating multiple pathways, such as NF-κB, JAK/STAT3 and PI3K/AKT; combination of an anti-IL6 antibody, or multiple pathway inhibitors with trastuzumab in HER2 positive breast cancer maybe a novel strategy to reverse trastuzumab resistance.
1. Introduction
HER2 gene amplification breast cancers like triple negative breast cancers (TNBCs) are the most virulent subtype of this malignancy.1 HER2 positive patients account for about 20–25% of breast cancer, and HER2 target therapy such as trastuzumab2,3 and lapatinib4 are initially effective in most HER2 positive patients with primary and metastatic diseases; however, approximately 15% of women diagnosed with early HER2-positive disease and >70% of HER2-overexpressing metastatic breast carcinomas show resistance to trastuzumab.3,5,6 It is urgent to find out new ways to improve the prognosis of these patients. Metastatic tumor relapses are characterized by rapidly proliferating, drug-resistant cancers, associated with a high mortality rate. An increasing body of evidence suggests that CAFs, the majority of tumor stromal cells in breast cancer, interact with tumor cells and promote cancer progression by secreting cytokines and activating multiple pathways.7 In this study, we sought to identify clinically targetable molecules or pathways driving the survival of trastuzumab-resistant cells induced by CAFs in HER2 positive patients.
Recent studies suggest that CAFs play important roles in breast cancer drug-resistance. Our group and other investigators showed that breast cancer cells co-cultured with CAFs were resistant to tamoxifen treatment.8,9 Moreover, CAFs are potent inducers of an epithelial-to-mesenchymal transition (EMT) and increasing cancer stem cells in mammary cells which have been associated with acquisition of tumor drug resistance and metastasis.10 Furthermore, CAFs can activate multiple pathways, such as the PI3K/AKT/mTOR pathway and the JAK/STAT3 pathway. The PI3K/AKT/mTOR pathway and other pathways were often detected to be activated in trastuzumab resistant cancer cells.11 PTEN (Phosphatase and Tensin Homolog) loss also took part in trastuzumab resistance by activating the IL6 inflammatory loop.12 These data suggest the possibility that CAFs in tumors may be involved in trastuzumab resistance in breast carcinomas by expanding cancer stem cells and activating multiple pathways.
PI3K/AKT/mTOR pathway inhibitors have been proposed and are being developed as anti-metastatic therapies in patients with breast cancer, such as everolimus,13 BEZ235 and BKM120.14 However, the impact of these inhibitors on drug-resistance induced by CAFs in HER2+ breast cancer has not yet been explored. We determined the roles of CAFs in trastuzumab resistance in HER2+ breast cancer cells; and tested the effect of combined trastuzumab with multiple pathway inhibitors in these cells. The results showed that PI3K/AKT/mTOR pathway inhibitors BEZ235, BKM120, JAK/STAT3 pathway inhibitor S3I-201, NF-κB pathway inhibitor bortezomib (BTZ), IL6 neutralizing antibody combined with trastuzumab can reverse trastuzumab resistance induced by CAFs. These studies are the first to our knowledge to demonstrate the role of CAFs in trastuzumab resistance and the efficacy of pathway inhibitors combined with trastuzumab in HER2 positive breast cancer. They provide a basis for future clinical studies to treat resistant HER2+ breast cancer patients with trastuzumab and multiple pathway inhibitors.
2. Materials and methods
Some reagents and antibodies are listed in Supplementary methods (ESI†).
2.1 Cell cultures and agents
Human breast cancer cell lines BT474 and SK-BR3 were obtained from American Type Culture Collection (Manassas, VA) and cultured in RPMI-1640 (Hyclone) supplement with 10% FBS (GIBCO) and 1% penicillin/streptomycin. They were cultured at 37 °C in a water-saturated 5% CO2 atmosphere.
2.2 RNA extraction and real-time quantitative PCR (qPCR)
Please see Supplementary methods for procedures (ESI†).
2.3 Western blot assays
Please see Supplementary methods for procedures (ESI†).
2.4 Isolation of primary fibroblasts
CAFs were isolated from human invasive mammary ductal carcinomas tissue center, non-cancer-associated fibroblasts (NAFs) from samples obtained from 3 reduction mammoplasty, in which only normal mammary tissue was detectable. All tissues were obtained from the Ruijin Hospital with approval of the hospital ethical committee and by the patient's written informed consent (Shanghai, China). The details of these patients are shown in Table S1 (ESI†). All fibroblasts were cultured in DMEM/F12 medium supplemented with 5% fetal bovine serum (FBS, GIBCO) and 5 μg mL−1 insulin (Tocris Bioscience, UK) (Supplementary methods, ESI†).
2.5 ELISA
Quantification of IL6 levels in the supernatants of fibroblasts or breast cancer cells was performed by ELISA assay according to the protocol in the quantikine ELISA kit (Cat#: DY206, R&D Systems). All experiments were done with 4 wells per experiment and repeated three times.
2.6 Proliferation assays
After a 24 h serum withdrawal, BT474 and SK-BR3 were digested and plated at 5000 cells per well into 96-well flat-bottomed cell culture plates with 5 replications in 100 μL complete culture medium for a night. Media were exchanged with 1% of FBS containing CM from CAFs, NAFs and normal DMEM/F12. After at least 48 h different concentrations of trastuzumab or vehicle (IgG) or various combinations of trastuzumab (10 μg mL−1), IL6 neutralizing antibody (100 ng mL−1), STAT3 inhibitor (S3I-201, 10 μmol L−1), PI3K inhibitors (BKM120, 1 μmol L−1, and BEZ-235, 1 μmol L−1), NF-κB inhibitor (Bortezomib, 10 nmol L−1), MEK inhibitor (U0126, 1 μmol L−1) were added into the CM. Five days later, 10 μL of cell counting kit-8 (CCK-8) (Cat#: CK04, Dojindo Molecular Technologies, Japan) reagent was added into each well and the plates were incubated in a 37 °C incubator with 5% CO2 for 3 h. Cell proliferation was determined by CCK-8 assay and read on a multi-well scanning spectrophotometer (Thermo) at A450 for wavelength correction. All experiments were repeated at least three times.
2.7 Luciferase reporter assays
Constructs of the PTEN promoter region at −1986/+14 were generated from the genomic DNA of SK-BR3 cells. The PTEN promoter was cloned into the pGL3 basic reporter gene vector and verified by sequencing. SK-BR3 and BT474 cells were maintained in RPMI 1640 containing 10% FBS and transfected using Lipofectamine™ 2000 Reagent (Invitrogen Corp, Carlsbad, CA) in 12-well plates. PTEN promoter constructs: Renilla plasmid DNA (200 ng: 20 ng) were co-transfected for 6–9 h and then incubated with IL6 (4 ng mL−1) for 36–48 h in fresh complete medium. Cells were then rinsed in cold PBS and lysed with the luciferase assay buffer. Luciferase activities were measured using a dual luciferase assay kit (Cat#: E1960, Promega, Wisconsin, USA) with a Berthold chemiluminometer (Berthold Detection Systems GmbH). The results were expressed as the ratio of firefly luciferase activity to Renilla luciferase activity. Data were expressed as the mean values and standard deviations from at least three independent transfections performed in triplicate.
2.8 Tumor sphere assay
Tumor sphere culture was performed as previously described.15,16 CM collected from CAFs and NAFs, or recombinant IL6 were added to ultralow attachment 6-well plates filled with 2 ml mammary epithelial growth medium (MEGM), and supplemented with B27 supplement (10 ng mL−1 EGF, 10 ng mL−1 bFGF, 10 μg mL−1 insulin) (Corning; Corning, NY) containing freshly plated single BC cells at the beginning of the tumor sphere formation assay. On day 10, numbers of tumor spheres (diameter ≥100 μm) were counted, and the tumor sphere efficacy was calculated based on the numbers of initially seeded cells.
For secondary sphere formation, first-passage tumor spheres from day 10 cultures were collected by gentle centrifugation (320g) and dissociated into single cells by incubation in a trypsin–EDTA solution (Invitrogen; Carlsbad, CA), before plating into new wells. Similar analysis was done as mentioned in the primary tumor sphere assay.
2.9 Flow cytometry
Single-cell suspensions prepared from the cell culture were stained with FITC-conjugated human CD44 antibody (Catalog #: 555742, BD Biosciences, Franklin Lakes, NJ), PE-conjugated CD24 antibody (Catalog #: 555574, BD Biosciences, Franklin Lakes, NJ) or isotype controls following the manufacturer's protocol. Flow cytometry assays were performed using a CyAn ADP cytometer (Dako; Carpinteria, CA) and analyzed using the FlowJo software (TreeStar; Ashland, OR).
2.10 Cell cycle analysis
BT474 and SK-BR3 single-cell suspensions were cultured in a 6-well plate at a density of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well for a night. Then media were exchanged for medium with 1% of FBS containing CM from CAFs, NAFs and Normal DMEM/F12 medium. At least 48 h later, different concentrations of trastuzumab or vehicle (IgG) or various combinations of trastuzumab (10 μg mL−1), IL6 neutralizing antibody (100 ng mL−1), STAT3 inhibitor (S3I-201, 10 μmol L−1), PI3K inhibitors (BKM120, 1 μmol L−1, and BEZ-235, 1 μmol L−1), NF-κB inhibitor (Bortezomib, 10 nmol L−1), MEK inhibitor (U0126, 1 μmol L−1) were added. After 24 h treatment, all cells were harvested and washed twice with cooled PBS and fixed in 75% ethanol for 2 h at 4 °C. The fixed cells were washed with cooled PBS and were then stained with the staining solution containing 0.05 μg mL−1 PI (Sigma-Aldrich), 1 μg mL−1 DNase-free RNase (Sigma-Aldrich) for 30 min at RT. Ten thousand events were acquired using a FACSCalibur analyzer (Becton–Dickinson, San Jose, CA, USA), and cell cycle data were determined using the Modfit software (Verity Software House, Topsham, ME, USA).
000 cells per well for a night. Then media were exchanged for medium with 1% of FBS containing CM from CAFs, NAFs and Normal DMEM/F12 medium. At least 48 h later, different concentrations of trastuzumab or vehicle (IgG) or various combinations of trastuzumab (10 μg mL−1), IL6 neutralizing antibody (100 ng mL−1), STAT3 inhibitor (S3I-201, 10 μmol L−1), PI3K inhibitors (BKM120, 1 μmol L−1, and BEZ-235, 1 μmol L−1), NF-κB inhibitor (Bortezomib, 10 nmol L−1), MEK inhibitor (U0126, 1 μmol L−1) were added. After 24 h treatment, all cells were harvested and washed twice with cooled PBS and fixed in 75% ethanol for 2 h at 4 °C. The fixed cells were washed with cooled PBS and were then stained with the staining solution containing 0.05 μg mL−1 PI (Sigma-Aldrich), 1 μg mL−1 DNase-free RNase (Sigma-Aldrich) for 30 min at RT. Ten thousand events were acquired using a FACSCalibur analyzer (Becton–Dickinson, San Jose, CA, USA), and cell cycle data were determined using the Modfit software (Verity Software House, Topsham, ME, USA).
2.11 Apoptosis analysis
All cells were subjected to the same treatment as mentioned above in cell cycle analysis. After 24 h treatment, cells were detached using EDTA-free trypsin and washed twice with cooled PBS. Cells were resuspended in 400 μL 1× buffer with 5 μL Annexin V and 5 μL PI (BD Pharmingen, San Diego, CA, USA) for 15 min on ice in the dark. Analyses were performed using the FACSCalibur analyzer (Becton–Dickinson).
2.12 Survival analysis
The 2014 version of the database including 88 HER2+ patients with enough information was analyzed for overall survival (OS) with 66% of IL6, STAT3, NF-κB and AKT gene expression as a cutoff. The results were presented as Kaplan–Meier plots and significance tested using log-rank tests. The analysis was performed according to the manufacture's instructions (http://kmplot.com/analysis/index.php?p).17
3. Results
3.1 Higher expression of α-SMA and lower expression of caveolin 1 (CAV1) in CAFs
It is well known that CAFs play important roles in cancer progression. But there were no identical markers to separate CAFs from non-cancer-associated fibroblasts (NAFs). However, increasing evidence suggests that compared to NAFs, CAFs were characterized by a higher expression of α-SMA, or loss of expression of CAV1.7 To study the interaction of CAFs and breast cancer cells, we first isolated CAFs from HER2 positive breast cancer tissue and non-cancer associated fibroblasts from patients with vice breast. Then we confirmed them using markers mentioned above. Surprisingly, α-SMA was strong and CAV1 was weak in CAF11, CAF23 and CAF64 compared with NAF1 (Fig. 1). These findings indicate that CAFs isolated from HER2+ breast cancer cells were really activated.
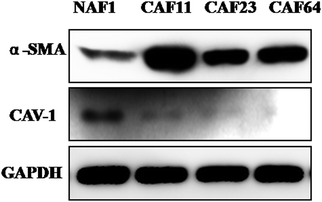 |
| | Fig. 1 Expression of CAF markers (α-SMA and CAV1) in isolated fibroblasts. Western blot analysis of lysates from CAF11, CAF23, CAF64 and NAF1 cells using anti-α-SMA and anti-CAV1 antibodies. GAPDH was used as a loading control. | |
3.2 Cancer-associated fibroblasts reduce sensitivity to trastuzumab in HER2+ breast cancer cells
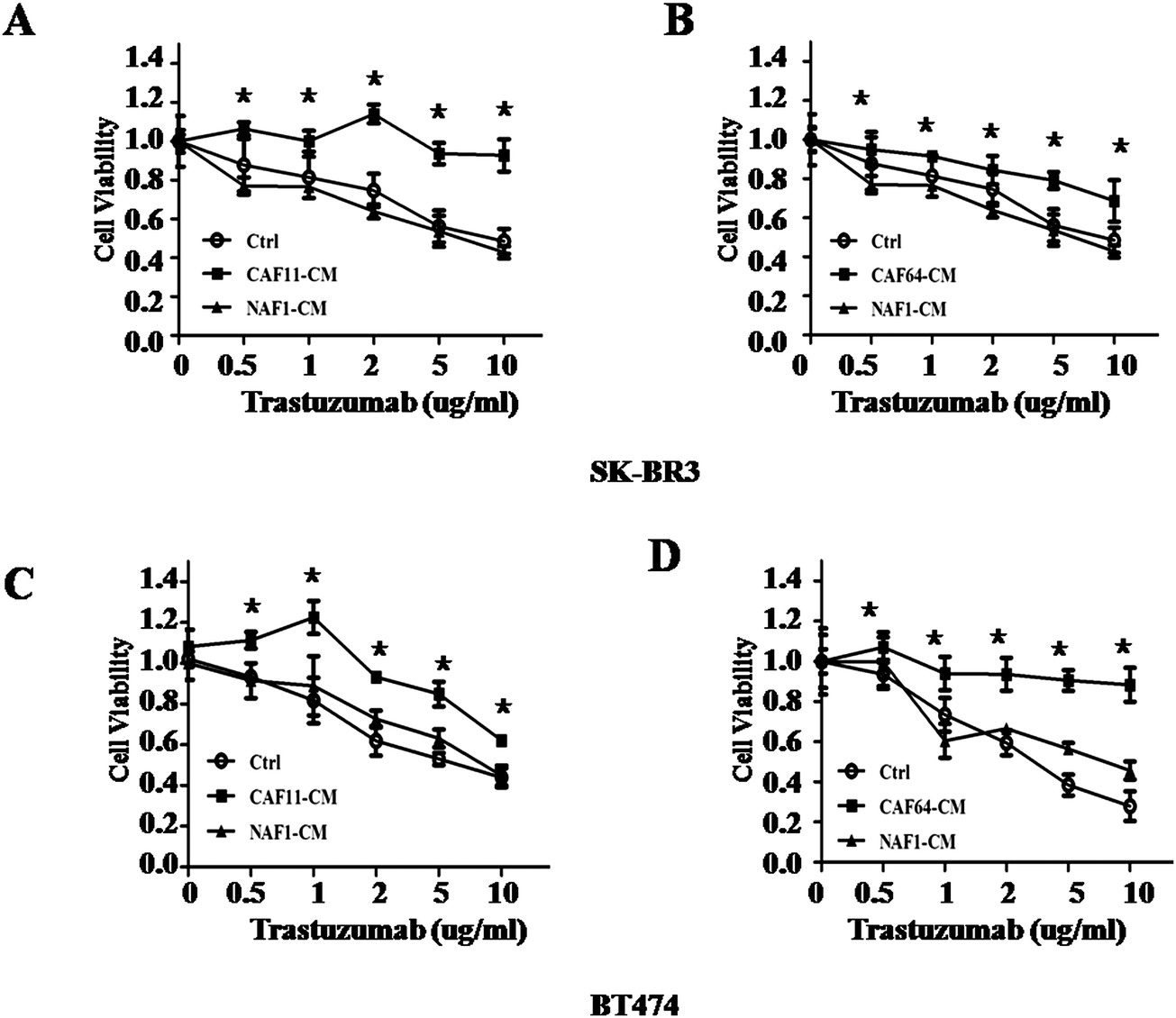
HER2 positive breast cancer is exquisitely sensitive to trastuzumab. However, at least 50% of these patients develop trastuzumab resistance during treatment.6 Since more and more studies suggested that CAFs involved in drug resistance, we asked whether CAFs in tumor can mediate trastuzumab resistance in HER2 positive breast cancer. To test this hypothesis, we first collected conditional medium (CM) from two cancer-associated fibroblasts (CAF11, CAF64) from HER2+ breast cancer and NAF1 from vice breast. Then we co-cultured BT474 and SK-BR3 with these media. Concomitantly, we observed that medium from CAFs can decrease the sensitivity to trastuzumab in BT474 and SK-BR3 cells compared with medium from NAF1 and control by CCK-8 assay (Fig. 2), indicating that fibroblasts in tumor were different from those in normal breast tissue and may participate in trastuzumab resistance.
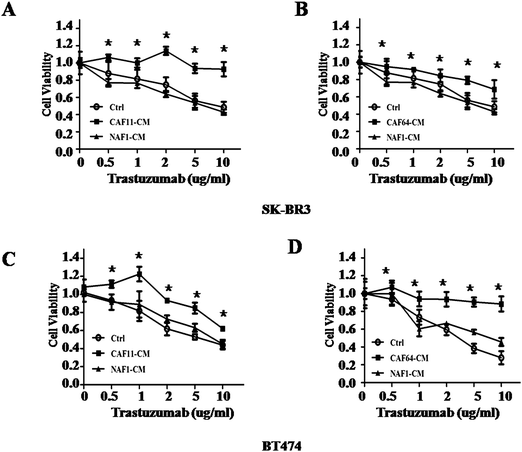 |
| | Fig. 2 Response to different concentrations of trastuzumab after culturing of SK-BR3 and BT474 cells with conditioned media from CAFs and NAFs. (A, B) Cell viability assay in SK-BR3 cells treated with different concentrations of trastuzumab after culturing with conditioned media (CM) from control, CAF11, CAF64, and NAF1 for 5 days. Three independent experiments were performed in triplicate. Data are presented as means ± s.d. * indicates p < 0.05. (C, D) Cell viability assay in BT474 cells treated with different concentrations of trastuzumab after culturing with CM from control, CAF11, CAF64, and NAF1 for 5 days. Three independent experiments were performed in triplicate. Data are presented as means ± s.d. * indicates p < 0.05. | |
3.3 IL6 is the major different cytokine secreted from CAFs and NAFs
As we know CAFs take part in cancer development and progression mainly by secreting cytokines and growth factors.7 Since IL6 has been reported to play important roles in cancer progression, we confirmed the results by ELISA in CAFs (Fig. 3A and B). The results showed that IL6 was really highly secreted from CAFs. Since some studies showed the existence of autocrine of IL6 in BT474 and SK-BR3 cells, we then compared the IL6 level among these two cell lines and CAFs. Interestingly, compared with CAFs, BT474 and SK-BR3 cells secrete less IL6 (Fig. 3C), which suggested that the paracrine way of IL6 may play more important roles in trastuzumab resistance. Moreover, recombined IL6 can partly induce trastuzumab resistance in BT474 and SK-BR3 cells (Fig. 3D). The data mentioned above indicated that IL6 may be the major factor contributing to trastuzumab resistance.
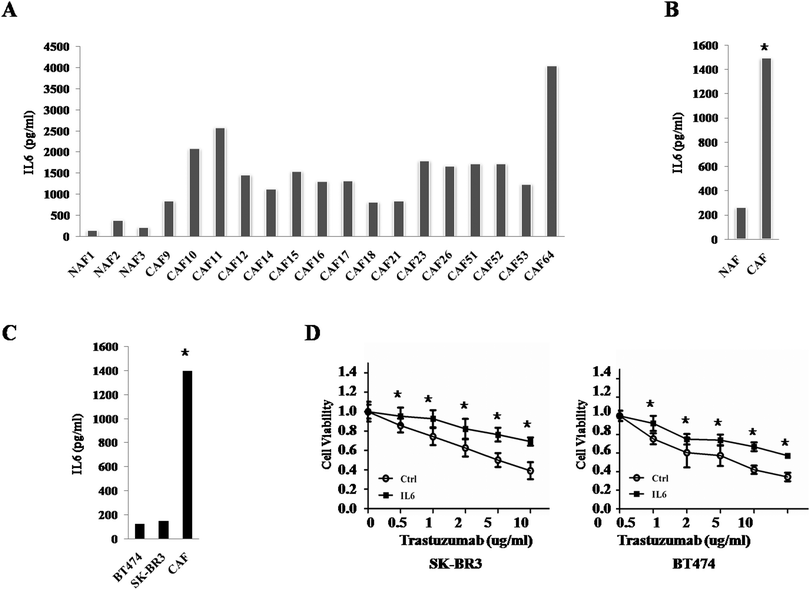 |
| | Fig. 3 IL6 is a key cytokine secreted from CAFs. (A) The ELISA results of IL6 secreted from CAFs and NAFs. The experiments were performed independently at least three times with similar results. Columns: mean of triplicate experiments; bars: s.d. (B) The ELISA results of average IL6 secreted from CAFs and NAFs. The experiments were performed independently at least three times with similar results. Columns: mean of triplicate experiments; bars: s.d. * indicates p < 0.05. (C) The ELISA results of IL6 secreted from BT474, SK-BR3 and CAFs. The experiments were performed independently at least three times with similar results. Columns: mean of triplicate experiments; bars: s.d. * indicates p < 0.05. (D) Cell viability assay in SK-BR3 (left) and BT474 (right) cells treated with different concentrations of trastuzumab after incubation in PBS or IL6 for 5 days. Three independent experiments were performed in triplicate. Data are presented as means ± s.d. * indicates p < 0.05. | |
3.4 CAF-CM and IL6 expand cancer stem cells (CSC) population
Cancer stem cells are one of the most important populations contributing to cancer recurrence and drug-resistance, and IL6 have been reported to increase CSC population by activating the JAK/STAT3 pathway,18 so we examined their changes in BT474 and SK-BR3 cells treated with IL6. The tumor sphere numbers were increased in both BT474 and SK-BR3 cells after 10 days of IL6 treatment (Fig. 4A). Moreover, CAF11-CM and CAF64-CM also increased the tumor sphere number (Fig. 4B) and subpopulation of CD44+/CD24− cells in these two cell lines after 10 days of culture (Fig. 4E and F). More importantly, after 72 hours treatment of trastuzumab, BT474 cells treated with IL6 compared with PBS still have almost 2-fold of the secondary tumor sphere number (Fig. 4C). Similar results were also found in SK-BR3 cells. Interestingly, these two cell lines cultured with CAF11-CM, CAF64-CM also have at least 3-fold of the secondary tumor sphere number compared with the control (Fig. 4D) and subpopulation of CD44+CD24− cells were also highly increased even after 72 hours treatment of trastuzumab. Furthermore, we tested other CSC-related gene expressions in cultured breast cancer cells with CAF-CM and IL6. RNA extracted from them showed that CD44 and ALDH1A1 gene expressions were upregulated after interaction with CAF-CM and IL6 in both cell lines (ESI,† Fig. S1). All these results suggested that CAF-CM and IL6 both can increase CSC population and then may decrease the sensitivity of trastuzumab in HER2 positive breast cancer cells.
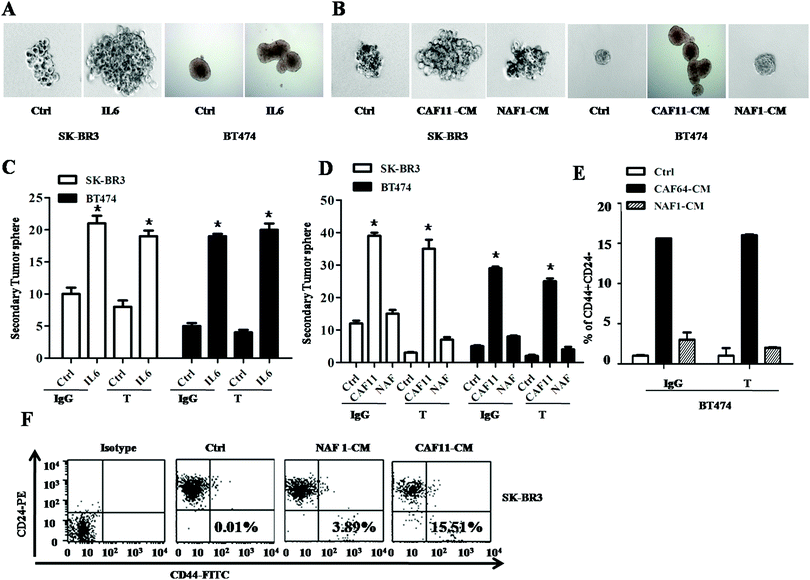 |
| | Fig. 4 IL6 secreted by CAFs expands cancer stem cells population. (A) Tumor sphere assay in SK-BR3 (left) and BT474 (right) cells treated with PBS or IL6 (4 ng mL−1) for 10 days. Three independent experiments were performed in triplicate. (B) Tumor sphere assay in SK-BR3 (left) and BT474 (right) cells treated with control, CAF11-CM or NAF1-CM for 10 days. Three independent experiments were performed in triplicate. (C) Secondary tumor sphere assay in SK-BR3 (left) and BT474 (right) cells cultured with PBS or IL6 (4 ng mL−1) upon IgG or trastuzumab (10 μg mL−1) treatment for 10 days. Three independent experiments were performed in triplicate. (D) Secondary tumor sphere assay in SK-BR3 (left) and BT474 (right) cells cultured with control, CAF11-CM or NAF1-CM upon IgG or trastuzumab (10 μg mL−1) treatment for 10 days. Three independent experiments were performed in triplicate. (E) Percentage of CD44+/CD24− population in BT474 cells cultured with control, CAF64-CM or NAF-CM upon IgG or trastuzumab (10 μg mL−1) treatment for 10 days. Three independent experiments were performed in triplicate. (F) Percentage of CD44+/CD24− population in SK-BR3 cells cultured with control, CAF11-CM or NAF-CM for 10 days by flow cytometry. Three independent experiments were performed in triplicate. | |
3.5 IL6 expands cancer stem cells population by activating the STAT3 pathway and downregulating PTEN through decreasing its transcriptional activity
To explore the mechanism of CSC expanded by IL6, we first tested CSC-related pathways. It has been reported that IL6 increased CSC population by activating the JAK/STAT3 pathway.18 So we tested this pathway and found that IL6 really increased the p-STAT3 level (Fig. 5A). More importantly, CAF23-CM also activated the JAK/STAT3 pathway in both BT474 and SK-BR3 cell lines (Fig. 5B). Moreover, to find out whether IL6 plays important roles in increasing CSC by activating these pathways, we tested the tumor sphere number and p-STAT3 protein expression after adding the IL6 neutralizing antibody to CAF11-CM and CAF64-CM. The results showed that upon addition of the IL6 neutralizing antibody, p-STAT3 expression and tumor sphere number were downregulated in BT474 and SK-BR3 cells compared with CAF-CM alone (Fig. 6C), which suggested that IL6 expands CSC population by activating the STAT3 pathway.
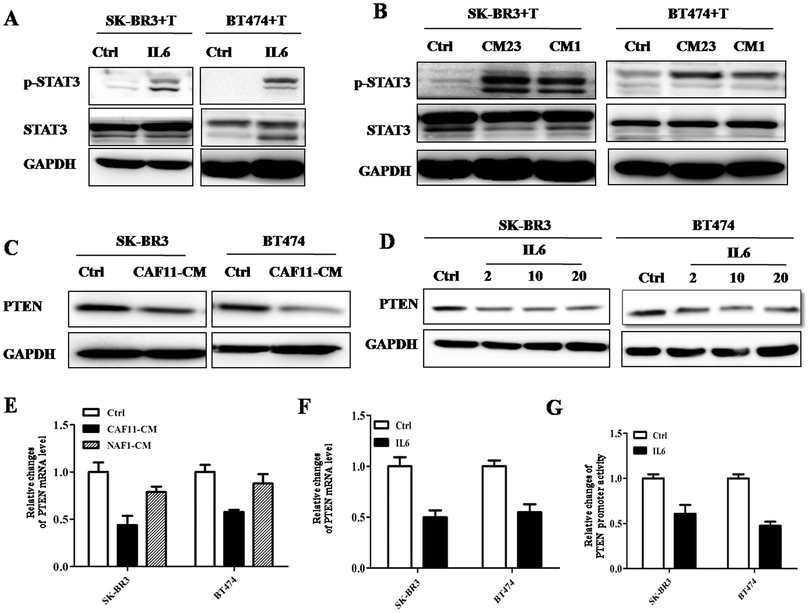 |
| | Fig. 5 IL6 activates the JAK/STAT3 pathway in HER2+ breast cancer cells. (A) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells cultured with control, CAF23-CM (CM23) or NAF1-CM (CM1) upon trastuzumab (10 μg mL−1) treatment for 24 h using indicated antibodies. GAPDH was used as a loading control. (B) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells cultured with PBS or IL6 (4 ng mL−1) upon trastuzumab (10 μg mL−1) treatment for 24 h using indicated antibodies. GAPDH was used as a loading control. (C) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells cultured with control or CAF11-CM for 24 h using anti-PTEN antibodies. GAPDH was used as a loading control. (D) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells cultured with different concentrations of IL6 (0, 0.2, 2, 4 ng mL−1) for 24 h using anti-PTEN antibodies. GAPDH was used as a loading control. (E) Quantitative real-time PCR (qRT-PCR) measuring PTEN mRNA levels in SK-BR3 (left) and BT474 (right) cells treated with control, CAF11-CM or NAF1-CM for 24 h. Data were normalized to GAPDH mRNA levels and expressed as fold change compared with controls (log2 scale). Data are shown as mean ± SE of 3 independent replicates for each condition. (F) Quantitative real-time PCR (qRT-PCR) measuring PTEN mRNA levels in SK-BR3 (left) and BT474 (right) cells treated with PBS or IL6 (4 ng mL−1) for 24 h. Data were normalized to GAPDH mRNA levels and expressed as fold change compared with controls (log2 scale). Data are shown as mean ± SE of 3 independent replicates for each condition. (G) PTEN promoter activity after transfection of the full length construct (−1086/+14) alone or together with IL6 (4 ng mL−1). pGL3-basic, control for promoter constructs. The results are expressed as a relative ratio of firefly luciferase to Renilla luciferase. Three independent experiments were performed in triplicate. | |
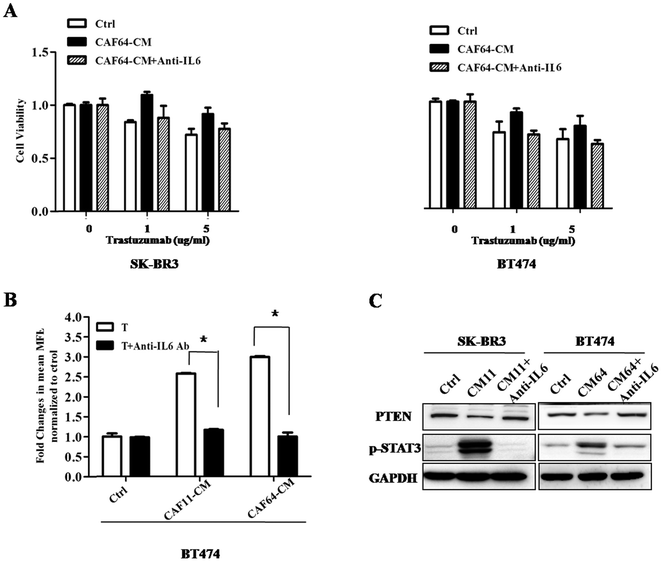 |
| | Fig. 6 IL6 neutralizing antibody decreased CSC population and restored trastuzumab sensitivity. (A) Cell viability assay in SK-BR3 (left) and BT474 (right) cells treated with different concentrations of trastuzumab after culturing with conditioned media (CM) from control, CAF64 or CAF64 combined with IL6 neutralizing antibody (100 ng mL−1) for 5 days. Three independent experiments were performed in triplicate. Data are presented as means ± s.d. * indicates p < 0.05. (B) Relative changes of the secondary tumor sphere in BT474 cells treated with trastuzumab (10 μg mL−1) alone or combined with IL6 neutralizing antibody (100 ng mL−1) after culturing with conditioned media (CM) from control, CAF11 or CAF64 for 10 days. Three independent experiments were performed in triplicate. (C) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells treated with trastuzumab (10 μg mL−1) after culturing with conditioned media (CM) from control, CAF11 (CM11), CAF64 (CM64) or combined with IL6 neutralizing antibody (100 ng mL−1) for 24 h using indicated antibodies. GAPDH was used as a loading control. | |
PTEN loss plays important roles in cancer progression and drug resistance.19 Moreover, one study suggested that PTEN loss also promotes CSC expanding.12 Therefore, we carried out further studies to find out whether IL6 affected PTEN expression. The results showed that PTEN protein expression was downregulated by CAF-CM and IL6 treatment in BT474 and SK-BR3 cells (Fig. 5C and D). We tested the mRNA level of PTEN in BT474 treated with IL6 or CAF-CM. Fig. 5E and F show that IL6 and CAF-CM both can downregulate the PTEN mRNA level, not only in BT474 but also in SK-BR3 cells. So, we performed the luciferase reporter assay to find out whether there were transcriptional activity changes in the PTEN promoter. The results showed that IL6 did decrease the transcriptional activity in the promoter of PTEN in both cell lines (Fig. 5G). The mechanisms in this process need further study.
3.6 Anti-IL6 treatment can rescue trastuzumab sensitivity by downregualting CSC
Since we found that CAFs induced trastuzumab resistance by increasing IL6, we want to answer whether blocking IL6 can reverse its sensitivity in HER2+ breast cancer. We used the IL6 neutralizing antibody to block its function and test its role in trastuzumab resistance. In Fig. 6A, it can be seen that SK-BR3 cultured with CAF64-CM and IL6 neutralizing antibody was more sensitive to trastuzumab compared with SK-BR3 cultured with CAF64-CM. Similar results were also observed in BT474. Furthermore, the tumor sphere number in BT474 also decreased after adding the IL6 neutralizing antibody to CAF64-CM (Fig. 6B). Meanwhile, PTEN expression was reversed and the JAK/STAT3 pathway was inhibited, too. These data support our previous hypothesis and that anti-IL6 treatment maybe a novel strategy to reverse trastuzumab resistance in HER2+ breast cancer cells.
3.7 Multiple activated pathways participate in trastuzumab resistance induced by CAFs
Since PI3K/AKT/mTOR pathway activation has been found to promote cancer progression, and mTOR inhibitor everolimus improved DFS in endocrine resistant patients,13 we want to find out whether this pathway also plays important roles in CAF induced trastuzumab resistance or there still exist other pathways. The results indicate that CAF11-CM and CAF64-CM from HER2 positive patients can activate the PI3K/AKT/mTOR pathway with or without trastuzumab treatment. More importantly, we also found that JAK/STAT3, NF-κB and ERK pathways were activated upon HER2-target treatment (Fig. 7A). More importantly, recombined IL6 also activated these multiple pathways even after trastuzumab treatment (Fig. 7B). These results suggest that IL6 mediates trastuzumab resistance not only by expanding CSC, but also by activating multiple pathways. So we wanted to test whether combined pathway inhibitors and trastuzumab can improve treatment response in trastuzumab resistant cells induced by CAFs. We first tested cell viability by treatment with combined trastuzumab and these pathway inhibitors: JAK/STAT3 inhibitor S3I-201, IL6 neutralizing antibody anti-IL6, PI3K/AKT pathway inhibitor BEZ235 and BKM120, NF-κB pathway inhibitor BTZ and MEK pathway inhibitor U0126. Fig. 7C shows that dual inhibitors of HER2 and one of these pathways can really improve trastuzumab activity, such as S3I-201, BEK235, BKM120 and BTZ. However, the MEK inhibitor U0126 did not have this effect. To find out the mechanisms, we tested the pathway status, apoptosis and cell cycle in these cells. As shown in Fig. 7D, every multiple pathway inhibitor can partly block its own pathway activation. However, adding anti-IL6, S3I-206, BEK235, BKM120 and BTZ compared with trastuzumab alone increased early or late apoptosis in SK-BR3 cells cultured with CAF64-CM, but U0126 had little effect in this field (Fig. S2, ESI†). We also tested cell cycle changes in BT474 and SK-BR3 cells upon the same treatment. The results showed that there were little changes in the G1 or G2/M phase, especially in the U0126 treated group. Other inhibitors, like BTZ and BKM120, almost play part of the roles in accumulation of cells in the G2/M phase fraction with concomitant reduction of cell numbers in the G1 phase, but not very obviously (Fig. S3, ESI†). These findings suggested that CAFs mediate trastuzumab resistance also by activating PI3K/AKT/mTOR, JAK/STAT3 and NF-κB pathways; adding these pathway inhibitors compared with trastuzumab alone had better treatment effect through increasing apoptosis.
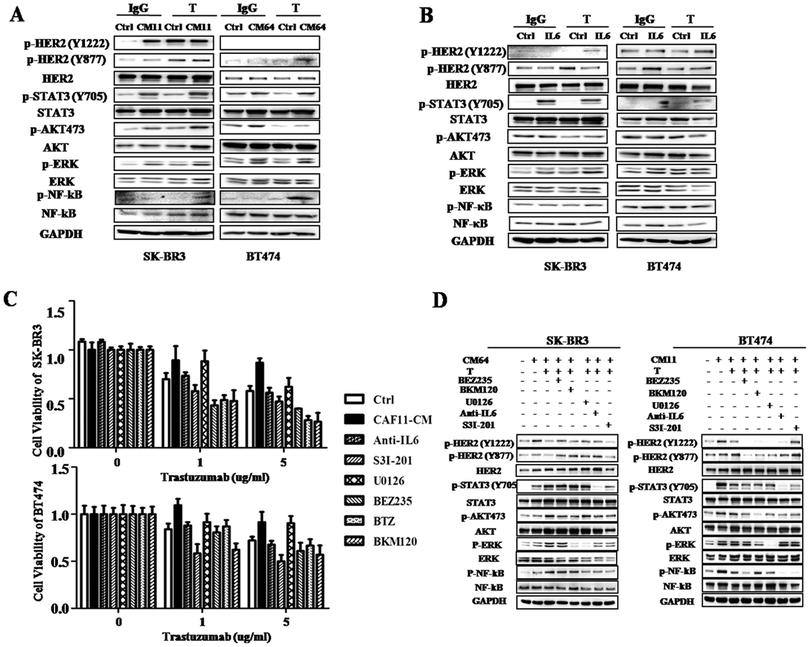 |
| | Fig. 7 CAF-CM activated multiple pathways and combined trastuzumab with inhibitors of these pathways restored trastuzumab resistance. (A) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells cultured with control or CM from CAF11 (CM11), CAF64 (CM64) and treated with trastuzumab (10 μg mL−1) or IgG for 24 h using indicated antibodies. GAPDH was used as a loading control. (B) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells cultured with PBS or IL6 (4 ng mL−1) and treated with trastuzumab (10 μg mL−1) or IgG for 24 h using indicated antibodies. GAPDH was used as a loading control. (C) Cell viability assay in SK-BR3 (left) and BT474 (right) cells cultured with control or CM from CAF11 and treated with a series of concentrations of trastuzumab with DMSO, IL6 neutralizing antibody (100 ng mL−1), STAT3 inhibitor S3I-201 (10 μM), BEZ235 (1 μM) or BKM120 (1 μM), NF-κB inhibitor Bortezomib (10 nM), MEK inhibitor U0126 (1 μM) for 5 days. Three independent experiments were performed in triplicate. Data are presented as means ± s.d. * indicates p < 0.05. (D) Western blot analysis of lysates from SK-BR3 (left) and BT474 (right) cells treated with CM from CAF64 (CM64) and treated with trastuzumab (10 μg mL−1), or trastuzumab (10 μg mL−1) combined with IL6 neutralizing antibody (100 ng mL−1), STAT3 inhibitor S3I-201 (10 μM), BEZ235 (1 μM) or BKM120 (1 μM), NF-κB inhibitor Bortezomib (10 nM), MEK inhibitor U0126 (1 μM) for 1 h using the indicated antibodies. GAPDH was used as a loading control. | |
3.8 Increasing IL6 gene expression indicates poor overall survival in HER2+ patients
To test our hypothesis in patients, we analyzed the gene expression of IL6, STAT3, NF-κB and AKT in HER2+ patients. The results showed that patients with IL6 and NF-κB gene overexpression had poor OS (n = 88, HR = 2.72, 95%CI, 1.18, 6.3, p = 0.015; HR = 2.37, 95%CI, 1.03, 5.48, p = 0.037) for 5 years follow-up (Fig. 8A and C), while patients with high expression of STAT3 and AKT genes (Fig. 8B and D) have poor OS, but do not reach the significant difference. These results may indicate that IL6 play important roles in HER2+ patients.
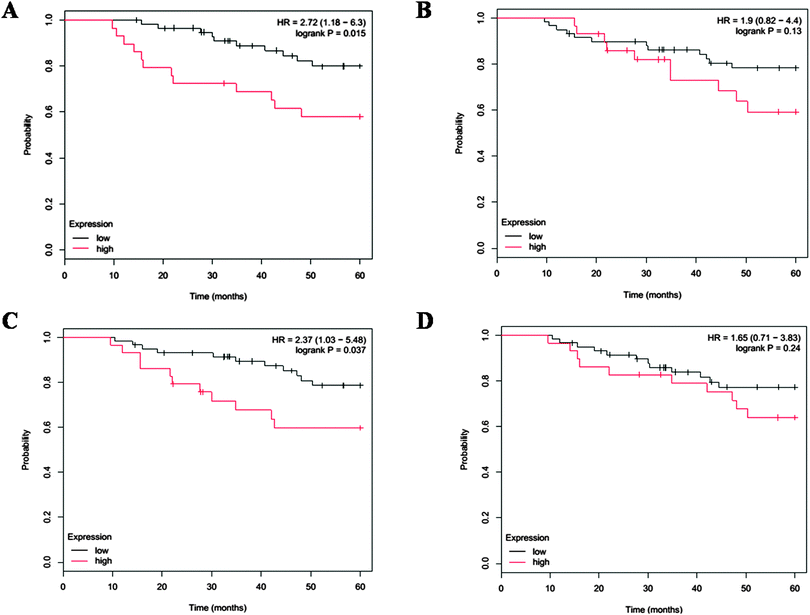 |
| | Fig. 8 Survival outcome in HER2+ patients. 5 year overall survival outcome affected by IL6 (A), STAT3 (B), NF-κB (C) and AKT (D) gene expression in 88 HER2+ patients. | |
4. Discussion
It has been proposed that tumor recurrences following treatment with anti-HER2 drugs are driven by a subpopulation of CSCs. CSCs are thought to possess intrinsic resistance to trastuzumab and other drugs compared with the bulk of tumor cell population.20,21 The tumor microenvironment has been reported to increase CSC population by secreting CCL2 in breast cancer and correlate with poor survival.10 In 2008, a stroma-derived prognostic predictor (SDPP) that stratifies disease outcome independently of standard clinical prognostic factors which highlights the importance of stromal biology in tumor progression was found.22 Therefore, to know more about the functional roles of stromal cells, such as cancer-associated fibroblasts, in breast cancer is an urgent problem to solve. To date, many studies showed that CAFs play roles in drug-resistance, such as chemoresistance,23 tamoxifen resistance8,9 and EGFR kinase inhibitor resistance.24 However, there were no data about their roles in trastuzumab resistance. In order to find out whether CAFs in HER2+ breast cancer patients participate in trastuzumab resistance, we examined the cell viability of BT474 and SK-BR3 cells cultured with CAF-CM with or without trastuzumab treatment. The results showed that CM from CAFs in tumor had the ability to decrease trastuzumab response. To test whether CSC play roles in this process, we also tested CSC-related gene expression in HER2+ breast cancer cells cultured with CAF-CM. RNA extracted from cultured BT474 and SK-BR3 cells showed enrichment of CSC-related genes, such as CD44 and ALDH1A1. Meanwhile, CD44+/CD24− cell population and tumor sphere numbers were also increased. In an effort to discover why CSC increased in this process, we examined several CSC-related pathways. The results suggested that the JAK/STAT3 pathway was activated in cultured BT474 and SK-BR3 cells even after trastuzumab treatment. These results concur with recent findings that the IL6 inflammatory loop participates in trastuzumab by expanding CSC population through JAK/STAT3 and NF-κB pathways.12 In this study, we found out that the major cytokine secreted from CAFs was also IL6, which has been reported to be correlated with expanding CSC population.12,18 Of note, IL6 is a pleiotropic cytokine and involves in a wide range of biological activities including inflammation, immune regulation, hematopoiesis and oncogenesis. Clinical studies have shown that high serum IL6 levels and high IL6 expression in tumor cells are indicative of poor prognosis and survival in breast and other kinds of cancer.25,26 Blockade of IL6 has also been reported to abrogate PTEN loss induced enrichment of CSCs and reverse trastuzumab resistance in a mouse model.12 Also, there are phase I/II clinical trials testing the effect of anti-IL6 therapy in prostate cancer,27 myeloma (NCT00911859) and ovarian cancer.25 These data suggest that IL6 may be a promising new target in cancer. Furthermore, our work also found that IL6 downregulated PTEN expression by affecting its transcriptional activity in the luciferase reporter assay which helps to increase the CSC population. Recombined IL6 also expanded the CSC population in BT474 and SK-BR3 cells. Also, adding IL6 neutralizing antibody compared with trastuzumab alone significantly reduced stem cell markers, and the tumor-initiating potential of BT474 and SK-BR3 cancer cells. However, our data do not exclude the autocrine cytokine changes in BT474 and SK-BR3 cells after culturing with CAFs. Taken together, these data suggest that upregulation of autocrine/paracrine IL6 signaling at tumor sites contributes to a tumor microenvironment that sustains CSCs in the primary cancer and also facilitates progression at metastatic sites.
In recent years, the progression in cancer treatment was targeting the activated signaling pathways. In BOLERO 2, everolimus combined with exemestane showed good response in endocrine resistant patients.13 Moreover, BOLERO-3 study showed that mTOR inhibition in combination with trastuzumab plus vinorelbine treatment significantly improved PFS compared to trastuzumab or vinorelbine alone in trastuzumab-resistant MBC patients.28 More interestingly, in our study, PI3K/AKT/mTOR pathway inhibitors BEZ235, BKM120; JAK/STAT3 inhibitors S3I-201; the NF-κB pathway inhibitor BTZ combined with trastuzumab also showed good effects to reverse trastuzumab resistance induced by CAFs. To find out the mechanisms, we did further study on the changes in signaling pathways status, apoptosis and cell cycle. The data suggested that these pathway inhibitors improved trastuzumab efficacy by increasing apoptosis in cancer cells. Besides, many preclinical and phase I/II studies showed good effect of BKM120, BEZ235 and BTZ in trastuzumab resistant cells.29–32
5. Conclusions
In summary, the results presented herein link anti-HER2 therapy, CAFs, paracrine IL6 signaling, the expansion of CSC, multiple signaling pathways activation with trastuzumab resistance (Fig. 9). We surmise that these data imply that anti-HER2 therapy in combination with systemic antagonists of IL6 signaling, either small molecular kinase inhibitor or therapeutic antibodies: PI3K/AKT pathway inhibitors BEZ235, BKM120; NF-κB pathway inhibitor BTZ may inhibit HER2+ breast tumor recurrences following trastuzumab treatment, and, as a result, improve the outcome of patients with this subtype of breast cancer.
 |
| | Fig. 9 Schematic representation showing the role of CAFs in mediating trastuzumab resistance. IL6 secreted from CAFs can increase CSC population by activating the JAK/STAT3 pathway and downregualting PTEN expression in HER2+ breast cancer cells, which induced trastuzumab resistance. Meanwhile, CAFs-CM also activate PI3K/AKT/mTOR, NF-κB pathways which may decrease apoptosis in HER2+ breast cancer cells and also contribute to trastuzumab resistance. | |
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
Research in the authors' laboratory is supported by the Doctor Innovation funding of Shanghai Jiao Tong University School of Medicine (BXJ201213), National Natural funding of China (81202087, 81202088, 81172520), Shanghai Municipal Science and Technology Commission funding (12ZR1446400, 12140901503).
References
- D. Pracella, S. Bonin, R. Barbazza, A. Sapino, I. Castellano, S. Sulfaro and G. Stanta, Dis. Markers, 2013, 35, 595–605 CrossRef PubMed.
- A. Goldhirsch, R. D. Gelber, M. J. Piccart-Gebhart, E. de Azambuja, M. Procter, T. M. Suter, C. Jackisch, D. Cameron, H. A. Weber, D. Heinzmann, L. Dal Lago, E. McFadden, M. Dowsett, M. Untch, L. Gianni, R. Bell, C. H. Kohne, A. Vindevoghel, M. Andersson, A. M. Brunt, D. Otero-Reyes, S. Song, I. Smith, B. Leyland-Jones and J. Baselga, Lancet, 2013, 382, 1021–1028 CrossRef CAS.
- C. L. Vogel, M. A. Cobleigh, D. Tripathy, J. C. Gutheil, L. N. Harris, L. Fehrenbacher, D. J. Slamon, M. Murphy, W. F. Novotny, M. Burchmore, S. Shak, S. J. Stewart and M. Press, J. Clin. Oncol., 2002, 20, 719–726 CrossRef CAS PubMed.
- A. Di Leo, H. L. Gomez, Z. Aziz, Z. Zvirbule, J. Bines, M. C. Arbushites, S. F. Guerrera, M. Koehler, C. Oliva, S. H. Stein, L. S. Williams, J. Dering, R. S. Finn and M. F. Press, J. Clin. Oncol., 2008, 26, 5544–5552 CrossRef CAS PubMed.
- F. J. Esteva, D. Yu, M. C. Hung and G. N. Hortobagyi, Nat. Rev. Clin. Oncol., 2010, 7, 98–107 CrossRef CAS PubMed.
- K. H. Lan, C. H. Lu and D. Yu, Ann. N. Y. Acad. Sci., 2005, 1059, 70–75 CrossRef CAS PubMed.
- Y. Mao, E. T. Keller, D. H. Garfield, K. Shen and J. Wang, Cancer Metastasis Rev., 2013, 32, 303–315 CrossRef PubMed.
- O. Pontiggia, R. Sampayo, D. Raffo, A. Motter, R. Xu, M. J. Bissell, E. B. Joffe and M. Simian, Breast Cancer Res. Treat., 2012, 133, 459–471 CrossRef CAS PubMed.
- M. P. Shekhar, S. Santner, K. A. Carolin and L. Tait, Am. J. Pathol., 2007, 170, 1546–1560 CrossRef CAS PubMed.
- A. Tsuyada, A. Chow, J. Wu, G. Somlo, P. Chu, S. Loera, T. Luu, A. X. Li, X. Wu, W. Ye, S. Chen, W. Zhou, Y. Yu, Y. Z. Wang, X. Ren, H. Li, P. Scherle, Y. Kuroki and S. E. Wang, Cancer Res., 2012, 72, 2768–2779 CrossRef CAS PubMed.
- S. Zhang, W. C. Huang, P. Li, H. Guo, S. B. Poh, S. W. Brady, Y. Xiong, L. M. Tseng, S. H. Li, Z. Ding, A. A. Sahin, F. J. Esteva, G. N. Hortobagyi and D. Yu, Nat. Med., 2011, 17, 461–469 CrossRef CAS PubMed.
- H. Korkaya, G. I. Kim, A. Davis, F. Malik, N. L. Henry, S. Ithimakin, A. A. Quraishi, N. Tawakkol, R. D'Angelo, A. K. Paulson, S. Chung, T. Luther, H. J. Paholak, S. Liu, K. A. Hassan, Q. Zen, S. G. Clouthier and M. S. Wicha, Mol. Cell, 2012, 47, 570–584 CrossRef CAS PubMed.
- J. Baselga, M. Campone, M. Piccart, H. A. Burris, 3rd, H. S. Rugo, T. Sahmoud, S. Noguchi, M. Gnant, K. I. Pritchard, F. Lebrun, J. T. Beck, Y. Ito, D. Yardley, I. Deleu, A. Perez, T. Bachelot, L. Vittori, Z. Xu, P. Mukhopadhyay, D. Lebwohl and G. N. Hortobagyi, N. Engl. J. Med., 2012, 366, 520–529 CrossRef CAS PubMed.
- X. Chen, M. Zhao, M. Hao, X. Sun, J. Wang, Y. Mao, L. Zu, J. Liu, Y. Shen and K. Shen, Mol. Cancer Res., 2013, 11, 1269–1278 CrossRef CAS PubMed.
- G. Dontu, W. M. Abdallah, J. M. Foley, K. W. Jackson, M. F. Clarke, M. J. Kawamura and M. S. Wicha, Genes Dev., 2003, 17, 1253–1270 CrossRef CAS PubMed.
- Y. Wang, Y. Yu, A. Tsuyada, X. Ren, X. Wu, K. Stubblefield, E. K. Rankin-Gee and S. E. Wang, Oncogene, 2011, 30, 1470–1480 CrossRef CAS PubMed.
- B. Gyorffy, A. Lanczky, A. C. Eklund, C. Denkert, J. Budczies, Q. Li and Z. Szallasi, Breast Cancer Res. Treat., 2010, 123, 725–731 CrossRef PubMed.
- L. L. Marotta, V. Almendro, A. Marusyk, M. Shipitsin, J. Schemme, S. R. Walker, N. Bloushtain-Qimron, J. J. Kim, S. A. Choudhury, R. Maruyama, Z. Wu, M. Gonen, L. A. Mulvey, M. O. Bessarabova, S. J. Huh, S. J. Silver, S. Y. Kim, S. Y. Park, H. E. Lee, K. S. Anderson, A. L. Richardson, T. Nikolskaya, Y. Nikolsky, X. S. Liu, D. E. Root, W. C. Hahn, D. A. Frank and K. Polyak, J. Clin. Invest., 2011, 121, 2723–2735 CAS.
- Y. Nagata, K. H. Lan, X. Zhou, M. Tan, F. J. Esteva, A. A. Sahin, K. S. Klos, P. Li, B. P. Monia, N. T. Nguyen, G. N. Hortobagyi, M. C. Hung and D. Yu, Cancer Cell, 2004, 6, 117–127 CrossRef CAS PubMed.
- X. Li, M. T. Lewis, J. Huang, C. Gutierrez, C. K. Osborne, M. F. Wu, S. G. Hilsenbeck, A. Pavlick, X. Zhang, G. C. Chamness, H. Wong, J. Rosen and J. C. Chang, J. Natl. Cancer Inst., 2008, 100, 672–679 CrossRef CAS PubMed.
- B. Martin-Castillo, C. Oliveras-Ferraros, A. Vazquez-Martin, S. Cufi, J. M. Moreno, B. Corominas-Faja, A. Urruticoechea, A. G. Martin, E. Lopez-Bonet and J. A. Menendez, Cell Cycle, 2013, 12, 225–245 CrossRef CAS PubMed.
- G. Finak, N. Bertos, F. Pepin, S. Sadekova, M. Souleimanova, H. Zhao, H. Chen, G. Omeroglu, S. Meterissian, A. Omeroglu, M. Hallett and M. Park, Nat. Med., 2008, 14, 518–527 CrossRef CAS PubMed.
- Y. Sun, J. Campisi, C. Higano, T. M. Beer, P. Porter, I. Coleman, L. True and P. S. Nelson, Nat. Med., 2012, 18, 1359–1368 CrossRef CAS PubMed.
- K. L. Mueller, J. M. Madden, G. L. Zoratti, C. Kuperwasser, K. List and J. L. Boerner, Breast Cancer Res., 2012, 14, R104 CrossRef CAS PubMed.
- J. Coward, H. Kulbe, P. Chakravarty, D. Leader, V. Vassileva, D. A. Leinster, R. Thompson, T. Schioppa, J. Nemeth, J. Vermeulen, N. Singh, N. Avril, J. Cummings, E. Rexhepaj, K. Jirstrom, W. M. Gallagher, D. J. Brennan, I. A. McNeish and F. R. Balkwill, Clin. Cancer Res., 2011, 17, 6083–6096 CrossRef CAS PubMed.
- R. Salgado, S. Junius, I. Benoy, P. Van Dam, P. Vermeulen, E. Van Marck, P. Huget and L. Y. Dirix, Int. J. Cancer, 2003, 103, 642–646 CrossRef CAS PubMed.
- G. Hudes, S. T. Tagawa, Y. E. Whang, M. Qi, X. Qin, T. A. Puchalski, M. Reddy, M. Cornfeld and M. Eisenberger, Invest. New Drugs, 2013, 31, 669–676 CrossRef CAS PubMed.
- M. O. Ruth O'Regan, F. Andre, M. Toi, G. H. M. Jerusalem, S. Wilks, C. Isaacs, B. Xu, N. Masuda, F. P. Arena, D. Aysel Yardley, Y. S. Yap, P. Mukhopadhyay, S. Douma, M. El-Hashimy, T. Taran, T. Sahmoud, D. E. Lebwohl and L. Gianni, J. Clin. Oncol., 2013, 31(suppl) Search PubMed , abstract 505.
- F. Cardoso, V. Durbecq, J. F. Laes, B. Badran, L. Lagneaux, F. Bex, C. Desmedt, K. Willard-Gallo, J. S. Ross, A. Burny, M. Piccart and C. Sotiriou, Mol. Cancer Ther., 2006, 5, 3042–3051 CrossRef CAS PubMed.
- T. Fujita, H. Doihara, K. Washio, K. Kawasaki, D. Takabatake, H. Takahashi, K. Tsukuda, Y. Ogasawara and N. Shimizu, Anti-Cancer Drugs, 2006, 17, 455–462 CrossRef CAS PubMed.
- B. N. Rexer, S. Chanthaphaychith, K. B. Dahlman and C. L. Arteaga, Breast Cancer Res., 2014, 16, R9 CrossRef PubMed . [Epub ahead of print].
- C. Saura, J. C. Bendell, G. Jerusalem, S. Su, Q. Ru, S. De Buck, D. Mills, S. Ruquet, A. Bosch, A. Urruticoechea, J. T. Beck, E. Ditomaso, D. W. Sternberg, C. Massacesi, S. Hirawat, L. Dirix and J. Baselga, Clin. Cancer Res., 2014, 20, 1935–1945 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4mb00710g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.