
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegiospecific synthesis of tetrasubstituted phthalocyanines and their liquid crystalline order†
Petru
Apostol
ab,
Ahmed
Bentaleb
ab,
Mbolotiana
Rajaoarivelo
ab,
Rodolphe
Clérac
ab and
Harald
Bock
*ab
aCNRS, CRPP, UPR 8641, F-33600 Pessac, France. E-mail: bock@crpp-bordeaux.cnrs.fr; Fax: +33-556845600; Tel: +33-556845673
bUniv. Bordeaux, UPR 8641, F-33600 Pessac, France
First published on 9th February 2015
Abstract
Metal-free and metal(II) all-endo-tetraalkoxy-phthalocyanines of C4h symmetry are synthesised regiospecifically from 3-(2-butyloctyloxy)phthalonitrile with lithium octanolate and subsequent metal ion exchange. The voluminous, yet not overly large, and racemically disordered alkoxy substituent not only renders the cyclotetramerisation regiospecific, but also leads to liquid crystalline self-assembly with attainable clearing temperatures and persisting columnar organisation at room temperature. A rare hexagonal mesophase with twelve columns per hexagonal unit cell is found in the metal-free homologue, whereas the metal complexes show rectangular mesophases. The clearing temperature increases with increasing axial component of the metal ion coordination sphere. At low temperature, significant antiferromagnetic exchange between magnetic centres is observed for the CoII homologue, whereas the magnetic centres are magnetically independent in the CuII derivative, in line with the observed higher clearing temperature in the CoII case that testifies of stronger interdisk interactions.
Introduction
Phthalocyanines are an immensely successful class of dyes that have been used as charge transport materials in organic electronic devices such as solar cells, including Tang and Van Slyke's seminal bilayer organic solar cell.1 When bearing a paramagnetic central metal ion, phthalocyanines may give rise to original magnetic properties such as single-molecule magnet (SMM) behaviour.2 They also are known to exhibit columnar liquid crystalline self-assembly if bearing four or eight flexible side chains,3 leading to a combination of liquid crystalline order with charge transport or magnetic properties which may give rise to pronounced anisotropic conductivity or magnetism.4Disk-shaped column-forming mesogens with a large central rigid core require long and numerous aliphatic side chains to obtain practical mesophase temperatures. If linear (non-branched) side chains are attached directly and exclusively to the outermost (exo) substitution positions, mesogenic phthalocyanines frequently exhibit a very high clearing temperature of more than 300 °C. At these high temperatures, the partial or total decomposition of these systems precludes homogeneous alignment of the columns by annealing in the vicinity of the clearing point. To lower this transition temperature to the isotropic liquid to a more workable value, eight large branched side chains have been introduced in some cases. Nevertheless this synthetic approach adds a vast amount of insulating alkyl periphery to the molecules and thus reduces considerably the part of the electronically relevant aromatic cores in the material.5 Whereas most mesogenic phthalocyanines bear eight or four side chains in the outermost (exo) substitution positions, the substitution of four of the eight bay (endo) hydrogens could be used to stabilise mesogens with moderate clearing temperatures and with a moderate relative weight of side chains. Substitution in these positions should entail the extent of the flat molecular interior and thus lead to reduced clearing temperatures without need for exceedingly large substituents. But to avoid variations in the π electron system between individual molecules, these four substituents should be introduced into well-defined positions, and mixtures of regioisomers should be avoided. This appealing strategy poses indeed a synthetic challenge. The cyclotetramerisation of 3-alkoxy-substituted phthalonitriles leads to a mixture of four regioisomers, of which the most symmetrical (1,8,15,22-substituted) isomer of C4h symmetry forms statistically in 12.5% yield, whilst the least symmetric (1,8,15,25-substituted) Cs isomer forms in 50% yield, if electronic and steric effects are negligible. Rager et al.6 showed that in the presence of templating transition metal ions such as NiII or CuII, the actual yields of C4h and Cs isomers from 3-alkoxyphthalonitriles are indeed roughly around these statistically expected values. On the other hand, they found that if 3-(2-ethylhexyloxy)phthalonitrile is tetramerised with lithium alcoholate in the absence of templating transition metal ions, the sterically favoured C4h isomer is formed in 87% relative yield, together with 11% of the statistically favoured Cs isomer, and 2% of the (1,8,18,25-substituted) C2V isomer, whilst the sterically most hindered (1,11,15,25-substituted) D2h isomer is not observed. Though the isomers could not be separated by recrystallisation nor by HPLC on commercially available stationary phases, the authors separated the isomers on specially prepared stationary HPLC phases based on phenylquinoline-grafted silica.7
Encouraged by the report of Leznoff et al.8 that with four bulky and rather rigid p-butylbenzyloxy substituents in endo positions, a poorly soluble and very high melting C4h isomer can be obtained exclusively, we conjectured that a moderate increase of bulkyness of Rager's racemically β-branched 3-alkoxy substituent could suppress the formation of the other three regioisomers altogether. Hence an electronically homogeneous metal-free phthalocyanine could thereafter be filled with divalent transition metal ions whilst exhibiting mesomorphic self-assembly. Although Rager et al.6 did not report liquid crystalline properties for their 1,8,15,22-tetrakis-(2-ethylhexyloxy)-phthalocyanine, we presumed that four such β-branched racemic alkyl substituents should efficiently favour mesophase formation, because they do so in a wide variety of tetrasubstituted polycyclic arenes that we explored in the past.9 Racemically branched alkyl chains are an efficient means of stabilising columnar mesophases by inducing a more evenly circular alkyl periphery. As a consequence, a better nanosegregation between aromatic column cores and aliphatic surrounding can be obtained, whilst hindering crystallisation due to the presence of a mixture of several stereoisomers (six in the C4h case: RRRR, RRRS, RRSS, RSRS, RSSS, SSSS, a priori with statistical abundances of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:4:2:4:1).
:4:4:2:4:1).
Results and discussion
To suppress the formation of non-C4h-symmetric isomers, we replaced 2-ethylhexanol by 2-butyloctanol in the reaction with 3-nitrophthalonitrile, and cyclised the so-obtained 3-(2-butyloctyloxy-)phthalonitrile to the corresponding phthalocyanine under transition-metal-free conditions similar to those used by Rager et al.6 To our delight, we obtained a product that showed a clean 1H-NMR spectrum (Fig. 1), indicating the sterically controlled exclusive formation of the C4h isomer. It is worth noting that the only other isomer that would give a similarly simple spectrum is the most sterically hindered D2h isomer, the only one that Rager et al. did not observe at all.6 | ||
| Fig. 1 1H-NMR spectrum of 3 in CD2Cl2 (resonance range of the aromatic protons). | ||
3-(2-Butyloctyloxy)phthalonitrile 2 was prepared by a nucleophilic substitution of the nitro group in 3-nitrophthalonitrile (1) with 2-butyl-1-octanol in DMF in the presence of K2CO3 for one week at room temperature. 1,8,15,22-Tetra(2-butyloctyloxy)-phthalocyanine 3 was synthesized from 2 with lithium octanolate in octanol under argon under slow temperature increase over 2 hours from 100 to 130 °C, followed by hydrolysis of the ensuing dilithium tetraalkoxy-phthalocyanine (Scheme 1). The 1H-NMR spectrum of 3 showed indeed only three signals in the aromatic region, one triplet and two doublets of equal intensity (Fig. 1), corresponding to the fourfold symmetry of 3. No transetherification leading to the partial replacement of 2-butyloctyl by octyl could be detected from the 1H- and 13C-NMR spectra (see ESI†). The phthalocyaninate complexes 4–7 of the divalent transition metal ions MII = ZnII, CuII, CoII and NiII were prepared by treating 3 with excess MII(CH3COO)2 in DMF for 12 hours at reflux under argon (Scheme 1) and purified by column chromatography in chloroform on silica gel.
 | ||
| Scheme 1 Synthesis of 1,8,15,22-tetra(2-butyloctyloxy)-phthalocyanine and its metal(II) complexes. (a) rac-2-Butyloctanol, K2CO3, DMF, 25 °C, 7 days; (b) lithium octanolate, octanol, 100–130 °C, 3 hours; then water; (c) M(OAc)2, DMF, reflux under Ar, 12 hours. Hex = n-hexyl. | ||
In the electronic absorption spectra of the phthalocyaninate complexes 4–7 (Fig. 2), the typical intense Q-band absorption is assigned to a π–π* transition from the HOMO of a1u symmetry to the eg symmetry LUMO. This results in a doubly degenerate first excited state of 1Eu symmetry.10 The peripheral endo-alkoxy substituents have no notable effect on the Q-band's position. As shown in Fig. 2, the split Q-band absorption of the metal-free phthalocyanine 3 appears at around 727 and 698 nm while the metallophthalocyanines give intense single Q-bands at about 700 nm, together with relatively more intense bands (hyperchromic effect) at 667 and 628 nm compared to the corresponding bands of the metal-free phthalocyanine.
 | ||
| Fig. 2 Absorption spectra (5 μM in CHCl3) of metal-free 1,8,15,22-tetraalkoxy-phthalocyanine 3 (black) and of the corresponding metal(II) phthalocyaninates 4 (Zn, yellow), 5 (Cu, red), 6 (Ni, green) and 7 (Co, blue). | ||
Metal-free 3 and its Zn, Ni and Co derivatives 4, 6 and 7 are obtained from recrystallization as crystalline powders, whereas the Cu derivative 5 separates from its cooled butanol solution as a wax. Whilst disk-shaped mesogens with non-branched alkyl chain derivatives usually show their columnar mesophase only upon heating and revert to the crystalline state on cooling to room temperature, many mesogens bearing racemically branched alkyl chains (such as used here) do not transit to the crystalline state when cooling from the temperatures of the liquid and liquid crystalline phases to room temperature. With some racemic-chain materials, a crystalline state can be obtained by crystallization from a solvent, whereas crystallization from the columnar liquid crystalline state of the undiluted material cannot be observed because crystal nucleation is inhibited by the high viscosity of the liquid crystalline state. With many other racemic chain materials, even precipitation from solution upon cooling does not yield a crystalline powder, but the material separates from solution directly in the waxy columnar liquid crystalline state.9 Here, the copper derivative 5 shows the latter behaviour. In the other cases we investigated whether, once melted upon heating, the columnar self-assembly prevails at ambient conditions. All compounds were characterised by differential scanning calorimetry (DSC) between 0 and 300 °C (Fig. 3, Table 1 and ESI†) and powder X-ray diffraction between 25 and 160 °C, (Fig. 4, Table 1 and ESI†) as well as by polarising optical microscopy at variable temperature (Fig. 5).
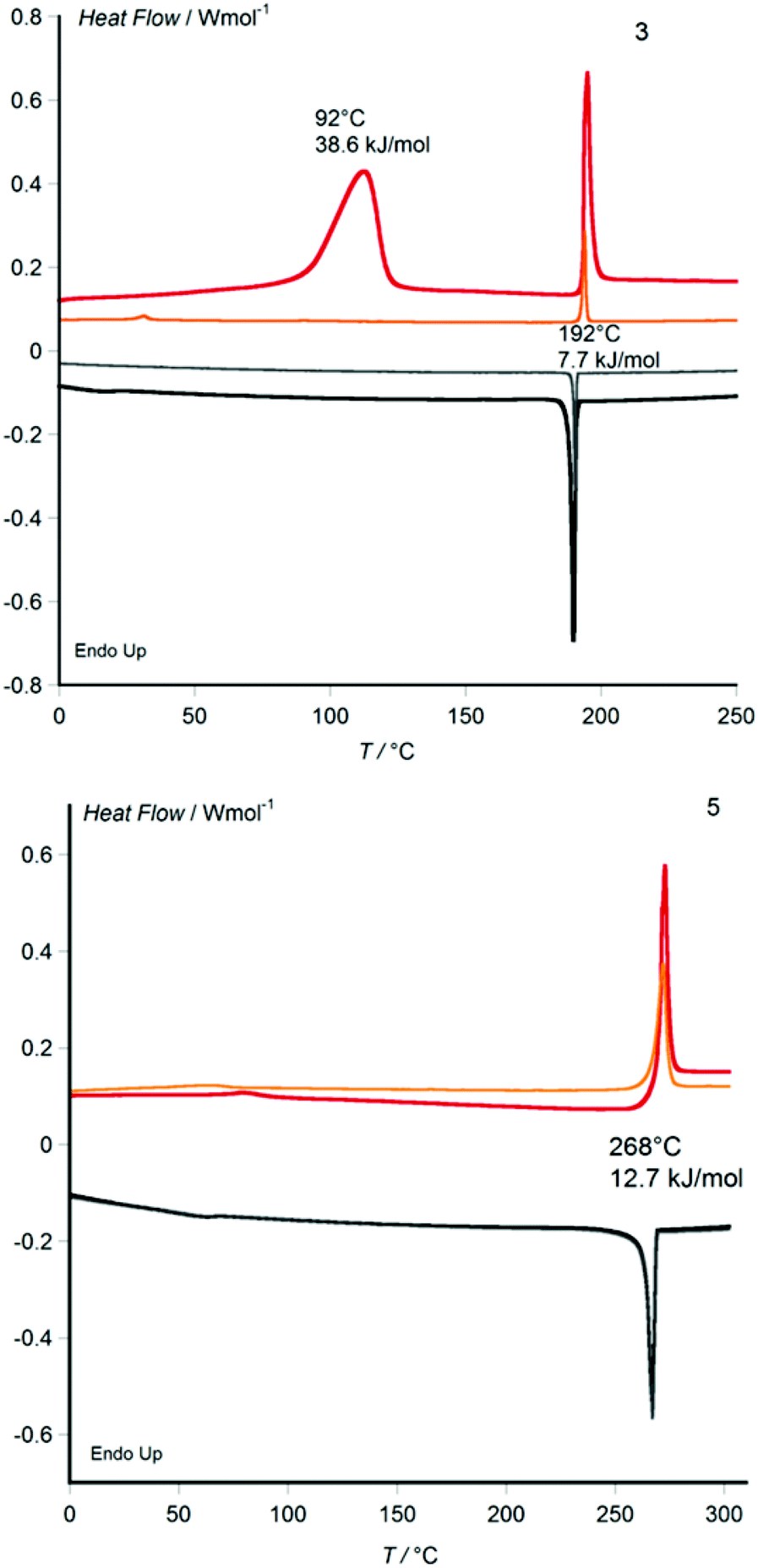 | ||
| Fig. 3 Differential scanning calorimetry (DSC) thermograms; top: metal-free phthalocyanine 3, bottom: CuII phthalocyanine 5. Onset temperatures of the melting and clearing transitions are given, with transition enthalpies. Scan rates: 10 K min−1 on first heating/cooling (red/black), 3 K min−1 on second heating/cooling (orange/grey). | ||
 | ||
| Fig. 4 Powder X-ray diffraction spectra at different temperatures upon stepwise cooling from 160 to 25 °C; top: metal-free phthalocyanine 3, bottom: CuII phthalocyanine 5. The Miller indices indicated in brackets correspond to a column lattice of hexagonal/rectangular symmetry. | ||
 | ||
| Fig. 5 Texture observed by polarising microscopy at room temperature after cooling through the isotropic liquid to liquid crystal phase transition: (a) metal-free phthalocyanine 3, (b) ZnII phthalocyanine 4, (c) CuII phthalocyanine 5, (d) NiII phthalocyanine 6, (e) CoII phthalocyanine 7. | ||
| Phase sequence (top: first heating of pristine sample, bottom: second heating) [°C] | Clearing enthalpy Hcol-iso [kJ mol−1] | 2D unit cell parameters (after initial heating to 160 °C) [Å] | |
|---|---|---|---|
| 3 (H2) | cr-84-colh-183-iso | 7.7 | 160 °C: a = 41.5 |
| colr,p-40-colr-183-iso | 25 °C: a = 35.4, b = 21.4 | ||
| 4 (Zn) | cr-96-colr-297-iso | 14.8 | 160 °C: a = 34.6, b = 24.1 |
| colr,p-80-colr-297-iso | 25 °C: a = 33.6, b = 23.2 | ||
| 5 (Cu) | colr,p-70-colr-268-iso | 12.7 | 160 °C: a = 34.8, b = 24.0 |
| colr,p-70-colr-268-iso | 25 °C: a = 33.8, b = 23.4 | ||
| 6 (Ni) | cr-63-colr-237-iso | 9.2 | 160 °C: a = 35.0, b = 23.9 |
| colr,p-45-colr-237-iso | 25 °C: a = 34.3, b = 23.3 | ||
| 7 (Co) | cr-54-colr-294-iso | 12.2 | 160 °C: a = 34.7, b = 23.9 |
| colr,p-42-colr-294-iso | 25 °C: a = 33.8, b = 23.2 |
With the exception of the CuII derivative 5 obtained as wax from solution, the DSC curves of all materials show a large-enthalpy melting transition on first heating which proves to be irreversible and does not reappear upon subsequent cooling and heating cycles. A reversible large-enthalpy transition marks the transition between mesophase and isotropic liquid (clearing transition), which occurs in all cases at temperatures below 300 °C, with a marked metal ion dependence. Whereas metal-free 3 clears at 192 °C, the ZnII and CoII derivatives 4 (297 °C) and 7 (292 °C) clear about one hundred degrees higher, with the CuII and NiII derivatives 5 (268 °C) and 6 (237 °C) in-between (Fig. 3). This can be coherently rationalised by the respective preferred coordination spheres of the four MII ions. The NiII metal ions with phthalocyanine ligands tend to adopt a square planar coordination sphere (confirmed by magnetic measurements that show the diamagnetic nature of 6), and therefore 6 shows only weak mesophase stabilisation by interdisk Ni–N interactions, expressed by an only moderate rise of the clearing temperature. In the case of the ZnII and CoII analogues, the metal ions prefer an octahedral coordination sphere that induces stronger interdisk Metal–N interactions leading to relatively high clearing points in 4 and 7. For the CuII complex, the Jahn–Teller effect should favour an elongated square bipyramidal environment, with interactions outside the phthalocyanine plane weaker than in the CoII or ZnII case but stronger than for the NiII analogue, yielding an intermediate clearing temperature for 5.
Besides the melting and clearing transition, metal-free 3 shows a reversible small-enthalpy transition at around 40 °C on the second heating cycle (Fig. 3). Similar weak, poorly expressed small-enthalpy transitions are detected between room temperature and 100 °C for all four MII derivatives 4–7 that could be the signature of either (i) a transition between two mesophases of different column lattices, (ii) a mesophase-to-mesophase transition involving only an increase of intermolecular order without change of column lattice, or (iii) a glass transition involving no structural order increase.
Powder X-ray diffraction (Fig. 4) reveals that the small-enthalpy transition at low temperature in metal-free 3 corresponds to a transition from a hexagonal mesophase at higher temperature to a rectangular phase at lower temperature, accompanied with the expected increased number of diffraction peaks in the rectangular phase due to the lowering of symmetry. The hexagonal mesophase is strikingly atypical because the main reflection is accompanied by a secondary peak corresponding to a lattice distance shorter by a factor  instead of the usual
instead of the usual  or
or  . Whilst density considerations confirm that the main peak corresponds as usual to the distance between neighbouring columns, the unusual closeness of the secondary (210) peak indicates that the main peak is the (200) peak instead of the usual (100) peak. Therefore the edges of the hexagonal 2D unit cell span over two instead of one column-to-column distance, and the unit cell contains 12 instead of 3 columns, implying that there are several distinct sets of columns in the lattice. Usually, hexagonal columnar mesophases consist of columns in which the molecular planes are on average perpendicular to the column axis, whereas the lower symmetry of rectangular columnar mesophases is a result of the tilt of the molecular planes out of the plane perpendicular to the column axis, breaking the circular symmetry of the columns. In addition, usually not all columns are tilted in the same direction, and most rectangular mesophases are made up of two sets of columns distinguished by their different tilt directions. Likewise, having several types of columns in a hexagonal mesophase can be explained by the tilting in some of the columns. Indeed Levelut and co-workers have identified in a chiral hexaalkoxy-triphenylene such a rare hexagonal mesophase with 12 columns per hexagonal unit cell (i.e. 4 columns in the lozenge-shaped minimal unit cell).11 In their case, at least 9 columns are tilted, three each in three symmetrically compensating tilt directions. A similar tilt arrangement can thus be presumed for the high temperature hexagonal mesophase of metal-free 3.
. Whilst density considerations confirm that the main peak corresponds as usual to the distance between neighbouring columns, the unusual closeness of the secondary (210) peak indicates that the main peak is the (200) peak instead of the usual (100) peak. Therefore the edges of the hexagonal 2D unit cell span over two instead of one column-to-column distance, and the unit cell contains 12 instead of 3 columns, implying that there are several distinct sets of columns in the lattice. Usually, hexagonal columnar mesophases consist of columns in which the molecular planes are on average perpendicular to the column axis, whereas the lower symmetry of rectangular columnar mesophases is a result of the tilt of the molecular planes out of the plane perpendicular to the column axis, breaking the circular symmetry of the columns. In addition, usually not all columns are tilted in the same direction, and most rectangular mesophases are made up of two sets of columns distinguished by their different tilt directions. Likewise, having several types of columns in a hexagonal mesophase can be explained by the tilting in some of the columns. Indeed Levelut and co-workers have identified in a chiral hexaalkoxy-triphenylene such a rare hexagonal mesophase with 12 columns per hexagonal unit cell (i.e. 4 columns in the lozenge-shaped minimal unit cell).11 In their case, at least 9 columns are tilted, three each in three symmetrically compensating tilt directions. A similar tilt arrangement can thus be presumed for the high temperature hexagonal mesophase of metal-free 3.
The presence of tilted columns in the high temperature mesophase has consequences on the alignment behaviour in thin films. Whereas usual non-tilted hexagonal columnar mesophases have a strong tendency for homeotropic alignment (with columns perpendicular to the film-substrate interfaces and with no apparent birefringence) when cooling through the transition from the isotropic liquid to the mesophase, 3 shows preferred planar alignment between glass plates, with typical fan-shaped birefringent domains (Fig. 5a). 3 shares this tendency to planar alignment in fan-shaped domains with the metal ion derivatives 4–7 (Fig. 5d), but depending on film thickness and cooling speed, also fingered, less birefringent domains and near-hexagonal overall growth patterns typical of a homeotropic or near-homeotropic alignment can be observed (Fig. 5b,c,e).
The metal-free derivative 3 is the only one to show a hexagonal mesophase, whereas all four metal complexes 4–7 directly form a rectangular mesophase upon cooling from the isotropic liquid, as testified by the presence of the two close (200) and (110) diffraction peaks in the powder diffractogram (Fig. 4 and ESI†).
In 3, the transition from the hexagonal mesophase to the rectangular phase is testified by the splitting of the single main diffraction peak at higher temperature into two close (200) and (110) peaks at lower temperature (inset Fig. 4a). Only part of the several high angle peaks appearing in the rectangular phase can be indexed in the frame of the rectangular column lattice, including the (300), (220), (310) and (120) peaks. The presence of weak additional high angle peaks indicates that this phase exhibits not only order in the two dimensions of the column lattice, but additionally also partial order in the third dimension. This, together with the very small transition enthalpy to the hexagonal mesophase, indicates that the rectangular phase is best addressed as a plastic crystalline phase (implying disorder at the atomic scale but order in three dimensions on the molecule-to-molecule scale).
In the metal derivatives 4–7, powder X-ray diffraction reveals that whilst the Bragg peaks of the rectangular column lattice of the high temperature phase are maintained, further high-angle peaks that are not indexable (in the frame of the bidimensional rectangular column lattice) appear on approaching room temperature. This indicates, as in the metal-free homologue 3, a transition from liquid crystalline to plastic crystalline order. In contrast to the concomitant symmetry change in 3, this small-enthalpy transition to plastic crystalline order proceeds in 4–7 without significant change to the column lattice (Fig. 4 and ESI†).
The magnetic properties of the metal-free 3 and its ZnII, CuII, NiII and CoII derivatives 4, 5, 6 and 7 have also been investigated. While the expected diamagnetic nature of 3 (χdia = −9.4(5) × 10−4 cm3 mol−1), 4 (−9.4(5) × 10−4 cm3 mol−1) and 6 (−8.8(5) × 10−4 cm3 mol−1) has been confirmed, the temperature dependence of the magnetic susceptibility χ for compound 5 and 7 reveals their paramagnetism (Fig. 6). At room temperature, the χT product estimated at 0.43 and 0.69 cm3 K mol−1 for 5 and 7 respectively corresponds well to the expected values for S = 1/2 CuII and CoII magnetic centres with g factors of 2.1(1) and 2.7(1) respectively.12
 | ||
| Fig. 6 Temperature dependence of the χT product at 1000 Oe for 5 and 7 between 1.8 and 300 K (with χ defined as the molar magnetic susceptibility equal to M/H). | ||
When lowering the temperature, the χT product decreases significantly for the CoII analogue suggesting the presence of significant antiferromagnetic interactions between S = 1/2 CoII sites (it is worth noting that the intrinsic magnetic anisotropy of the CoII metal ion can also be partially responsible for the observed decrease). On the other hand, the CuII derivative displays a Curie-like paramagnetism indicating the quasi-absence of magnetic interactions between S = 1/2 CuII magnetic centres. The fit of the experimental data to the Curie–Weiss law confirms this tendency with Weiss constants of −0.06 and −0.90 K for 5 and 7 respectively (while Curie constants are estimated at 0.43 and 0.69 cm3 K mol−1 respectively). This result is well in line with the discussion of the clearing points (vide supra) that support stronger interdisk Metal–N interactions in the CoII complex 7 (thus mediating strong magnetic interactions) than in 5 due to the expected Jahn–Teller distortion of the CuII square bipyramidal coordination sphere.
Conclusions
In summary, the introduction of a moderately voluminous racemic β-branched 3-(2-butyloctyl)oxy substituent in the phthalonitrile precursor allows the regiospecific formation of an all-endo-tetraalkoxy-phthalocyanine and its transition metal ion complexes that show columnar liquid crystalline phases with clearing transition temperatures below 300 °C. Whilst the metal-free homologue shows an unusual hexagonal columnar mesophase with a twelve-column unit cell at elevated temperatures, the ZnII, CuII, NiII and CoII derivatives form rectangular columnar mesophases. Their clearing temperatures follow the order Ni < Cu < Co ≈ Zn, in coherence with their preferential coordination spheres that pass from planar to octahedral in this order, implying increasing interdisk metal–nitrogen interactions within the columnar stack. As inferred by powder X-ray diffraction, the columnar order is maintained upon cooling to ambient conditions. All five homologues exhibit a small enthalpy liquid crystal to plastic crystal transition upon cooling to room temperature, which in the case of the metal-free homologue includes a transition from hexagonal to rectangular column lattice symmetry. The combination of attainable clearing temperatures with room temperature columnar stacking and a relatively high content of conjugated core within the molecular mass make these materials potentially useful as uniformly orientable charge transporters in organic electronic devices. In addition, the rational control of the intra-column magnetic interactions between metal ion sites could possibly lead to original cooperative magnetic behaviour at low temperatures.13 Therefore the choice of paramagnetic centres coordinating these mesogenic all-endo-tetraalkoxy-phthalocyanine ligands and the introduction of well designed magnetic linkers between metal ions could be optimised and lead to new orientable one-dimensional magnetic systems.Experimental
NMR spectra were recorded on a JEOL ECS-40 spectrometer, IR spectra with a Nicolet 6700 FT-IR spectrometer and UV/visible spectra with a Unicam UV 4 spectrometer. Differential scanning calorimetry (DSC) was performed on a Perkin-Elmer DSC7 apparatus, and polarising optical microscopy on a Olympus BH-2 microscope. Mass spectra were performed on a QStar Elite mass spectrometer (Applied Biosystems) in positive mode. Powder X-ray diffraction data were collected on a Rigaku Nanoviewer (XRF microsource generator, MicroMax 007HF), with a 1200 W rotating anode coupled to a confocal Max-Flux® Osmic mirror (Applied Rigaku Technologies, Austin, USA) and a MAR345 image plate detector (MARResearch, Norderstedt, Germany); samples were put in glass capillaries which were exposed to the X-ray beam in a high temperature oven (ambient to 250 °C). The detector was placed at a distance of 309 mm providing access to 2θ angles in the 0.5° to 29° range. Magnetic susceptibility measurements were performed on a Quantum Design SQUID magnetometer MPMS-XL housed at the Centre de Recherche Paul Pascal at temperatures between 1.8 and 300 K and dc magnetic fields ranging from −7 to 7 T. The measurements were carried out on freshly filtered polycrystalline samples (9.54, 23.18, 22.90, 17.68 and 15.18 mg for 3, 4, 5, 6 and 7 respectively) introduced in a sealed polyethylene bag (3 × 0.5 × 0.02 cm). Prior to the experiments, the field-dependent magnetisation was measured at 100 K on each sample in order to detect the presence of any bulk ferromagnetic impurities. In fact, paramagnetic or diamagnetic materials should exhibit a perfectly linear dependence of the magnetisation that extrapolates to zero at zero dc field. The samples appeared to be free of any significant ferromagnetic impurities. The magnetic data were corrected for the sample holder and the intrinsic diamagnetic contributions.3-(2-Butyloctyloxy)phthalonitrile (2)
3-Nitrophthalonitrile 1 (24.0 g, 0.144 mol), K2CO3 (60 g, 0.56 mol) and racemic 2-butyl-1-octanol (40.3 ml, 0.833 g mL−1, 0.180 mol) were thoroughly stirred in anhydrous DMF (250 mL) for 7 days at room temperature. After adding water and extracting three times with ethyl acetate, the solvent was evaporated in vacuo. The residue was chromatographed in DCM on silica gel and crystallized from ca. 1500 mL of a 2:1 water–ethanol mixture. Yield: 13.35 g (31%) of white solid. 1H NMR (400 MHz, CD2Cl2, TMS): δ = 7.61 (t, J = 8 Hz, 1H), 7.31 (d, J = 8 Hz, 1H), 7.22 (d, J = 8.5 Hz, 1H), 4.0 (d, J = 5.5 Hz, 2H), 1.84 (hept, J = 6 Hz, 1H), 1.48–1.38 (m, 4H), 1.34–1.23 (m, 12H), 0.88 ppm (t, J = 7.0, 3H), 0.86 ppm (t, J = 7.0, 3H); 13C NMR (100 MHz, CD2Cl2, TMS): δ = 162.7, 135.1, 125.4, 117.5, 117.4, 116.2, 113.8, 105.3, 73.3, 38.2, 32.4, 31.6, 31.3, 30.2, 29.5, 27.3, 23.5, 23.2, 14.42, 14.37 ppm; IR (ATR): ![[small nu, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0ce.gif) = 3081 (w), 2955 (vs), 2925 (s), 1855 (s), 2230 (m, CN) cm−1; elemental analysis calcd (%) for C20H28N2O: C 76.88, N 8.96, H 9.03; found C 76.70, N 9.05, H 9.10.
= 3081 (w), 2955 (vs), 2925 (s), 1855 (s), 2230 (m, CN) cm−1; elemental analysis calcd (%) for C20H28N2O: C 76.88, N 8.96, H 9.03; found C 76.70, N 9.05, H 9.10.
1,8,15,22-Tetra-(2-butyloctyloxy)-phthalocyanine (3)
Lithium (0.40 g, 0.057 mol) was heated in 1-octanol (70 ml) at 170 °C until all the metal was dissolved. After cooling to 100 °C, 3-(2-butyloctyloxy)phthalonitrile, 2, (2.0 g, 6.40 mmol) was added and the temperature was raised in small steps over 2 hours to 130 °C whereupon the mixture became green. After stirring for a further hour at 130 °C, the dark green solution was cooled at room temperature, poured into water and extracted with chloroform. The solvent was evaporated and the residue was purified by column chromatography in chloroform on silica gel followed by recrystallization from butanol. Yield: 0.83 g (43%) of blue-green powder. 1H NMR (400 MHz, CD2Cl2, TMS): δ = 9.05 (d, J = 7.5 Hz, 4H), 8.10 (t, J = 7.5 Hz, 4H), 7.67 (d, J = 7.5 Hz, 4H), 4.59 (d, J = 5.5 Hz, 8H), 2.46 (hept, J = 6 Hz, 4H), 2.15 (m, 8H), 1.96 (m, 8H), 1.71–1.61 (m, 16H), 1.50–1.24 (m, 32H), 0.96 (t, J = 7.5 Hz, 12H), 0.79 (t, J = 7.5 Hz, 12H), −0.39 ppm (s, 2H); 13C NMR (100 MHz, CD2Cl2, TMS): δ = 157.4, 150.4, 140.0, 131.3, 123.8, 115.8, 113.1, 72.7, 39.3, 32.6, 32.0, 31.7, 30.6, 29.9, 27.6, 23.9, 23.3, 14.6, 14.4 ppm; IR (ATR): = 3292 (w) 2952 (w), 2922 (vs), 2853 (s), 1586 (s), 1494 (m), 1458 (m), 1333 (s), 1264 (s), 1225 (s), 1142 (s), 1058 (s), 1010 (s), 931 (w), 864 (w), 797 (w), 743 (w), 726 (s), 705 (s) cm−1; UV-Vis (CHCl3): λmax = 727(146000), 696(131000), 662(42800), 627(32200) nm(M−1 cm−1); magnetic susceptibility measurements: diamagnetic constant of about −9.3 × 10−4 cm3 K mol−1 that compares well with the expected theoretical value (MW × 10−6 cm3 K mol−1); elemental analysis calcd (%) for C80H114N8O4: C 76.75, N 8.95, H 9.18; found 76.45, N 9.05, H 9.48.
Zinc 1,8,15,22-Tetra-(2-butyloctyloxy)-phthalocyaninate (4)
Compound 3 (0.50 g, 0.40 mmol) was refluxed with Zn(OAc)2 (0.60 g, 2.73 mmol) in DMF (50 ml) for 12 hours under argon. After cooling to room temperature, the dark green solution was poured into water and extracted with chloroform, purified by column chromatography in chloroform on silica gel followed by recrystallization from butanol/ethanol. Yield: 0.38 g (72%), of blue-green solid. IR (ATR, cm−1): = 2952 (w), 2921 (vs), 2853 (s), 1585 (s), 1490 (m), 1459 (m), 1338 (s), 1270 (s), 1250 (s), 1133 (s), 1080 (s), 1042 (s), 947 (w), 876 (w), 798 (w), 750 (w), 735 (s); UV-Vis (CHCl3): λmax = 704(220000), 673(36600), 632(41400) nm(M−1 cm−1); MS: m/z [M + H]+ calcd 1313.8, found 1314.1; magnetic susceptibility measurements: diamagnetic constant of about −9.4 × 10−4 cm3 K mol−1 that compares well with the expected theoretical value (MW × 10−6 cm3 K mol−1); elemental analysis calcd (%) for C80H112ZnN8O4: C 73.06, N 8.52, H 8.58; found C 72.95, N 8.54, H 8.75.
Copper 1,8,15,22-Tetra-(2-butyloctyloxy)-phthalocyaninate (5)
Compound 3 (0.22 g, 176 μmol) was refluxed with Cu(OAc)2·H2O (0.60 g, 3 mmol) in DMF (50 ml) for 12 hours under argon. The complex was purified by column chromatography as described above for the zinc complex and recrystallization from butanol. Yield: 0.18 g (73%) of dark green waxy solid. IR (ATR, cm−1): = 2952 (w), 2921 (vs), 2854 (s), 1590 (s), 1490 (m), 1461 (m), 1330 (s), 1270 (s), 1250 (s), 1138 (s), 1080 (s), 1060 (s), 941 (w), 886 (w), 797 (w), 750 (w), 739 (s); UV-Vis (CHCl3): λmax = 707(225000), 675(39400), 635(46000) nm(M−1 cm−1); MS: m/z [M + H]+ calcd 1312.8, found 1313.1; magnetic susceptibility measurements: paramagnetic behaviour with an effective moment at 300 K of 1.8 μB (χT = 0.42 cm3 K mol−1 and thus g = 2.1(1)) indicating an S = 1/2 species; elemental analysis calcd (%) for C80H112CuN8O4: C 73.16, N 8.53, H 8.59; found C 73.05, N 8.20, H 8.83.
Nickel 1,8,15,22-Tetra-(2-butyloctyloxy)-phthalocyaninate (6)
Compound 3 (0.50 g, 0.40 mmol) was refluxed with Ni(OAc)2 (0.60 g, 3.39 mmol) in DMF (50 ml) for 12 hours under argon. The complex was purified by column chromatography as described above for the zinc complex and recrystallization from butanol. Yield: 0.48 g (92%), of green solid. IR (ATR, cm−1): = 2953 (w), 2922 (vs), 2854 (s), 1597 (s), 1493 (m), 1459 (m), 1332 (s), 1270 (s), 1250 (s), 1143 (s), 1080 (s), 1060 (s), 956 (w), 899 (w), 794 (w), 750 (w), 735 (s); UV-Vis (CHCl3): λmax = 700(200000), 668(39600), 627(38600) nm(M−1 cm−1); MS: m/z [M + H]+ calcd 1307.8, found 1308.1; magnetic susceptibility measurements: diamagnetic constant of about −8.8 × 10−4 cm3 K mol−1 that compares well with the expected theoretical value (MW × 10−6 cm3 K mol−1); elemental analysis calcd (%) for C80H112NiN8O4: C 73.43, N 8.56, H 8.62; found C 73.11, N 8.72, H 9.00.
Cobalt 1,8,15,22-Tetra-(2-butyloctyloxy)-phthalocyaninate (7)
Compound 3 (0.50 g, 0.40 mmol) were refluxed with Co(OAc)2·4H2O (0.70 g, 2.81 mmol) in DMF (50 ml) for 12 hours under argon. The complex was purified by column chromatography as described above for the zinc complex and recrystallization from butanol. Yield: 0.35 g (67%) of blue solid. IR (ATR): = 2952 (w), 2921 (vs), 2854 (s), 1593 (s), 1491 (m), 1459 (m), 1331 (s), 1270 (s), 1250 (s), 1172 (s), 1060 (s), 1045 (s), 956 (w), 897 (w), 795 (w), 750 (w), 735 (s) cm−1; UV-Vis (CHCl3): λmax = 694(163000), 625(36600) nm(M−1 cm−1); MS: m/z [M + H]+ calcd 1308.8, found 1309.1; magnetic susceptibility measurements: paramagnetic behaviour with an effective moment at 300 K of 2.3 μB (χT = 0.69 cm3 K mol−1 and thus g = 2.7(1)) indicating a low spin S = 1/2 species; elemental analysis calcd (%) for C80H112CoN8O4: C 73.41, N 8.56, H 8.62; found C 72.91, N 8.72, H 8.95.
Acknowledgements
We are grateful to the Région Aquitaine, ANR, Capes-Cofecub and Erasmus Mundus (Action 2, Strand 1/EMA2) for financial support and to Fabien Durola and Mathieu Rouzières for their help and advice.Notes and references
- C. W. Tang and S. A. Van Slyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS PubMed.
- (a) M. Gonidec, F. Luis, A. Vilchez, J. Esquena, D. B. Amabilino and J. Veciana, Angew. Chem., Int. Ed., 2010, 49, 1623 CrossRef CAS PubMed; (b) S. Sakaue, A. Fuyuhiro, T. Fukuda and N. Ishikawa, Chem. Commun., 2012, 48, 5337 RSC; (c) F. Branzoli, P. Carretta, M. Filibian, G. Zoppellaro, M. J. Graf, J. R. Galan-Mascaros, O. Fuhr, S. Brink and M. Ruben, J. Am. Chem. Soc., 2009, 131, 4387 CrossRef CAS PubMed; (d) N. Ishikawa, M. Sugita, T. Ishikawa, S.-Y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694 CrossRef CAS PubMed; (e) N. Ishikawa, M. Sugita and W. Wernsdorfer, J. Am. Chem. Soc., 2005, 127, 3650 CrossRef CAS PubMed.
- (a) N. B. McKeown, Phthalocyanine Materials: Synthesis, Structure and Function, Cambridge University Press, 1998 Search PubMed; (b) C. Piechocki, J. Simon, A. Skoulios, D. Guillon and P. Weber, J. Am. Chem. Soc., 1982, 104, 5245 CrossRef CAS; (c) N. Kumaran, P. A. Veneman, B. A. Minch, A. Mudalige, J. E. Pemberton, D. F. O'Brien and N. R. Armstrong, Chem. Mater., 2010, 22, 2491 CrossRef CAS; (d) G. Schweicher, G. Gbabode, F. Quist, O. Debever, N. Dumont, S. Sergeyev and Y. H. Geerts, Chem. Mater., 2009, 21, 5867 CrossRef CAS; (e) L. Sosa-Vargas, F. Nekelson, D. Okuda, M. Takahashi, Y. Matsuda, Q.-D. Dao, Y. Hiroyuki, A. Fujii, M. Ozaki and Y. Shimizu, J. Mater. Chem. C 10.1039/c4tc02557a.
- (a) C. Deibel, D. Janssen, P. Heremans, V. De Cupere, Y. Geerts, M. L. Benkhedir and G. J. Adriaenssens, Org. Electron., 2006, 7, 495 CrossRef CAS PubMed; (b) T. Inabe and H. Tajima, Chem. Rev., 2004, 104, 5503 CrossRef CAS PubMed; (c) S. Sergeyev, W. Pisula and Y. H. Geerts, Chem. Soc. Rev., 2007, 36, 1902 RSC; (d) H. Xu, R. Chen, Q. Sun, W. Lai, Q. Su, W. Huang and X. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- S. Sergeyev, E. Pouzet, O. Debever, J. Levin, J. Gierschner, J. Cornil, R. Gomez Aspec and Y. H. Geerts, J. Mater. Chem., 2007, 17, 1777 RSC.
- C. Rager, G. Schmid and M. Hanack, Chem. – Eur. J., 1999, 5, 280 CrossRef CAS.
- M. Sommerauer, C. Rager and M. Hanack, J. Am. Chem. Soc., 1996, 118, 10085 CrossRef CAS.
- C. C. Leznoff, M. Hu, C. R. McArthur and Y. Qin, Can. J. Chem., 1994, 72, 1990 CrossRef CAS.
- (a) T. Hassheider, S. A. Benning, H.-S. Kitzerow, M.-F. Achard and H. Bock, Angew. Chem., Int. Ed., 2001, 40, 2060 CrossRef CAS; (b) S. Saïdi-Besbes, E. Grelet and H. Bock, Angew. Chem., Int. Ed., 2006, 45, 1783 CrossRef PubMed; (c) H. Bock, M. Rajaoarivelo, S. Clavaguera and E. Grelet, Eur. J. Org. Chem., 2006, 2889 CrossRef CAS; (d) S. Alibert-Fouet, I. Seguy, J.-F. Bobo, P. Destruel and H. Bock, Chem. – Eur. J., 2007, 13, 1746 CrossRef CAS PubMed; (e) J. Kelber, M.-F. Achard, B. Garreau de Bonneval and H. Bock, Chem. – Eur. J., 2011, 17, 8145 CrossRef CAS PubMed; (f) J. Kelber, H. Bock, O. Thiebaut, E. Grelet and H. Langhals, Eur. J. Org. Chem., 2011, 707 CrossRef CAS.
- M. Gouterman and G. H. Wagniere, J. Mol. Spectrosc., 1963, 11, 108 CrossRef CAS.
- A. M. Levelut, P. Oswald, A. Ghanem and J. Malthete, J. Phys., 1984, 45, 745 CAS.
- R. L. Carlin, Magnetochemistry, Springer-Verlag, Berlin Heidelberg, 1986 Search PubMed.
- (a) C. Coulon, H. Miyasaka and R. Clérac, Struct. Bonding, 2006, 122, 163 CrossRef CAS; (b) C. Coulon, V. Pianet, M. Urdampilleta and R. Clérac, Struct. Bonding DOI:10.1007/430_2014_154.
Footnote |
| † Electronic supplementary information (ESI) available: DSC, powder X-ray diffraction and NMR data. See DOI: 10.1039/c5dt00076a |
| This journal is © The Royal Society of Chemistry 2015 |