
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Spectroscopic study on uranyl carboxylate complexes formed at the surface layer of Sulfolobus acidocaldarius†
Thomas
Reitz
*ab,
Andre
Rossberg
a,
Astrid
Barkleit
a,
Robin
Steudtner
a,
Sonja
Selenska-Pobell
a and
Mohamed L.
Merroun
c
aHelmholtz-Center Dresden-Rossendorf, Institute of Resource Ecology, Bautzener Landstrasse 400, 01328 Dresden, Germany. E-mail: thomas.reitz@ufz.de; Fax: +49 345 5585449; Tel: +49 345 5585409
bHelmholtz-Center for Environmental Research, Department of Soil Ecology, Theodor-Lieser-Strasse 4, 06120 Halle, Germany
cDepartamento de Microbiología, Universidad de Granada, Campus Fuentenueva s/n 18071, Granada, Spain
First published on 11th November 2014
Abstract
The complexation of U(VI) at the proteinaceous surface layer (S-layer) of the archaeal strain Sulfolobus acidocaldarius was investigated over a pH range from pH 1.5 to 6 at the molecular scale using time-resolved laser-induced fluorescence spectroscopy (TRLFS) and U LIII-edge extended X-ray absorption fine structure (EXAFS). The S-layer, which represents the interface between the cell and its environment, is very stable against high temperatures, proteases, and detergents. This allowed the isolation and purification of S-layer ghosts (= empty cells) that maintain the size and shape of the cells. In contrast to many other microbial cell envelope compounds the studied S-layer is not phosphorylated, enabling the investigation of uranyl carboxylate complexes formed at microbial surfaces. The latter are usually masked by preferentially formed uranyl phosphate complexes. We demonstrated that at highly acidic conditions (pH 1.5 to 3) no uranium was bound by the S-layer. In contrast to that, at moderate acidic pH conditions (pH 4.5 and 6) a complexation of U(VI) at the S-layer via deprotonated carboxylic groups was stimulated. Titration studies revealed dissociation constants for the carboxylic groups of glutamic and aspartic acid residues of pKa = 4.78 and 6.31. The uranyl carboxylate complexes formed at the S-layer did not show luminescence properties at room temperature, but only under cryogenic conditions. The obtained luminescence maxima are similar to those of uranyl acetate. EXAFS spectroscopy demonstrated that U(VI) in these complexes is mainly coordinated to carboxylate groups in a bidentate binding mode. The elucidation of the molecular structure of these complexes was facilitated by the absence of phosphate groups in the studied S-layer protein.
Introduction
Microorganisms, and in particular their cell wall structures, are known to effectively adsorb uranium and other heavy metals from even low concentrated solutions.1,2 The high metal complexation ability is primarily based on two facts: the high surface-to-volume ratio and the large number of metal binding ligands, which are present at the organic cell surface polymers, e.g. peptidoglycan, lipopolysaccharides, proteins, and glycolipids. These ligands primarily include charged functional groups, such as phosphoryl, carboxyl, hydroxyl, amine and sulfhydryl groups. Recent studies suggested that uranyl, initial coordinated to these functional groups, can subsequently serve as nucleation sites for further uranium precipitation.3,4 Therefore, a deeper knowledge on the complexation mechanisms at microbial surfaces is fundamental for understanding and modelling uranium behavior in the environment. In order to determine the structural key compounds for the complexation of U(VI), selected components of prokaryotic cell walls and membranes have been purified and their interactions with U(VI) have been investigated. Spectroscopic studies on lipopolysaccharides, which represent the main components of the cell walls of Gram-negative bacteria demonstrated that U(VI) was predominantly complexed via phosphoryl groups within a pH range of 2.5 to 9.5 A corresponding predominant binding of U(VI) to phosphoryl groups have also been described for glycerol-1-phosphate6 as well as o-phosphoethanolamine (NH3CH2CH2OPO3−) and 1,2-dimyristoyl-sn-glycero-3-phosphate (DMGP)7 that represent the polar head and the non-polar tail of phospholipids, the major components of cell membranes. While the latter studies consistently revealed that phosphoryl groups are the main U(VI) binding site provided by microbial surfaces, some studies also demonstrate that carboxyl groups are involved in the uranyl complexation. In 2009 Barkleit and colleagues described three different uranyl carboxylate complexes formed at phosphate-free peptidoglycan, the main component of the cell walls of Gram-positive bacteria.8 The outermost cell envelope structure of many prokaryotic organisms, however, includes a proteinaceous surface layer (S-layer). These highly ordered and self-assembling monomolecular protein layers, if present, forms the permeable interface between the cell and its environment and are among the most abundant (up to 20%) cellular proteins.9 In Gram-positive bacteria the S-layer is usually anchored to peptidoglycan and in Gram-negative bacteria mostly to the lipopolysaccharides of their outer membrane. In many archaea, as in the case of the here studied strain Sulfolobus acidocaldarius, the S-layer is even the only cell wall structure.10 A high potential for U(VI) complexation by S-layer proteins has already been described for a Bacillus isolate.11 The high U(VI) binding capacity was attributed by the authors to the considerably high content of carboxylated amino acid residues and to the unusual fact that this S-layer is phosphorylated, which is rather uncommon for the S-layers studied up to date.9In this work the S-layer of S. acidocaldarius was studied, which consists of assembled protein subunits (SlaA) that forms a p3-symmetry (Fig. S1†). The S-layer is fasten to the cells via an anchoring protein (SlaB) which is integrated into the cytoplasmic membrane12 (Fig. S1†). In contrast to the before mentioned Bacillus species, this S-layer is not phosphorylated. Instead, each of the SlaA protein subunits comprises 18 aspartic acid and 27 glutamic acid residues, i.e. each SlaA protein theoretically provides a total number of 45 binding-capable, carboxylic groups. Moreover, amine side chains of arginine, histine and lysine in the SlaA protein additionally provide 41 potential binding sites. The main goal of this study was to determine the molecular structure of uranyl complexes formed at organic macromolecules of microbial surfaces. The investigation of these complexes is possible due to the natural absence of phosphorylated sites in the S-layer of S. acidocaldarius. The strain had originally been isolated from geothermal springs and it grows optimally at 79 °C and at pH between 2 and 3.13 Representatives of this species are, however, rather adaptive and have been found in a range of different habitats. Their presence was demonstrated, for instance, also at moderate temperatures in heavy metal contaminated acidic soils.14
This study continues the investigation of the interactions between isolated microbial cell envelope compounds and U(VI) on a molecular level. The results should improve the understanding and modelling U(VI)–microbe interactions as well as biogeochemical U(VI) behaviour in nature.
Materials and methods
Preparation of the S-layer
S. acidocaldarius (strain DSM 639) was cultivated according to our studies on whole cells.15 Preparation of the S-layer was performed in the following way: For cell lysis, the biomass was suspended in 10 mM HEPES buffer (pH 7), containing 2 mM EDTA and 0.15% SDS, and stirred for one hour at room temperature.16 After addition of 4 mM MgCl2, released DNA was digested by DNase I. To achieve complete removal of the cytoplasmic membrane and the SlaB protein, the cell lysate was treated with 2% SDS and stirred overnight. After centrifugation of the suspension (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g, 30 min), the lower dark part of the resulting pellet was discarded, whereas the upper white part was suspended in HEPES buffer (pH 7) containing 2 mM EDTA and 2% SDS and incubated for 60 min at 60 °C. After that the suspension was centrifuged and the upper part of the resulting pellet, consisting of the purified S-layer was suspended in distilled water. SDS was removed by eight wash steps with distilled water. The S-layer suspension was stored at 4 °C and microscopically checked for microbial contamination before use.
000g, 30 min), the lower dark part of the resulting pellet was discarded, whereas the upper white part was suspended in HEPES buffer (pH 7) containing 2 mM EDTA and 2% SDS and incubated for 60 min at 60 °C. After that the suspension was centrifuged and the upper part of the resulting pellet, consisting of the purified S-layer was suspended in distilled water. SDS was removed by eight wash steps with distilled water. The S-layer suspension was stored at 4 °C and microscopically checked for microbial contamination before use.
Biochemical analyses
The protein content of the S-layer suspension was analyzed by using denaturing sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE).17 For complete solubilization, the S-layers were incubated in sodium carbonate buffer (20 mM, pH 10) for 20 min at 45 °C18 and subsequently heated (95 °C) for 5 min in SDS sample buffer.17 The proteins were separated on a 10% separation gel. Because of the low content of basic amino acids (0.3 mol% arginine, 0.3 mol% histidine and 2.2 mol% lysine)19 staining of the gel was performed with colloidal Coomassie Brilliant Blue G-250,20 which increases staining sensitivity.The pH stability of the isolated S-layer ghosts was studied in two buffer systems from pH 1 to 12 with a step size of 0.5 – Sörensen's citrate buffer (0.1 M citric acid, 0.2 M NaOH; pH 1 to 4, adjusted with 0.1 M HCl) and Theorell–Stenhagen-buffer (33 mM citric acid, 33 mM phosphoric acid, 57 mM boric acid, 34.4 mM NaOH; pH 2 to 12, adjusted with 0.1 M HCl). 200 μg each of the purified S-layer ghosts were suspended in 2 ml of the buffer solutions. The suspensions were incubated for 48 hours on a rotary shaker at room temperature and afterwards absorption spectra of all samples were recorded in the range from 245 to 400 nm using a UV/Vis spectrometer (TIDAS 100, J&M Analytik AG, Esslingen, Germany) with the accompanying software TidasDAQ. The integration time was set to 250 ms and 20 single spectra were accumulated for each sample. Background was recorded by measuring a buffer solution without S-layer ghosts and automatically subtracted from the spectra of all samples. For comparison, untreated S-layer ghosts suspended in distilled water and SlaA-protein monomers were measured as well. The latter were produced by a dissociation of the S-layer ghosts at 60 °C in 0.1 M phosphate buffer (pH 9).16
Potentiometric titration experiments were carried out under nitrogen atmosphere at 25 °C. About 75 mg S-layer ghosts were suspended in carbonate-free deionized water, supplemented with sodium perchlorate to assure an ionic strength of 0.1 M. Titration was started at pH 5.5, the self-contained pH of the suspension, and carried out up to pH 10 with carbonate-free 10 mM NaOH (Titrisol, Merck, Darmstadt, Germany) and subsequently down to pH 3 with 10 mM HCl (Titrisol, Merck, Darmstadt, Germany) and vice versa (first down, then up, respectively). Titration solutions were adjusted to I = 0.1 M with NaClO4. The pH was measured with a BlueLine 16 pH electrode (Schott, Mainz, Germany). The electrode was calibrated for each experiment with NIST/PTB standard buffers (pH 4.008, 6.865 and 9.180, Schott). The minimum drift was set to 0.5 mV min−1, and a delay time of at least 60 s at each titration point was maintained before measuring the pH. The dynamic titration procedure was performed with an automatic titrator (TitroLine alpha plus, Schott, Mainz, Germany), monitored by the accompanied software (TitriSoft 2.11, Schott, Mainz, Germany), and analyzed with the Software Hyperquad2006.21
Treatments with U(VI)
Parallel portions of the S-layer ghosts were rinsed twice with 0.1 M NaClO4 solutions at pH 1.5, 3, 4.5, and 6, respectively, and after that suspended in the respective NaClO4 solution. The pH of the suspensions was checked and, if necessary, adjusted to the desired values. After the complete removal of the washing solution, uranyl nitrate diluted in 0.1 M NaClO4 with the corresponding pH was added. The final concentration of the S-layer ghosts was 0.5 g L−1 and that of uranium was 5 × 10−4 M, beside at pH 6, where it was reduced to 5 × 10−5 M to prevent the formation and precipitation of poorly soluble uranyl hydroxide species. Samples were incubated at room temperature for 48 hours on a rotary shaker. Subsequently, the S-layer ghosts were removed from the solutions by centrifugation (30000g, 30 min) and the unbound uranium in the supernatant was measured by ICP-MS using an Elan 9000 system (Perkin Elmer, Waltham, MA, USA). Control reactions without S-layer were treated in the same way to exclude abiotic uranium removal from the solution, due to precipitation and/or chemical sorption at the used test vials. The amount of accumulated uranium was normalized to the dry biomass, which was determined by weighting parallel samples after drying for 48 hours at 70 °C.
Time-resolved laser-induced fluorescence spectroscopy (TRLFS)
After the contact with U(VI) for 48 hours the S-layer ghosts were washed twice with 0.1 M NaClO4 at pH 1.5, 3, 4.5, and 6, respectively, and subsequently suspended in the corresponding washing solution. Initial measurements of the suspensions at room temperature were conducted in a quartz micro cuvette by using a Nd-YAG laser system with an excitation wavelength of 410 nm.15For luminescence measurements at cryogenic conditions, 3 ml each of the S-layer suspensions were filled into plastic cuvettes, subsequently sealed with parafilm and placed horizontally in a freezer (−20 °C) over night. Immediately before measurement, the frozen sample was removed from the cuvette and transferred in a specifically designed sample holder (metal block with one hole to insert the sample and three windows for laser irradiation). Temperature of the measuring system was decreased to 153 K using a cryostat (RDK 10-320) with an ultra-pump station (PT50 KIT/DN 40 KF), a compression unit (RW2) and a low temperature controller LTC60 (complete system: Oerlikon Leybold, Dresden, Germany). A Nd-YAG laser system (Inlite II, Continuum Electro-Optics Inc., Santa Clara, CA, USA), providing a wavelength of 266 nm, was used for luminescence excitation in the samples. The central wavelength of the spectrograph was 515 nm and spectra were recorded between 429 and 600 nm using an ICCD-camera (model 7467-0008, Princeton Instruments, Trenton, NJ, USA). The spectrograph was calibrated using a mercury lamp with known emission lines. 51 U(VI) luminescence spectra (each calculated by averaging three single measurements) were collected from each sample. The first spectrum was recorded immediately after U(VI) excitation and the following 50 after defined delay times. For both samples two series of measurements differing in the delay time steps (3 and 10 μs) were performed, i.e. the luminescence decay was recorded over a period of 150 μs and 1 ms, respectively. Deconvolution of the emission spectra and luminescence lifetime calculations were performed using Origin 7.5 (OriginLab Corporation, Northampton, MA, USA) including the PeakFit module 4.0.
X-ray absorption spectroscopy (XAS)
For XAS measurements, 50 mg of the S-layer ghosts were treated with U(VI) for 48 hours. After the contact with U(VI) the S-layer ghosts were washed twice with 0.1 M NaClO4 at pH 4.5 and 6, respectively. Subsequently, the samples were dried for three days at 35 °C in a vacuum oven (VT 6025, Heraeus-Instruments Vacutherm, Hanau, Germany), powdered and finally mounted on Kapton tape. This sample preparation based on a soft desiccation and previous studies proved that this method did not change the structural parameters of the formed uranium complexes.22 Six layers of sample-covered tape were stuck on the top of each other and subsequently shrink-wrapped. The XAS measurements were performed at the Rossendorf Beamline (ROBL) at the European Synchrotron Radiation Facility (ESRF), Grenoble, France.23 Samples were measured at room temperature in fluorescence mode using a Si(111) double-crystal monochromator and a 13-element germanium fluorescence detector. The energy was calibrated by measuring the yttrium K-edge transmission spectrum of a Y foil and defining the first inflection point as 17038 eV. Six U LIII-edge fluorescence spectra of each cell sample were recorded and averaged. Subsequently dead-time correction was applied. For XANES analysis the pre-edge background was subtracted and the absorption coefficient was normalized to equal intensity at 17230 eV so that all spectra could be plotted on the same scale. The EXAFS oscillations were isolated from the raw, averaged data by removal of the pre-edge background, approximated by a first-order polynomial, followed by μ0-removal via spline fitting techniques and normalization using a Victoreen function. The ionization energy for the U LIII electron, E0, was arbitrarily defined as 17185 eV for all averaged spectra. The EXAFS spectra were analyzed according to standard procedures using the program EXAFSPAK.24 The theoretical scattering phases and amplitude functions were calculated from a structural model of uranyl triacetate via the software FEFF8.2.25 The amplitude reduction factor was held constant at 1.0 for FEFF8.2 calculations and EXAFS fits. The shift in threshold energy, ΔE0, was varied as a global parameter in the fits.
Results and discussion
Purity and stability of the S-layer ghosts
The purified S-layer ghosts of S. acidocaldarius were checked microscopically to ensure the absence of non-lysed cells or other contaminations. The S-layer ghosts had a translucent appearance, indicating that they were free of intracellular compounds (Fig. S2†). However, the S-layer ghosts maintained the shape and size of whole cells, which supports the exoskeleton function of the S-layer that was recently postulated.26 These ghosts could be only mechanically disrupted (e.g. via sonication) in S-layer sheets, which are comparable to those usually yielded from bacteria without mechanical disintegration.12The protein composition of the isolated S-layer ghosts was checked by using denaturing SDS-PAGE. Only one protein band was observed in the lane of the purified S-layer ghosts (Fig. S2†). This band represented the SlaA-protein monomers. The molecular mass of about 150 kDa was well in line with the molecular weight of 151.04 kDa, calculated from the primary structure of the SlaA-protein.10,19
The pH stability of the S-layer ghosts was studied, as the maintenance of the S-layer in its polymeric form during the U(VI) sorption experiments is of importance for drawing conclusion on the U(VI) complexation by whole cells. Dissociation into SlaA-protein monomers might enable complexation of U(VI) at additional functional groups which were masked in the polymeric form. Stability of the S-layer ghosts was checked after an incubation at the different pH conditions for 48 hours according to the incubation time used for the U(VI) sorption experiments. UV/Vis spectroscopy demonstrated that the S-layer ghosts were stable at all acidic conditions as well as at neutral and slightly basic pH conditions up to pH 8. The spectra of these samples correspond very well to that recorded from native S-layer ghosts, suspended in distilled water (Fig. 1). Intact S-layers show a protein-specific absorption at around 277 nm and an additional light absorption at lower wavelength. The latter was caused by a light scattering at the crystal structure of the S-layer lattice. At pH 8.5 the S-layer ghost polymers started to dissociate into the SlaA monomers, indicated by a reduced absorption below 277 nm. At pH 9 and above the S-layer ghosts were fully dissociated into SlaA-protein monomers, which exhibited exclusively the peak at 277 nm (Fig. 1). Consistently, the spectra recorded from these samples (pH 9 to pH 12) show a high similarity to that of SlaA-protein monomers, which were produced from the S-layer ghosts via incubation in 0.1 M phosphate buffer (pH 9) at 60 °C according to Michel and co-workers.16 An extension of the incubation time from 48 to 60 hours did not change the results regarding the stability of the S-layer ghosts (not shown). These findings indicate that the S-layer ghosts were stable at the experimental conditions used for the U(VI) binding studies, namely room temperature, pH range from 1.5 to 6, and incubation time of 48 hours.
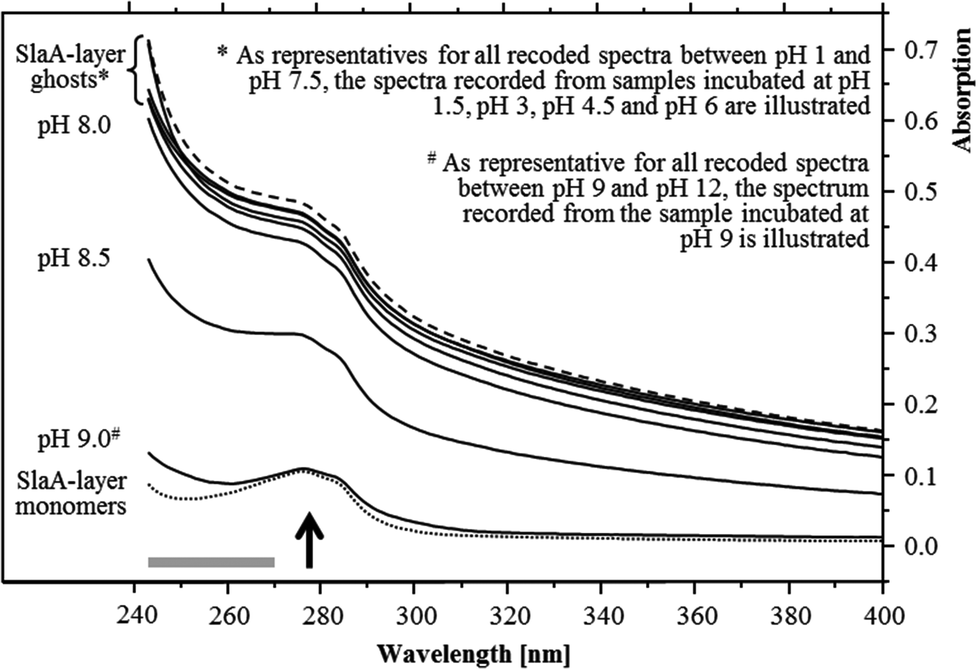 | ||
| Fig. 1 UV/Vis spectra recorded from the S-layer ghosts after incubations at different pH values for 48 hours at room temperature. The preservation of the polymeric structure is exemplary shown by the samples incubated at pH 1.5, 3, 4.5 and 6 together with untreated S-layer ghosts suspended in water (dashed spectrum). The SlaA-monomers (dotted spectrum) were obtained by incubation at 60 °C in 0.1 M phosphate buffer (pH 9). The arrow indicates the S-layer protein-specific absorption peak and the grey bar the region of the spectrum, where the absorption is increased due to a light scattering at the polymeric paracrystalline S-layer lattice. | ||
Potentiometric titration
Potentiometric titration, covering a pH from 3 to 10, was performed to determine the dissociation constants of chargeable groups at the S-layer protein. Titration curves and fitting results are provided in the ESI (Fig. S3 and S4†). Basically the degree of dissociation of chargeable groups is a function of pH and the ionic strength of the surrounding electrolyte solution. The obtained data revealed the presence of three ionizable groups with pKa values of 4.78, 6.31, and 8.03 (Table 1). According to the amino acid composition of the SlaA protein27 the two lower dissociation constants were assigned to the carboxylic groups of glutamic and aspartic acid.5,28 The second pKa value of 6.31 is unusually high for carboxylic groups. However, this high pKa could have been caused by an electrostatic potential, which was built up due to nearby charged groups in the mature structure of the SlaA protein. This electrostatic effect is known to change the dissociation constants of chargeable groups.29 Comparable high pKa values for carboxylic groups have already been described in the literature.30,31 For the sake of completeness we have to mention that this pKa could also be assigned to the imidazole ring of histidine.28 However, we calculated a site density of 11.8 ± 0.2 mol molSlaA protein−1 for this pKa which exclude the assignment to histidine as only four histidine residues are present in the SlaA protein.27| pKa | Site density/mol molSlaA−1 | Proposed site |
|---|---|---|
| 4.78 ± 0.08 | 6.83 ± 0.66 | Carboxyl (glutamic acid/aspartic acid) |
| 6.31 ± 0.04 | 11.8 ± 0.2 | |
| 8.03 ± 0.09 | 5.74 ± 0.48 | Hydroxyl/amine |
With respect to the pH range used for the uranium sorption studies mainly the carboxylic groups with a pKa of 4.78 ± 0.08 were involved in the U(VI) complexation at pH 4.5 and, in particular, at pH 6. The carboxylic groups with a dissociation constant of 6.31 might only played a minor role in the U(VI) complexation at pH 6. According to the primary structure of the mature S-layer protein,27 18 aspartic acid residues and 27 glutamic acid residues provided a total number of 45 binding-capable, carboxylic groups per SlaA protein. However, the potentiometric titration data suggested, that only about 19 of these groups were available in the native form of the S-layer ghosts polymers. The third pKa of (8.03 ± 0.09) most likely concerned amine groups and/or hydroxyl groups,28 which may also contribute to the U(VI) complexation at the studied conditions, as high-affinity functional groups for H+ can participate in binding of a metal ions together with low-affinity carboxylate groups even at pH lower than their pKa.
U(VI) sorption at the S-layer
No U(VI) removal from the aqueous solutions by the S-layer ghosts was observed in the samples incubated at pH 1.5 and pH 3. In contrast to that, U(VI) was complexed by the S-layer ghosts at pH 4.5 (2.1 ± 0.6 mg gS-layer−1) and pH 6 (5.5 ± 1.1 mg gS-layer−1). These values corresponded to a binding capacity of (1.3 ± 0.4) and (3.5 ± 0.7) mol uranium per mol of SlaA protein at pH 4.5 and 6, respectively. With regard to the total amount of the binding-capable carboxylic groups determined in the titration studies (6.83 ± 0.66 mol molSlaA-protein−1 for the groups with a pKa of 4.78) and considering that at pH 6 all of them existed in their deprotonated form, the stoichiometric ratio between complexed U(VI) and deprotonated carboxylic groups was approximately 1:2. This finding could be explained on the one hand by an unfavorable placement of some carboxylic groups in the highly ordered S-layer lattice, which were inaccessible for U(VI). On the other hand, more than one carboxylic group per uranyl ion could have been involved in the U(VI) complexation. The significantly lower amount of U(VI) bound to the S-layer at pH 4.5 could be explained with the pKa value of 4.78, which means that only few carboxylic groups are deprotonated at this pH.
TRLFS
Although U(VI) complexation by the S-layer ghosts at pH 4.5 and 6 was demonstrated by the sorption studies, we were not able to confirm this result by TRLF spectroscopic measurements at room temperature. Both samples showed very low luminescence properties and the recorded spectra did not allow for the detection of any protein-bound U(VI) (not shown). This likely was caused by the low amount of U(VI) bound to the S-layer and/or due to a quenching of the uranyl luminescence by carboxylate functional groups which is caused by an energy dissipation.32In order to reduce static quenching effects and therewith increase the sensitivity of this spectroscopic method we performed additional TRLF measurements at cryogenic conditions (153 K). As evident from the spectra presented in Fig. 2 clearly distinguishable luminescence emission maxima were observed. They corresponded well to those of uranyl acetate complexes published by Vogel and colleagues33 (Table 2). In addition, in both samples a small but distinct luminescence peak around 463 nm was detected, which was also considered as indicative for uranyl carboxylate complexes (Table 2). The U(VI) luminescence decay was fitted with a bi-exponential and tri-exponential decay function, respectively. From the sample incubated at pH 4.5, luminescence lifetimes of (23.7 ± 1.3) μs and of (234 ± 15) μs were calculated (Table 3). The lifetime of (23.7 ± 1.3) μs was attributed to the formed uranyl carboxylate complexes. This assignment was confirmed by a lifetime fit of the data between 460 and 466 nm, i.e. around the first emission peak which was specific for uranyl carboxylate complexes. A corresponding lifetime of (23.0 ± 2.4) μs was also calculated for the complexes formed at pH 6, indicating the formation of identical uranyl carboxylate complexes at both pH values. One should mention that some aquatic uranyl carbonate complexes, which might be formed in the solution at pH 6, can also generate an emission band between 460 and 465 nm. These aquatic uranyl carbonate complexes, however, have significantly longer luminescence lifetimes of 800 to 1000 μs at cryogenic conditions,34,35 and were, according to calculations with EQ3/6, not formed at pH 4.5. The extremely long luminescence lifetimes of (234 ± 15) μs and (141 ± 6) μs detected in the samples treated at pH 4.5 and 6, respectively, were attributed to one or a mixture of uranyl hydrolytic species as the emission maxima after longer delay times are shifted to higher wavelength, i.e. towards positions, which are typical for uranyl hydrolytic species (not shown). This assignment was also supported by lifetimes of more than 100 μs that have been reported for aquatic U(VI) hydroxide species by using cryo-TRLFS.34 In addition, a third luminescence lifetime of (4.21 ± 0.44) μs was calculated from the sample incubated at pH 6, which may have resulted from uranyl carboxylate complexes formed at the carboxylic groups with a dissociation constant of pKa = 6.31, as these groups likely started to deprotonate at pH 6.
 | ||
| Fig. 2 Luminescence spectra recorded at low temperature (153 K) after excitation (266 nm) of the uranium complexes formed at the S-layer of S. acidocaldarius at pH 4.5 and 6. For a better comparison the position of the calculated peak maxima of the sample incubated at pH 4.5 are pointed out by dotted lines. | ||
| Sample | Emission peak below 470 nm | Main luminescence emission maximaa | Reference | ||
|---|---|---|---|---|---|
| a Error of emission bands is ±0.5 nm | |||||
| Uranyl ion | |||||
| UO22+ (pH 1.5) | 488.9 | 510.5 | 534.0 | This work | |
| SlaA-layer ghosts | |||||
| Uranyl complexes formed at pH 4.5 (153 K) | 463.8 | 492.9 | 513.7 | 536.2 | This work |
| Uranyl complexes formed at pH 6 (153 K) | 462.4 | 494.7 | 514.4 | 535.9 | This work |
| Whole cells | |||||
| Uranyl complexes formed at pH 4.5 | 499.3 | 520.0 | 542.7 | 38 | |
| Uranyl complexes formed at pH 6 | 502.6 | 523.1 | 545.4 | (Reitz, unpublished) | |
| Uranyl carboxylate complexes | |||||
| Uranyl acetate | 462.9 | 494.6 | 514.3 | 535.9 | 33 |
| R-COO-UO2+/(R-COO)2-UO2 | 466.0 | 498.1 | 518.0 | 539.0 | 8 |
| pH 4.5 (153 K) | pH 6 (153 K) | Proposed complexes | |
|---|---|---|---|
| Lifetime 1 (τ1) | 23.7 ± 1.3 μs | 23.0 ± 2.4 μs | Uranyl carboxylate complexes |
| Lifetime 2 (τ2) | 234 ± 15 μs | Aquatic uranyl hydroxides | |
| Lifetime 3 (τ3) | 4.21 ± 0.44 μs | Uranyl carboxylate complexes | |
| Lifetime 4 (τ4) | 141 ± 6 μs | Aquatic uranyl hydroxides |
XAS studies
In order to obtain information regarding the molecular structure of the uranyl carboxylate complexes, which were formed at pH 4.5 and 6, we performed X-ray absorption spectroscopy. The X-ray absorption near edge structure (XANES) region (±10 eV around the absorption edge) provided information about the oxidation state of uranium. The absorption edge, i.e. white line, of the XANES spectra of both samples was located at ∼17166 eV and therewith corresponded well to the edge position of the U(VI) reference solution (Fig. S5†). In addition, a peak located at 17188 eV, arising from the multiple scattering contribution of the two axial oxygen atoms of U(VI),36 was observed in both spectra (Fig. S5†). These findings clearly demonstrated that uranium was present in both samples as U(VI).
The extended X-ray absorption fine structure (EXAFS) region (from 50 eV above the absorption edge) comprised information about the local environment of the uranium atoms and therewith about the molecular structure of the formed complexes. The isolated U LIII-edge k3-weighted EXAFS spectra and their corresponding Fourier transforms (FT) are shown in Fig. 3. Quantitative fitting of the spectra was performed by using the theoretical phase and amplitude functions calculated with the FEFF8.2 code from a structural model of uranyl triacetate. The best calculated fits for both samples are also shown in Fig. 3 and the underlying structural parameters are summarized in Table 4. During the fitting procedure, the coordination number (N) of the U–Oax single scattering (SS) path was held constant at two. N of the U–Oax multiple scattering (MS) path was fixed to the same value and the radial distance (R) as well as the Debye–Waller (σ2) factor were linked twice to those of the U–Oax SS path.
 | ||
| Fig. 3 Uranium LIII-edge k3-weighted EXAFS spectra (left) and the corresponding Fourier transforms (right), along with the best shell fits, of the uranium complexes formed at the S-layer of S. acidocaldarius at pH 4.5 and pH 6 as well as those of the two model compounds uranyl succinate and uranyl triacetate. | ||
| Sample | Shell | N | R (Å) |
σ
2d (Å2) |
ΔE0 (eV) |
|---|---|---|---|---|---|
| a Standard deviations as estimated by EXAFSPAK are given in parenthesis. b Errors in coordination numbers are ±25%. c Errors in distance are ±0.02 Å. d Debye–Waller factor. e Parameter fixed for calculation. f Fit included the U–Oax multiple scattering path and its radial distance (R) and Debye–Waller factor (σ2) were linked twice to R and σ2 of the U–Oax path. g Coordination number (N) linked to the N of U–C1 path. | |||||
| pH 4.5 | U–Oaxf | 2.0e | 1.77(1) | 0.0021(1) | 2.9(4) |
| U–Oeq | 5.0(4) | 2.42(1) | 0.014(1) | ||
| U–C | 2.8(3) | 2.90(1) | 0.0042e | ||
| U–Cdis | 2.8g | 4.35(1) | 0.00645e | ||
| pH 6 | U–Oaxf | 2.0e | 1.77(1) | 0.0026(1) | 3.2(5) |
| U–Oeq | 4.4(4) | 2.44(1) | 0.011(1) | ||
| U–C | 2.5(3) | 2.90(1) | 0.0042e | ||
| U–Cdis | 2.5g | 4.34(1) | 0.00645e | ||
| Uranyl succinate37 | U–Oaxf | 2.0e | 1.776(2) | 0.0014(1) | 3.5(5) |
| pH 4.46 | U–Oeq | 5.0(4) | 2.449(5) | 0.0089(9) | |
| U–C | 2.6(3) | 2.888(8) | 0.0042e | ||
| U–Cdis | 2.6g | 4.35(1) | 0.00645e | ||
| Uranyl triacetate | U–Oaxf | 2.0e | 1.78(1) | 0.0022(1) | 4.9(4) |
| U–Oeq | 4.8(3) | 2.47(1) | 0.0064(6) | ||
| U–C | 3.1(3) | 2.89(1) | 0.0038e | ||
| U–Cdis | 3.1g | 4.38(1) | 0.0038(1) | ||
As already suggested by the results of the TRLFS studies, the EXAFS spectra recorded from both samples corresponded to each other, which confirmed the formation of structurally similar uranium complexes at both pH values. In addition, the EXAFS spectra were well in line with those of uranyl triacetate and uranyl succinate37 indicating that structural similar U(VI) complexes have been formed at the S-layer. The most prominent peak of both FTs located at R + Δ ∼ 1.3 Å was assigned to the single backscattering mode (U–Oax) of the two axial oxygen atoms of U(VI). The multiple scattering path (U–Oax–U–Oax) of this axial oxygen shell had an intensity maximum in the FT at R + Δ ∼ 2.9 Å. The second peak of both FTs was attributed to the scattering contribution of the equatorial oxygen atoms. Fitting results of both investigated samples revealed four to five equatorial oxygen atoms at a radial distance of 2.42 to 2.44 Å. In addition, a shell (U–C) containing two to three carbon atoms was fitted at a radial distance of 2.90 Å. This distance is typical for uranyl complexed by the two oxygen atoms of a carboxylate group in a bidentate binding mode. A corresponding complexation mode was already suggested by the EXAFS studies of the uranium complexes formed at the surface of whole cells of S. acidocaldarius at pH 4.538 as well as at a Bacillus S-layer.11 However, in both cases the EXAFS spectra were rather complex and strongly dominated by U(VI) coordination to phosphoryl groups in a monodentate binding mode. Hence, a proper determination of the structural parameters of the U(VI) complexes formed at carboxylate groups was not possible.
A second shell of carbon atoms (U–Cdis) related to the carbon atom bearing the carboxylic group was fitted at radial distances of 4.35 Å and 4.34 Å in the samples incubated at pH 4.5 and 6, respectively. The U–Cdis backscattering contribution considered in the shell fit included the SS path as well as the MS paths, U–Cdis–C and U–C–Cdis–C. During the fitting procedure, the coordination numbers of these two MS paths were linked twice and once to that of the U–Cdis SS path, respectively. The radial distance of both MS paths was linked to that of the U–Cdis SS path.
As mentioned above, the obtained structural parameters of both samples are well in line with respect to the calculated errors. The only difference was observed in the U–Oeq shell. At pH 6 a radial distance of 2.44 Å and a Debye–Waller factor of 0.011 Å2 were calculated for this shell and agree very well to the structural parameters calculated for the recently investigated uranyl succinate complex (Table 4).37 In the latter complex the uranyl ion is bound bidentately to two to three carboxylic groups. The calculated structural parameters of the uranyl carboxylate complexes formed at pH 6 (R = 2.44 Å, σ2 = 0.011 Å2) can be explained by a split U–Oeq shell comprising four equatorial oxygen atoms which are bidentately bound to carbon (characteristic distance ∼2.47 Å39) and one or two equatorial oxygen atoms which exists in their hydrated form (characteristic distance 2.41–2.42 Å40).
In contrast to that, the lower radial distance (2.42 Å) calculated for the U–Oeq shell of the U(VI) complexes formed at pH 4.5 suggested that U(VI) was bound to only one carboxylate group. This assumption was supported by highly similar structural data (R = 2.41 Å, σ2 = 0.011 Å2) obtained for the U–Oeq shell of 1:1 uranyl acetate complex.37 However, the high coordination number calculated for the U–C shell (N = 2.8) was not in agreement with this complexation model. This implicated an additional, unknown complexation mode of the uranyl ion to the carboxylate groups at pH 4.5. Infrared and Raman spectroscopic studies of uranyl acetate complexes revealed that U(VI) can be bound to carboxylate groups in a bidentate, and additionally in a pseudo-bridging complexation mode.41 The U–Oeq distance of such pseudo-bridged structures was estimated to be 2.35–2.40 Å42 and could therewith explain both, the lower radial distance and the higher Debye–Waller factor of the U–Oeq shell obtained from the shell fit of the sample incubated at pH 4.5. As EXAFS do not allow to distinguish between uranyl complexation via oxygen of carboxylic groups and nitrogen atoms of amine groups, another explanation for the EXAFS data could be a combined carboxyl and amino coordination of the uranyl ion. This binding mode has already been postulated for the uranium complexation by peptidoglycan.8 However, the involvement of amine groups in the U(VI) complexation cannot be supported by further experimental data and has also so far not been described for U(VI)–microbe interactions.
The XAS studies demonstrated that at moderate acidic conditions U(VI) formed consistently inner-sphere complexes at the carboxylate groups of the S-layer, predominantly in a bidentate binding mode. Moreover, the data suggested an additional, yet unknown complexation mode at pH 4.5.
Conclusions
In this study, the complexation of U(VI) by the S-layer of S. acidocaldarius was investigated. Whereas the S-layer did not bound uranium at highly acidic conditions (pH 1.5 and 3), uranium was bound by deprotonated carboxylic groups of the S-layer at pH 4.5 and pH 6.The formed uranyl carboxylate complexes showed no luminescence at room temperature. However, at cryogenic conditions we obtained emission maxima which correspond well to those of uranyl triacetate. This finding demonstrated that the use of cryogenic techniques opens up new possibilities to investigate the U(VI) complexation by ligands of organic biopolymers. EXAFS studies revealed that the complexation of U(VI) at the S-layer predominantly occurred only via two to three carbon atoms per uranyl ion, which were coordinated in a bidentate binding mode.
The studied S-layer ghosts of S. acidocaldarius may also become a subject of biotechnological interest as a U(VI) biosorbent for remediation of uranium-contaminated liquids. Due to its extreme stability, in particular against acidic pH, which is common for uranium-contaminated sites, the S-layer has a sufficient advantage compared to bacterial S-layer proteins, which mostly originate from neutrophilic species and disintegrate at acidic pH.
References
- J. R. Lloyd and L. E. Macaskie, in Biochemical Basis of Microbe-Radionuclide Interactions, Elsevier Science, 2002 Search PubMed.
- K. Pedersen, J. Nucl. Radiochem. Sci., 2005, 6, 11–15 CrossRef CAS.
- M. J. Beazley, R. J. Martinez, P. A. Sobecky, S. M. Webb and M. Taillefert, Geomicrobiol. J., 2009, 26, 431–441 CrossRef CAS.
- T. Reitz, A. Rossberg, A. Barkleit, S. Selenska-Pobell and M. L. Merroun, PLoS One, 2014, 9(8), e102447 Search PubMed.
- A. Barkleit, H. Moll and G. Bernhard, Dalton Trans., 2008, 2879–2886 RSC.
- A. Koban and G. Bernhard, Polyhedron, 2004, 23, 1793–1797 CrossRef CAS PubMed.
- A. Koban and G. Bernhard, J. Inorg. Biochem., 2007, 101, 750–757 CrossRef CAS PubMed.
- A. Barkleit, H. Moll and G. Bernhard, Dalton Trans., 2009, 5379–5385 RSC.
- P. Messner, C. Schäffler, E. M. Egelseer and U. B. Sleytr, Prokaryotic Cell Wall Compounds, Springer Verlag, 2010 Search PubMed.
- H. König, R. Rachel and H. Claus, Archaea: Molecular and Cellular Biology, ASM Press, 2007 Search PubMed.
- M. L. Merroun, J. Raff, A. Rossberg, C. Hennig, T. Reich and S. Selenska-Pobell, Appl. Environ. Microbiol., 2005, 71, 5532–5543 CrossRef CAS PubMed.
- A. Veith, A. Klingl, B. Zolghadr, K. Lauber, R. Mentele, F. Lottspeich, R. Rachel, S. V. Albers and A. Kletzin, Mol. Microbiol., 2009, 73, 58–72 CrossRef CAS PubMed.
- T. D. Brock, K. M. Brock, R. T. Belly and R. L. Weiss, Arch. Mikrobiol., 1972, 84, 54–68 CAS.
- S. N. Groudev and V. I. Groudeva, FEMS Microbiol. Rev., 1993, 11, 261–268 CrossRef CAS PubMed.
- T. Reitz, M. L. Merroun, A. Rossberg and S. Selenska-Pobell, Radiochim. Acta, 2010, 98, 249–257 CrossRef CAS.
- H. Michel, D. C. Neugebauer and D. Oesterhelt, Electron microscopy at molecular dimensions, 1980, pp. 27–35 Search PubMed.
- U. K. Laemmli, Nature, 1970, 227, 680–685 CrossRef CAS.
- D. W. Grogan, J. Microbiol. Methods, 1996, 26, 35–43 CrossRef CAS.
- H. Claus, E. Akca, T. Debaerdemaeker, C. Evrard, J. P. Declercq, J. R. Harris, B. Schlott and H. Konig, Can. J. Microbiol., 2005, 51, 731–743 CrossRef CAS PubMed.
- V. Neuhoff, N. Arold, D. Taube and W. Ehrhardt, Electrophoresis, 1988, 9, 255–262 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef CAS.
- M. Merroun, C. Hennig, A. Rossberg, T. Reich and S. Selenska-Pobell, Radiochim. Acta, 2003, 91, 583–591 CrossRef CAS.
- W. Matz, N. Schell, G. Bernhard, F. Prokert, T. Reich, J. Claußner, W. Oehme, R. Schlenk, S. Dienel, H. Funke, F. Eichhorn, M. Betzl, D. Pröhl, U. Strauch, G. Hüttig, H. Krug, W. Neumann, V. Brendler, P. Reichel, M. A. Denecke and H. Nitsche, J. Synchrotron Radiat., 1999, 6, 1076–1085 CrossRef CAS.
- G. N. George and I. J. Pickering, A Suite of Computer Programs for Analysis of X-ray Absorption Spectra, http://ssrl.slac.stanford.edu/~george/exafspak/manual.pdf, 2000 Search PubMed.
- A. L. Ankudinov, B. Ravel, J. J. Rehr and S. D. Conradson, Phys. Rev. B: Condens. Matter, 1998, 58, 7565–7576 CrossRef CAS.
- H. Engelhardt, J. Struct. Biol., 2007, 160, 115–124 CrossRef CAS PubMed.
- E. Peyfoon, B. Meyer, P. G. Hitchen, M. Panico, H. R. Morris, S. M. Haslam, S. V. Albers and A. Dell, Archaea, 2010 Search PubMed.
- R. L. Thurlkill, G. R. Grimsley, J. M. Scholtz and C. N. Pace, Protein Sci., 2006, 15, 1214–1218 CrossRef CAS PubMed.
- A. van der Wal, W. Norde, A. J. B. Zehnder and J. Lyklema, Colloids Surf. B, 1997, 9, 81–100 CrossRef.
- J. Davoodi, W. W. Wakarchuk, R. L. Campbell, P. R. Carey and W. K. Surewicz, Eur. J. Biochem./FEBS, 1995, 232, 839–843 CAS.
- M. D. Joshi, A. Hedberg and L. P. McIntosh, Protein Sci., 1997, 6, 2667–2670 CrossRef CAS PubMed.
- M. Ahmad, A. Cox, T. J. Kemp and Q. Sultana, J. Chem. Soc., Perkin Trans., 1975, 2, 1867–1872 RSC.
- M. Vogel, A. Günther, A. Rossberg, B. Li, G. Bernhard and J. Raff, Sci. Total Environ., 2010, 409(2), 384–395 CrossRef CAS PubMed.
- R. Steudtner, T. Arnold, G. Geipel and G. Bernhard, J. Radioanal. Nucl. Chem., 2010, 284(2), 421–429 CrossRef CAS PubMed.
- Z. M. Wang, J. M. Zachara, W. Yantasee, P. L. Gassman, C. X. Liu and A. G. Joly, Environ. Sci. Technol., 2004, 38, 5591–5597 CrossRef CAS.
- E. A. Hudson, P. G. Allen, L. J. Terminello, M. A. Denecke and T. Reich, Phys. Rev. B: Condens. Matter, 1996, 54, 156–165 CrossRef CAS.
- C. Lucks, A. Rossberg, S. Tsushima, H. Foerstendorf, A. C. Scheinost and G. Bernhard, Inorg. Chem., 2012, 51, 12288–12300 CrossRef CAS PubMed.
- T. Reitz, M. L. Merroun, A. Rossberg, R. Steudtner and S. Selenska-Pobell, Radiochim. Acta, 2011, 99, 543–553 CrossRef CAS.
- M. A. Denecke, T. Reich, M. Bubner, S. Pompe, K. H. Heise, H. Nitsche, P. G. Allen, J. J. Bucher, N. M. Edelstein and D. K. Shuh, J. Alloys Compd., 1998, 271, 123–127 CrossRef.
- H. A. Thompson, G. E. J. Brown and G. A. Parks, Am. Mineral, 1997, 82, 483–496 CAS.
- F. Quiles and A. Burneau, Vib. Spectrosc., 1998, 18, 61–75 CrossRef CAS.
- J. F. Mosselmans, E. Bailey and P. Schofield, J. Synchrotron Radiat., 2001, 8, 660–662 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4dt02555e |
| This journal is © The Royal Society of Chemistry 2015 |