
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Uranium triamidoamine chemistry
Benedict M.
Gardner
and
Stephen T.
Liddle
*
School of Chemistry, University of Nottingham, University Park, Nottingham, NG7 2RD, UK. E-mail: stephen.liddle@nottingham.ac.uk
First published on 2nd June 2015
Abstract
Triamidoamine (Tren) complexes of the p- and d-block elements have been well-studied, and they display a diverse array of chemistry of academic, industrial and biological significance. Such in-depth investigations are not as widespread for Tren complexes of uranium, despite the general drive to better understand the chemical behaviour of uranium by virtue of its fundamental position within the nuclear sector. However, the chemistry of Tren–uranium complexes is characterised by the ability to stabilise otherwise reactive, multiply bonded main group donor atom ligands, construct uranium–metal bonds, promote small molecule activation, and support single molecule magnetism, all of which exploit the steric, electronic, thermodynamic and kinetic features of the Tren ligand system. This Feature Article presents a current account of the chemistry of Tren–uranium complexes.
 Benedict M. Gardner | Ben Gardner completed his MSci and PhD degrees at the University of Nottingham, UK, the latter in 2012 under the supervision of Steve Liddle, where he investigated the organometallic chemistry of uranium supported by sterically demanding Tren ligands. He has since been undertaking postdoctoral research with Steve Liddle investigating low-valent uranium activation chemistry of small molecules. He has 14 publications to date. |
 Stephen T. Liddle | Steve Liddle obtained his BSc (Hons) in 1997 and PhD in 2000 from Newcastle University. After postdoctoral fellowships at the Universities of Edinburgh, Newcastle, and Nottingham, he took up a Royal Society University Research Fellowship at Nottingham where he is currently a Professor of Inorganic Chemistry. His research interests include metal–ligand multiple bonding, metal–metal bonds, small molecule activation, and single molecule magnetism of the f-block elements, with a particular focus on uranium. He has published over 140 papers, reviews, and book chapters to date. |
Introduction
The chemistry of N-alkyl, -aryl or -silyl substituted triamidoamine (Tren) ligands, Fig. 1, is well known, given their significance as supporting ligands for intermediates for ceramic materials and semiconductors, biomimetic models, extraction agents, superbases and homogeneous catalysts.1 In particular, transition metal–Tren complexes, of which a search of the Cambridge Structural Database returns ∼400 results,2 are significant as they are able to capture otherwise reactive moieties, as demonstrated by the wide array of examples of Tren-complexes featuring multiple bonds between a metal ion and a main group donor atom.3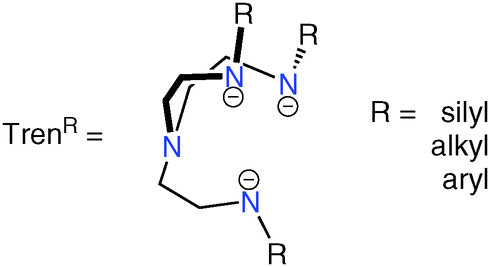 | ||
| Fig. 1 The generic Tren–ligand framework. | ||
Terminal metal–carbon double and triple bonds have been stabilised in the form of tantalum alkylidene and tungsten carbyne complexes featuring Tren frameworks (I and II, Fig. 2).4 Multiply-bonded transition metal–nitrogen fragments have been the focus of much attention due to their biological significance. In particular, it has been shown that a number of parent or substituted imides, nitrides, diazenides and hydrazenides can all be accessed; of note was the landmark report of the first molecular catalytic reduction of dinitrogen to ammonia employing sterically demanding Tren-derivatives in complexes of molybdenum(III) in which dinitrogen adducts are implicated (e.g.III, Fig. 2).5 A handful of Tren–transition metal complexes featuring multiply bound heavier pnictides in the form of phosphinidenes, phosphidos, arsenidos and stibidos have been reported (IV–VII, Fig. 2), demonstrating the versatility of Tren at stabilising unusual and reactive multiply-charged main group ligands.5k,7 Such systems have established routes to metal-bound main group fragments and terminal Tren–transition metal–pnitctide/chalcogenide complexes featuring M![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O/Se/Te/PS/AsS multiple bonds (e.g.VIII, IX, Fig. 2) that would otherwise be challenging to directly construct.4b,5k,6
O/Se/Te/PS/AsS multiple bonds (e.g.VIII, IX, Fig. 2) that would otherwise be challenging to directly construct.4b,5k,6
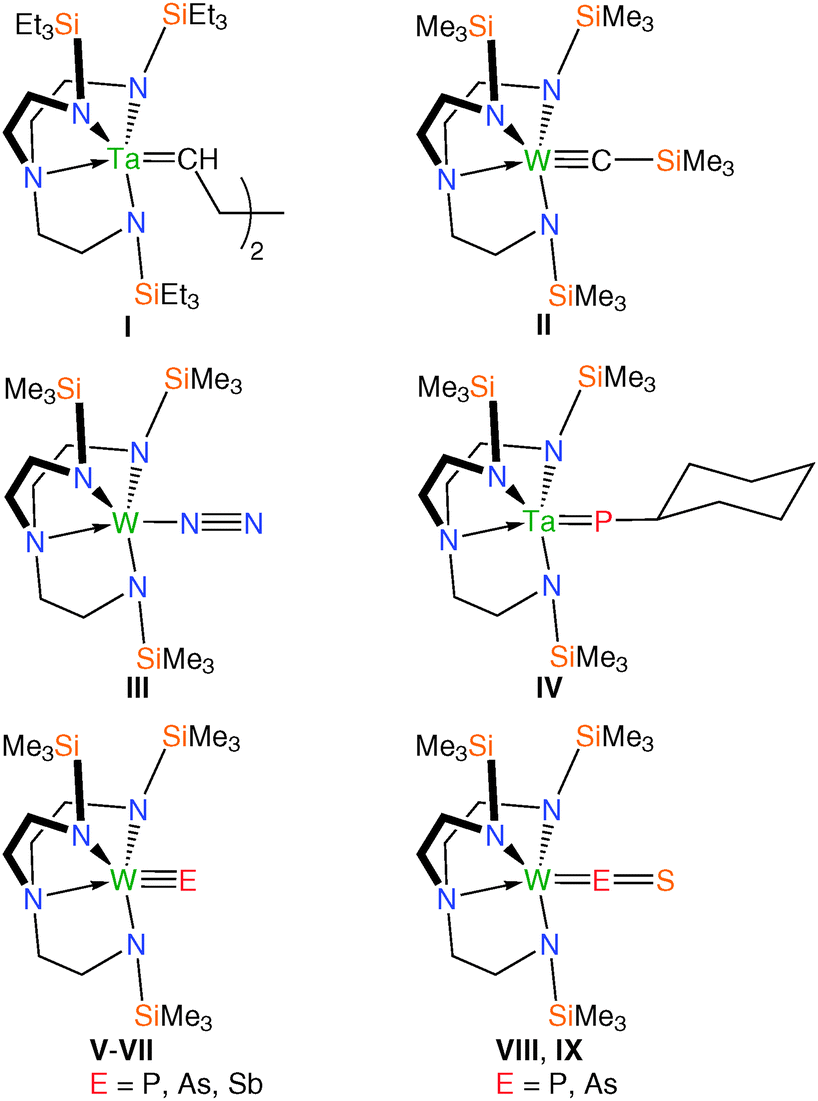 | ||
| Fig. 2 Selected examples of Tren–transition metal complexes supporting novel linkages. | ||
Reports of Tren–uranium compounds have demonstrated that the [U(TrenR)] fragment is robust, stabilising reactive functionalities yet permitting further reactivity in the auxiliary coordination sphere. The chemical reactivity profile of Tren–uranium compounds complements structurally related uranium tris(pyrrolyl-α-methyl)amine7 and uranium polyaryloxide or polyamide systems, which are increasingly well explored.8–10 As this Feature Article will describe, the use of Tren ligands can lead to novel types of reactivity at uranium, ascribed to a combination of the chelate effect, facial coordination of the ligating atoms and tuneable steric demands via variation of the amido substituents.11 Furthermore, it is becoming apparent that the tertiary amine centre of Tren can play a role in the electronic stabilisation of multiply bonded main group donor atoms that reside trans to the amine centre.
This Feature Article will systematically guide the reader through the Tren–uranium chemistry published in the primary scientific literature with an emphasis on structurally characterised compounds. It is organised firstly by common precursor compounds, then by the group number of the principal donor atom in the ligand under consideration that is coordinated to uranium, then small molecule activation chemistry before lastly covering uranium–transition metal systems (sub-organised by group number). The field of Tren–uranium chemistry can justifiably be described as burgeoning simply by the diverse array of fragments from across the p- and d-block that TrenR–uranium has been shown to stabilise in recent years.
Tren–uranium precursor complexes
Halide derivatives
The Tren ligand is conveniently introduced into the coordination sphere of uranium via salt metathesis of uranium tetrachloride with the appropriate trilithiated Tren proligand, [Li3TrenR].5b,12 This gives the pale green Tren–uranium(IV) chloride complexes 1–3 (Scheme 1).12b,13 Conversion of 2 into the dark green bromo congener 4 can be achieved by treatment with Me3SiBr,13b and the pale green iodo complexes 5–7 analogously with Me3SiI.13b,14 Complexes 1–6 are all structurally characterised, and are mononuclear in the solid state. TrenTMS–uranium(IV) halide complexes 1 and 5 crystallise as THF solvates with one molecule of coordinated THF per uranium that can be removed by sublimation (180 °C, 10−6 mbar) to afford yellow 8 and 9, respectively, each of which exhibit dimeric solid state structures with bridging halide centres.15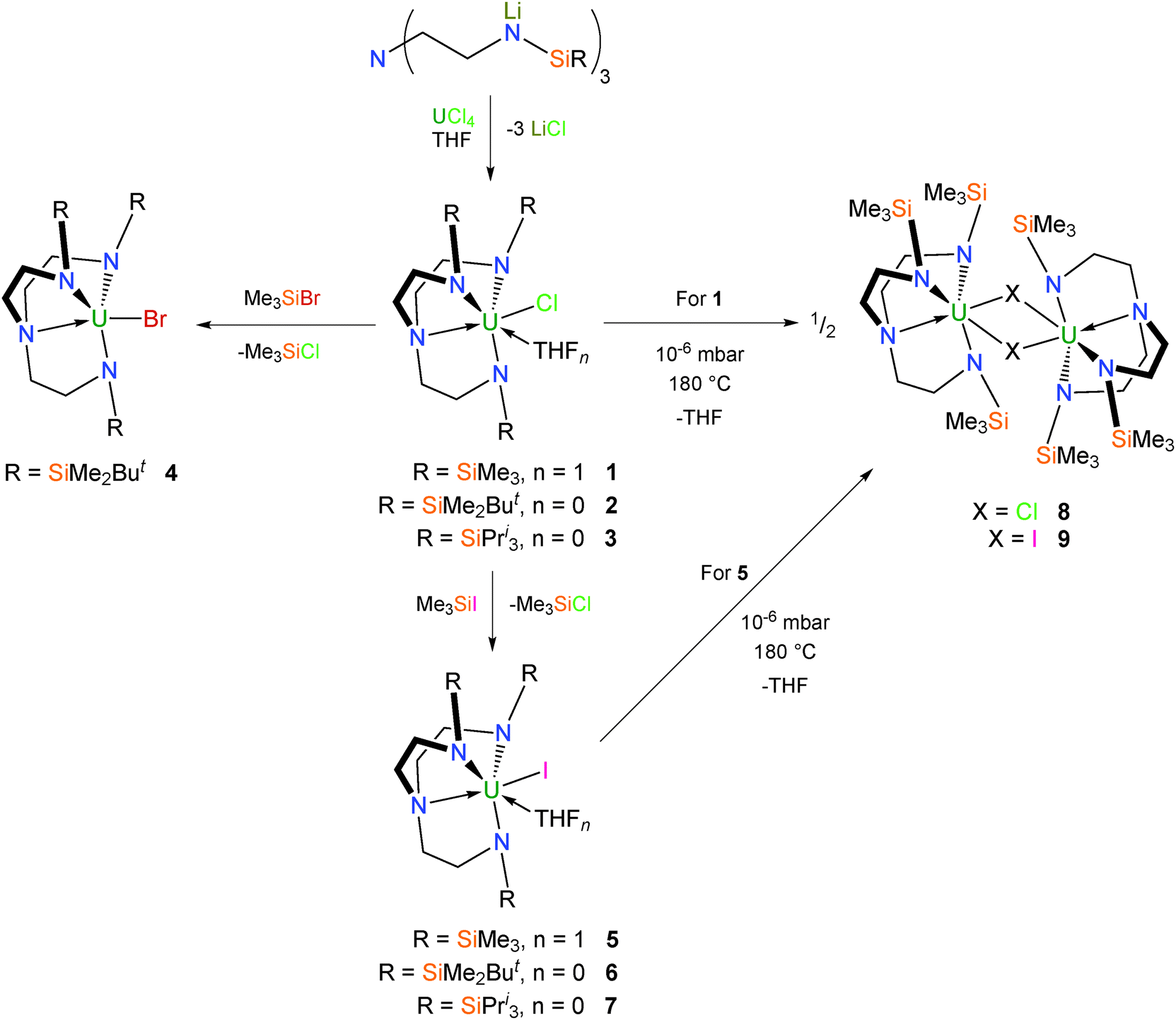 | ||
| Scheme 1 Synthesis of 1–9. | ||
Uranium(III) derivatives
Reduction of 2 over a potassium mirror affords the dark purple mixed-valent uranium(III/IV) bridging chloride complex [{U(TrenDMBS)}2(μ-Cl)] (10) (Scheme 2),16 as determined by a single crystal X-ray diffraction (XRD) experiment. Careful sublimation of 10 at 120 °C and 10−6 mbar affords a dark purple solid characterised as the trivalent complex [U(TrenDMBS)] (11), and if heated up to 180 °C pale green 3 is recovered. It has also been shown that 11 can be independently prepared from 6 by reduction over a potassium mirror.17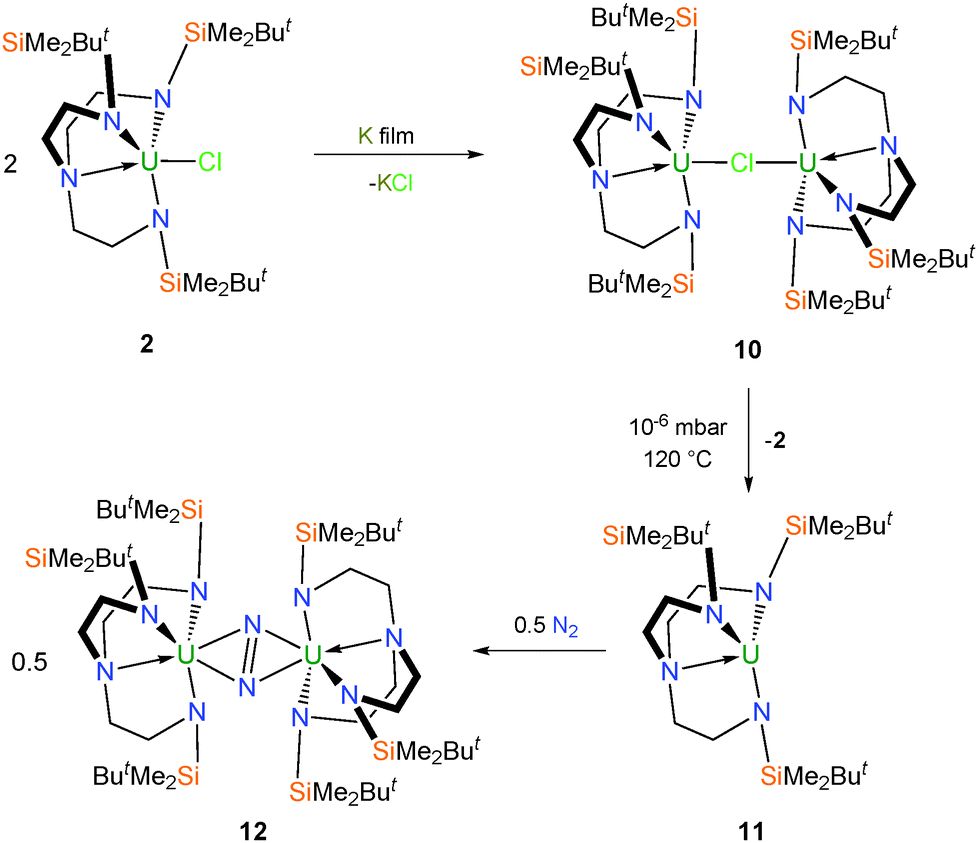 | ||
| Scheme 2 Synthesis of 10–12. | ||
Very early on the potential novelty of Tren–uranium complexes was highlighted by the observation that freeze–thaw degassed solutions of 11 placed under an atmosphere of dinitrogen change colour from purple to red. The identity of the new complex was confirmed by a structural determination, performed on dark red crystals grown from pentane, to be the novel side-on bridging dinitrogen complex [{U(TrenDMBS)}2(μ-η2:η2-N2)] (12), which was the first f-block dinitrogen complex.18 Inspection of the characterisation data for 12 initially suggested a UIII–N2–UIII bonding picture with no increase in valency for the uranium centres and a neutral N2 ligand; however, with advances in computational and spectroscopic techniques it was later suggested that in fact the solid state data for 12 were somewhat misleading and a more likely bonding situation is that of a reduced N2 unit coordinated to uranium(IV) centres consistent with U → N2 backbonding.18,19 This has since been supported by additional examples of diuranium–dinitrogen complexes that have been characterised by Raman spectroscopy, which is regarded as the best probe of the dinitrogen unit and thus uranium oxidation state in this context.20
Treatment of trivalent 11 with neutral Lewis bases such as pyridine and hexamethylphosphoramide [(Me2N)3PO, HMPA] produces colour changes from purple to orange and black, respectively, affording [U(TrenDMBS)(C5H5N)] (13) and [U(TrenDMBS){OP(NMe2)3}] (14), Scheme 3. The molecular structure of 14 was confirmed by a single crystal XRD study, and although 13 was not structurally characterised, its analytical data support its proposed formulation.17
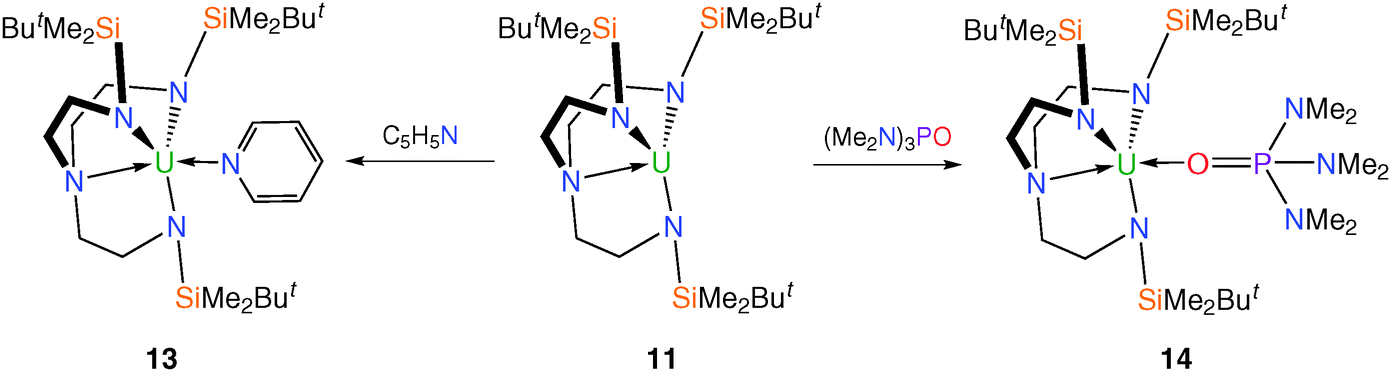 | ||
| Scheme 3 Synthesis of 13 and 14. | ||
The HMPA molecule in 14 coordinates through the oxygen atom; the NMR and absorption spectra for 13 and 14 are typical of uranium(III) species. Given the extreme sensitivity to air and moisture of 11, and indeed its highly reactive nature generally as exemplified by the formation of 12, a significant increase in stability was observed for the adducts 13 and 14.
Analogously to 11, [U(TrenTIPS)] (15) was prepared in a straightforward manner from potassium reduction of 3 (Scheme 4) although notably the reduction proceeds cleanly to completion from U(IV) to U(III) without the formation of mixed valent species.12b Surprisingly given the expected coordinative unsaturation, the solid state structure of 15 has been shown not to feature any U⋯HC agostic interactions and reveals a well-defined axial steric ‘pocket’. In contrast to 11, there is no evidence that 15 reacts with dinitrogen, presumably because the steric bulk of the TrenTIPS precludes side-on binding of dinitrogen.
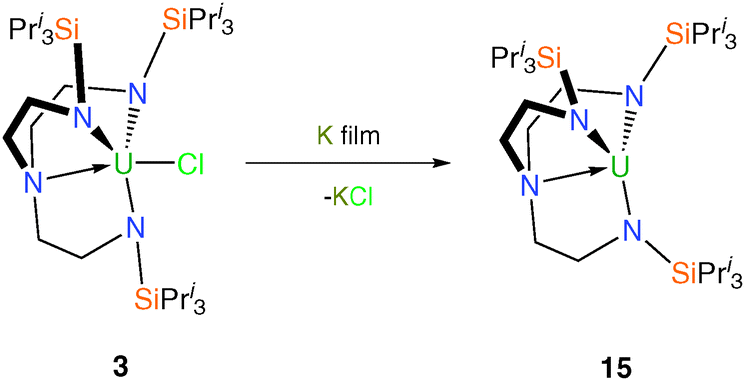 | ||
| Scheme 4 Synthesis of 15. | ||
Amide derivatives
Tren–uranium amide complexes have found utility as precursors to a range of novel Tren–uranium complexes, usually as protonolysis reagents. Treatment of 1 with one equivalent of lithium dicyclohexylamide or lithium bis(trimethylsilyl)amide afforded – after workup and recrystallisation from hexane – yellow crystals of solvent-free [U(TrenTMS)(NR2)] (R = cyclohexyl, 16; R = SiMe3, 17), Scheme 5.15b Single-crystal XRD studies confirmed the molecular structures of 16 and 17, which were found to be as expected with the NR2 units adopting trigonal planar geometries suggesting likely π-base character.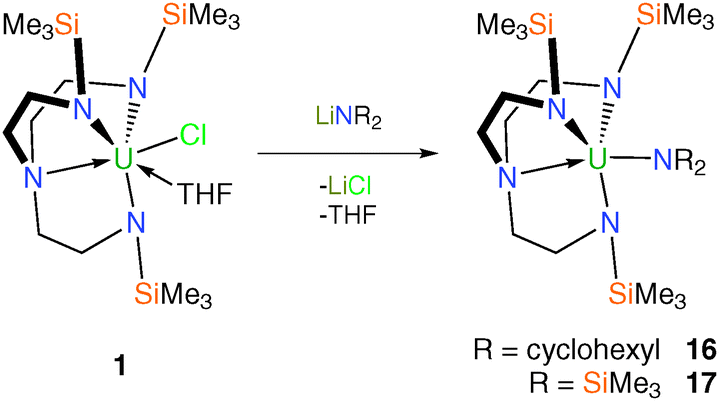 | ||
| Scheme 5 Synthesis of 16 and 17. | ||
Tren–uranium – group 13 derivatives
Borate derivatives
The single example of a Tren–uranium tetrahydroborato derivative was reported in 1995.21 It was prepared in THF by treatment of the corresponding uranium(IV) chloride, 8, with one equivalent of lithium tetrahydroborate per uranium and crystallised from pentane. A single crystal XRD study determined the structure to be [U(TrenTMS){(μ-H)3BH}(THF)] (18), Fig. 3, which includes a coordinated molecule of THF. Whilst the hydride atoms of the tetrahydroborate ligand were not located by the diffraction experiment, the assignment of the (μ-H)3BH coordination mode is supported by the U–B distance of 2.68(2) Å and IR data.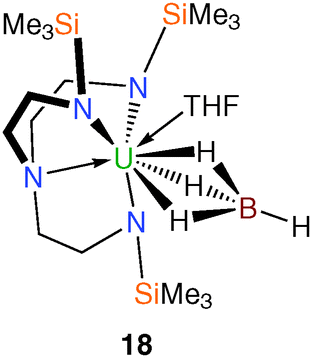 | ||
| Fig. 3 Molecular structure of 18. | ||
Tetraarylborate complexes can be more desirable borate synthetic precursors than tetrahydroborates in view of the reduced capacity for coordination of the aryl units to the uranium centre relative to borohydrides; the former are out-competed by Lewis bases such as THF and therefore should be removed more easily than the latter. Since it is known that Tren–uranium halide complexes do not react with tetraarylborate sources such as KBPh4, amine or alkane elimination methodologies have been employed to access Tren–uranium tetraarylborates.15b Either of the two TrenTMS–uranium(IV) amide complexes [U(TrenTMS)(NR2)] (R = cyclohexyl, trimethylsilyl)15b can be treated with [Et3NH][BPh4] to afford, after work up, the target separated ion pair complex [U(TrenTMS)(THF)2][BPh4] (19) as a free-flowing green powder from hexanes in 95% yield (Scheme 6).
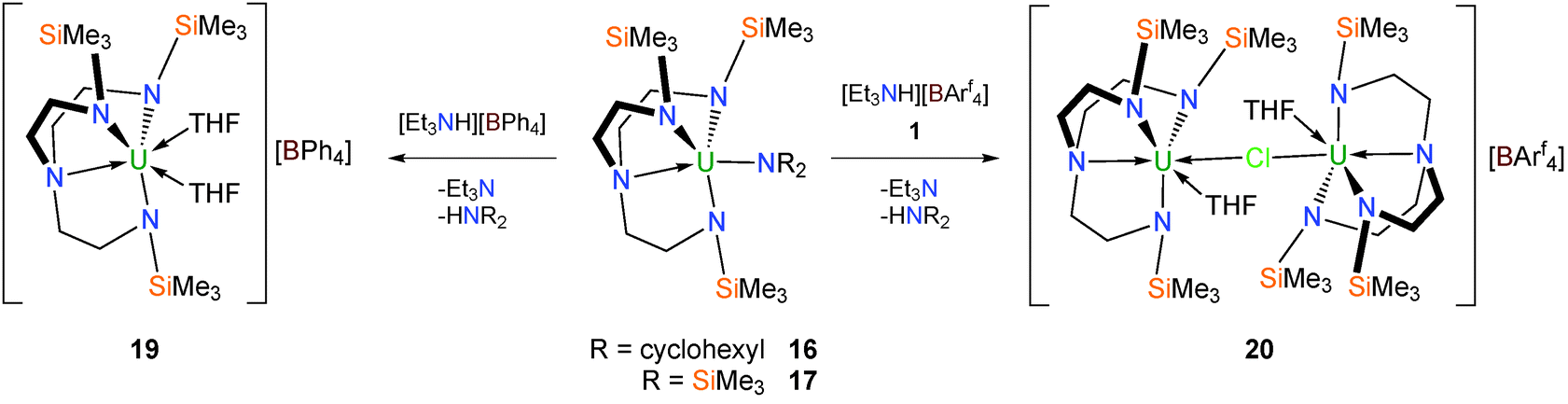 | ||
| Scheme 6 Synthesis of 19 and 20. Arf = 3,5-(CF3)2–C6H3. | ||
A single crystal XRD experiment was conducted on a yellow-green crystal of 19 grown from toluene, which confirmed it to be a separated ion pair complex, with no contacts between the cation and anion, incorporating two molecules of coordinated THF. The cationic nature of the uranium-containing fragment in 19 is supported by inspection of the U–Namide bond lengths; the mean U–Namide distance of 2.238(3) Å compares to the mean U–Namide distance of 2.253(8) Å in 514a and is consistent with the cationic nature of the TrenTMS–uranium(IV) fragment.
Due to the potential for the tetraphenylborate anion to engage in side reactions22 the more robust [BArf4]− [Arf = 3,5-(CF3)2–C6H3] anion was investigated as an alternative non-coordinating anion via the target complex [U(TrenTMS)(THF)2][BArf4]. Analogously to 19, treatment of the amides with [Et3NH][BArf4] in THF (Scheme 6) afforded an oily yellow-brown product to which was added one equivalent of 1, in the anticipation that a coordinated THF molecule would be displaced and the second uranium centre would bind via a bridging chloride to give an isolable complex. Accordingly the cationic separated ion pair complex [{U(TrenTMS)(THF)}2(μ-Cl)][BArf4] (20) was isolated as pale green crystals, confirmed by a structural determination.15b The dinuclear uranium(IV) cationic component consists of two essentially identical [U(TrenTMS)(THF)]+ units bridged by a chloride anion whose U–Cl bond distances are not equivalent [2.887(2) and 2.918(2) Å], which suggests that the chloride is not equally associated with the uranium centres.
Tren–uranium tetraphenylborate complexes were targeted via alkyl precursors, namely [U{N(CH2CH2NSiMe2But)2(CH2CH2NSiMeButCH2)}]23 (21) and [U{N(CH2CH2NSiPri3)2(CH2CH2NSiPri2C[H]MeCH2)}] (22).24 Treatment of 21 and 22 with [Et3NH][BPh4] yielded the cationic complexes [(TrenDMBS)U(MeCN)2][BPh4] (23) and [(TrenTIPS)U(THF)][BPh4] (24), after addition of donor solvents respectively, as a pale green solids in near-quantitative yield (Scheme 7).
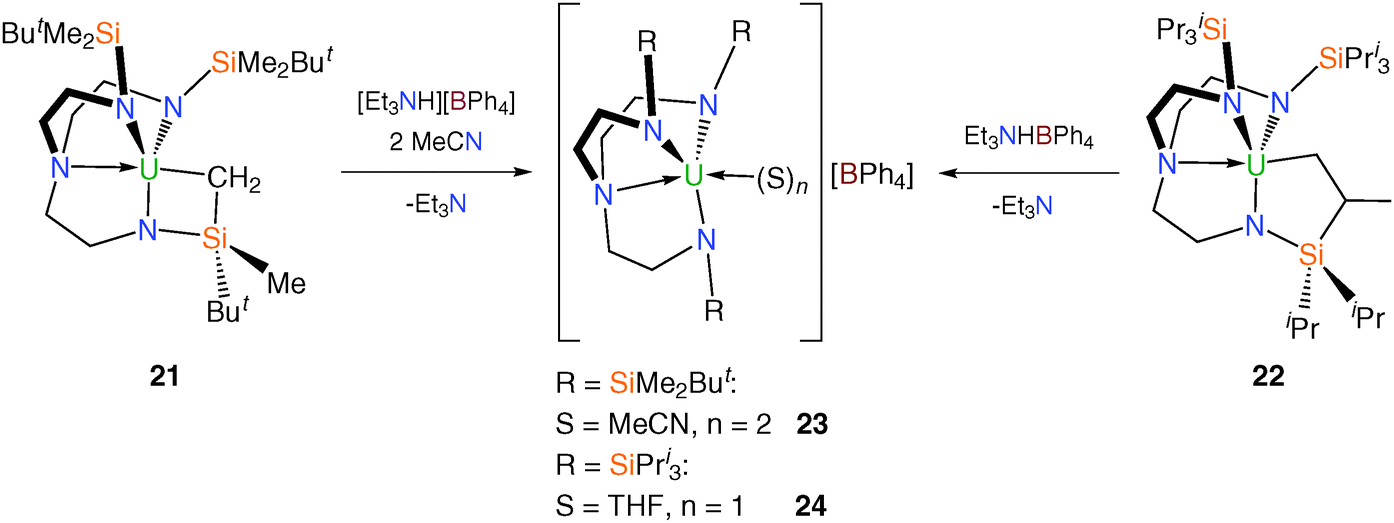 | ||
| Scheme 7 Synthesis of 23 and 24. | ||
Following work-up, the green-brown solid produced from the reaction of [U{N(CH2CH2NSiMe2But)2(CH2CH2NSiMeButCH2)}], 21, with triethylammonium tetraphenylborate appeared to be consistent with the formation of “[U(TrenDMBS)(THF)][BPh4]” by inspection of its 1H NMR spectrum, but persistent impurities precluded any further analysis and slowly cooled saturated solutions of this solid in THF afforded oily material. However, dissolution in a toluene–acetonitrile mix resulted in the formation of a yellow-brown solution which upon cooling yielded green-brown crystals suitable for a single crystal X-ray diffraction study, revealing the bis(acetonitrile) complex 23. The solid state structure of 23 consists of a [U(TrenDMBS)(NCMe)2]+ cation exhibiting the expected contraction of the U–N bond distances [U–Namide 2.220(6) Å (av.), U–Namine 2.577(6) Å and U–Nnitrile 2.595(7) Å (av.)] relative to many comparable neutral Tren–uranium systems. The structure of 23 also features a non-coordinated tetraphenylborate anion.25
The solid state structure of 24 could not be ascertained by XRD due its oily nature, however it could be deduced from the 1H NMR spectrum and the elemental microanalysis data for 24 that one molecule of coordinated THF is present in the complex and there is no evidence of any contacts to the uranium centre from the tetraphenylborate anion.24
Gallyl derivatives
In 2009 the isolation and characterisation of the first actinide–group 13 complex to exhibit both σ- and π-components in the metal–metal bond was reported.13a,26 Prepared from an equimolar mixture of 1 and [K{Ga(NArCH)2}(TMEDA)] (Ar = 2,6-iPr2–C6H3; TMEDA = Me2NCH2CH2NMe2)27 in THF, the target Tren–uranium(IV) gallyl complex [U(TrenTMS){Ga(NArCH)2}(THF)] (25), Fig. 4, was characterised by a single crystal XRD study that revealed two crystallographically independent molecules of 25 in the asymmetric unit, each with slightly different U–Ga bond lengths [3.221(2) and 3.298(2) Å].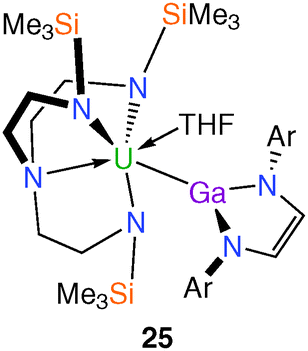 | ||
| Fig. 4 Molecular structure of 25. | ||
Both U–Ga bond distances are slightly longer than the sum of the covalent radii of U and Ga (3.18 Å),28 which may be a consequence of the high steric demands of the gallyl and TrenTMS components. Although undoubtedly a weak and highly polarised U–Ga bond, a DFT study of a closely related model complex revealed not only a σ-interaction but also a π-component to the uranium–gallium bond; the latter interaction is characterised by donation of nitrogen lone pairs into the vacant gallyl p-orbital with subsequent donation to uranium. Additionally, 25 represents a model for the as yet unknown isolobal [U(IV)–CO−˙] fragment.
Tren–uranium – group 14 complexes
Alkyl, acetylide, and cyclometallate derivatives
The monomeric metallacyclic uranium(IV) alkyl complex [U{N(CH2CH2NSiMe2But)2(CH2CH2NSiMeButCH2)}] (21), Fig. 5,23,29 was initially prepared by treatment of 2 with a range of lithium, potassium, magnesium and zinc alkyls in pentane to give brown solutions from which extremely air-sensitive orange-brown crystals of 21 could be isolated. However, optimum yields (ca. 70%) were later obtained by the reaction of the iodide derivative 6 with stoichiometric quantities of diethylzinc, neopentyllithium or benzylpotassium in toluene. The molecular structure of 21 was determined by a single crystal XRD experiment, which revealed a U–C bond, which at 2.752(11) Å is amongst the longest U–C σ-bonds reported and this is probably a consequence of its incorporation within a highly strained four-membered [U–N–Si–C] metallacyclic ring.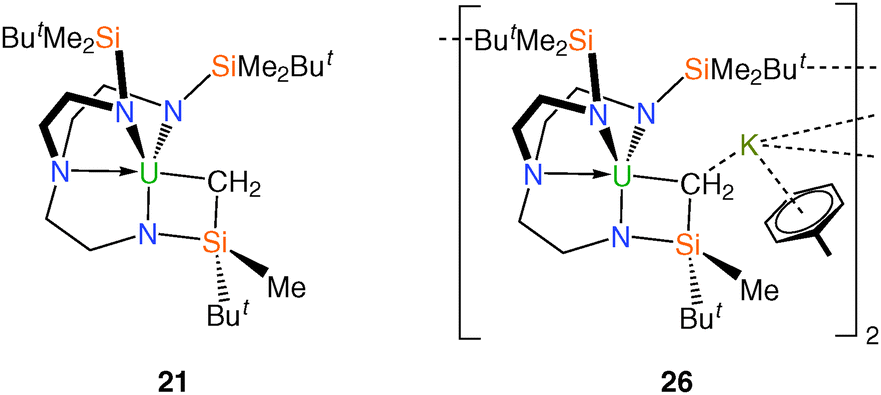 | ||
| Fig. 5 Structures of 21 and 26. | ||
The attempted reduction of 6 in toluene by a potassium film unexpectedly afforded the bimetallic metallacyclic anion [{(K[η6-C6H5Me])(U[N{CH2CH2NSiMe2But}2{CH2CH2NSiMeButCH2}])}2] (26), Fig. 5, which was remarkable as 6 is routinely reduced to trivalent 11 by a potassium film in pentane.18a The solid state structure of 26 reveals two metallacyclic anions bridged by two K cations, related to one another by a crystallographic centre of inversion. Each potassium ion is bound by a toluene ligand that coordinates in an η6 fashion and the metallacyclic U–C distance of 2.575(10) Å is significantly shorter than that observed in 21 but remains at the upper end of known U–C bond distances. It was proposed that 26 was formed via the in situ potassium reduction of 21, which itself is produced within the reaction mixture from small quantities of benzylpotassium (that form by the reaction of the K film with toluene solvent) reacting with the starting material 6.
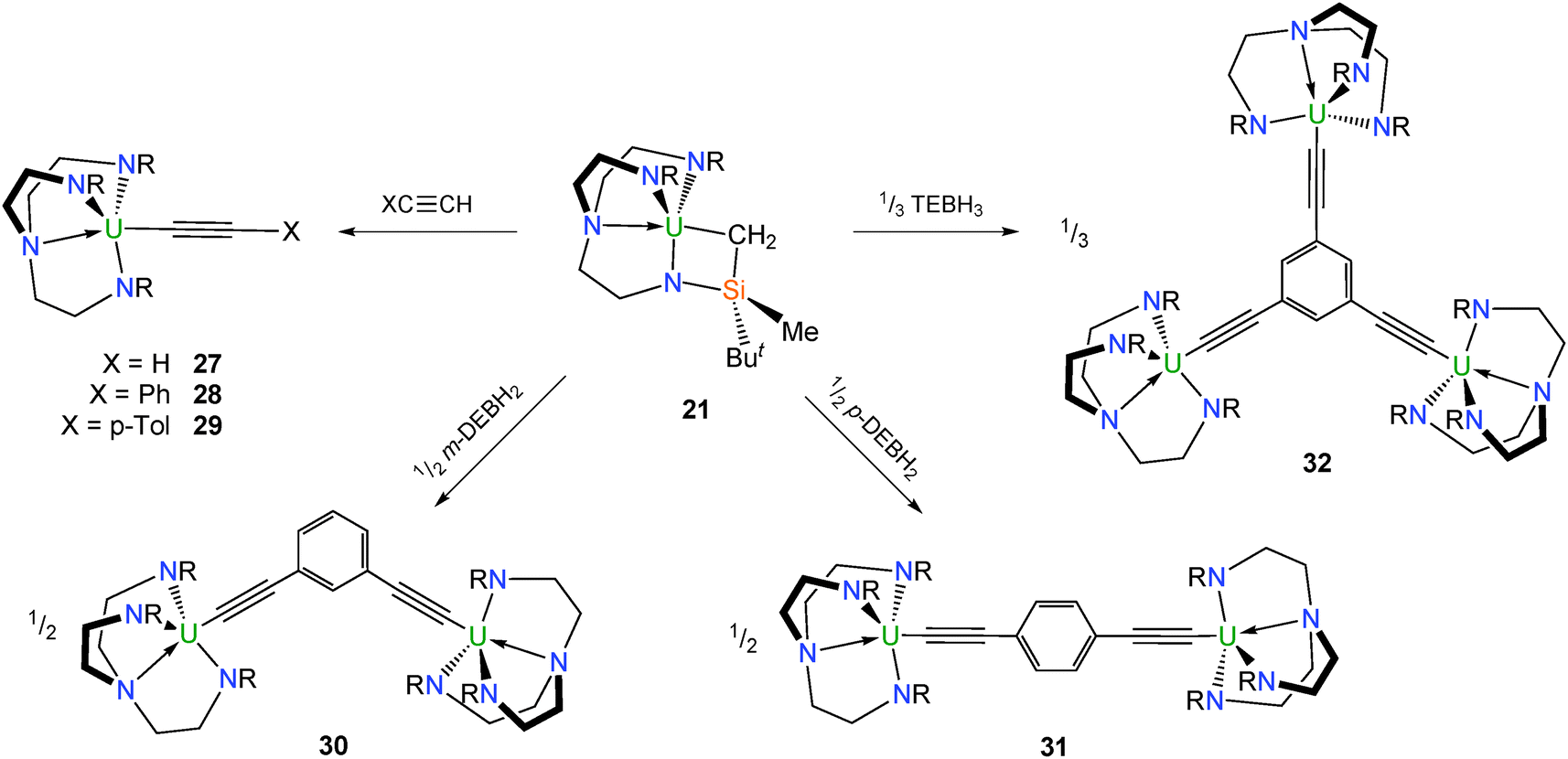
Metallacycle 21 reacts with a variety of acetylenes to afford mono-, di- and trimetallic TrenDMBS–uranium(IV) acetylide complexes (Scheme 8). Complex 21 reacts with stoichiometric quantities of the acetylenes HCCX (X = H, Ph, p-tolyl) to afford the respective mononuclear acetylide complexes [U(TrenDMBS)(C2X)] [X = H (27), Ph (28), p-tolyl (29)], although structural characterisation is lacking for 27.23,30 It was stated in one publication30 that 28 could not be prepared from 2 and LiCCPh as initially reported23 – with the lithium ‘ate’ complex [U(TrenDMBS)(C2Ph)2(μ-Li)(THF)] being the only isolable product – instead a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 21 and phenylacetylene was required to synthesise 28. The U–C–C bond angles in 28 and 29 of 160.9(4) and 156.4(6)°, respectively, are significantly more acute than the reported range for structurally characterised terminal f-element alkynyls (170–176°), which was ascribed to a geometric distortion to maximise U–C π-interactions.
:1 mixture of 21 and phenylacetylene was required to synthesise 28. The U–C–C bond angles in 28 and 29 of 160.9(4) and 156.4(6)°, respectively, are significantly more acute than the reported range for structurally characterised terminal f-element alkynyls (170–176°), which was ascribed to a geometric distortion to maximise U–C π-interactions.
 | ||
| Scheme 8 Synthesis of 27–32. R = SiMe2But; m-DEBH2 = 1,3-diethynylbenzene; p-DEBH2 = 1,4-diethynylbenzene; TEBH3 = 1,3,5-triethynylbenzene. | ||
Treatment of 21 with meta- and para-diethynylbenzene as well as triethynylbenzene in the appropriate stoichiometry afforded the bright green Tren–uranium arylacetylides [{U(TrenDMBS)}2(μ-κ2-1,3-(C2)2–C6H4)] (30), [{U(TrenDMBS)}2(μ-κ2-1,4-(C2)2–C6H4)] (31) and [{U(TrenDMBS)}3(μ-κ3-1,3,5-(C2)3–C6H3)] (32), respectively (Scheme 8).30 X-ray structural studies on 30–32 confirmed the individual cluster connectivities, however in the case of 32 a full analysis was precluded by poor data quality. A detailed magnetometric analysis of all three of the polynuclear compounds identified magnetic singlet ground states at low temperature, although these data were suggestive of weak ferromagnetic communication between the uranium centres in 30–32.
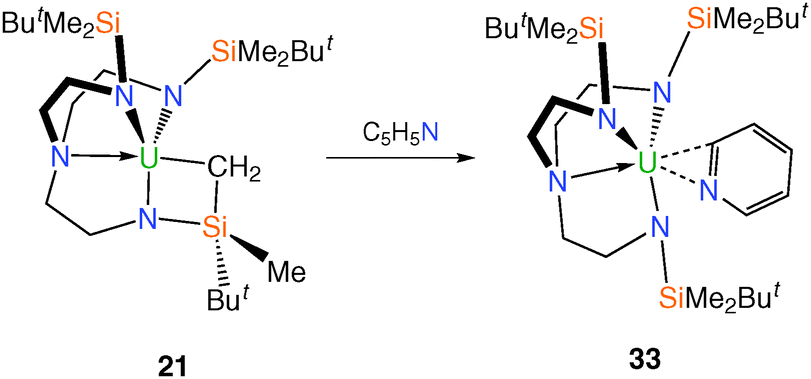
Further reactivity of 21 was disclosed in reports23,29 that metallacyclic 21 undergoes an acid–base reaction with pyridine, affording the TrenDMBS–uranium(IV) pyridyl complex 33, Scheme 9. Complex 33 was structurally characterised and confirmed the pyridyl unit to be bound in a planar η2 coordination mode.
 | ||
| Scheme 9 Synthesis of 33. | ||
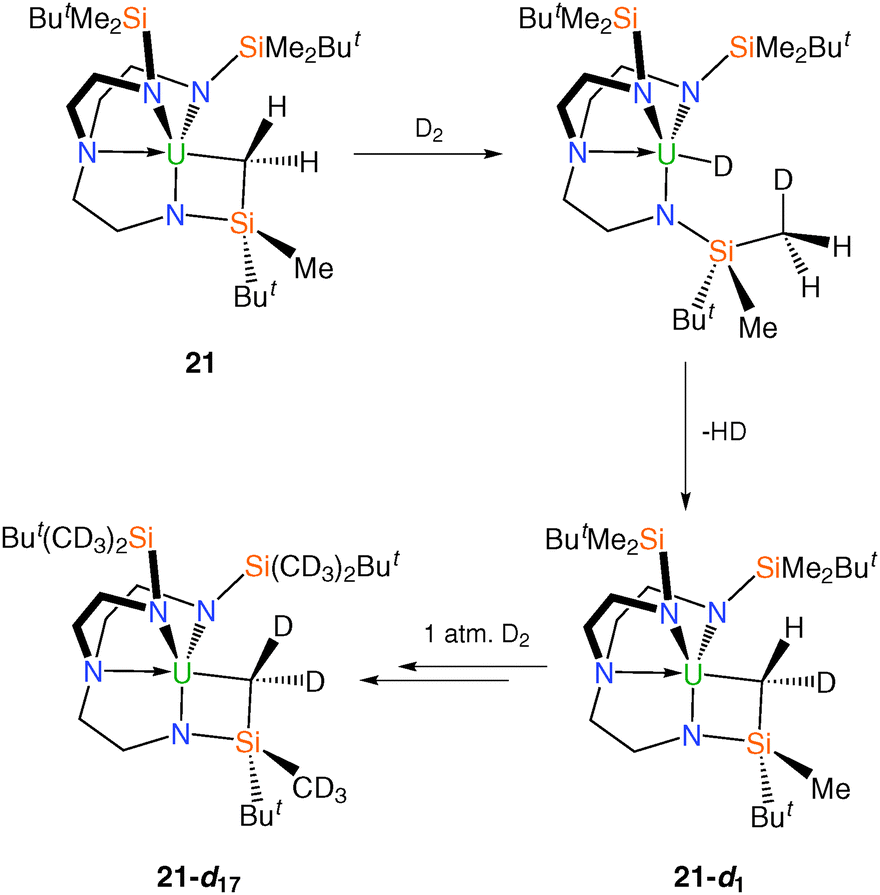
Given the documented ability for metalated alkyl groups in uranium complexes to undergo facile deuteration in solution,10f a d8-toluene solution of 21 was exposed to D2 at room temperature, Scheme 10. It was reported that under these conditions over a period of a few hours deuteration of all SiMe2 groups as well as the metallacyclic CH2 unit was observed. tert-Butyl CH3 groups and methylene CH2 groups were not deuterated, ascribed to the absence of α-Si atoms for these units.29
 | ||
| Scheme 10 Selective H/D exchange process for 21. | ||
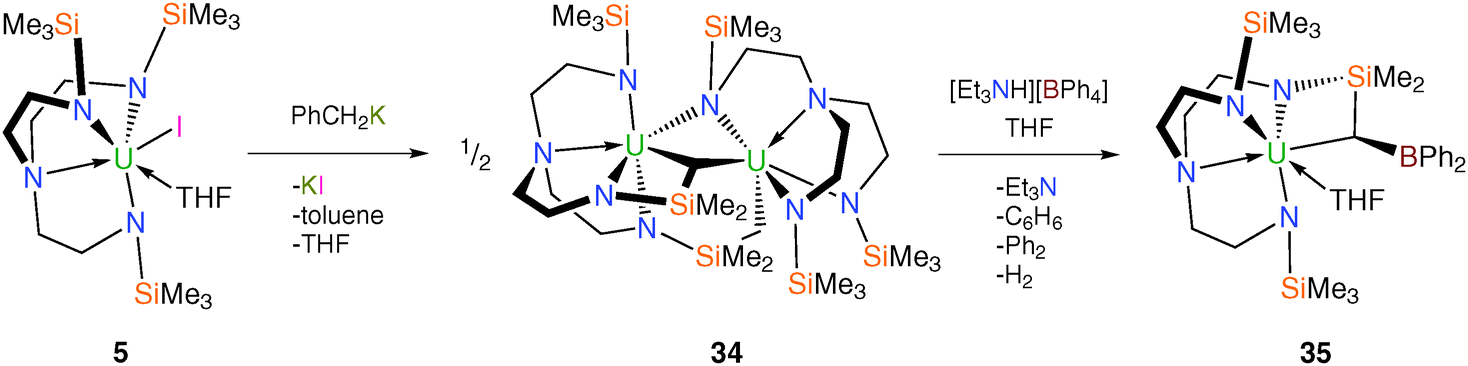
Subsequently, cyclometallation chemistry at uranium has been explored using the less sterically demanding TrenTMS ligand system.22 Treating 5 with benzylpotassium was anticipated to give the TrenTMS–uranium(IV) ‘tuck-in’ metallacycle “[U{N(CH2CH2NSiMe3)2(CH2CH2NSiMe2CH2)}(THF)]” both by analogy to 21 and with reference to the wider literature.10f,23 A green toluene solution of 5 reacts with KCH2Ph to give a dark yellow turbid suspension, suggesting KI elimination. Following work-up, yellow crystals were isolated from hexanes and a single crystal XRD study revealed them to be the unusual dinuclear tuck-in-tuck-over tuck-over TrenTMS–uranium(IV) dialkyl complex [U{N(CH2CH2NSiMe3)(CH2CH2NSiMe2CH2)2}U(TrenTMS)] (34), Scheme 11.
 | ||
| Scheme 11 Synthesis of 34 and 35. | ||
The molecular structure of 34 is bimetallic and consists of two uranium(IV) units in a tuck-in-tuck-over tuck-over triamidedialkyl–triamide coordination mode, which was unprecedented for Tren ligands. Two four-membered metallacycles are formed – [U–N–Si–C] and [U–N–U–C] – that feature markedly different bite angles [67.13(13)° and 81.89(12)°, respectively] reflecting the difference between the constituents and their relative location within the structure.
The synthesis of the anticipated separated ion pair TrenTMS–uranium(IV) complex [U(TrenTMS)(THF)2][BPh4] (19) was attempted by treatment of 34 with one molar equivalent per U of [Et3NH][BPh4] in THF. However the molecular structure of the uranium-containing product was determined to be the mononuclear BPh2-functionalised metallacyclic tuck-in TrenTMS–uranium(IV) complex [U{N(CH2CH2NSiMe3)2(CH2CH2NSiMe2C[H]BPh2)}(THF)] (35), Scheme 11. The boron centre is trigonal planar and the B–Calkyl bond distance of 1.493(11) Å is short inferring partial multiple B–C bond character, which was supported by a DFT study that revealed a B–C Mayer bond order of 1.33, consistent with the presence of a B–C π-bond which is perturbed by the polarising uranium centre. The reaction between 34 and [Et3NH][BPh4] was monitored using variable temperature 1H NMR spectroscopy and GC-MS, which revealed the presence of hydrogen, benzene and biphenyl as identifiable products of the reaction. The isolation of 35 represents the first example of double dearylation of BPh4− in a molecular context, adding to the debate against the use of this anion in homogeneous catalysts.22
Cyclometallation chemistry has been explored with the sterically encumbered TrenTIPS ligand in the context of divergent reactivity patterns for Tren–uranium(IV) and thorium(IV) systems.31 Whilst 7, upon treatment with KCH2Ph, affords the orange-red cyclometallated Tren–uranium complex [U{N(CH2CH2NSiPri3)2(CH2CH2NSiPri2C[H]MeCH2)}] (22) at temperatures well below ambient, the equivalent reaction with the colourless thorium(IV) analogue [Th(TrenTIPS)(I)] gives a colourless, isolable η1-benzyl complex. This benzyl requires heating to afford the thoracyclic analogue of 22, Scheme 12.
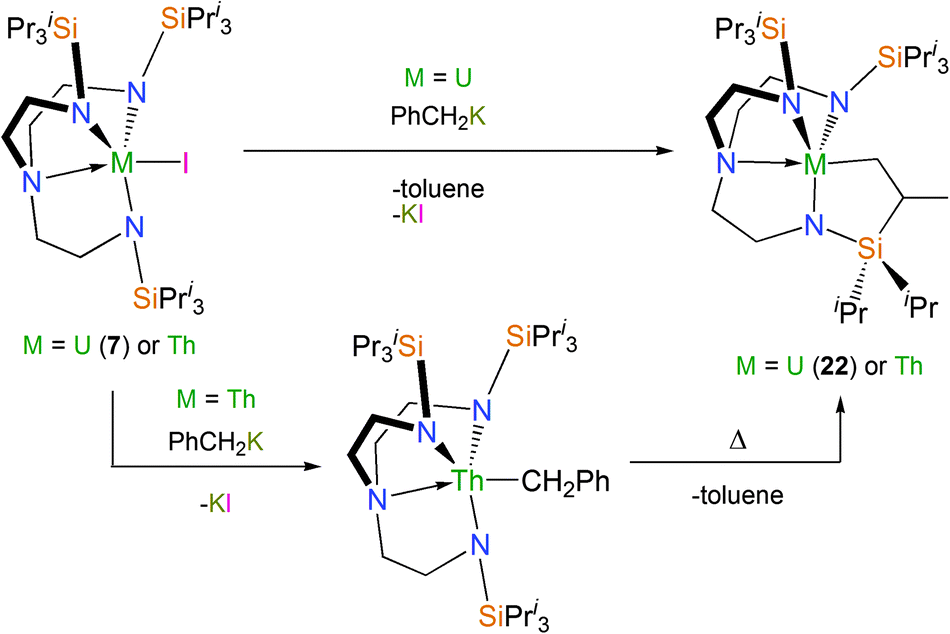 | ||
| Scheme 12 Synthesis of 22. | ||
The origin of the inversion of the above reactivity trend was investigated by a DFT study. This showed that the greater f-orbital participation for uranium compared to thorium facilitates the σ-bond metathesis transition state. For thorium the transition state is more ionic and so the benzyl intermediate can be isolated experimentally, whereas no such benzyl complex was observed for the uranium case, even when probed using low-temperature 1H NMR spectroscopy.
Ylide derivatives
The synthesis of terminal actinide alkylidene complexes remains a major synthetic target despite extensive investigations into phosphorus ylide complexes with significant U–C multiple bond character.9d,32 Given the synthetic accessibility of, for instance, a tantalum alkylidene using methylene trimethylphosphorane, such a complex was targeted utilising the TrenDMBS ligand scaffold. It was reported17 that the addition of CH2PMe3 to a purple solution of 11 afforded a dark green solution from which a dark green solid was isolated. The analytical data supported the proposed formulation of the Tren–uranium methylene(trimethylphosphorane) complex [U(TrenDMBS)(CH2PMe3)] (36a), Fig. 6, although it lacks structural characterisation. The electronic absorption spectrum of 36a is strongly suggestive of a CH2PMe3 adduct of uranium(III).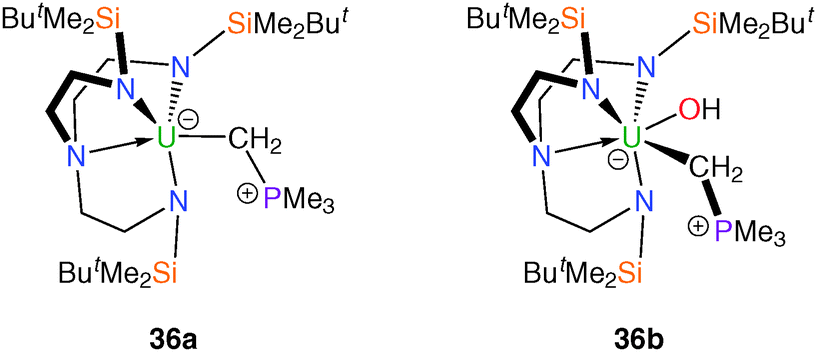 | ||
| Fig. 6 Structures of 36a and 36b. | ||
During attempts to structurally characterise 36a a small sample of dark green crystals was isolated and shown by X-ray diffraction to be the uranium(IV) methylene(trimethylphosphorane) hydroxide complex 36b, Fig. 6. Due to the low yield additional analytical data are unavailable, however the structure of 36b does have some unusual structural features, namely the highly extended U–C distance [2.706(12) Å], which is comparable to that found in the highly strained metallacycle 21, and the presence of the first terminal U–OH linkage.
Cyclopentadienyl derivatives
Cyclopentadienyl (Cp) complexes of uranium are well known but only one Tren–uranium–Cp derivative has been structurally characterised.21 [U(TrenTMS)(η5-C5Me5)] (37), Fig. 7, was prepared from [Na(C5Me5)] and half a molar equivalent of 8. The product 37 was purified by sublimation and a single crystal XRD experiment confirmed the anticipated structure, with the C5Me5− ring exhibiting the expected η5 coordination mode. The room-temperature 1H NMR spectrum of 37 is indicative of average C3v molecular symmetry, but it was stated that the ground-state geometry would be close to that in the solid state structure.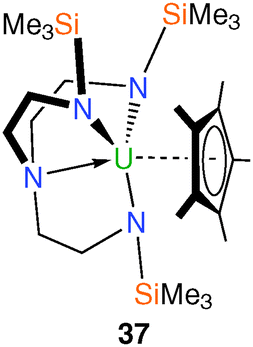 | ||
| Fig. 7 Structure of 37. | ||
Tren–uranium – group 15 complexes
Amide derivatives
The synthesis of [U(TrenDMBS)(NEt2)] (38) demonstrates how alternative methodologies can be employed to access Tren–uranium amide complexes.13b The diethylamide complex 38 is prepared from either 2 or [{U(NEt2)4}2] as the source of uranium (Scheme 13). The solid state structure of 38 was obtained via an XRD experiment, which revealed a monomeric complex with the overall structural features typical for Tren–uranium(IV) complexes and it is similar to complexes 16 and 17.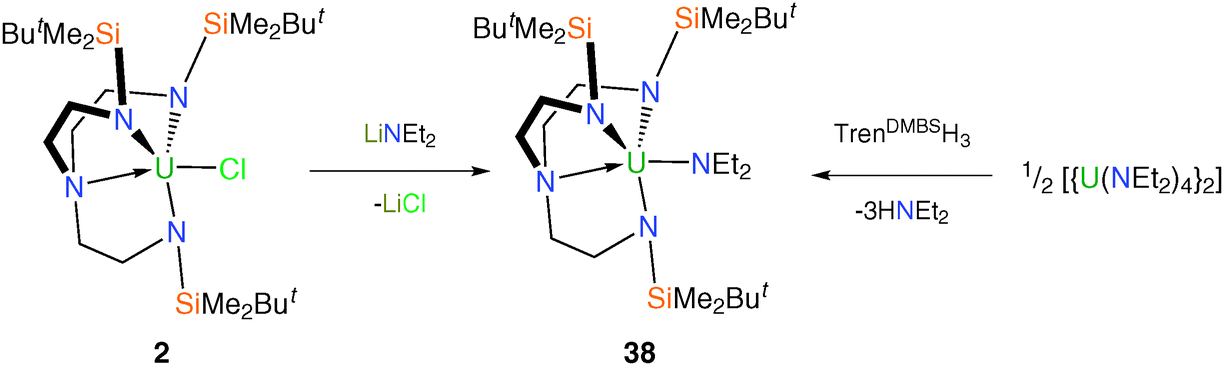 | ||
| Scheme 13 Synthesis of 38. | ||
Following its previously reported inadvertent production in varying yields,14b it was shown that the parent terminal amide complex [U(TrenTIPS)(NH2)] (39) can be prepared in high yield on multi-gram scales. Compound 39 represents an attractive precursor from which to target U–N multiple bonds via subsequent deprotonation methods, bearing in mind the successful isolation of a terminal molybdenum carbide complex [Mo(C){N(R)Ar}3][K(benzo15C5)2] [R = C(CD3)2CH3, Ar = C6H3Me2-3,5], from a methylidyne precursor by deprotonation and alkali metal sequestration.33 The parent amide 39 was synthesised by salt elimination from the reaction of 3 with NaNH2 in THF (Scheme 14). A single crystal XRD study performed on a yellow crystal of 39 revealed a monomeric structure with a terminal U–NH2 bond distance of 2.228(4) Å, which is closely comparable to the only other structurally characterised example of a U–NH2 linkage [2.194(5) Å (av.)].34
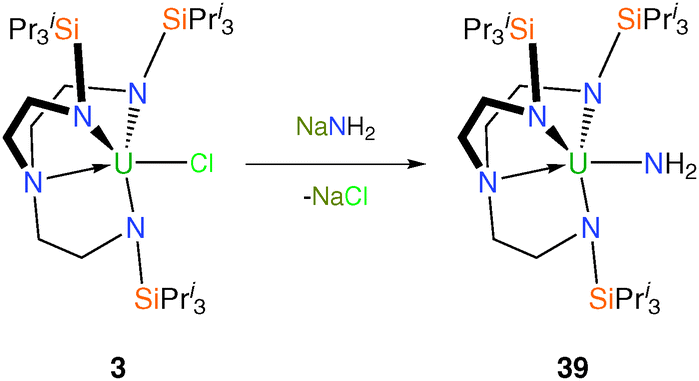 | ||
| Scheme 14 Synthesis of 39. | ||
Imido derivatives
The first report of a Tren–uranium imido complex came in 2002 when it was demonstrated that dark purple trivalent complex 11 reacts with trimethylsilylazide, Me3SiN3, with a colour change to dark red and evolution of gas.17 Although the characterisation data for the isolated product supported the formulation as [U(TrenDMBS)(NSiMe3)] – the imido product of a two-electron oxidation at uranium – no corroborative structural data are available. Along similar lines, dark purple 15 was shown to react with Me3SiN3 or AdN3 (Ad = 1-adamantyl) evolving nitrogen gas to afford, after work-up and crystallisation, red-brown and dark brown crystals, respectively, of the terminal Tren–uranium(V) imidos [U(TrenTIPS)(NSiMe3)] (40a) and [U(TrenTIPS)(NAd)] (40b), Scheme 15.14b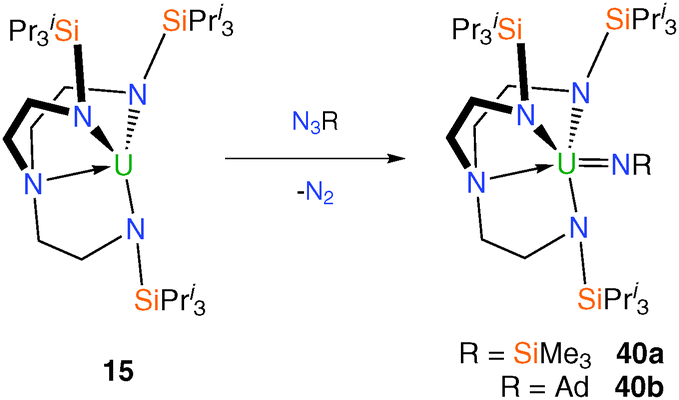 | ||
| Scheme 15 Synthesis of 40a and 40b. | ||
Single crystal XRD experiments confirmed the expected structures of the two products. The terminal uranium(V) imido complexes 40a and 40b are the product of two-electron oxidations at uranium, facilitated by nitrogen evolution, and the UNimido bond distances of 1.954(3) and 1.946(13) Å for 40a and 40b, respectively, are typical of terminal uranium(V) imido complexes.9d,35 Complex 40a could be viewed as a nitride precursor since the polarised Me3Si–N bond could in theory be cleaved to generate a nitride, however attempts to this end were not successful.
Until very recently there were no reports of f-element terminal parent imido linkages (LnMNH), presumably due to the requirement for significant kinetic stabilisation at the metal centre, usually conferred by large R groups installed on the imido nitrogen; for metals toward the bottom of the periodic table this issue would be compounded by their large ionic radii and would certainly be at its most acute for actinide centres. Deprotonation of the parent amide 39 with LiBut or MCH2Ph (M = Na, K, Rb, Cs) affords the dimeric alkali metal-bridged uranium(V) imido complexes [{U(TrenTIPS)(μ-NH)(μ-M)}2] [M = Li–Cs (41a–e)] as pale-pink crystalline solids after workup and crystallisation, Scheme 16.36 The molecular structures of 41a–e were determined by single crystal XRD, which revealed in each case dimeric structures constructed around a centrosymmetric M2N2 four-membered ring. The uranium–imido bonds in 41a–e span the range 2.042(3) to 2.135(3) Å and are significantly shorter than the U–NH2 bond length of 2.228(4) Å in 39, reflecting the build-up of imido character of 41a–e compared with the amide character of 39.
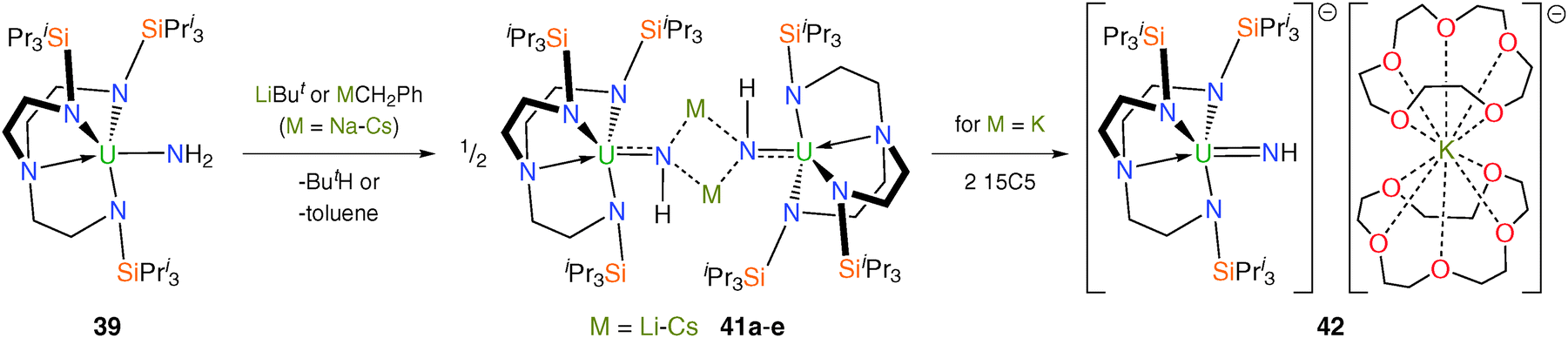 | ||
| Scheme 16 Synthesis of 41a–e and 42. | ||
Treatment of 41c with 2 equivalents of 15-crown-5 ether (15C5) and stirring of the resulting oil in hexanes affords a brown solid. The solid-state structure of [U(TrenTIPS)(NH)][K(15C5)2] (42) was determined by single crystal XRD, which confirmed the separated ion pair (SIP) formulation. The U–Nimido bond length in 42 was found to be comparable to those in 41a–e at 2.034(3) Å, though this was attributed to the anionic nature of [U(TrenTIPS)(NH)]− partially offsetting the expected U–Nimido bond contraction upon abstraction of an alkali metal from the bridging imido to give a terminal UNH unit. Theoretical studies of the near-linear UN–H group [172(3)°] in 42 show the presence of a threefold σ2π4 bonding combination, supporting the assertion that 42 represents a protected nitride given that the proton bound to the imide nitrogen may in principle be removable.
Nitride derivatives
Until 2012, an isolable molecule under ambient conditions containing the uranium nitride linkage as a terminal unit had evaded all attempts to prepare it for decades even though it represents a fundamental synthetic target for the study of metal–ligand multiple bonding and f-orbital participation. Previous reports of terminal uranium nitride species were restricted to elegant matrix isolation studies or mass spectrometric observations;11b molecular uranium nitride complexes prepared under ambient conditions included polymetallic compounds with bridging nitride units, covalently-bound nitridoboranes, or transient species that undergo C–H insertion reactions with ancillary ligands under photolytic conditions.11b The key advance came with the report of a terminal uranium nitride supported by the bulky TrenTIPS ligand framework.12b Trivalent 15 undergoes a two-electron oxidation upon treatment with sodium azide to afford the dimeric, sodium-bridged Tren–uranium(V) nitride complex [{U(TrenTIPS)(μ-N)(μ-Na)}2] (43). Subsequent addition of two equivalents of 12-crown-4 ether (12C4) per sodium affords the terminal SIP uranium nitride [U(TrenTIPS)(N)][Na(12C4)2] (44), or one equivalent of 15-crown-5 ether (15C5) per sodium produces the capped nitride [U(TrenTIPS)(μ-N)(μ-Na)(15C5)]14b (45), Scheme 17. The success of this approach to give 44 rests on the combination of the sterically demanding Tren ligand preventing the nitride from bridging to another uranium centre, stabilisation of the nitride during installation by the sodium, but straightforward abstraction of the sodium ion due to its ionic bonding to the nitride and appropriate size-matching to a crown ether.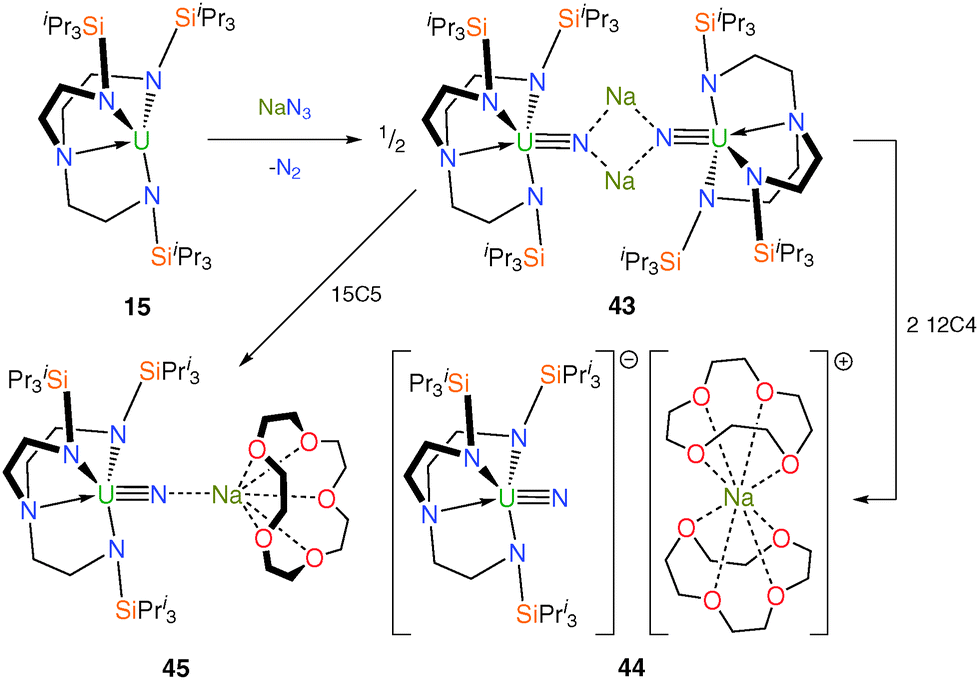 | ||
| Scheme 17 Synthesis of 43–45. | ||
Single crystal XRD experiments confirmed the molecular structures of 43, 44 and 45, and revealed very short U–Nnitride bond distances of 1.883(4), 1.825(15) and 1.810(5) Å, respectively, with the latter two being indistinguishable but as expected showing a moderate contraction of ca. 0.06 Å relative to 43 either upon complete encapsulation of the alkali metal and removal from the nitride centre or upon removal of one sodium per nitride and inclusion of a capping 15C5. This contraction was ascribed to the removal of polarising Na+ cations from the nitride atom resulting in a higher charge density for the terminal or Na-capped nitride centres in 44 and 45, respectively, relative to the disodium-bridged 43. Upon formation of 43, it is postulated that the coordinated alkali metal centres help to stabilise the high charge density on the nitride moieties, minimising deleterious side reactions, despite the likely weak nature of the sodium–nitride interactions in 43. Electronic absorption spectroscopy and variable temperature magnetometric measurements support the assignment of the +5 oxidation state for uranium in 43–45. A computational study revealed the expected threefold σ2π4 molecular orbital description of the U![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N triple bonds with Mayer bond orders of 2.21, 2.91 and 2.45 for the uranium–nitride linkages in 43, 44 and 45, respectively, reflecting the effect of the number of coordinated sodium ions located on the nitride centre in each case. For 44, the molecular orbitals representing the U–N σ bond are higher in energy than those representing the π interactions and this is the same as for the uranyl dication, but the reverse of what would be expected based on observations in UC and UN double bonds. This can be explained by considering an antibonding interaction at short U–N distances between the σ-orientated N 2pz orbital and the annular lobes of the U 6d and 5f orbitals (with the UN bond orientated along the z-axis).12b
N triple bonds with Mayer bond orders of 2.21, 2.91 and 2.45 for the uranium–nitride linkages in 43, 44 and 45, respectively, reflecting the effect of the number of coordinated sodium ions located on the nitride centre in each case. For 44, the molecular orbitals representing the U–N σ bond are higher in energy than those representing the π interactions and this is the same as for the uranyl dication, but the reverse of what would be expected based on observations in UC and UN double bonds. This can be explained by considering an antibonding interaction at short U–N distances between the σ-orientated N 2pz orbital and the annular lobes of the U 6d and 5f orbitals (with the UN bond orientated along the z-axis).12b
The terminal nitride 44 reacts with excess water in the presence of three equivalents of the reductant cobaltocene, CoCp2, to produce ammonia confirming the existence of a basic nitride unit. Additionally, 44 was shown to react with Me3SiCl producing the Tren–uranium(V) imido complex [U(TrenTIPS)(NSiMe3)] (40a), eliminating [Na(12C4)2][Cl], thus demonstrating nucleophilic character.12b
Attempts to oxidise 43 with mild oxidants such as AgPF6 resulted in decomposition to, for instance, [U(TrenTIPS)(F)], but that when 44 was treated with half a molar equivalent of iodine, I2, elimination of [Na(12C4)2][I] was observed, Scheme 18, and red crystals of the uranium(VI)–nitrido complex [U(TrenTIPS)(N)] (46) were isolated.14b
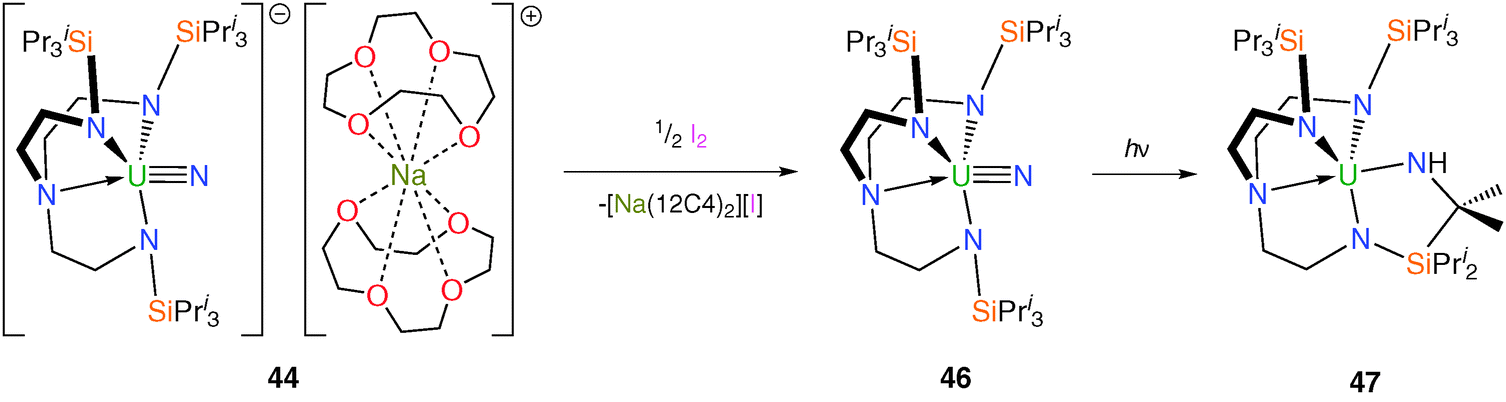 | ||
| Scheme 18 Preparation of 46 and 47. | ||
The uranium(VI)–nitride bond length in 46 was crystallographically determined to be 1.799(7) Å, which is statistically invariant to the U–Nnitride bonds in 44 and 45 and this feature is attributed to the removal of a non-bonding 5f electron upon oxidation; the very short uranium–Namine distance [2.465(5) Å] is ascribed to a consequence of the inverse-trans-influence.37 Thus, as mentioned in the introduction, the amine may play a vital electronic role in the successful isolation of these terminal uranium nitrides by providing stabilisation to trans multiply bonded ligands. FTIR and NMR spectroscopic data also supported the formulation of a diamagnetic 5f0 uranium(VI) nitride. DFT studies on 46 again reveal a σ2π4 UN threefold bonding manifold and a Mayer bond order of 2.92 for the uranium–nitride linkage, which is all but identical to that for 44 (2.91) and underlines the minor effect on the UN interaction upon removal of the non-bonding 5f electron. Surprisingly, a topological analysis of the electron density for the terminal uranium–nitride linkages in 44 and 46 suggested a comparable degree of covalency to terminal group 6 nitrides.
Photolysis for ca. twenty minutes or exposure to sunlight for several days of toluene solutions of 46 resulted in C–H activation and insertion of the nitride into an isopropyl C–H bond to afford the secondary amide [U{(N[H]CMe2SiPri2NCH2CH2)N(CH2CH2NSiPri3)2}] (47), Scheme 18, underlining the highly reactive nature of the uranium(VI) nitride species under photolytic conditions. What is noticeable about the observed reaction chemistry of 44 and 46 is that despite the removal of a formally non-bonding 5f electron upon oxidation of 44 to 46, which is suggested by the X-ray and theoretical data not to have much of an impact on the bonding, the relative photochemical reactivity of 44 and 46 are profoundly different. Under photolytic conditions 44 does not decompose whereas 46 does, which can be attributed to the more oxidising nature of uranium(VI) compared to (V) since the mechanism of this photochemical C–H activation requires reduction of uranium. However, under normal conditions 44 is generally more reactive than 46 which suggests a weaker uranium–nitride linkage in the former compared to the latter.
The reactivity of Tren–uranium nitrides towards CO has been investigated,38 owing to its ambiphilicity, industrial and environmental significance, and also that carbonylation of transition metal nitride complexes is rare.39 As shown in Scheme 19, the red Tren–uranium(VI) nitride 46 undergoes reductive carbonylation when treated with carbon monoxide to afford green crystals of the Tren–uranium(IV) cyanate complex [U(TrenTIPS)(NCO)] (48), which mirrors reactivity seen for a handful of transition metal nitrides.39 The solid state structure of 48 is unremarkable, and features a virtually linear U–NCO unit [U–N–C = 173° (av.)]. The outcome of a reduction of 48 with KC8 is dependent on the reaction conditions although a nitride product – arising from reductive decarbonylation – is never isolated. In the absence of crown ethers, the reaction of 48 with KC8 results in the extrusion of KNCO and trivalent 15 is isolated from the reaction; however when two equivalents of benzo-15C5 are present, the uranium(III) cyanate SIP complex [U(TrenTIPS)(NCO)][K(B15C5)2] (49) is isolated as dark green crystals. In contrast to 48, the U–NCO unit in 49 is bent [U–N–C = 138° (av.)] in the solid state, likely by virtue of the attenuation of U–Ncyanate π-interactions due to the increased electron density on the U(III) centre in 49 relative to the U(IV) centre in 48.
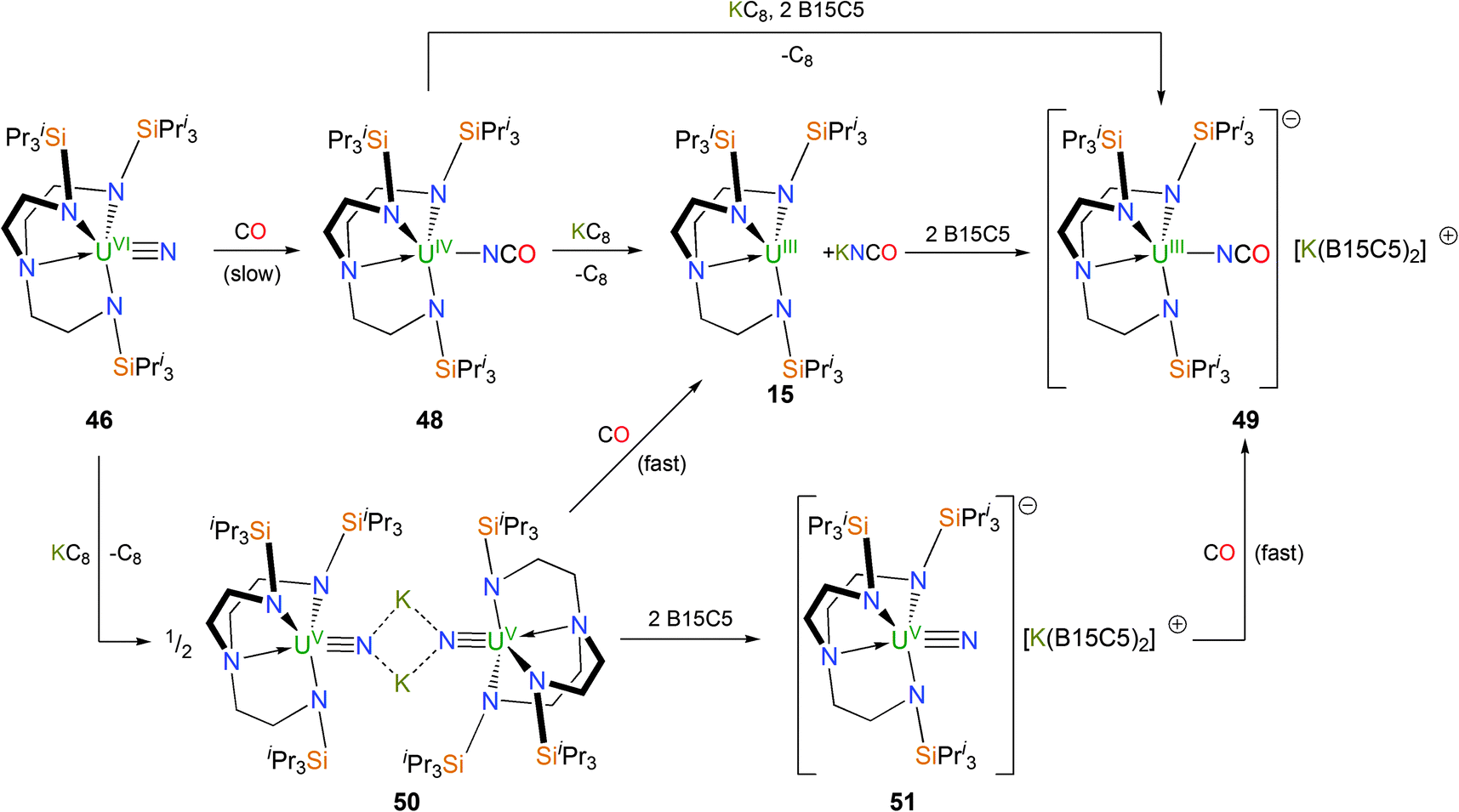 | ||
| Scheme 19 Synthesis of 48–51. B15C5 = benzo-15-crown-5. | ||
The bridging uranium(V) nitride [{U(TrenTIPS)(μ-N)(μ-K)}2] (50) also undergoes similar reductive carbonylation chemistry under an atmosphere of CO, affording 15 and KNCO. The SIP nitride complex [U(TrenTIPS)(N)][K(B15C5)2] (51) was prepared from 50 by treatment with 2 equivalents of benzo-15C5 and reacted with CO to afford 49. Both the reductive carbonylation reactions of 50 to produce 15 and KNCO as well as that of 51 to produce 49 were observed to proceed much more rapidly than that of 46 affording 48, which underscores the divergent reactivity for these uranium(V) and uranium(VI) nitrides. This discrepancy was examined by a DFT investigation, and it was found that in either case the reaction can be described as nucleophilic attack of the nitride to the incoming CO molecule in a [2+1]-cycloaddition reaction and the difference in observed rates can be explained by U–CO pre-coordination. The barrier to the transition state is higher in the case of the uranium(VI) species due to the smaller size of uranium(VI) relative to uranium(V), which requires the CO molecule to approach closer to the metal atom; a process that is energetically costly.
Photochemistry of azide, diazomethane, and isocyano derivatives
Metal azide complexes are known to be attractive precursors to nitride complexes as they can undergo photolytic denitrification to afford a nitride compound that is the product of a two-electron oxidation.39b,c No isolable uranium nitride complex has been accessed in this manner, although the photochemical conversion of uranium(IV) azides into C–H bond activated products, likely via a uranium(VI) nitride intermediate, has been studied.40 This methodology was tested using TrenTIPS, whereby the azide precursor [U(TrenTIPS)(N3)] (52) was synthesised via salt metathesis from 3 and sodium azide, Scheme 20.14b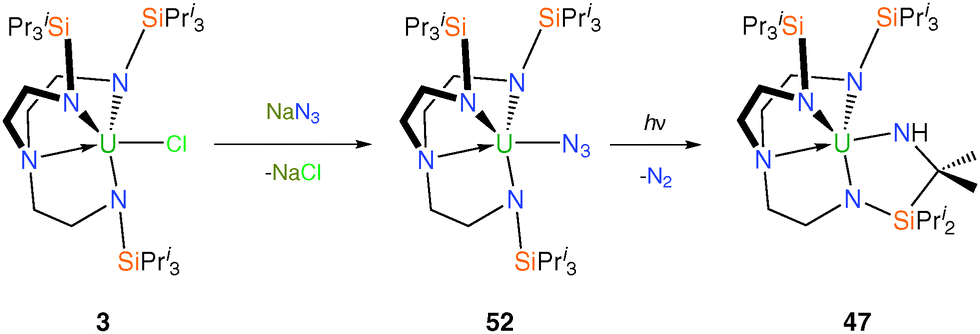 | ||
| Scheme 20 Synthesis and photolysis of 52 to give 47. | ||
The solid state structure of 52 was confirmed by a single crystal XRD experiment, which revealed a monomeric Tren–uranium(IV) azide. No reaction was observed when a toluene solution of 52 was heated and extended reaction times result in quantitative decomposition. Photolysis of 52 in toluene results in C–H activation to afford the secondary amide 47, which suggested that a transient uranium(VI) nitride could have been formed but decomposed under the harsh photolytic conditions required to promote N2 evolution; a rationale supported by the observed photolytic activation of 46 (see Scheme 18).
Metal–diazomethane (LnM–CR2N2) complexes represent attractive precursors for metal alkylidene species.41 Additionally, reports of organometallic actinide photochemistry are exceedingly rare in contrast to that for the d-block,42 although they suggest that f-block-diazoalkane reactivity trends are different to those for the transition metals. In an attempt to access a Tren–uranium alkylidene of the form “[U(TrenDMBS)(CHSiMe3)]”, it was reported that a dark purple pentane solution of 11 reacted with Me3SiCHN2 to afford a dark red solution, but no evolution of gas was observed.17 Dark red crystals formulated as the uranium hydrazido complex [U(TrenDMBS){N2CH(SiMe3)}] (53) were isolated, however no structural data were provided, although the identity of 53 was supported by its EI-mass spectrum and 1H NMR spectrum. No evidence of the target alkylidene complex was obtained after refluxing a d6-benzene solution of 53 or irradiation with ultraviolet light.
A more recent report utilised TrenTMS to access a novel isocyano(trimethylsilyl)amide complex 54via salt elimination using Me3SiCN2Li (obtained from the lithiation of Me3SiCHN2 with n-butyllithium), Scheme 21.43 Crystallographic refinement of N-bound versus C-bound disorder models for the SiMe3 group in the N(SiMe3)NC unit led to the assignment of the N-silyl isomer exclusively.
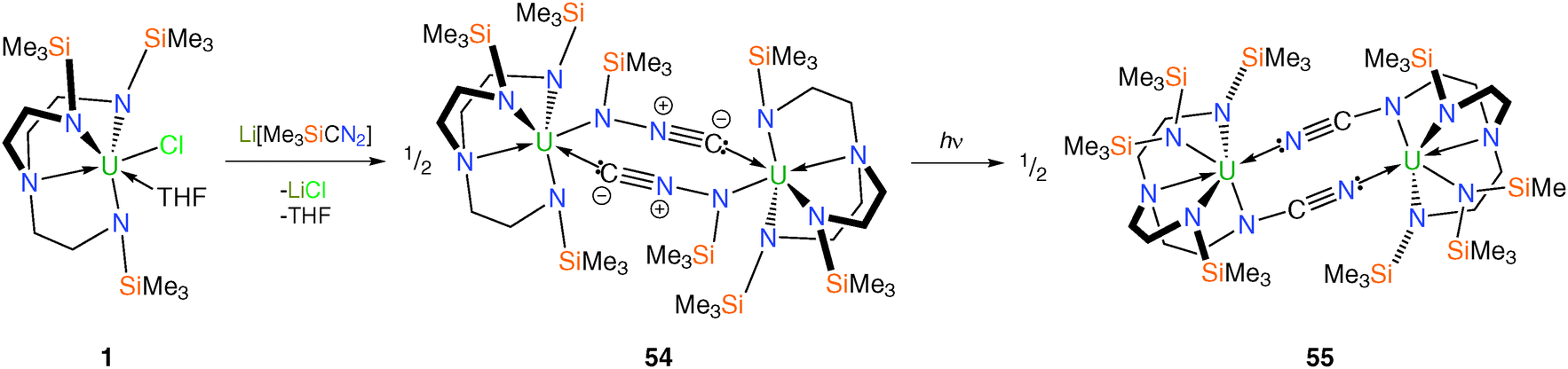 | ||
| Scheme 21 Synthesis of 54 and 55. | ||
Given the documented reactivity of diazomethane derivatives, a toluene solution of 54 was heated at 110 °C for three days. However, even after this time no reaction was observed and heating the solution to higher temperatures eventually resulted in the quantitative decomposition of 54 and the formation of unidentifiable products. Photolysis of a toluene solution of 54 at room temperature, however, resulted in a gradual colour change of the solution from green-yellow to brown. The molecular structure of the photolysis product 55 was determined by an XRD experiment is illustrated in Scheme 21.
With the assumption that [LiC(N2)SiMe3] has a C-bound trimethylsilyl group, as is preferred thermodynamically over the N-bound form,44 a 1,3-silyl shift from C to N is required during the formation of 54. The production of 55 is less straightforward, requiring N–Si and N–N bonds to be broken and C–N and N–Si bonds to be formed. The photolytic transformation of 54 to 55, involving multiple bond-cleavage and -capture, is not thermally accessible and was without precedent in diazoalkane chemistry.
Phosphinidene derivatives
Reports of terminal metal phosphinidene complexes, LnMPR, remain rare relative to those for metal imides, carbenes and alkylidenes. In 2014, it was shown that the parent phosphide could be installed onto a TrenTIPS–uranium(IV) fragment to afford the first PH2− complex of uranium (Scheme 22).24 Yellow [U(TrenTIPS)(PH2)] (56) was structurally characterised and features a U–P bond distance of 2.883(2) Å which is slightly longer than comparable reported complexes such as in [U(C5Me5)2{P(SiMe3)2}(Cl)] [U–P = 2.789(4) Å].45
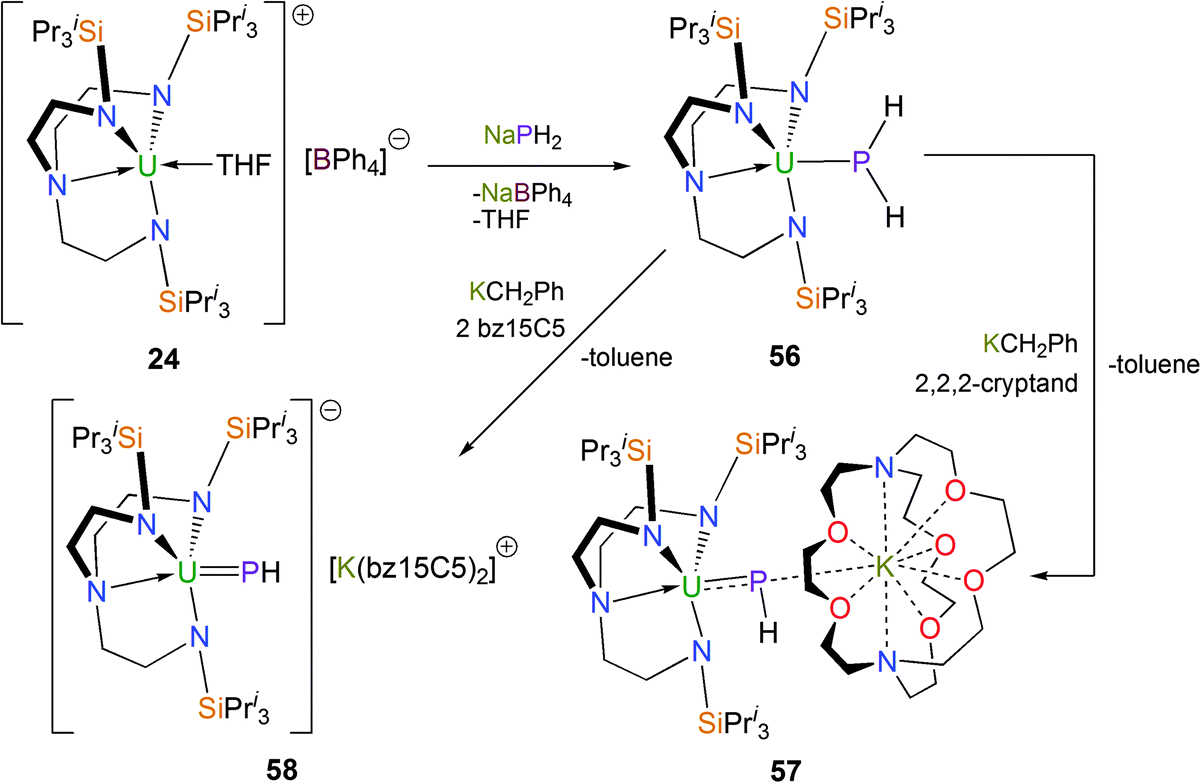 | ||
| Scheme 22 Synthesis of 56–58. B15C5 = benzo-15-crown-5. | ||
When 56 is treated with benzylpotassium and 2,2,2-cryptand, the bridging phosphinidiide [U(TrenTIPS)(μ-PH)(μ-K)(2,2,2-cryptand)] (57) was isolated as black crystals. A single crystal XRD study demonstrated a contracted UPH distance of 2.661(2) Å relative to 56 and revealed that the potassium ion is coordinated by the P atom and by the cryptand, although the long P–K distance of 3.575(2) Å suggests the P–K interaction should be regarded as weak.
Treatment of 56 with KCH2Ph and two equivalents of benzo-15-crown-5 ether (benzo-15C5) furnished the uranium(IV) terminal parent phosphinidene complex [U(TrenTIPS)(PH)] [K(benzo-15C5)2] (58) as black crystals. Complex 58 was structurally authenticated by a single crystal XRD study revealing a UP distance of 2.613(2) Å, which is around 0.05 Å shorter than the UP distance in 57, representing the first metal-stabilised terminal parent phosphinidene. The sum of the double bond covalent radii of U and P is 2.36 Å, so the UP distance in 58 lies midway between the sum of the covalent single and double bond radii values and within the range of the few reported uranium phosphinidene and phosphinidiide complexes – 2.562(3) Å in [U(η5-C5Me5)2(OPMe3)(P-2,4,6-But3C6H2)]46 and 2.743(1) Å in [{U(η5-C5Me5)2(OMe)}2(μ-PH)].47 Compound 58 has a calculated Mayer bond index of 1.92, which is as expected and Natural Bond Orbital (NBO) analysis identifies σ- and π-bonding interactions in the UP double bond.
Tren–uranium – group 16 complexes
Oxo derivatives
The ubiquity of the uranyl ion [UO2]2+ within aqueous and non-aqueous uranium chemistry imparts particular significance to the study of oxo complexes.48 The prerequisite facial coordination mode and steric constraint of the axial coordination environment of Tren complexes has meant that Tren–uranium frameworks have proven attractive targets towards the isolation of novel uranium O-atom containing compounds as well as the elusive cis-uranyl fragment.49During attempts to grow crystals of trivalent 11 suitable for an XRD experiment, a small quantity of black crystals were isolated, and a structural characterisation determined the structure to be the bimetallic bridging oxo complex [{U(N[CH2CH2NSiButMe2]2[μ-NSiMeButCH2])}2(μ-O)] (59), Fig. 8.23 Complex 59 is dinuclear and features two metalated methylsilyl groups and a bridging oxo unit, each of which bridge the two uranium centres. Formally, the uranium centres in 59 can be assigned as uranium(V) based on charge balance arguments, as the U–N bond distances in 59 are not normally diagnostic of +4 or +5 oxidation states of uranium due to the predominantly ionic bonding regime. No further characterisation data are available to confirm the assignment. Compound 59 is believed to have formed via the ingress of air into a solution of 11 leading to oxo-abstraction.
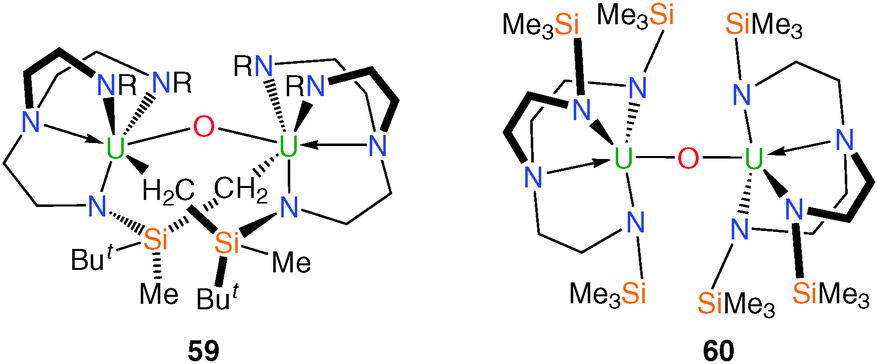 | ||
| Fig. 8 Structures of 59 and 60. R = SiMe2But. | ||
In the course of efforts to synthesise molecular heterobimetallic complexes featuring unsupported U–Mn bonds, it was reported that [KMnCp2] reacted with [U(TrenTMS)(THF)2][BPh4] (19) under a variety of conditions to eliminate KBPh4 and MnCp2, which were separated. From the remaining material, a crop of yellow plates was isolated and identified as the bridging oxo complex [{U(TrenTMS)}2(μ-O)] (60), Fig. 8.15b The uranium(IV) centres in 60 are bridged by an oxo group that exhibits a linear geometry by virtue of its position on a crystallographic centre of inversion. Complex 60 is considered the product of oxo abstraction, the origin of which is ascribed to coordinated or bulk THF solvent and it was proposed that, following salt elimination, a putative [U(TrenTMS)(MnCp2)] complex is formed, but that this decomposes via homolytic bond cleavage yielding MnCp2 and, ultimately, 60. The tetravalent bridging oxo complex [{U(TrenDMBS)}2(μ-O)] has also been reported17 and spectroscopically characterised but no structural data are not available.
The trivalent complex 15 was treated with the oxo atom transfer reagent trimethylamine-N-oxide, Me3NO, Scheme 23, which afforded the pentavalent Tren–uranium oxo complex [U(TrenTIPS)(O)] (61) as red crystals via a formal two-electron oxidation process.50 In the single crystal XRD structure U–Namine bond distance is highly contracted at 2.482(6) Å, which is ascribed to the inverse trans influence of the oxo ligand as is the case for 46. Although EPR silent, 61 displays complex magnetic behaviour and it was determined via a raft of variable temperature SQUID (Superconducting Quantum Interference Device) measurements that it exhibites single molecule magnetism (slow relaxation of the molecular magnetisation). Complex 61 was the first monometallic uranium(V) single molecule magnet, the origin of which was ascribed to the strong axial ligand field in 61 giving rise to a large magnetic anisotropy. Whilst the energy barrier of this complex to relaxation of the magnetization breaks no records at 15.3 cm−1 (22 K), it does exhibit slow relaxation up to a blocking temperature of 3.5 K and at scanning frequencies as low as 10 Hz which suggests this is fertile territory with respect to discovering novel magnetic phenomena.51
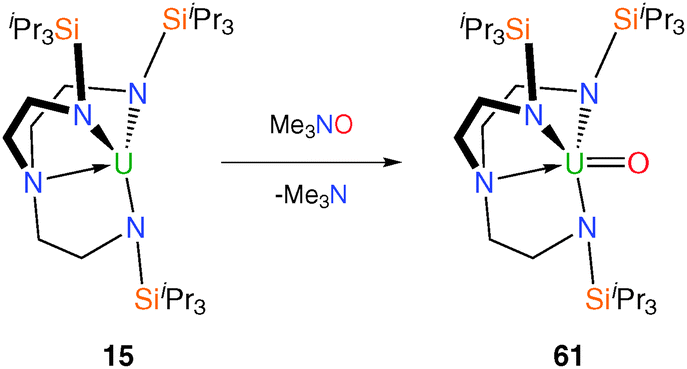 | ||
| Scheme 23 Synthesis of 61. | ||
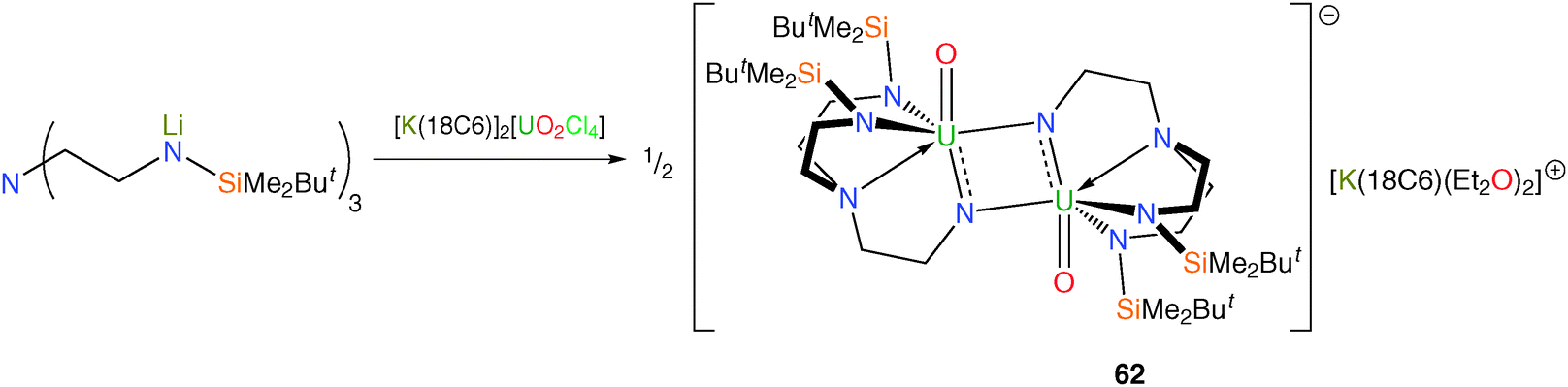 | ||
| Scheme 24 Synthesis of 62. | ||
In pursuit of the elusive cis-uranyl fragment, use of the Tren framework has been studied due to the geometric constraints imposed by the face-capping nature of this chelating ligand.52 A yellow suspension of the uranyl chloride complex [K(18-crown-6)]2[UO2Cl4] was treated with the ligand transfer reagent [Li3TrenDMBS] in THF. Following workup and crystallisation from diethyl ether, red crystals were isolated and subjected to an X-ray structural determination. The molecular structure was found to be the mixed-valent uranium(V/VI) oxo-imido dimer [U(O){μ-NCH2CH2N(CH2CH2NSiButMe2)2}]2[K(18C6)(Et2O)2] (62), Scheme 24.
The generation of 62 results from the activation of both the uranyl fragment and the Tren ligand, with nominal loss of one O atom and one silyl group per molecule of starting material as well as a one electron reduction overall since the product is U(V/VI). Inspection of the structural parameters supports the mixed-valence formulation, with notably long UO and UN bonds arising from electron-rich uranium centres, with the conclusion that the extra electron is delocalised around the two imido fragments.
Alkoxide derivatives
Several neutral and anionic Tren–uranium(IV) alkoxides and aryloxides are known. [U(TrenTMS)(OR)] [R = But, 63a; t-C4F9, 63b; Ph, 63c; 2,6-But2-4-Me-C6H2, 63d] and [U(TrenTMS)(OR)(OR′)(μ-Li)(thf)] [R, R′ = But, 63e; R = But, R′ = OPh, 63f; R, R′ = OPh, 63g] were synthesised from 1 and the appropriate lithium alkoxide or aryloxide in the requisite stoichiometry.21,23,53 The reactions to produce 63 proceed straightforwardly and the characterisation data supported their proposed formulations, however structural authentication was only obtained for 63a, 63b and 63e. The attempted oxidation of the anionic ‘ate’ complexes 63e–g with ferrocenium hexafluorophosphate [FeCp2][PF6] afforded brown solids assumed to be [U(TrenTMS)(OR)(OR′)] on the basis of NMR, IR and microanalysis data, but again no structural data are available to support their pentavalent assignments.Small molecule activation
Reductive homologation of carbon monoxide
Given the documented ability for the TrenTIPS–uranium(III) complex 15 to activate azide (N3−) and form a bridging nitride complex,12b the reductive capacity of other Tren–uranium(III) complexes with varying steric demands has been investigated; this may highlight contrasting reactivities such as the reactivity of dinitrogen with 11 to give 12 but no such reactivity with 15. It has been demonstrated that the TrenDMBS–uranium(III) complex 11 can reductively homologate CO under ambient conditions to selectively produce the diuranium(IV)–ethynediolate complex [{U(TrenDMBS)}2(μ-η1:η1-OCCO)] 64, Scheme 25,54a which in uranium chemistry is generally a rare transformation for CO effected by only a handful of complexes.10a,54b Upon thermolysis, 64 undergoes Si–N bond insertion and oxo-abstraction affording 65, which is in contrast to the only other reported example of uranium-coordinated ethynediolate reactivity where an ethynediolate inserts into a ligand C–H bond.10a The insertion product 65 is afforded quantitatively, which emphasises the silicophilic nature of the (C2O2)2− unit in 64.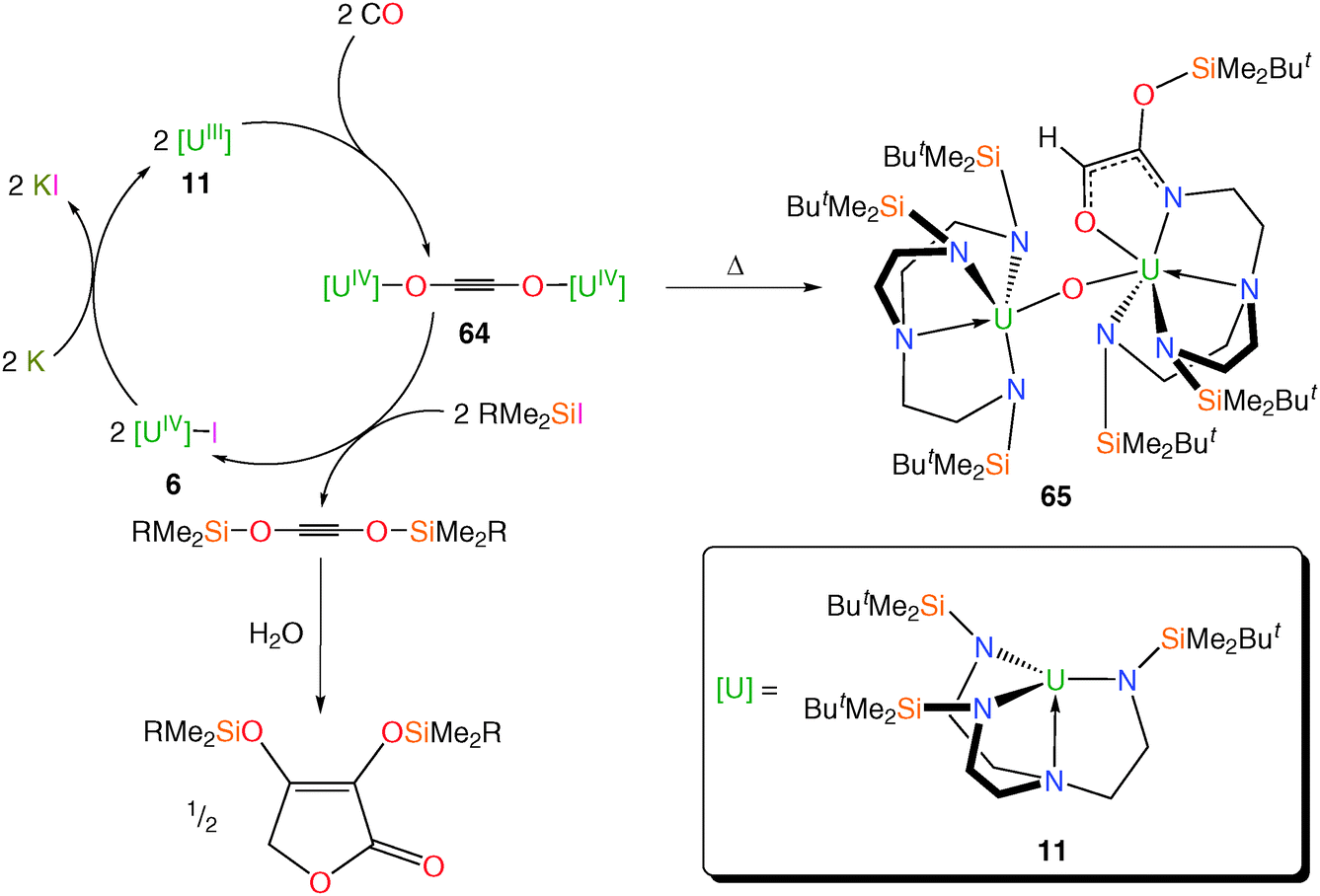 | ||
| Scheme 25 Synthetic cycle for the reductive homologation and functionalisation of CO and thermolysis of 64 to 65. R = Me, Ph; Cp* = η5-C5Me5. | ||
Treatment of 64 with RMe2SiI liberated the functionalised acetylenes “(RMe2SiOC)2” (R = Me, Ph) in high yield alongside 6, which can be recycled and re-used, closing the synthetic cycle via reduction with potassium to regenerate 11. A DFT study suggested that the pre-organised nature of the TrenDMBS–uranium unit may be important to this uniquely straightforward liberation chemistry, probably arising from the minimal ligand reorganisation energies required and which may have significant application in the design of future catalytic cycles for CO activation. The C2-bis(ether)acetylenes that are produced undergo conversion to C4-furanones upon treatment with water, both of which are precursors to industrially relevant diols and furans.
Tren–uranium–metal bonds
Manganese isocarbonylate derivatives
Due to its widespread utility in the construction of p-block – and transition metal – manganese linkages the pentacarbonylmanganese fragment [Mn(CO)5] has been utilised in attempts to access elusive unsupported U–Mn bonded complexes.15b It transpired that none of the manganese carbonylate compounds discussed below contain U–Mn bonds, but they are included here because they illustrate important lessons in the drive to isolate uranium–metal bonds. Treatment of the SIP complex [U(TrenTMS)(THF)2][BPh4] (19) with [KMn(CO)5] in THF or DME (DME = 1,2-dimethoxyethane) affords the manganese carbonylate complexes [U(TrenTMS)(THF)(μ-OC){Mn(CO)4}] (66) or [U(TrenTMS)(DME)][Mn(CO)5] (67), respectively (Scheme 26) with no evidence for the formation of U–Mn bonds.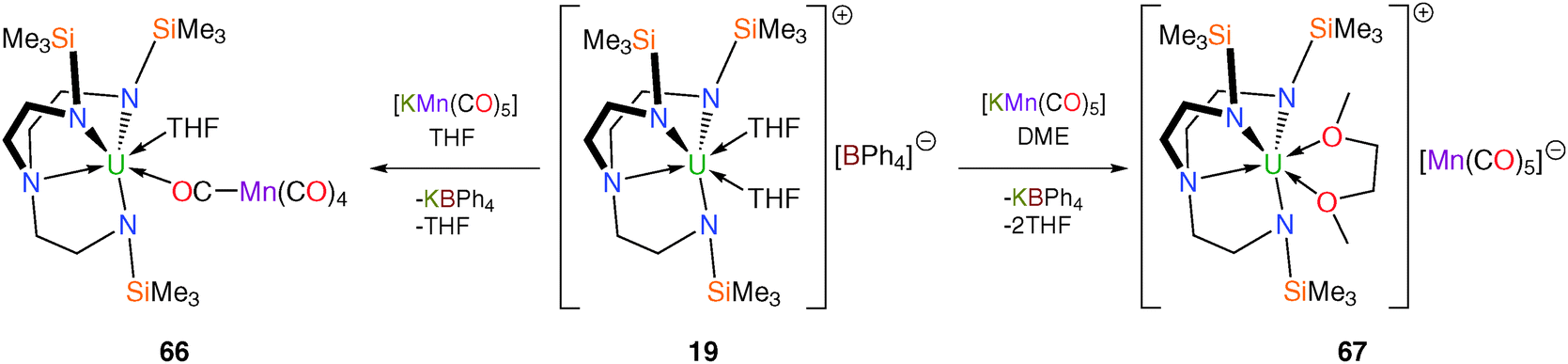 | ||
| Scheme 26 Synthesis of 66 and 67. | ||
Both 66 and 67 were structurally characterised, revealing the [Mn(CO)5]− unit to be directly coordinated to the uranium(IV) centre in 66 but displaced by the chelating DME molecule in 67. The Mn centre in 66 adopts a distorted square pyramidal geometry whereas that in 67 assumes an axially elongated, trigonal bipyramidal geometry by virtue of the solvent separated ion pair formulation for the latter.
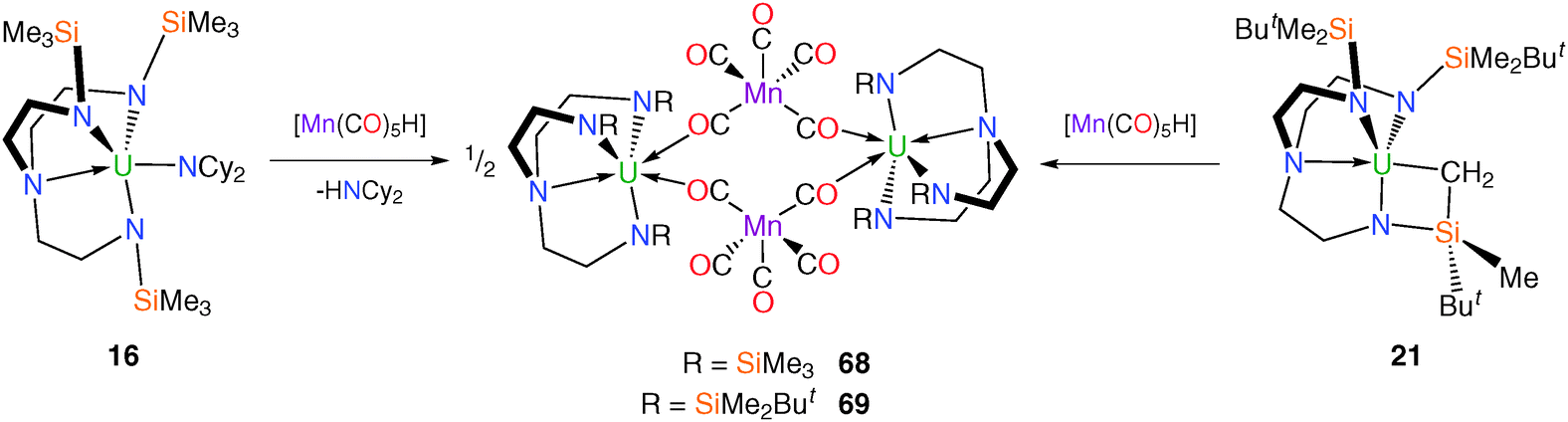
Attempts to access U–Mn bonded species by avoiding coordinating solvents, for instance via amine or alkane elimination methods produced diuranium doubly-bridging complexes, even when the more sterically encumbered TrenDMBS ligand is employed to disfavour such oligomerisation. When treated with manganese pentacarbonyl hydride, [Mn(CO)5H], [U(TrenTMS)(NCy2)] (16) and [U{N(CH2CH2NSiMe2But)2(CH2CH2NSiMeButCH2)}] (21) undergo amine and alkane elimination, respectively, to afford [{U(TrenTMS)(μ-OC)2Mn(CO)3}2] (68) or [{U(TrenDMBS)(μ-OC)2Mn(CO)3}2] (69), in each case (Scheme 27).
 | ||
| Scheme 27 Synthesis of 68 and 69. | ||
Both complexes were structurally characterised and exhibited very similar structural features with minor differences ascribed to the greater steric demands of TrenDMBS relative to TrenTMS. IR data for the two complexes revealed the expected isocarbonyl stretching bands at 1731 and 1734 cm−1 for 68 and 69, respectively.
Rhenocene derivatives
The aforementioned uranium–manganese complexes underscore the difficulties in isolating metal–metal bonds when carbonyl groups are present since via backbonding carbonyls can carry an appreciable charge and make them better donors to uranium than the manganese centres. As an alternative to carbonyl-containing anionic manganese fragments, construction of a U–Mn bond featuring the bis(η5-cyclopentadienyl)manganese (manganocene) anion was attempted as the absence of carbonyl co-ligands should circumvent the problem of isocarbonyl formation. However, as shown in Fig. 8, [KMnCp2] reacts with [U(TrenTMS)(THF)2][BPh4] (19) under a variety of conditions to produce the bridging oxo complex [{U(TrenTMS)}2(μ-O)] (60).15b As a result, the rhenium analogue of the manganocene anion, [ReCp2]−, has been targeted construct Tren–uranium–metal bonds as it was anticipated that U–Re bonds should be more stable than U–Mn bonds on the basis of improved Re 5d orbital overlap with the U valence orbitals relative to that for 3d manganese. Accordingly, it has been found that both salt- and alkane-elimination methods can be used to access unsupported U–Re complexes, either via the reaction of 5 with pale yellow [KReCp2] or 21 with yellow rhenocene hydride, [ReCp2H], to produce the dark red complexes [U(TrenTMS)(ReCp2)] (70) or [U(TrenDMBS)(ReCp2)] (71), respectively (Scheme 28).14a,55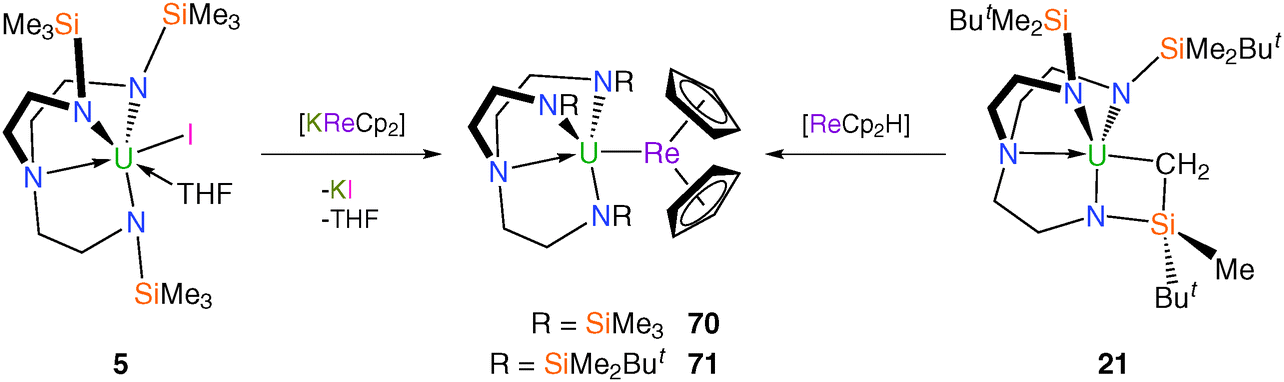 | ||
| Scheme 28 Synthesis of 70 and 71. | ||
The heterobimetallic complex 70 was the first structurally authenticated uranium–transition metal bond, with a U–Re bond distance of 3.0475(4) Å, ca. 0.42 Å shorter than the sum of the covalent radii of uranium and rhenium (3.47 Å).28 Complex 71 could not be prepared by salt elimination, presumably due to the increased steric bulk of the TrenDMBS ligand, so was accessed via the treatment of the alkyl complex 21 with rhenocene hydride. Complex 71 was characterised by single crystal XRD, revealing a U–Re bond of 3.0479(6) Å, which is virtually identical to the U–Re bond length in 70. DFT analysis of 70 and 71 describes the bonding as polarised-covalent, notably with σ- and very weak π-bonding components in the U–Re bonding interactions.55a
Cyclopentadienyl ruthenium dicarbonyl derivatives
Analogously to 70 and 71, and to earlier reports of [Th(η5-C5Me5)(I){RuCp(CO)2}] and [LuCp2(THF){RuCp(CO)2}],56 Tren–uranium–ruthenium complexes were targeted using the [RuCp(CO)2]− anion, the heavier congener of the ubiquitous [FeCp(CO)2]− fragment. Complexes 5 and 6 both react with [KRuCp(CO)2] to afford orange crystals suitable for single crystal XRD studies, which confirmed the anticipated bimetallic structures of [U(TrenTMS){RuCp(CO)2}] (72) and [U(TrenDMBS){RuCp(CO)2}] (73), Scheme 29.57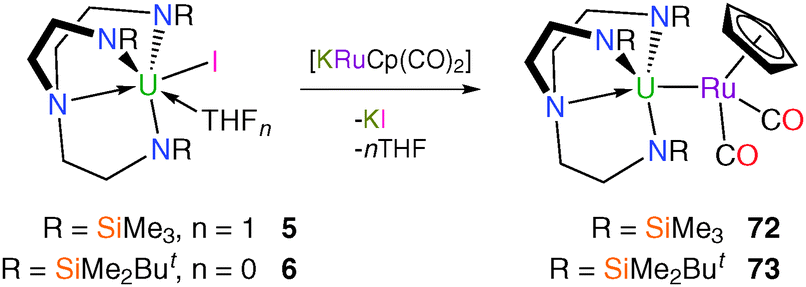 | ||
| Scheme 29 Synthesis of 72 and 73. | ||
Heterobimetallic complexes 72 and 73 were the first structurally authenticated examples of uranium–ruthenium bonds, with U–Ru bond distances of 3.0925(3) and 3.0739(2) Å, respectively. Similarly to 70 and 71, the uranium–metal bonds in 72 and 73 are approximately 0.33 and 0.35 Å shorter than the sum of the covalent radii of uranium and ruthenium (3.42 Å),28 respectively, although unlike 71, 73 could be prepared via salt elimination. The U–Ru bond length of 3.0739(2) Å in 73 is very slightly shorter than that in 72, which was surprising considering the increased steric demands of TrenDMBS relative to TrenTMS. A theoretical study of 72 and 73 suggested that the bonding is predominantly electrostatic in nature.57
Summary & conclusions
This Feature Article provides an up-to-date account of the diverse array of Tren–uranium chemistry presented in the scientific literature. It is now clear that the marriage of uranium with triamidoamine ligands is a profitable one, Fig. 9. The Tren ligand certainly imparts kinetic and thermodynamic stability, by virtue of being a quadridentate ligand and forming relatively strong uranium–amide bonds. There is mounting evidence that the trialkylamine, which often resides trans to a novel ligand, is implicated in electronic stabilisation of such linkages by the inverse-trans-influence. The steric variation of the N-R groups is also key, not only in terms of optimising the crystallinity of target complexes, but also by controlling the coordination environment at uranium. When the SiMe3 group is used, solvent may coordinate to uranium, however moving to the bulky SiPri3 group generally precludes solvent coordination and generates a well-defined pocket to stabilise reactive groups. Perhaps the clearest demonstration of the effect of the steric bulk of the N-R groups is in reactivity studies; 11 reacts with dinitrogen and carbon monoxide, whereas the much bulkier 15 does not, but in contrast 15 has so far proven to be the only triamidoamine uranium complex capable of stabilising a terminal nitride linkage. Tren ligands are also capable of stabilising uranium over all commonly accessible oxidation states (III–VI), which is important for reactivity in stoichiometric and catalytic transformations as well as stabilising reactive ligand fragments. Lastly, Tren uranium complexes have been shown to exhibit single molecule magnetism, which exploits the strong axial crystal field that results from an axial amine-multiply bonded ligand combination; given the single molecule magnetism of uranium is burgeoning this holds promise. Although the complexes described herein are highly air and moisture sensitive, the straightforward ability to control the steric and electronic properties of Tren–uranium fragments by variation of the N-substituents holds much promise for future endeavours. It is therefore likely that Tren–uranium chemistry will continue to deliver exciting developments in the future.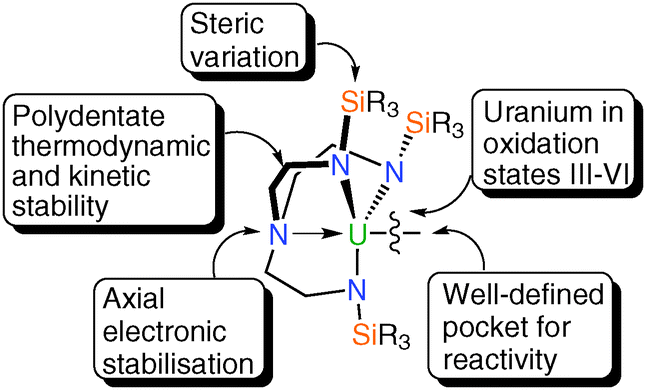 | ||
| Fig. 9 Summary of design features of uranium triamidoamine complexes. | ||
Acknowledgements
We thank the Royal Society, the European Research Council, the Engineering and Physical Sciences Research Council, the University of Nottingham, the National Nuclear Laboratory, the UK EPSRC National EPR service, and COST for generously supporting this work.References
- (a) P. L. Shutov, S. S. Karlov, K. Harms, A. V. Churakov, J. Lorberth and G. S. Zaitseva, Eur. J. Inorg. Chem., 2004, 2123–2129 CrossRef CAS PubMed; (b) B. Chatelet, H. Gornitzka, V. Dufaud, E. Jeanneau, J.-P. Dutasta and A. Martinez, J. Am. Chem. Soc., 2013, 135, 18659–18664 CrossRef CAS PubMed; (c) N. S. Sickerman, R. M. Henry, J. W. Ziller and A. S. Borovik, Polyhedron, 2013, 58, 65–70 CrossRef CAS PubMed; (d) M. Forcato, F. Lake, M. Mba Blazquez, P. Renner, M. Crisma, L. H. Gade, G. Licini and C. Moberg, Eur. J. Inorg. Chem., 2006, 1032–1040 CrossRef CAS PubMed; (e) X. Liu, P. Ilankumaran, I. A. Guzei and J. G. Verkade, J. Org. Chem., 2000, 65, 701–706 CrossRef CAS; (f) J. Lee and Y. Kim, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2012, 68, 3317 Search PubMed; (g) N. Thirupathi, X. Liu and J. G. Verkade, Inorg. Chem., 2002, 42, 389–397 CrossRef; (h) P. D. Raytchev, A. Martinez, H. Gornitzka and J.-P. Dutasta, J. Am. Chem. Soc., 2011, 133, 2157–2159 CrossRef CAS PubMed; (i) X.-D. Liu and J. G. Verkade, Inorg. Chem., 1998, 37, 5189–5197 CrossRef CAS; (j) P. L. Shutov, S. S. Karlov, K. Harms, A. V. Churakov, J. A. K. Howard, J. Lorberth and G. S. Zaitseva, Eur. J. Inorg. Chem., 2002, 2784–2788 CrossRef CAS; (k) J. V. Kingston, A. Ellern and J. G. Verkade, Angew. Chem., Int. Ed., 2005, 44, 4960–4963 CrossRef CAS PubMed; (l) V. R. Chintareddy, A. Ellern and J. G. Verkade, J. Org. Chem., 2010, 75, 7166–7174 CrossRef CAS PubMed; (m) D. Shetty, S. Y. Choi, J. M. Jeong, L. Hoigebazar, Y.-S. Lee, D. S. Lee, J.-K. Chung, M. C. Lee and Y. K. Chung, Eur. J. Inorg. Chem., 2010, 5432–5438 CrossRef CAS PubMed; (n) J. Pinkas, T. Wang, R. A. Jacobson and J. G. Verkade, Inorg. Chem., 1994, 33, 4202–4210 CrossRef CAS; (o) W. Plass, J. Pinkas and J. G. Verkade, Inorg. Chem., 1997, 36, 1973–1978 CrossRef CAS PubMed; (p) C. Lensink, S. K. Xi, L. M. Daniels and J. G. Verkade, J. Am. Chem. Soc., 1989, 111, 3478–3479 CrossRef CAS; (q) X. Liu, Y. Bai and J. G. Verkade, J. Organomet. Chem., 1999, 582, 16–24 CrossRef CAS; (r) Y. Wan and J. G. Verkade, Organometallics, 1994, 13, 4164–4166 CrossRef CAS; (s) R. J. Warr, A. N. Westra, K. J. Bell, J. Chartres, R. Ellis, C. Tong, T. G. Simmance, A. Gadzhieva, A. J. Blake, P. A. Tasker and M. Schröder, Chem. – Eur. J., 2009, 15, 4836–4850 CrossRef CAS PubMed.
- F. H. Allen, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 380–388 CrossRef PubMed.
- (a) B. P. Johnson, G. Balázs and M. Scheer, Coord. Chem. Rev., 2006, 250, 1178–1195 CrossRef CAS PubMed; (b) M. Scheer, Coord. Chem. Rev., 1997, 163, 271–286 CrossRef CAS; (c) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- (a) J. S. Freundlich, R. R. Schrock and W. M. Davis, Organometallics, 1996, 15, 2777–2783 CrossRef CAS; (b) S. W. Seidel, R. R. Schrock and W. M. Davis, Organometallics, 1998, 17, 1058–1068 CrossRef CAS.
- (a) D. V. Yandulov and R. R. Schrock, Science, 2003, 301, 76–78 CrossRef CAS PubMed; (b) Z. Duan, A. A. Naiini, J.-H. Lee and J. G. Verkade, Inorg. Chem., 1995, 34, 5477–5482 CrossRef CAS; (c) Z. Duan, V. G. Young and J. G. Verkade, Inorg. Chem., 1995, 34, 2179–2185 CrossRef CAS; (d) K. W. Hellmann, L. H. Gade, O. Gevert, P. Steinert and J. W. Lauher, Inorg. Chem., 1995, 34, 4069–4078 CrossRef CAS; (e) R. R. Schrock, Acc. Chem. Res., 1997, 30, 9–16 CrossRef CAS; (f) C. C. Cummins, R. R. Schrock and W. M. Davis, Organometallics, 1992, 11, 1452–1454 CrossRef CAS; (g) D. V. Yandulov and R. R. Schrock, Inorg. Chem., 2005, 44, 1103–1117 CrossRef CAS; (h) M. B. O’Donoghue, W. M. Davis and R. R. Schrock, Inorg. Chem., 1998, 37, 5149–5158 CrossRef; (i) C. C. Cummins, R. R. Schrock and W. M. Davis, Inorg. Chem., 1994, 33, 1448–1457 CrossRef CAS; (j) M. Scheer, J. Müller, M. Schiffer, G. Baum and R. Winter, Chem. – Eur. J., 2000, 6, 1252–1257 CAS; (k) N. C. Mösch-Zanetti, R. R. Schrock, W. M. Davis, K. Wanninger, S. W. Seidel and M. B. O’Donoghue, J. Am. Chem. Soc., 1997, 119, 11037–11048 CrossRef; (l) X. Liu, J. R. Babcock, M. A. Lane, J. A. Belot, A. W. Ott, M. V. Metz, C. R. Kannewurf, R. P. H. Chang and T. J. Marks, Chem. Vap. Deposition, 2001, 7, 25–28 CrossRef CAS; (m) D. V. Yandulov and R. R. Schrock, Can. J. Chem., 2005, 83, 341–357 CrossRef CAS; (n) V. Ritleng, D. V. Yandulov, W. W. Weare, R. R. Schrock, A. S. Hock and W. M. Davis, J. Am. Chem. Soc., 2004, 126, 6150–6163 CrossRef CAS PubMed; (o) D. V. Yandulov, R. R. Schrock, A. L. Rheingold, C. Ceccarelli and W. M. Davis, Inorg. Chem., 2003, 42, 796–813 CrossRef CAS PubMed; (p) N. C. Smythe, R. R. Schrock, P. Müller and W. W. Weare, Inorg. Chem., 2006, 45, 7111–7118 CrossRef CAS PubMed; (q) P. L. Hill, G. P. A. Yap, A. L. Rheingold and E. A. Maatta, J. Chem. Soc., Chem. Commun., 1995, 737–738 RSC; (r) M. R. Reithofer, R. R. Schrock and P. Müller, J. Am. Chem. Soc., 2010, 132, 8349–8358 CrossRef CAS PubMed; (s) W. H. Harman, M. F. Lichterman, N. A. Piro and C. J. Chang, Inorg. Chem., 2012, 51, 10037–10042 CrossRef CAS PubMed.
- (a) R. Gupta, C. E. MacBeth, V. G. Young and A. S. Borovik, J. Am. Chem. Soc., 2002, 124, 1136–1137 CrossRef CAS PubMed; (b) C. E. MacBeth, R. Gupta, K. R. Mitchell-Koch, V. G. Young, G. H. Lushington, W. H. Thompson, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2004, 126, 2556–2567 CrossRef CAS PubMed; (c) V. Christou and J. Arnold, Angew. Chem., Int. Ed. Engl., 1993, 32, 1450–1452 CrossRef PubMed; (d) Z. Shirin, B. S. Hammes, V. G. Young and A. S. Borovik, J. Am. Chem. Soc., 2000, 122, 1836–1837 CrossRef CAS; (e) W. Plass and J. G. Verkade, J. Am. Chem. Soc., 1992, 114, 2275–2276 CrossRef CAS; (f) D. C. Lacy, R. Gupta, K. L. Stone, J. Greaves, J. W. Ziller, M. P. Hendrich and A. S. Borovik, J. Am. Chem. Soc., 2010, 132, 12188–12190 CrossRef CAS PubMed; (g) P. L. Larsen, R. Gupta, D. R. Powell and A. S. Borovik, J. Am. Chem. Soc., 2004, 126, 6522–6523 CrossRef CAS PubMed; (h) C. C. Cummins, R. R. Schrock and W. M. Davis, Angew. Chem., Int. Ed. Engl., 1993, 32, 756–759 CrossRef PubMed; (i) G. Balázs, J. C. Green and M. Scheer, Chem. – Eur. J., 2006, 12, 8603–8608 CrossRef PubMed; (j) N. C. Zanetti, R. R. Schrock and W. M. Davis, Angew. Chem., Int. Ed. Engl., 1995, 34, 2044–2046 CrossRef CAS PubMed; (k) M. Scheer, J. Müller and M. Häser, Angew. Chem., Int. Ed. Engl., 1996, 35, 2492–2496 CrossRef CAS PubMed; (l) M. Scheer, J. Muller, G. Baum and M. Haser, Chem. Commun., 1998, 1051–1052 RSC; (m) G. Balázs, M. Sierka and M. Scheer, Angew. Chem., Int. Ed., 2005, 44, 4920–4924 CrossRef PubMed.
- A. J. Lewis, U. J. Williams, J. M. Kikkawa, P. J. Carroll and E. J. Schelter, Inorg. Chem., 2011, 51, 37–39 CrossRef PubMed.
- (a) I. Castro-Rodriguez and K. Meyer, Chem. Commun., 2006, 1353–1368 RSC; (b) O. P. Lam, C. Anthon and K. Meyer, Dalton Trans., 2009, 9677–9691 RSC; (c) O. P. Lam and K. Meyer, Polyhedron, 2012, 32, 1–9 CrossRef CAS PubMed; (d) H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., John Wiley & Sons, Inc., 2014, vol. 58, pp. 303–416 Search PubMed.
- (a) D. E. Smiles, G. Wu and T. W. Hayton, Inorg. Chem., 2014, 53, 12683–12685 CrossRef CAS PubMed; (b) L. P. Spencer, P. Yang, S. G. Minasian, R. E. Jilek, E. R. Batista, K. S. Boland, J. M. Boncella, S. D. Conradson, D. L. Clark, T. W. Hayton, S. A. Kozimor, R. L. Martin, M. M. MacInnes, A. C. Olson, B. L. Scott, D. K. Shuh and M. P. Wilkerson, J. Am. Chem. Soc., 2013, 135, 2279–2290 CrossRef CAS PubMed; (c) W. W. Lukens, N. M. Edelstein, N. Magnani, T. W. Hayton, S. Fortier and L. A. Seaman, J. Am. Chem. Soc., 2013, 135, 10742–10754 CrossRef CAS PubMed; (d) T. W. Hayton, Chem. Commun., 2013, 49, 2956–2973 RSC; (e) J. L. Brown, G. Wu and T. W. Hayton, Organometallics, 2013, 32, 1193–1198 CrossRef CAS; (f) J. L. Brown, S. Fortier, R. A. Lewis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2012, 134, 15468–15475 CrossRef CAS PubMed; (g) S. Fortier, J. L. Brown, N. Kaltsoyannis, G. Wu and T. W. Hayton, Inorg. Chem., 2012, 51, 1625–1633 CrossRef CAS PubMed; (h) S. Fortier, N. Kaltsoyannis, G. Wu and T. W. Hayton, J. Am. Chem. Soc., 2011, 133, 14224–14227 CrossRef CAS PubMed.
- (a) P. L. Arnold, Z. R. Turner, R. M. Bellabarba and R. P. Tooze, Chem. Sci., 2011, 2, 77–79 RSC; (b) J. L. Stewart and R. A. Andersen, Polyhedron, 1998, 17, 953–958 CrossRef CAS; (c) J. L. Stewart and R. A. Andersen, New J. Chem., 1995, 19, 587–595 CAS; (d) A. Zalkin, J. G. Brennan and R. A. Andersen, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1988, 44, 1553–1554 CrossRef; (e) R. A. Andersen, A. Zalkin and D. H. Templeton, Inorg. Chem., 1981, 20, 622–623 CrossRef CAS; (f) S. J. Simpson, H. W. Turner and R. A. Andersen, Inorg. Chem., 1981, 20, 2991–2995 CrossRef CAS; (g) R. A. Andersen, Inorg. Chem., 1979, 18, 1507–1509 CrossRef CAS; (h) H. W. Turner, R. A. Andersen, A. Zalkin and D. H. Templeton, Inorg. Chem., 1979, 18, 1221–1224 CrossRef CAS.
- (a) B. M. Gardner and S. T. Liddle, Eur. J. Inorg. Chem., 2013, 3753–3770 CrossRef CAS PubMed; (b) D. M. King and S. T. Liddle, Coord. Chem. Rev., 2014, 266–267, 2–15 CrossRef CAS PubMed.
- (a) P. Roussel, N. W. Alcock and P. Scott, Inorg. Chem., 1998, 37, 3435–3436 CrossRef CAS; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS PubMed.
- (a) S. T. Liddle, J. McMaster, D. P. Mills, A. J. Blake, C. Jones and W. D. Woodul, Angew. Chem., Int. Ed., 2009, 48, 1077–1080 CrossRef CAS PubMed; (b) P. Roussel, N. W. Alcock, R. Boaretto, A. J. Kingsley, I. J. Munslow, C. J. Sanders and P. Scott, Inorg. Chem., 1999, 38, 3651–3656 CrossRef CAS PubMed.
- (a) B. M. Gardner, J. McMaster, W. Lewis and S. T. Liddle, Chem. Commun., 2009, 2851–2853 RSC; (b) D. M. King, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2013, 5, 482–488 CrossRef CAS PubMed.
- (a) P. Scott and P. B. Hitchcock, Polyhedron, 1994, 13, 1651–1653 CrossRef CAS; (b) B. M. Gardner, W. Lewis, A. J. Blake and S. T. Liddle, Inorg. Chem., 2011, 50, 9631–9641 CrossRef CAS PubMed.
- P. Roussel, P. B. Hitchcock, N. Tinker and P. Scott, Chem. Commun., 1996, 2053–2054 RSC.
- P. Roussel, R. Boaretto, A. J. Kingsley, N. W. Alcock and P. Scott, J. Chem. Soc., Dalton Trans., 2002, 1423–1428 RSC.
- (a) P. Roussel and P. Scott, J. Am. Chem. Soc., 1998, 120, 1070–1071 CrossRef CAS; (b) N. Kaltsoyannis and P. Scott, Chem. Commun., 1998, 1665–1666 RSC.
- (a) P. Roussel, P. Scott and N. D. Tinker, J. Alloys Compd., 1998, 271–273, 150–153 CrossRef CAS; (b) P. Roussel, W. Errington, N. Kaltsoyannis and P. Scott, J. Organomet. Chem., 2001, 635, 69–74 CrossRef CAS.
- S. M. Mansell, J. H. Farnaby, A. I. Germeroth and P. L. Arnold, Organometallics, 2013, 32, 4214–4222 CrossRef CAS.
- P. Scott and P. B. Hitchcock, J. Chem. Soc., Dalton Trans., 1995, 603–609 RSC.
- B. M. Gardner, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2009, 131, 10388–10389 CrossRef CAS PubMed.
- R. Boaretto, P. Roussel, N. W. Alcock, A. J. Kingsley, I. J. Munslow, C. J. Sanders and P. Scott, J. Organomet. Chem., 1999, 591, 174–184 CrossRef CAS.
- B. M. Gardner, G. Balazs, M. Scheer, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 4484–4488 CrossRef CAS PubMed.
- B. M. Gardner, W. Lewis, A. J. Blake and S. T. Liddle, Organometallics, 2015 DOI:10.1021/om501177s.
- B. Vlaisavljevich, P. Miro, C. J. Cramer, L. Gagliardi, I. Infante and S. T. Liddle, Chem. – Eur. J., 2011, 17, 8424–8433 CrossRef CAS PubMed.
- R. J. Baker, R. D. Farley, C. Jones, M. Kloth and D. M. Murphy, J. Chem. Soc., Dalton Trans., 2002, 3844–3850 RSC.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverria, E. Cremades, F. Barragan and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- R. Boaretto, P. Roussel, A. J. Kingsley, I. J. Munslow, C. J. Sanders, N. W. Alcock and P. Scott, Chem. Commun., 1999, 1701–1702 RSC.
- B. S. Newell, A. K. Rappé and M. P. Shores, Inorg. Chem., 2010, 49, 1595–1606 CrossRef CAS PubMed.
- B. M. Gardner, P. A. Cleaves, C. E. Kefalidis, J. Fang, L. Maron, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Sci., 2014, 5, 2489–2497 RSC.
- (a) O. J. Cooper, D. P. Mills, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, Chem. – Eur. J., 2013, 19, 7071–7083 CrossRef CAS PubMed; (b) D. P. Mills, O. J. Cooper, F. Tuna, E. J. L. McInnes, E. S. Davies, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2012, 134, 10047–10054 CrossRef CAS PubMed; (c) O. J. Cooper, D. P. Mills, J. McMaster, F. Moro, E. S. Davies, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 2383–2386 CrossRef CAS PubMed; (d) D. P. Mills, F. Moro, J. McMaster, S. J. van, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Chem., 2011, 3, 454–460 CAS; (e) O. J. Cooper, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Dalton Trans., 2010, 39, 5074–5076 RSC; (f) R. C. Stevens, R. Bau, R. E. Cramer, D. Afzal, J. W. Gilje and T. F. Koetzle, Organometallics, 1990, 9, 694–697 CrossRef CAS; (g) R. E. Cramer, S. Roth, F. Edelmann, M. A. Bruck, K. C. Cohn and J. W. Gilje, Organometallics, 1989, 8, 1192–1199 CrossRef CAS; (h) R. E. Cramer, M. A. Bruck, F. Edelmann, D. Afzal, J. W. Gilje and H. Schmidbaur, Chem. Ber., 1988, 121, 417–420 CrossRef CAS PubMed; (i) M. Ephritikhine, C. R. Chim., 2013, 16, 391–405 CrossRef CAS PubMed; (j) J.-C. Tourneux, J.-C. Berthet, T. Cantat, P. Thuery, N. Mezailles, F. P. Le and M. Ephritikhine, Organometallics, 2011, 30, 2957–2971 CrossRef CAS; (k) J.-C. Tourneux, J.-C. Berthet, P. Thuery, N. Mezailles, F. P. Le and M. Ephritikhine, Dalton Trans., 2010, 39, 2494–2496 RSC; (l) T. Cantat, T. Arliguie, A. Noel, P. Thuery, M. Ephritikhine, F. P. Le and N. Mezailles, J. Am. Chem. Soc., 2009, 131, 963–972 CrossRef CAS PubMed.
- J. C. Peters, A. L. Odom and C. C. Cummins, Chem. Commun., 1997, 1995–1996 RSC.
- G. Zi, L. Jia, E. L. Werkema, M. D. Walter, J. P. Gottfriedsen and R. A. Andersen, Organometallics, 2005, 24, 4251–4264 CrossRef CAS.
- (a) M. Ephritikhine, Dalton Trans., 2006, 2501–2516 RSC; (b) T. W. Hayton, Dalton Trans., 2010, 39, 1145–1158 RSC; (c) I. Korobkov and S. Gambarotta, Inorg. Chem., 2010, 49, 3409–3418 CrossRef CAS PubMed; (d) I. Castro-Rodriguez, K. Olsen, P. Gantzel and K. Meyer, J. Am. Chem. Soc., 2003, 125, 4565–4571 CrossRef CAS PubMed; (e) T. W. Hayton, J. M. Boncella, B. L. Scott, P. D. Palmer, E. R. Batista and P. J. Hay, Science, 2005, 310, 1941–1943 CrossRef CAS PubMed; (f) C. R. Graves, B. L. Scott, D. E. Morris and J. L. Kiplinger, J. Am. Chem. Soc., 2007, 129, 11914–11915 CrossRef CAS PubMed; (g) I. Castro-Rodriguez, H. Nakai and K. Meyer, Angew. Chem., Int. Ed., 2006, 45, 2389–2392 CrossRef CAS PubMed.
- D. M. King, J. McMaster, F. Tuna, E. J. L. McInnes, W. Lewis, A. J. Blake and S. T. Liddle, J. Am. Chem. Soc., 2014, 136, 5619–5622 CrossRef CAS PubMed.
- (a) R. G. Denning, J. Phys. Chem. A, 2007, 111, 4125–4143 CrossRef CAS PubMed; (b) N. Kaltsoyannis, Inorg. Chem., 2000, 39, 6009–6017 CrossRef CAS; (c) B. Kosog, H. S. La Pierre, F. W. Heinemann, S. T. Liddle and K. Meyer, J. Am. Chem. Soc., 2012, 134, 5284–5289 CrossRef CAS PubMed.
- P. A. Cleaves, D. M. King, C. E. Kefalidis, L. Maron, F. Tuna, E. J. L. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2014, 53, 10412–10415 CrossRef CAS PubMed.
- (a) J. S. Silvia and C. C. Cummins, J. Am. Chem. Soc., 2008, 131, 446–447 CrossRef PubMed; (b) B. L. Tran, M. Pink, X. Gao, H. Park and D. J. Mindiola, J. Am. Chem. Soc., 2010, 132, 1458–1459 CrossRef CAS PubMed; (c) B. Askevold, J. T. Nieto, S. Tussupbayev, M. Diefenbach, E. Herdtweck, M. C. Holthausen and S. Schneider, Nat. Chem., 2011, 3, 532–537 CrossRef CAS PubMed; (d) A. F. Cozzolino, J. S. Silvia, N. Lopez and C. C. Cummins, Dalton Trans., 2014, 43, 4639–4652 RSC.
- R. K. Thomson, T. Cantat, B. L. Scott, D. E. Morris, E. R. Batista and J. L. Kiplinger, Nat. Chem., 2010, 2, 723–729 CrossRef CAS PubMed.
- M. Dartiguenave, M. Joëlle Menu, E. Deydier, Y. Dartiguenave and H. Siebald, Coord. Chem. Rev., 1998, 178–180, 623–663 CrossRef.
- (a) H. Vos (ed.), Coord. Chem. Rev., 2008, 252, 2445–2612 CrossRef PubMed; (b) D. Hanss, M. E. Walther and O. S. Wenger (ed.), Coord. Chem. Rev., 2010, 254, 2584–2592 CrossRef CAS PubMed.
- B. M. Gardner, D. Patel, W. Lewis, A. J. Blake and S. T. Liddle, Angew. Chem., Int. Ed., 2011, 50, 10440–10443 CrossRef CAS PubMed.
- W. J. Evans, E. Montalvo, T. M. Champagne, J. W. Ziller, A. G. DiPasquale and A. L. Rheingold, J. Am. Chem. Soc., 2008, 130, 16–17 CrossRef CAS PubMed.
- S. W. Hall, J. C. Huffman, M. M. Miller, L. R. Avens, C. J. Burns, D. S. J. Arney, A. F. England and A. P. Sattelberger, Organometallics, 1993, 12, 752–758 CrossRef CAS.
- D. S. J. Arney, R. C. Schnabel, B. C. Scott and C. J. Burns, J. Am. Chem. Soc., 1996, 118, 6780–6781 CrossRef CAS.
- M. R. Duttera, V. W. Day and T. J. Marks, J. Am. Chem. Soc., 1984, 106, 2907–2912 CrossRef CAS.
- (a) P. L. Arnold, J. B. Love and D. Patel, Coord. Chem. Rev., 2009, 253, 1973–1978 CrossRef CAS PubMed; (b) A.-C. Schmidt, F. W. Heinemann, W. W. Lukens and K. Meyer, J. Am. Chem. Soc., 2014, 136, 11980–11993 CrossRef CAS PubMed.
- (a) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS; (b) B. Levason (ed.), Coord. Chem. Rev., 2014, 266–267, 1–194 Search PubMed.
- D. M. King, F. Tuna, J. McMaster, W. Lewis, A. J. Blake, E. J. L. McInnes and S. T. Liddle, Angew. Chem., Int. Ed., 2013, 52, 4921–4924 CrossRef CAS PubMed.
- S. T. Liddle and J. Van Slageren, Actinide Single Molecule Magnets, in Lanthanides and Actinides in Molecular Magnetism, ed. R. A. Layfield and M. Murugesu, Wiley-VCH, Weinheim, Germany, 2015, p. 315 Search PubMed.
- P. B. Duval, C. J. Burns, W. E. Buschmann, D. L. Clark, D. E. Morris and B. L. Scott, Inorg. Chem., 2001, 40, 5491–5496 CrossRef CAS PubMed.
- P. Roussel, P. B. Hitchcock, N. D. Tinker and P. Scott, Inorg. Chem., 1997, 36, 5716–5721 CrossRef CAS PubMed.
- (a) B. M. Gardner, J. C. Stewart, A. L. Davis, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9265–9270 CrossRef CAS PubMed; (b) A. S. Frey, F. G. N. Cloke, P. B. Hitchcock, I. J. Day, J. C. Green and G. Aitken, J. Am. Chem. Soc., 2008, 130, 13816–13817 CrossRef CAS PubMed.
- (a) D. Patel, D. M. King, B. M. Gardner, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Commun., 2011, 47, 295–297 RSC; (b) B. M. Gardner, J. McMaster, F. Moro, W. Lewis, A. J. Blake and S. T. Liddle, Chem. – Eur. J., 2011, 17, 6909–6912 CrossRef CAS PubMed.
- (a) R. S. Sternal, C. P. Brock and T. J. Marks, J. Am. Chem. Soc., 1985, 107, 8270–8272 CrossRef CAS; (b) I. P. Beletskaya, A. Z. Voskoboynikov, E. B. Chuklanova, N. I. Kirillova, A. K. Shestakova, I. N. Parshina, A. I. Gusev and G. K. I. Magomedov, J. Am. Chem. Soc., 1993, 115, 3156–3166 CrossRef CAS.
- B. M. Gardner, D. Patel, A. D. Cornish, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Chem. – Eur. J., 2011, 17, 11266–11273 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |